

Abstract
Tumor blood vessels are frequently inefficient in their design and function, leading to high interstitial fluid pressure, hypoxia, and acidosis in the tumor microenvironment (TME), rendering tumors refractory to the delivery of chemotherapeutic agents and immune effector cells. Here we identified the NOTCH antagonist delta-like 1 homologue (DLK1) as a vascular pericyte-associated antigen expressed in renal cell carcinomas (RCC), but not in normal kidney tissues in mice and humans. Vaccination of mice bearing established RCC against DLK1 led to immune-mediated elimination of DLK1+ pericytes and to blood vessel normalization (i.e., decreased vascular permeability and intratumoral hypoxia) in the TME, in association with tumor growth suppression. After therapeutic vaccination, tumors displayed increased prevalence of activated VCAM1+CD31+ vascular endothelial cells (VECs) and CXCL10, a type-1 T cell recruiting chemokine, in concert with increased levels of type-1 CD8+ tumor-infiltrating lymphocytes (TIL). Vaccination against DLK1 also yielded (i) dramatic reductions in Jarid1B+, CD133+, and CD44+ (hypoxia-responsive) stromal cell populations, (ii) enhanced tumor cell apoptosis, and (iii) increased NOTCH signaling in the TME. Coadministration of a γ-secretase inhibitor (N-[N-(3,5-Difluorophenacetyl-l-alanyl)]-(S)-phenylglycine t-butyl ester (DAPT)) that interferes with canonical NOTCH signaling resulted in the partial loss of therapeutic benefits associated with lentivirus encoding full-length murine (lvDLK1)-based vaccination.
Introduction
The vasculature of solid tumors is structurally and functionally “abnormal”, being composed of an irregular network of blood vessels characterized by aberrant coverage of endothelial tubes and by a loosely attached and largely immature population of mural cells (i.e., smooth muscle cells and pericytes).1,2 In contrast to mature pericyte-vascular endothelial cell (VEC) collaboration found in normal tissues that orchestrates blood vessel integrity/stability,3 in tumors, this interaction is deranged leading to a high-degree of vascular permeability, high interstitial fluid pressure, hypoxia, and acidosis.1,4
Renal cell carcinoma (RCC) is highly vascularized and generally considered to represent an immunogenic form of cancer.5,6,7 Current treatment options mediate only transient efficacy in a minority of RCC patients, with frequent development of progressive disease that is refractory to conventional chemo-/radiotherapy.8,9,10,11 Vaccines targeting tumor-associated antigens have also thus far demonstrated only modest curative value.12 The limited perfusion of tumor blood vessels likely contributes to the muted benefits of these treatment approaches by preventing the efficient delivery of chemotherapeutic agents and antitumor T cells into the tumor microenvironment (TME).13,14 As a consequence, the development of novel therapies that can “normalize” the tumor vasculature (by coordinately improving blood vessel perfusion, reducing tumor hypoxia, and allowing for improved and sustained delivery of anticancer agents into the TME) remains a high priority.14,15,16,17,18
To achieve the goal of tumor vascular normalization via immunization, we and others have recently advocated the use of vaccine formulations capable of promoting specific type-1 CD8+ T cell (aka Tc1) recognition of tumor-associated vascular cell (i.e., pericytes and VEC) antigens,13,14,15 including delta-like 1 homologue (DLK1).14 DLK1, aka preadipocyte factor-1 (Pref-1), is a transmembrane member of the EGF-like family of proteins, which includes NOTCH receptors and their ligands.19,20,21 The extracellular domain of DLK1 contains six EGF-like repeats and a tumor necrosis factor-α–converting enzyme cleavage site, but it lacks the delta/serrate/LAG-2 domain found in canonical NOTCH ligands.20 As a consequence, while DLK1 binds NOTCH1, it fails to promote NOTCH activation, and indeed both the membrane-bound and tumor necrosis factor-α–converting enzyme-cleaved extracellular domain forms of DLK1 serve as functional inhibitors of NOTCH signaling.19,20,21 DLK1 has been reported to inhibit a broad range of NOTCH-dependent differentiation pathways including normal adipogenesis, muscular and neuronal differentiation, bone differentiation, and hematopoiesis.20 In the cancer setting, the functional impact of DLK1 modulation cannot be intuitively assumed, since NOTCH activation has been reported to either promote or suppress tumor development/progression based on the balance of its contextual influences on the myriad of cell populations located within the evolving TME.19,20,21
In this report, we investigated the therapeutic impact of active vaccination against DLK1 in a murine model of RCC (i.e., RENCA tumor cells transplanted subcutaneously (s.c.) into syngenic BALB/c mice), where the DLK1 antigen is preferentially expressed by blood vessel-associated pericytes in the progressively growing TME. We show that DLK1 peptide- or gene-based vaccines are both immunogenic and therapeutic against established RCC, with treatment benefits linked to CD8+ T cell-mediated “normalization” of tumor-associated blood vessels (based on criteria established by Jain et al. (i.e., reduction in blood vessel numbers and extent of arborization, loss of hypoxia, and reduced vascular permeability)).16,17 Responder tumors were highly infiltrated by CD8+ tumor-infiltrating lymphocytes (TIL) that localized within the perivascular (pericyte-enriched) space. Residual pericytes lacked expression of DLK1 and were tightly approximated to CD31+ VEC. Consistent with the vaccine-induced, immune-mediated eradication of tumor-associated DLK1 protein expression, increased NOTCH signaling was evidenced within the therapeutic TME. These results are consistent with the ability of DLK1-based vaccines to promote therapeutic CD8+ T cell-dependent vascular normalization in the RCC microenvironment, supporting the clinical translation of such approaches in the setting of RCC and other forms of solid cancer.
Results
RCC-associated pericytes differentially express the DLK1 antigen
In a previous report,14 we identified several melanoma-associated vascular antigens, including DLK1, which may represent promising therapeutic vaccine targets. Before assessing the therapeutic potential of DLK1 peptide- and gene-based vaccines in the setting of RCC, we first investigated the pattern of DLK1 expression in the TME and tumor uninvolved kidneys of BALB/c mice harboring established syngenic RENCA (an RCC line established from a spontaneously arising renal adenocarcinoma of BALB/c origin)22 tumors. After enzymatic digestion of tissues, tumor- and kidney-derived pericytes and VEC were isolated via flow sorting from single-cell suspensions (Figure 1a) and their extracted mRNA (along with mRNA from the cultured RENCA cell line) was analyzed by real-time PCR for DLK1 (and housekeeping control HPRT1) transcript content (Figure 1b). We observed that pericytes sorted from RCC tumors were uniquely enriched for DLK1 transcripts (Figure 1b) when compared with normal kidney vascular cells or RENCA tumor cells, suggesting that DLK1 may represent a general tumor pericyte-associated antigen. Immunofluorescence microscopy performed on day 21 RENCA tumor sections confirmed that DLK1 protein was coexpressed by NG2+ (a general marker of pericytes in both normal and tumor tissue)23 pericytes that were closely approximated to CD31+ VEC in situ (Figure 1c).
Figure 1.

DLK1 is differentially expressed by RENCA tumor-associated pericytes. The spontaneously arising renal cortical adenocarcinoma RENCA (106 tumor cells) was injected s.c. into female BALB/c mice and allowed to progress for 21 days after which animals were euthanized and tumors and normal kidneys harvested. (a) Tissues were processed into single-cell suspensions and sorted by flow cytometry based on forward versus side scatter profiles, DAPI exclusion (to reject dead cells), a CD45neg phenotype (i.e., non-leukocytic), and then selectively into CD146+CD34neg pericytes and CD146+CD34+ VEC populations based on published assignments of these cell lineage-restricted phenotypes.48,49 (b) mRNA was then isolated from flow-sorted pericytes and VEC, and analyzed for DLK1 transcript expression by real-time PCR. Relative mRNA expression was normalized to housekeeping HPRT1 mRNA expression. (c) Day 21 RENCA tumor tissue sections were analyzed for expression of CD31 (blue), NG2 (green), and DLK1 (red) by immunofluorescence microscopy. Metamorph quantitation (Materials and Methods) was performed on 10 high power field (HPF) of the fluorescent images, with 28.1 ± 4.4% of tumor-associated NG2+ pericytes coexpressing the DLK1 marker. The analysis also revealed that the majority (i.e., 58.9 ± 7.6%) of DLK1+ cells coexpressed the NG2 marker within the TME. All data are representative of three independent experiments performed. DAPI, 4′,6-diamidino-2-phenylindole; FSC, forward scatter; SSC, side scatter; TME, tumor microenvironment; VEC, vascular endothelial cell.
Treatment of RENCA tumor-bearing mice with a DLK1 peptide-based vaccine is therapeutic and associated with specific type-1 CD8+ T cell (Tc1) activation and recruitment into the TME
We have previously demonstrated that vaccine formulations composed of interleukin-12 (IL-12) gene-modified dendritic cells (DCs) (i.e., DC.IL12) pulsed with major histocompatibility complex class I-presented peptides promote robust CD4+ T helper cell-independent priming of antigen-specific CD8+ T cells in vivo.14,24 Using this approach, we analyzed the impact of treating BALB/c mice bearing established s.c. RENCA tumors with a DLK1 peptide (a pooled equimolar mixture of the DLK1158–166, DLK1161–169, and DLK1259–270 peptides)-based vaccine. As depicted in Figure 2a, mice treated with the DLK1 peptide-based vaccine, but not a control vaccine (i.e., DC.IL12, no peptide) or phosphate-buffered saline (PBS), exhibited a significant reduction in the growth of RENCA tumors (Figure 2a; P < 0.05 (analysis of variance) on days >13). On day 21 (i.e., 7 days after the booster immunization), CD8+ splenocytes were isolated and analyzed for secretion of interferon-γ (IFN-γ) in response to stimulation with specific DLK1 peptides presented by syngenic DC in vitro. We noted elevated levels of IFN-γ secretion from CD8+ T cells isolated from mice treated with the DC.IL12 + DLK1 peptide vaccine (versus mice treated with DC.IL12 only or PBS) after stimulation with individual DLK1 peptides, indicating that the vaccine induced poly-specific, anti-DLK1 CD8+ T cell responses in vivo (Figure 2b).
Figure 2.

DC/DLK1 peptide-based vaccines are both immunogenic and therapeutic in the murine RENCA model of RCC. BALB/c mice were inoculated with RENCA tumor cells s.c. on the right flank on day 0. (a) After randomizing for similar mean tumor size per treatment cohort (n = 5), mice were injected s.c. on their left flank on days 7 and 14 (post-tumor inoculation) with PBS, 106 DC.IL12 or 106 DC.IL12 pre-pulsed with equimolar mix (10 μmol/l each) of the three synthetic DLK1 peptides. Tumor growth (mean ± SD) was then monitored over time. (b) On day 21 post-tumor inoculation, splenic CD8+ T cells were isolated from each cohort and co-cultured with syngenic DC pre-pulsed with individual DLK1 peptides for 24 hours, at which time, IFN-γ ELISA were performed on the harvested cell-free supernatants. (c,d) Day 21 tumors were fixed, sectioned and analyzed by immunofluorescence microscopy; CD31 (green in c,d), CXCL10 (red in c), VCAM1 (red in d). The percentage of VCAM1 co-localization with CD31 is depicted as a yellow signal in d and was quantitated using Metamorph software as described in Materials and Methods. Histograms to the right of images reflect mean fluorescence intensity quantitation of the indicated markers (±SD) from three independent fields per slide as described in Materials and Methods. Data are representative of three independent experiments performed. *P < 0.05 versus control treatments (analysis of variance). DC, dendritic cell; IFN, interferon; PBS, phosphate-buffered saline; RCC, renal cell carcinoma.
Since therapeutic type-1 CD8+ T cells preferentially express a VLA-4+CXCR3+ phenotype,25,26 we next determined whether specific vaccination resulted in the altered expression of VLA-4 and CXCR3 ligands, VCAM-1 and CXCL10, respectively in the TME. A coordinate immunofluorescence microscopy analysis of the TME after DLK1 peptide-based vaccination versus control treatment revealed fewer CD31+ tumor blood vessels (Figure 2c), and these vessels contained VEC enriched in the activated VCAM1+ phenotype (Figure 2d). We also observed that these same tumors contained elevated levels of CXCL10/IP-10 chemokine protein expression versus control tumors (Figure 2c), suggesting that the DLK1-based vaccination induces a proinflammatory TME that is competent to recruit type-1 T effector cells.
Vaccination with a recombinant lentivirus encoding murine DLK1 cDNA is therapeutic in the RENCA model of RCC
Clinical trials implementing synthetic tumor peptide-based vaccines have needed to restrict patient accrual to those individuals expressing relevant human leukocyte antigen class I (peptide-presenting) allotypes. To develop a more universal immunization platform, we next engineered a genetic vaccine that would theoretically allow for virally transduced host antigen-presenting cells to cross-prime a more comprehensive anti-DLK1 T effector cell repertoire. Given the reported superiority of lentiviral-based vaccines to promote prolonged antigen-specific CD8+ T cell responses after a single administration in vivo,27 we first constructed a recombinant lentivirus encoding full-length murine DLK1 (lvDLK1) and a negative control virus (lvNEG; Supplementary Figure S1).
To assess the therapeutic efficacy of specific genetic vaccination against the full-length DLK1 antigen, BALB/c mice bearing established day 7 RENCA tumors received a single intradermal injection of lvDLK1 or control lvNEG at a site distal to tumor (i.e., contralateral). Animals treated with lvDLK1 exhibited significant reductions in tumor growth compared with animals treated with lvNEG (Figure 3a). As was the case for DLK1 peptide-based vaccines, immunofluorescence microscopy analysis of tumor sections supported decreased vascularity and loss of (DLK1+) vascular pericytes (Figure 3b), and increased presence of the CXCR3 ligand chemokine, CXCL10, and VCAM1+CD31+ VEC in the TME of mice treated with lvDLK1 versus lvNEG (Figure 3c,d). Enhanced expression of CXCL10 and VCAM1 in the TME was associated with greater numbers of CD8+ TIL in mice receiving LvDLK1-based vaccines (Figure 3e). These findings suggest that immune targeting of DLK1 via a single administration of lvDLK1 can effectively limit tumor growth and induce a proinflammatory TME promoting the improved recruitment of TIL.
Figure 3.
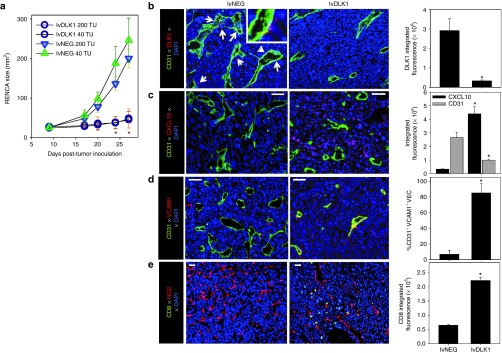
Recombinant lvDLK1-based vaccines are therapeutic and promote a type-1–polarized TME. (a–d) BALB/c mice were inoculated s.c. with RENCA tumor cells in the right flank on day 0. (a) After cohort (n = 5) randomization for similar mean tumor size on day 10 post-tumor inoculation, mice were treated i.d. in the left flank with 40 or 200 transduction units (TU) of lvDLK1 or control virus, lvNEG. Tumor size was then monitored longitudinally. (b–e) On day 27 post-tumor inoculation, mice were euthanized, with harvested tumors fixed, sectioned and analyzed by immunofluorescence microscopy for expression of (b) CD31 (green) and DLK1 (red) with white arrows indicating DLK1+ cells, (c) CXCL10, (d) co-localization of VCAM1 with CD31, and (e) CD8+ TIL (green) and NG (red). Histograms to the right of images reflect mean fluorescence intensity quantitation of the indicated markers (±SD) from three independent fields per slide as described in Materials and Methods. Data are representative of three independent experiments performed. *P < 0.05 versus control treatments (analysis of variance). i.d., intradermal.
Vaccination with lvDLK1 normalizes the RENCA vasculature
It has been suggested that the tumor-associated vasculature of mice deficient in immature pericytes appears “normal” with minimal arborization and reduced vascular permeability,18 supporting therapeutic strategies to selectively reduce or eradicate immature vascular pericytes within tumor sites. Given the ability of our lvDLK1-based genetic vaccine to reduce the content of DLK1+ (immature) pericytes in the tumor stroma, we sought further evidence supporting therapeutic vascular remodeling as a consequence of treatment with this modality. We noted that RENCA tumors harvested from mice treated with lvDLK1 appeared “anemic” when compared to control tumors (Figure 4a), a subjective index that was subsequently confirmed based on an analysis of hemoglobin content in tumor lysates (Figure 4b). When we analyzed tumors for expression of NG2 using immunofluorescence microscopy, we observed that animals vaccinated with lvDLK1 exhibited tumors with significant reductions in numbers of NG2+ pericytes in their TME versus tumors from animals vaccinated with lvNEG (Figure 4c,d). Residual tumor pericytes in lvDLK1-treated animals were tightly approximated to CD31+ VEC, unlike the randomly distributed pattern of pericytes detected in the stroma of tumors isolated from control mice. To investigate changes in tumor vascular permeability, vaccinated animals received intravenous injections of two fluorescently labeled probes, tomato lectin-FITC to bind/mark the vascular endothelium and small 20 nm (red) FluoSpheres to determine vessel leakiness into tissue. When compared with controls, the tumor blood vessels in mice vaccinated with lvDLK1 displayed a simple tubular architecture devoid of extensive branching (Figure 5a). Furthermore, while the perivascular stroma of tumors in control animals was littered with the red FluoSpheres, these probes were virtually undetected in tumors harvested from lvDLK1-vaccinated mice, consistent with diminished vascular permeability in the TME of these latter animals (Figure 5a). These data suggest that immunization against DLK1 allows for the immunotherapeutic “normalization” (i.e., reduction in blood vessel numbers and arborization, reduced vascular permeability) of tumor blood vessels in vivo.
Figure 4.

Recombinant lvDLK1-based vaccines promote normalization of the tumor vasculature. Mice bearing day 10 RENCA tumors were treated with 200 TU of lvDLK1 or lvNEG as outlined in Figure 3. On day 27 post-tumor inoculation, mice were euthanized and the (a) tumors were resected and evaluated macroscopically and (b) for hemoglobin content. In c and d, tumor sections were analyzed by immunofluorescence microscopy for expression of CD31 (green) and NG2 (red). In e, 6 μm sections were imaged by wide field microscopy, while in d, 30 μm sections were imaged by confocal microscopy to generate 3D reconstructions. For e, mean data ± SD of three independent fields per slide in c is reported for each group from one representative experiment of three performed. (f) The percentage of NG2 fluorescence signal overlapping CD31 fluorescence signal was calculated using Metamorph software as described in Materials and Methods, and is reported as mean ± SD of three independent fields per slide. *P < 0.05 for lvDLK1 versus lvNEG (t-test).
Figure 5.

Recombinant lvDLK1-based vaccination reduces tumor vascular permeability resulting in the development of apoptotic “dead zones” in the TME. In repeated experiments as outlined in Figure 3, (a) treated mice received intravenous injections of tomato lectin-FITC to label vascular endothelium (green) and 20 nm FluorSpheres to assess vascular permeability (red) on day 24 post-tumor inoculation. Whole tumor tissue was then imaged immediately by confocal microscopy at a depth of 17 μm. *P < 0.05 for lvDLK1 versus lvNEG (t-test). (b) On the same day, unlabeled mice were euthanized, with tumors resected, fixed, sectioned, and analyzed for expression of CD31 (green) and apoptotic nuclear staining with TUNEL reagent (red). Histograms to the right of images reflect mean fluorescence intensity quantitation of the indicated markers (±SD) from three independent fields per slide as described in Materials and Methods. Data are representative of three independent experiments performed. *P < 0.05 for lvDLK1 versus lvNEG (t-test).
Therapeutic vaccination with lvDLK1 results in increased cellular apoptosis in the treated TME
Given the apparent trimming of vascular branches in the RENCA TME, and reduction in vascular permeability after vaccination with lvDLK1 (but not lvNEG), we hypothesized that plasma nutrients required for sustaining tumor cell viability would be limited to regions adjacent to the remaining normalized blood vessel network. TUNEL analyses revealed that indeed, the level of cellular apoptosis in the TME of lvDLK1-treated mice was substantially increased when compared with tumors isolated from control treated animals (Figure 5b). Furthermore, virtually all apoptotic events (i.e., “dead zones”) in RENCA tumors isolated from lvDLK1-vaccinated mice were located in tissue regions >~60 μm away from residual CD31+ blood vessels in planar tissue imaging analyses (Figure 5b).
Therapeutic vaccination with lvDLK1 results in reduced hypoxia and a lower incidence of cell populations expressing hypoxia-responsive markers in the TME
Hypoxia frequently occurs in solid cancers as a consequence of inefficient perfusion of oxygen into tumors by “aberrant” blood vessels,16,28 resulting in reduced recruitment and function of TIL, increased prevalence of immunosuppressive cells/modulators, dysregulated angiogenesis, and the accumulation of “stem-like” cell populations (i.e., cancer stem cells/tumor initiating cells, cells undergoing epithelial-to-mesenchymal transition) in the TME.29,30 To investigate changes in hypoxia within tumors after vaccination with lvDLK1 versus lvNEG, we injected mice intraperitoneally (i.p.) with pimonidazole (a hypoxia marker that undergoes reductive activation and then conjugates to thiol-containing proteins specifically in hypoxic cells, allowing for immunohistochemical detection of tissue regions exhibiting low (<1.3%) O2 tension).31 Using this imaging technology, we found that tumors isolated from mice receiving lvDLK1 vaccines had a very low hypoxic index when compared to tumors culled from control animals (Figure 6a). Given this large reduction in TME hypoxia postvaccination with lvDLK1, we next investigated treatment impact on expression of hypoxia-responsive gene products associated with immature vascular stromal cells (i.e., RGS514) and/or stem-like cell populations (i.e., Jarid1B aka histone demethylase lysine demethylase 5b; CD133, CD44).32,33,34 Immunofluorescence microscopy analysis of day 27 tumor sections revealed that the expression of these markers was coordinately reduced in RENCA tumors after host vaccination with lvDLK1 (Figure 6b–f). When taken together, these data indicate that vaccination with lvDLK1 results in the recovery of normoxia in the TME in association with the conditional alteration in the phenotype (and presumably function) of a range of stromal cell subpopulations in vivo.
Figure 6.

Recombinant lvDLK1-based vaccines promote normoxia in the TME in association with the loss of cells bearing stem cell-like phenotypes. Mice bearing day 10 RENCA tumors were treated with 200 TU of lvDLK1 or lvNEG as outlined in Figure 3. (a) On day 21, mice were injected intraperitoneally with the hypoxia probe pimonidazole hydrochloride and euthanized, with tumors resected, sectioned, and analyzed by HRP immunohistochemistry. (b–e) Day 21 tumor-bearing mice that did not receive pimonidazole hydrochloride were euthanized, with tumors harvested, fixed, sectioned and analyzed by immunofluorescence microscopy for expression of (b) CD31 and RGS5, (c) Jarid1b, (d) CD133, and (e) CD44. (f) Histograms to the right of panel b–e images reflect mean fluorescence intensity quantitation of the indicated markers (±SD) from three independent fields per slide as described in Materials and Methods. Data are representative of three independent experiments performed. *P < 0.05 for lvDLK1 versus lvNEG (t-test). TME, tumor microenvironment; TU, transduction unit.
Loss of DLK1 expression in the TME after therapeutic vaccination with lvDLK1 leads to increased locoregional activation of NOTCH
Since lvDLK1-based vaccination leads to loss of DLK1 expression in the TME (Figure 3) and DLK1 represents a functional inhibitor of NOTCH signaling,20 we hypothesized that this therapeutic vaccine would promote enhanced canonical NOTCH signaling in therapeutic RENCA TME. As shown in Figure 7a,b, RENCA tumors isolated from lvDLK1-treated (but not control) mice contained cells strongly expressing cytoplasmic/nuclear Hes1 protein, a NOTCH transcriptional target required for the tumor suppressor action of activated NOTCH.19,35 Hes1+ cells included both CD31+VEC and non-VEC stromal cell populations in the TME (Figure 7a). Corollary gene array analyses also supported the enhanced transcription of numerous NOTCH target genes (including the canonical NOTCH ligands (DLL1, DDL3, DLL4, and Jag1/2) and the NOTCH1-4 receptors, among others), but not control β2-microglobulin, in lvDLK1- versus lvNEG-treated tumors (Figure 7c).
Figure 7.

Treatment with lvDLK1 vaccines results in NOTCH activation in the TME, which is partially responsible for the antitumor effectiveness of this treatment strategy. (a) Tumor sections were isolated as described in Figure 3 and evaluated by fluorescence microscopy using specific antibodies against CD31 (green) and Hes1 (red). DAPI counterstaining was used to image cell nuclei (blue). White arrows in image insets indicate Hes1+CD31+ VEC. (b) Mean fluorescence intensity quantitation of Hes1 protein expression (±SD) from three independent fields per slide is reported as described in Materials and Methods. Data are representative those obtained in three independent experiments performed. *P < 0.05 (t-test). (c) mRNA transcripts of NOTCH target genes were analyzed using an reverse transcription-PCR gene array. The ratio of transcript levels for a given gene product among total tumor mRNA isolated from lvDLK1- versus lvNEG-treated mice is reported. Negative control transcript = β2-microglobulin (beta 2-m). (d) Established day 8 s.c. RENCA tumors were treated with 200 TU of lvNEG or lvDLK1 (i.e., VAC) as described in the Figure 3a legend and Materials and Methods. DAPT (in DMSO; depicted as small gray ovals labeled “D” on the x-axis) or vehicle control DMSO was then provided as indicated for three consecutive days/week/cycle for two cycles beginning on day 12 post-tumor inoculation. Tumor size was then monitored longitudinally. *P < 0.05 for lvDLK1 + DAPT treatment versus lvDLK1 treatment; also P < 0.05 for the lvDLK1 + DAPT and lvDLK1 treatments versus lvNEG control treatment on days ≥15 post-tumor inoculation (analysis of variance). DAPI, 4′,6-diamidino-2-phenylindole; TME, tumor microenvironment; TU, transduction unit; VAC, vaccination.
Therapeutic benefits associated with lvDLK1-based genetic vaccination are partially dependent on canonical NOTCH signaling
To determine the importance of canonical NOTCH signaling on the antitumor efficacy of genetic vaccination against DLK1, we immunized BALB/c mice bearing established s.c. RENCA tumors with lvNEG or lvDLK1 as described in Figure 3a, with cohorts of lvDLK1-vaccinated animals injected i.p. with the γ-secretase inhibitor DAPT (N-[N-(3,5-Difluorophenacetyl-L-alanyl)]-(S)-phenylglycine t-butyl ester; which inhibits the generation of the NOTCH intracellular domain required for downstream NOTCH signaling events, ref. 36) or vehicle control DMSO. As shown in Figure 7d, administration of DAPT partially suppressed the antitumor action of lvDLK1-based therapeutic vaccination.
Discussion
The major finding in this report is that DLK1 is a tumor pericyte-associated antigen that can be immunologically targeted via specific peptide- or gene-based vaccination in vivo, leading to the effective “normalization” of the vasculature in the TME and a drastic reduction in solid tumor (i.e., RENCA) growth in vivo. Effective therapeutic vaccination resulted in the activation of type-1 (IFN-γ–producing) DLK1-specific CD8+ T cells in the periphery and the improved recruitment of CD8+ T cells into/around residual blood vessels in the TME. Therapeutically normalized blood vessels in RENCA tumors exhibit a simplified conduit design with tightly approximated (abluminal) NG2+DLK1negRGS5neg mature pericyte populations that appear improved in their structural integrity based on a reduction in vascular permeability. RENCA tumors in DLK1-vaccinated mice became normoxic and displayed a dramatic increase in the rate of apoptotic death in regions of the tumor that were further away from residual blood vessels. The relationship between loss of hypoxia and promotion of tumor cell apoptosis in the therapeutic TME may not be intuitively obvious. We hypothesize that consistent with the paradigm of Jain,17 therapeutic vascular normalization results in the coordinate loss of vascular permeability and hypoxia in the TME. In turn, loss of hypoxia (and hypoxia-responsive genes such as HIF-1α) has been associated with enhanced rates of tumor cell apoptosis37 and reduced expression of a broad range of tumor growth and survival/antiapoptotic gene products.38 As such, under normoxic conditions post-therapy (as in the case of lvDLK1-based vaccination), tumor cells most removed from blood vessels may be rendered most susceptible to undergo apoptosis based on limited access to pro-survival/growth factor gradients emanating from normalized blood vasculature. Overall, our findings support a model in which specific immune effector T cells may serve as regulators of the “angiogenic switch”14,15,16,17,18 by monitoring and controlling the status of DLK1+ pericytes within the TME.
Vaccination against DLK1 also induced a proinflammatory TME based on the acquisition of activated VCAM1+ VEC and concomitant production of the CXCR3 ligand chemokine CXCL10, responsible for recruiting type 1 TIL. We hypothesize that an initial wave of DLK1-reactive type-1 TIL results in perivascular secretion of IFN-γ and tumor necrosis factor-α in the TME, leading to locoregional upregulation of IFN-γ/tumor necrosis factor-α–responsive gene products such as VCAM-1 and CXCL10.39 Such alterations in the TME would then be expected to foster improved uptake of tumor debris (i.e., apoptotic bodies) by recruited/activated antigen-presenting cells and the corollary reiterative cross-priming of an expanded, protective T cell repertoire reactive against both tumor- and tumor vascular-associated antigens14 that may be directed into the proinflammatory TME.
Interestingly, a recent report by Reis et al.39 suggests that the conditional activation of the Wnt/β-catenin/NOTCH signaling pathway can lead to vascular normalization, as indicated by reduced vascular density and improved mural cell attachment, in intracranial murine glioma models. Our data support such a paradigm, with specific vaccination resulting in removal of DLK1 expression (and NOTCH antagonism)20 in the TME. Such immune pressure improved NOTCH signaling based on a dramatic increase in the intratumoral expression of Hes1 protein and the transcriptional activation of multiple NOTCH target genes. The transcriptional profiling also supports differentially increased expression of Frzd2, Frzd4, Frzd7, and β-catenin (Ctnnb1) in RENCA tumors harvested from lvDLK1-vaccinated mice supporting the coactivation of canonical Wnt/β-catenin signaling40 in the therapeutic TME, consistent with the model proposed by Reis et al.39 As such, our data suggest that vaccination against DLK1 (as an integral transmembrane protein or via its shed extracellular domain)41 may derepress canonical NOTCH/Wnt/β-catenin signaling in endothelial cells (and other stromal cell populations) within the TME, thereby promoting vascular quiescence/normalization.20,39,42 Vaccination against DLK1 may also improve type-1 functionality of tumor-associated macrophages and DC (i.e., enhanced IL-12p70 and CXCL10 production) and T cells.43 Indeed, we observed that the functional antagonism of NOTCH signaling in vivo (based on administration of the γ-secretase inhibitor DAPT) partially ablated the antitumor benefits associated with lvDLK1-based therapeutic vaccination, suggesting a supporting role for canonical NOTCH signaling in treatment outcome. Future studies will investigate the potential role of Wnt/β-catenin signaling in therapeutic benefit associated with DLK1-based vaccines by applying specific inhibitors of these pathways in our therapeutic model.
The TME of progressively growing, control RENCA tumors was enriched in cells expressing markers known to contain HRE in their promoter regions, such as CD44, CD133, and Jarid1B,32,33,34 that have been previously linked to cell populations with “stem-like” characteristics.29,30 Notably, the “normalized” TME after therapeutic vaccination with lvDLK1 was normoxic and largely devoid of cells expressing these hypoxia-responsive antigens. Although the most simplistic reason for this change reflects the transcriptional silencing of these gene products in the TME of lvDK1-vaccinated animals, it is also conceivable that the therapeutic TME is poor in recruiting cells bearing these phenotypic markers, and/or that the vaccine evoked corollary cross-priming14,24 of cytotoxic CD8+ T cell responses capable of eradicating CD44+, CD133+, and Jarid1B+ target cells in effectively treated RENCA tumors. With regard to the latter scenario, we currently plan to longitudinally evaluate the reactivity of the evolving therapeutic CD8+ T cell repertoire against peptide epitopes derived from the CD44, CD133, and Jarid1B (as well as alternate “stem cell”-associated/hypoxia-responsive markers such as ALDH1, Oct4, and Nanog)34 antigens in RENCA-bearing mice treated with DLK1 peptide/gene-based vaccines.
The antiangiogenic action mediated by the DLK1 vaccine-induced CD8+ T cell repertoire would be anticipated to differ, and likely complement, that of alternative pharmacological antiangiogenic treatment modalities such as antivascular endothelial growth factor antibodies (i.e., bevacizumab) and small molecule tyrosine kinase inhibitors.11,44,45 In most cases, tumors treated with these agents rapidly become drug-refractory due to their adoption of compensatory growth/progression pathways. As such, DLK1-based vaccines could represent a logical second-line approach in the many cases of developed resistance to bevacizumab, sunitinib or similar antiangiogenic drugs. DLK1-based vaccines may also represent effective co-first line therapeutic agents, since the specific activation, recruitment and function of anti-DLK1 T effector cells in the TME would be anticipated to be improved by the coadministration of antiangiogenic tyrosine kinase inhibitor that reduce suppressor cell populations (most notably in RCC patients) and activate a proinflammatory TME in vivo.26,46,47 Based on these expectations, we plan to evaluate the comparative therapeutic efficacy of combined sunitinib + lvDLK1 vaccination treatment in our existing s.c. RENCA model, as well as, in an orthotopic RCC model using RENCA.luc (RENCA cells transduced with luciferase cDNA) to allow for vital bioluminescence monitoring of tumor growth and metastasis. Although we have not observed signs of off-target autoimmune pathology as a consequence of DLK1-targeted vaccination (i.e., inhibition of cutaneous wound healing,14 tissue vasculitis; data not shown) to date, these new models will provide us with additional opportunities to investigate potential combination treatment-associated toxicities in future.
Consistent with our findings in the RENCA model, pericytes from freshly isolated human RCC (but not patient-matched normal adjacent kidney tissue) also differentially (over)express the DLK1 antigen in situ (Supplementary Figure S2). When coupled with the knowledge that anti-DLK1 CD8+ T cell responses can be developed from human cancer patients after in vitro sensitization,24 we believe that DLK1-based vaccines (as single agents or in combination approaches) represent attractive candidates for clinical translation in the setting of RCC and alternate well-vascularized forms of solid cancer.
Materials and Methods
Mice. Female 6–8 weeks old BALB/c mice (The Jackson Laboratory, Bar Harbor, ME) were maintained in a pathogen-free animal facility, with all animal work performed in accordance with an Institutional Animal Care and Use Committee-approved protocol.
Tumor cells. The mouse RCC line RENCA derived from a spontaneous renal cortical adenocarcinoma in BALB/cCr mice (CRL-2947; American Type Culture Collection, Manassas, VA)22 was cultured as previously reported.15
Stromal cell isolation. Human RCC tumor and adjacent (patient-matched) normal kidney specimens were obtained with written-consent under an Institutional Review Board-approved protocol. Murine RCC tumors and tumor uninvolved kidneys were harvested 21 days after s.c. injection of 106 RENCA cells into syngenic BALB/c recipient animals. Tissues were dissected/minced, then enzymatically digested, with pericytes and VEC isolated by flow sorting as previously described,14 with one modification. Specifically, murine cells were labeled with anti-mouse CD34-FITC (eBioscience, San Diego, CA), anti-mouse CD146-PE (BD-Biosciences, San Diego, CA), and anti-mouse CD45-APC (BD-Biosciences) before flow sorting into pericyte (CD146+CD34negCD45neg) and VEC (CD146+CD34+CD45neg) populations.48,49 In all cases, cells were >95% pure for the stated phenotype.
Real-time PCR. Messenger RNA was isolated from pericytes and VEC using the RNeasy Plus Micro kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA was then generated using High Capacity RNA-to-cDNA kit (Applied Biosystems, Carlsbad, CA) and real-time PCR performed using Fast SYBR Green Master Mix (Applied Biosystems) with primer pairs for human or mouse HPRT1 (Qiagen), human DLK1 (Applied Biosystems) or mouse DLK1 (forward primer: TGTGACCCCCAGTATGGATT, reverse primer: CCAGGGGCAGTTACACACTT). Reactions were performed in duplicate in a 96-well reaction plate on a StepOnePlus real-time PCR thermocycler (Applied Biosystems) using cycling conditions of 95 °C for 20 minutes, then 35 cycles of 95 °C for 3 minutes and 60 °C for 30 minutes.
In vitro generation of bone marrow-derived DC and DC.IL12. DC were generated in 5-day rIL-4 + rGM-CSF–supplemented cultures from bone marrow precursors isolated from the tibias/femurs of BALB/c mice infected with recombinant adenovirus encoding mouse IL-12p70 (yielding DC.IL12), as previously described.24
Synthetic peptides. The H-2d class I-presented DLK1158–166 (CPPGFSGNF; presented by H-2Ld), DLK1161–169 (GFSGNFCEI; presented by H-2Kd), DLK1259–270 (TILGVLTSLVVL; containing overlapping DLK1259–267 and DLK1262–270 sequences presented by H-2Kd) peptide were synthesized as previously described.14
Recombinant lentiviral vector production. Genes encoding mDLK1 and the reverse sequence of mRGS5 (as a negative control) were cloned into the pLenti6/V5 D-TOPO vector downstream of the cytomegalovirus promoter using the Lentiviral Directional TOPO Expression Kit (Invitrogen, Grand Island, NY). To determine insert presence in the plasmid, expression of the V5 tag was detected by immunofluorescence using an anti-V5 FITC antibody (Invitrogen) and by western blot using an anti-V5 HRP antibody (Invitrogen). In the initial production of the lentiviruses, 293FT cells (Invitrogen) were transfected with plasmid DNA pLenti-DLK1 (or pLenti-NEG) using ViraPower Packaging Mix (Invitrogen) combined with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 48 hours, lentivirus was collected and concentrated using a Fast-Trap Virus Purification and Concentration kit (Millipore, Billerica, MA). Lentiviral (lvDLK1 and lvNEG) titers, reported in transduction units, were determined by quantitating blasticidin (Invitrogen)-resistance in HT-1080 cells (kindly provided by Dr Chuanyue Wu, University of Pittsburgh, Pittsburgh, PA) according to the manufacturer's instructions. Expanded lentiviral production was performed by the University of Pittsburgh Cancer Institute Lentiviral Vector Core Facility. Lentivirus quality was assessed by infecting HT-1080 cells for 24 hours and monitoring cells for coordinate V5 protein expression (western blot) and cell-surface expression of DLK1 (flow cytometry using an anti-DLK1-PE conjugated antibody; Adipogen, San Diego, CA).
Animal therapy experiments. BALB/c mice received s.c. injection of 106 RENCA tumor cells (right flank) on day 0. Six days later, the animals were randomized into cohorts of five mice with comparable mean tumor sizes. On days 7 and 14 after tumor implantation, mice were treated with 100 μl s.c. injections (left flank) of PBS, 106 DC.IL12 or 106 DC.IL12 that had been pre-pulsed for 2 hours at 37 °C with an equimolar (10 μmol/l) mixture of the DLK1158–166, DLK1161–169, and DLK1259–270 peptides. For lentivirus vaccination experiments, randomized BALB/c mice bearing established (day 10; right flank) s.c. RENCA tumors received a single left flank intradermal injection of lvDLK1 or negative control lvNEG at a dose of 4 × 104 or 2 × 105 transduction units in a total volume of 50 μl PBS. For all animal experiments, tumor size was assessed every 3–4 days and recorded in mm2, as determined by the product of orthogonal measurements taken using vernier calipers. Data are reported as mean tumor area ± SD. To determine the impact of canonical NOTCH signaling on vaccine efficacy, tumor-bearing animals vaccinated with lvDLK1 or lvNEG were injected i.p. with the γ-secretase inhibitor DAPT (10 mg/kg/day in 50 μl DMSO (Sigma-Aldrich, St Louis, MO) on three consecutive days (followed by 4 days without injections) per week schedule, for 2 weeks beginning on day 12 post-tumor inoculation) or vehicle control (DMSO).
Evaluation of specific CD8+ T cell responses in vitro. Spleens were harvested from three mice per group 7 days after the second DC injection. Splenocytes were then stimulated in vitro for 5 days with syngenic DC pulsed with an equimolar (10 μmol/l) mix of the three DLK1 peptides applied in the vaccine. Responder CD8+ T cells were then isolated using magnetic bead cell sorting (Miltenyi Biotec, Auburn, CA) and co-cultured with syngenic DC pulsed with individual DLK1 peptides for 72 hours, 37 °C and 5% CO2, at which time cell-free supernatants were analyzed for mIFN-γ content using a cytokine-specific ELISA (BD-Biosciences).
Fluorescent imaging of tumors. Tumor tissue samples were prepared and sectioned as previously reported.15 Six micron tissue sections were analyzed for expression of CD31 (BD-Biosciences), VCAM1 (R&D Systems, Minneapolis, MN), CXCL10 (R&D Systems), NG2 (Millipore), DLK1 (Santa Cruz Biotechnology, Santa Cruz, CA), RGS5, Jarid1b (all from Abcam, Cambridge, MA), CD133 (BD-Biosciences), CD44 (Abcam), and Hes1 (Millipore) by immunofluorescence microscopy, with wide field images collected with fixed illumination conditions using a cooled CCD camera (Olympus Magnafire; Olympus, Center Valley, PA). Using Metamorph software (Molecular Devices, Downingtown, PA), images were thresholded to delineate signal above background and individual structures measured as the integration of pixel number (total number of positive pixels in the structure above background) multiplied by the brightness of each pixel in gray scales. This product provides the integrated pixel intensity of positive structures and is reported as the mean integrated fluorescence intensity ± SD. For the analysis of activated VEC in the TME, cellular identity was first defined using co-localization of specific markers (cells staining for both CD31 and VCAM-1) using image overlay and manual counting. We found this method was essential to ensure accuracy in cell identification in tissue with complex morphologies. To perform the quantification images were overlaid with Metamorph software and co-localized structures that could be defined as cells were counted. For analysis of cellular apoptosis, tissue sections were labeled using TUNEL kit (Roche, Indianapolis, IN) as per the manufacturer's instructions, followed by incubation with secondary anti-streptavidin Cy3 antibody (Jackson Immunoresearch, West Grove, PA). Some sections were analyzed by confocal microscopy to generate 30 μm 3D reconstructions of images. For the vascular permeability imaging, animals received retro-orbital intravenous injections of FITC-labeled tomato lectin (Sigma-Aldrich) and red 20 nm FluoSpheres (Invitrogen), followed by cardiac perfusion of PBS and 4% paraformaldehyde. Tumors were then immediately resected and imaged by confocal microscopy to generate 17 μm 3D reconstructions. In all depicted tissue images, white ruler insets indicate 50 μm (low magnification images) or 10 μm (high magnification images).
Hemoglobin quantitation. The amount of hemoglobin contained in tissues was quantitated using the Drabkin method50 and reported as μg hemoglobin per mg wet weight of tissue.
Measurement of tumor hypoxia using pimonidazole. BALB/c mice bearing established (treated or untreated) day 21 s.c. RENCA tumors were injected i.p. with 60 mg/kg pimonidazole hydrochloride (Hypoxyprobe; HPI, Burlington, MA) 30 minutes before euthanasia and tumor harvest and 6 μm tissue sections prepared and analyzed by immunohistochemistry as previously reported.15
RNA purification and reverse transcription-PCR array. Total RNA was isolated from bulk single-cell suspensions of day 21 tumors harvested from lvNEG- or lvDLK1-treated mice using Trizol reagents (Invitrogen). Total RNA was further purified using the RNeasy Plus Mini Kit (Qiagen) including the gDNA Eliminator spin column. The purity and quantity of the total RNA was assessed using Nanodrop ND-1000 (CelBio SpA, Milan, Italy). Total RNA (1 μg) was reversed transcribed into cDNA using the RT2 First Strand Kit (Qiagen) and the cDNA added to RT2 SYBR Green ROX qPCR Mastermix (Qiagen) and used for quantitative PCR using the RT2 Profiler PCR Array (96-well) for Mouse Notch Signaling Pathway (Qiagen) all according to the manufacturer's instructions. Reactions were performed on a StepOnePlusTM Real-Time PCR thermocycler (Applied Biosystems) using the recommended cycling conditions. All mRNA expression levels were normalized to the expression of GAPDH.
Statistical analysis. Comparisons between groups were performed using a two-tailed Student's t-test or one-way analysis of variance with Tukey's post-hoc analysis, as indicated. All data were analyzed using SigmaStat software, version 3.5 (Systat Software, Chicago, IL). Differences between groups with a P value <0.05 were considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Production of recombinant lvDLK1 and control lvNEG lentiviruses. Figure S2. DLK1 is differentially (over)expressed by human RCC-associated pericytes.
Acknowledgments
The authors thank Adriana Larregina (University of Pittsburgh) for her careful review and useful discussions provided during the preparation of this manuscript. This work was supported by National Institutes of Health (NIH) grants P01 CA109688 and R01s CA114071, CA143075, and CA169118 (to W.J.S.). This project used the UPCI Vector and Lentiviral Core Facilities supported by the University of Pittsburgh's NIH CCSG award P30 CA047904. The authors declared no conflict of interest.
Supplementary Material
References
- Gerhardt H, Semb H. Pericytes: gatekeepers in tumour cell metastasis. J Mol Med. 2008;86:135–144. doi: 10.1007/s00109-007-0258-2. [DOI] [PubMed] [Google Scholar]
- Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002;160:985–1000. doi: 10.1016/S0002-9440(10)64920-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaengel K, Genové G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- Fukumura D, Duda DG, Munn LL, Jain RK. Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation. 2010;17:206–225. doi: 10.1111/j.1549-8719.2010.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke JH, Tubbs R, Connelly B, Pontes E, Montie J. Tumor-infiltrating lymphocytes in patients with renal-cell carcinoma. Ann N Y Acad Sci. 1988;532:387–394. doi: 10.1111/j.1749-6632.1988.tb36356.x. [DOI] [PubMed] [Google Scholar]
- Muul LM, Spiess PJ, Director EP, Rosenberg SA. Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. J Immunol. 1987;138:989–995. [PubMed] [Google Scholar]
- Lokich J. Spontaneous regression of metastatic renal cancer. Case report and literature review. Am J Clin Oncol. 1997;20:416–418. doi: 10.1097/00000421-199708000-00020. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol. 2006;24:5601–5608. doi: 10.1200/JCO.2006.08.5415. [DOI] [PubMed] [Google Scholar]
- Escudier B, Roigas J, Gillessen S, Harmenberg U, Srinivas S, Mulder SF, et al. Phase II study of sunitinib administered in a continuous once-daily dosing regimen in patients with cytokine-refractory metastatic renal cell carcinoma. J Clin Oncol. 2009;27:4068–4075. doi: 10.1200/JCO.2008.20.5476. [DOI] [PubMed] [Google Scholar]
- Najjar YG, Rini BI. Novel agents in renal carcinoma: a reality check. Ther Adv Med Oncol. 2012;4:183–194. doi: 10.1177/1758834012443725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich I, Scheffrahn I, Bartling S, Weis J, von Felbert V, Middleton M, et al. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J Exp Med. 2010;207:491–503. doi: 10.1084/jem.20091846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic L, Butterfield LH. Melanoma cancer vaccines and anti-tumor T cell responses. J Cell Biochem. 2007;102:301–310. doi: 10.1002/jcb.21473. [DOI] [PubMed] [Google Scholar]
- Ahmed F, Steele JC, Herbert JM, Steven NM, Bicknell R. Tumor stroma as a target in cancer. Curr Cancer Drug Targets. 2008;8:447–453. doi: 10.2174/156800908785699360. [DOI] [PubMed] [Google Scholar]
- Zhao X, Bose A, Komita H, Taylor JL, Chi N, Lowe DB, et al. Vaccines targeting tumor blood vessel antigens promote CD8(+) T cell-dependent tumor eradication or dormancy in HLA-A2 transgenic mice. J Immunol. 2012;188:1782–1788. doi: 10.4049/jimmunol.1101644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komita H, Zhao X, Taylor JL, Sparvero LJ, Amoscato AA, Alber S, et al. CD8+ T-cell responses against hemoglobin-beta prevent solid tumor growth. Cancer Res. 2008;68:8076–8084. doi: 10.1158/0008-5472.CAN-08-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin YH, Chi HC, Liao CH, Liao CJ, Wu SM, et al. Thyroid hormone regulation of miR-21 enhances migration and invasion of hepatoma. Cancer Res. 2013;73:2505–2517. doi: 10.1158/0008-5472.CAN-12-2218. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature. 2008;453:410–414. doi: 10.1038/nature06868. [DOI] [PubMed] [Google Scholar]
- Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- Falix FA, Aronson DC, Lamers WH, Gaemers IC. Possible roles of DLK1 in the Notch pathway during development and disease. Biochim Biophys Acta. 2012;1822:988–995. doi: 10.1016/j.bbadis.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Begum A, Kim Y, Lin Q, Yun Z. DLK1, delta-like 1 homolog (Drosophila), regulates tumor cell differentiation in vivo. Cancer Lett. 2012;318:26–33. doi: 10.1016/j.canlet.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GP, Hrushesky WJ. A murine renal cell carcinoma. J Natl Cancer Inst. 1973;50:1013–1025. doi: 10.1093/jnci/50.4.1013. [DOI] [PubMed] [Google Scholar]
- Stallcup WB. The NG2 proteoglycan: past insights and future prospects. J Neurocytol. 2002;31:423–435. doi: 10.1023/a:1025731428581. [DOI] [PubMed] [Google Scholar]
- Zhao X, Bose A, Komita H, Taylor JL, Kawabe M, Chi N, et al. Intratumoral IL-12 gene therapy results in the crosspriming of Tc1 cells reactive against tumor-associated stromal antigens. Mol Ther. 2011;19:805–814. doi: 10.1038/mt.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Zhu X, Vasquez C, Nishimura F, Dusak JE, Huang J, et al. Preferential expression of very late antigen-4 on type 1 CTL cells plays a critical role in trafficking into central nervous system tumors. Cancer Res. 2007;67:6451–6458. doi: 10.1158/0008-5472.CAN-06-3280. [DOI] [PubMed] [Google Scholar]
- Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, et al. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. 2011;129:2158–2170. doi: 10.1002/ijc.25863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhang J, Donahue C, Falo LD., Jr Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity. 2006;24:643–656. doi: 10.1016/j.immuni.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Yasui H, Batra S, Kinoshita Y, Bernardo M, Munasinghe JP, et al. Simultaneous imaging of tumor oxygenation and microvascular permeability using Overhauser enhanced MRI. Proc Natl Acad Sci USA. 2009;106:17898–17903. doi: 10.1073/pnas.0908447106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- Cabarcas SM, Mathews LA, Farrar WL. The cancer stem cell niche–there goes the neighborhood. Int J Cancer. 2011;129:2315–2327. doi: 10.1002/ijc.26312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque JP, Winkler IG, Hendy J, Williams B, Helwani F, Barbier V, et al. Hematopoietic progenitor cell mobilization results in hypoxia with increased hypoxia-inducible transcription factor-1 alpha and vascular endothelial growth factor A in bone marrow. Stem Cells. 2007;25:1954–1965. doi: 10.1634/stemcells.2006-0688. [DOI] [PubMed] [Google Scholar]
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Ma Y, Liu J, Trope CG, Holm R, Nesland JM, et al. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer. 2012;12:201. doi: 10.1186/1471-2407-12-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–4652. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it's NOTCH what you think. J Exp Med. 2011;208:1931–1935. doi: 10.1084/jem.20111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha B, Jyothi Prasanna S, Chandrasekar B, Nandi D. Gene modulation and immunoregulatory roles of interferon gamma. Cytokine. 2010;50:1–14. doi: 10.1016/j.cyto.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Nagao M, Yamada Y, Narikiyo M, Ueno M, Miyagishi M, et al. Small interfering RNA expression vector targeting hypoxia-inducible factor 1 alpha inhibits tumor growth in hepatobiliary and pancreatic cancers. Cancer Gene Ther. 2006;13:131–140. doi: 10.1038/sj.cgt.7700871. [DOI] [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC. HIF1a and HIF2a: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S, et al. Endothelial Wnt/ß-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J Exp Med. 2012;209:1611–1627. doi: 10.1084/jem.20111580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno K, Hirata H, Hinoda Y, Dahiya R. Frizzled homolog proteins, microRNAs and Wnt signaling in cancer. Int J Cancer. 2013;132:1731–1740. doi: 10.1002/ijc.27746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sul HS. Ectodomain shedding of preadipocyte factor 1 (Pref-1) by tumor necrosis factor alpha converting enzyme (TACE) and inhibition of adipocyte differentiation. Mol Cell Biol. 2006;26:5421–5435. doi: 10.1128/MCB.02437-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fukuhara S, Sako K, Takenouchi T, Kitani H, Kume T, et al. Angiopoietin-1/Tie2 signal augments basal Notch signal controlling vascular quiescence by inducing delta-like 4 expression through AKT-mediated activation of beta-catenin. J Biol Chem. 2011;286:8055–8066. doi: 10.1074/jbc.M110.192641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Cabezas B, Naranjo-Gómez M, Bastos-Amador P, Requena-Fernández G, Pujol-Borrell R, Borràs FE. Ligation of Notch receptors in human conventional and plasmacytoid dendritic cells differentially regulates cytokine and chemokine secretion and modulates Th cell polarization. J Immunol. 2011;186:7006–7015. doi: 10.4049/jimmunol.1100203. [DOI] [PubMed] [Google Scholar]
- Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10:992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
- Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6:734–745. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674–6682. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–2522. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treviño-Villarreal JH, Cotanche DA, Sepúlveda R, Bortoni ME, Manneberg O, Udagawa T, et al. Host-derived pericytes and Sca-1+ cells predominate in the MART-1- stroma fraction of experimentally induced melanoma. J Histochem Cytochem. 2011;59:1060–1075. doi: 10.1369/0022155411428078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan M, Corselli M, Chen WC, Péault B. Perivascular cells for regenerative medicine. J Cell Mol Med. 2012;16:2851–2860. doi: 10.1111/j.1582-4934.2012.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungsøyr L, Støa KF. Spectrophotometric determination of hemoglobin oxygen saturation: the method of Drabkin & Schmidt as modified for its use in clinical routine analysis. Scand J Clin Lab Invest. 1954;6:270–276. doi: 10.3109/00365515409134863. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.