

Abstract
Oncolytic viruses are structurally and biologically diverse, spreading through tumors and killing them by various mechanisms and with different kinetics. Here, we created a hybrid vesicular stomatitis/measles virus (VSV/MV) that harnesses the safety of oncolytic MV, the speed of VSV, and the tumor killing mechanisms of both viruses. Oncolytic MV targets CD46 and kills by forcing infected cells to fuse with uninfected neighbors, but propagates slowly. VSV spreads rapidly, directly lysing tumor cells, but is neurotoxic and loses oncolytic potency when neuroattenuated by conventional approaches. The hybrid VSV/MV lacks neurotoxicity, replicates rapidly with VSV kinetics, and selectively targets CD46 on tumor cells. Its in vivo performance in a myeloma xenograft model was substantially superior to either MV or widely used recombinant oncolytic VSV-M51.
Introduction
Vesicular stomatitis virus (VSV), of the Rhabdoviriade family, has potent tumor activity against human tumor xenografts and syngeneic murine tumors.1,2,3,4,5,6,7 However, VSV is promiscuous and is neurotoxic when inoculated into research animals.8 To attenuate the virus for use in cancer therapy, two strategies both involving induction of the host antiviral interferon (IFN) response were used. One method involves mutating or deleting methionine 51 in the VSV matrix protein (VSV-M51) such that the mutant M protein can no longer block mRNA export from the nucleus, thereby permitting host-cell production of antiviral proteins, including type I IFN.9,10 The VSV-M51 virus triggers antiviral responses in infected normal tissues, including neurons, restricting viral spread and further tissue damage.11,12 Another VSV attenuation strategy involves engineering an additional transcription unit encoding IFN-β into the VSV genome such that VSV-IFN-β–infected cells will produce high levels of secreted IFN-β, protecting neighboring cells from virus infection.13 These viruses demonstrated potent antitumor activities, including complete responses of syngeneic tumors in immunocompetent hosts after one intravenous dose of virus.2
However, the premise of tumor cells, in contrast to normal cells, have defective IFN induction or response pathways and thus are highly susceptible to VSV oncolysis is not entirely correct. Increasingly, reports show that there are some cancer cell lines that are IFN defective and some that have retained fully functional IFN response pathways for a tumor specific tumor type.3,11,13,14,15,16 In these IFN sensitive tumors, IFN-inducing VSV has diminished antitumor activity and have more limited viral spread.1,15
The measles virus (MV) Edmonston strain, infect the cells through CD46 (Membrane cofactor protein, a transmembranal protein involved in the regulation of complement activation)17 in a receptor density dependent manner.18 Although all the nucleated cells express this receptor, CD46 is overexpressed on many types of cancer cells;17 High-CD46–expressing cancers include thyroid, breast, ovarian, endometrial, lung, colorectal, pancreatic, gastric, skin, cervical cancers, hepatoma, melanoma and hematological malignancies such as leukemia and lymphoma). Therefore, MV preferentially induces extensive cytopathic effects (CPEs) inCD46 high cancer cells and causes minimal damage in CD46-low normal cells. Oncolytic MVs, carcinoembryonic antigen–expressing MV (MV-CEA) and MV expressing the NIS reporter gene (MV-NIS), have been safely given to >50 patients in Phase I clinical trials against ovarian cancer, multiple myeloma, glioma, mesothelioma and squamous cell carcinoma.19 In ovarian cancer, carcinoembryonic antigen–expressing MV was used at doses ranging from 103 to 109 TCID50. No dose-limiting toxicity was found highlighting its safety profile but viral replication, as measured by virally encoded soluble carcinoembryonic antigen, was rather modest.20 As the safety profile and preferential infection for cancer cells by MV is dependent on the Hemagglutinin (H) and Fusion (F) glycoproteins, we hypothesized that by combining MV envelope proteins with the rapid VSV replication machinery, a safe but more potent oncolytic virus might be obtained.
Hence, as a new strategy to attenuate VSV infection without inducing IFN antiviral responses, increase VSV specificity for tumor cells and to increase the potency of MV, we have generated a “virus amalgam” comprising a VSV core with the MV entry and fusion machinery (VSV-FH). We previously showed that the infection of a replicative defective VSV lacking the glycoprotein gene (VSV-ΔG) can pseudotyped by transfection of producer cells with plasmids encoding MV-FH or MV-FH retargeted to different cellular receptors through single-chain antibodies.21 The infection of these pseudotyped VSV was specific for cells expressing the corresponding receptors. Contrary to the pseudotyped VSV-ΔG, the VSV-FH virus described in this present report, is fully replicative and encodes the MV-F and MV-H genes in the full-length infectious genome of VSV deleted for the G protein. This VSV-FH virus exhibits a unique CPE of syncytial formation (due to the expression of MV glycoproteins) and shows higher production of viral particles than MV, although conserving the specific tropism for cells expressing MV receptors. Moreover, VSV-FH is not neurotoxic in transgenic mice expressing measles-receptor CD46. Compared with MV or VSV-M51, the VSV-FH hybrid has superior activity against human multiple myeloma tumors.
Results
VSV-FH has highly attenuated neurotoxicity
VSV is neurovirulent in mice and causes paralysis, seizures, and loss in body condition.22 One of the main goals in amalgamating VSV with MV is to ablate the neurotoxicity associated with VSV-G interaction with neuronal cells. The G glycoprotein (1.6 kb) at position 4 of the full-length infectious cDNA clone of VSV was removed and replaced by MV-Fusion (F, 1.8 kb) and MV-Hemagglutinin (H, 2 kb) at positions 4 and 5, respectively (Figure 1a). As shown in the immunoblots, this new replication-competent VSV-FH expresses measles F and H glycoproteins as well as VSV N/P and M proteins (Figure 1b). Visualization of VSV-FH by electron microscopy (Figure 1c) reveals that VSV-FH presents a bullet shape, similar to parental VSV. There appears to be F and H glycoprotein complexes studded on the surface of the bullet-shaped VSV-FH virions but further work would be required to more clearly define these structures.
Figure 1.

Physical and biochemical characterization of VSV-FH hybrid virus. (a) Schematic representation of VSV and VSV-FH genomes. VSV intergenic regions and restriction sites for MV-F and MV-H of VSV-FH are shown below the representation of VSV-FH genome. (b) Immunoblots of virions using the indicated antibodies. (c) Transmission electron microscopy of purified virions. Arrows indicate the magnified area (shown below). Bar = 100 nm.
To test if this hybrid virus is less neurotoxic than VSV, CD46 transgenic mice, which express human CD46 (MV-receptor) with the same tissue specificity as in humans,23 were used. The brains of these mice express CD4624 and potentially can be infected by VSV-FH. Animals were given high doses of 107 TCID50 VSV or VSV-FH intravenously. All mice given VSV (n = 6) succumbed to neurotoxicity by day 6 and the survival curve was significantly different from VSV-FH (P = 0.0003, Figure 2a). These mice lost weight, had ungroom scruffy coats and also showed clinical signs of neurotoxicity (tremors, head tilt, limb paralysis, or circling), and were euthanized. The presence of VSV in the brain was confirmed by immunofluorescence using polyclonal antibodies against VSV envelope proteins (Figure 2b). In contrast, mice given VSV-FH (n = 7) did not exhibit weight loss and continued to gain weight similar to the saline control mice (n = 4) (Figure 2c). In contrast, both VSV and VSV-FH viruses were neurotoxic when given intravenously to CD46 transgenic mice lacking a functional type I IFN receptor (Ifnartm-CD46Gemice).25 Hence, host innate defense is important in protecting the animals from VSV neurovirulence (Figure 2d).
Figure 2.
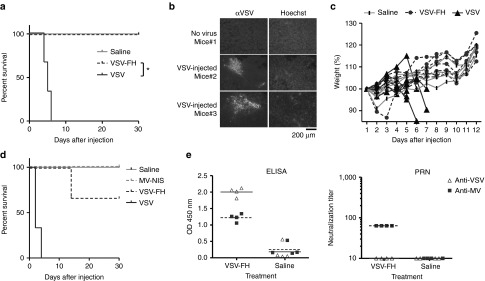
Neurovirulence study of viruses in CD46 transgenic mice. CD46+ transgenic mice were given 107 TCID50 of VSV-FH (n = 7) or VSV (n = 6). Mice were euthanized when neurotoxic symptoms were observed. (a) Survival curves of treated mice. *Statistically significant difference (Logrank test, P = 0.0003). (b) Presence of VSV in treated mice. Brain sections from VSV-treated mice were analyzed by immunohistochemistry. VSV proteins were detected using a polyclonal antibody directed to VSV structural proteins and an Alexa-conjugated secondary antibody. (c) Percent body weight change from baseline at the start of experiment. Mice were weighed at the indicated days after treatment, starting at the day of treatment. (d) Survival curves of Ifnartm-CD46Ge mice treated with VSV-FH. CD46 transgenic mice lacking functional α/β IFN receptor were given 107 TCID50 of VSV-FH or MV-NIS (n = 3) or 106 TCID50 VSV (n = 3). Mice were euthanized when neurotoxic symptoms were observed.(e) VSV- or MV-specific antibodies in serum of CD46+ transgenic mice at day 30 after virus. Titers were measured by MV or VSV specific ELISA assays and by plaque-reduction neutralization (PRN) assay. MV, measles virus; VSV, vesicular stomatitis virus.
At the end of the study (30 days), CD46 transgenic mice were euthanized and VSV- or MV-specific antibodies were determined by enzyme-linked immunoassay (ELISA) and by plaque-reduction neutralization assay on Vero cells (Figure 2e). Mice injected with VSV-FH were seropositive against both MV and VSV proteins. However, as shown in the plaque-reduction neutralization (amount of sera to achieve a complete neutralization of the CPEs at 300 TCID50 units of the indicated virus) assay (Figure 2d) these antibodies were able to neutralize only MV but not VSV.
VSV-FH is more potent than MV in vivo
MV-NIS is being evaluated in a phase I clinical trial in patients with relapsed or recurrent myeloma after intravenous delivery.26,27 Conversely, VSV-M51 expressing NIS (VSV-M51-NIS) has been previously shown to be active against IFN nonresponsive 5TGM1 murine model of myeloma.28 To test the efficacy of VSV-FH in a clinically relevant model, SCID mice bearing disseminated KAS 6/1 human multiple myeloma were treated with three doses of VSV-FH, VSV-M51-NIS, or MV-NIS at 107 TCID50/dose (Figure 3a). Repeat dosing of VSV-FH or MV-NIS delayed myeloma disease progression and prolonged the survival of mice compared with the control group (P = 0.0016 for MV-NIS, P < 0.0001 for VSV-FH). Importantly, VSV-M51-NIS treatment, did not result in an increase in the survival of mice compared with control group (P = 0.6680).
Figure 3.

Comparative study of antitumor activities against human multiple myeloma after intravenous delivery of the viruses. (a) Survival curves of mice with systemic multiple myeloma. A disseminated model of multiple myeloma was established in mice as described in materials and methods. Mice were treated with three doses of 107 TCID50 of the indicated viruses. *Statistically significant difference between VSV-FH and saline (Logrank test, P < 0.0001) or VSV-M51-NIS (P = 0.0124). Arrows indicate virus injection days (post-KAS 6/1 implantation). (b) Mice bearing subcutaneous plasmacytomas of human multiple myeloma were treated with one dose of the following viruses: Saline (n = 8), MV (n = 6), VSV-FH (n = 7), or VSV-M51-NIS (n = 8). Plasmacytoma volumes were measured three times per week and plotted on a logarithmic scale. Error bars represent SEM. (c) Survival curves of mice with subcutaneous multiple myeloma. *Significant difference between VSV-FH and saline (P = 0.001), MV-NIS (P = 0.0354), or VSV-M51-NIS (P = 0.0011). MV, measles virus; VSV, vesicular stomatitis virus.
To dissect the relative activity of the viruses, SCID mice with subcutaneous KAS 6/1 plasmacytomas (tumor diameter 0.4–0.5 cm) were given one single intravenous dose of 107 TCID50 of either VSV-M51-NIS or MV-NIS, or tenfold lower dose of VSV-FH (106 TCID50). VSV-FH was not only able to control tumor growth more rapidly but also significantly debulked the tumor very early after treatment (Figure 3b). The Kaplan–Meier survival curve of mice treated with VSV-FH was significantly prolonged compared with saline control mice (P = 0.001) or MV-NIS (P = 0.035) or VSV-M51-NIS (P = 0.001) (Figure 3c).
VSV-FH is able to kill CD138+ cells derived from patients with myeloma
We next evaluated the antitumor potency of VSV-FH further in cells harvested from the bone marrow aspirates of patients with myeloma (Figure 4a). MV, VSV-FH, and VSV-M51 showed good oncoselectivity for CD138+ malignant plasma cells (myeloma) cells and did not infect CD138− cells from normal bone marrow. Importantly, the VSV-FH virus does not induce the production of type I IFN-α or -β in the human myeloma cells. In contrast, VSV-M51 induced robust IFN-α or -β production in the human KAS6/1 cells (Figure 4b) which could potentially inhibit viral spread in vivo and limit its oncolytic activity (Figure 3).
Figure 4.
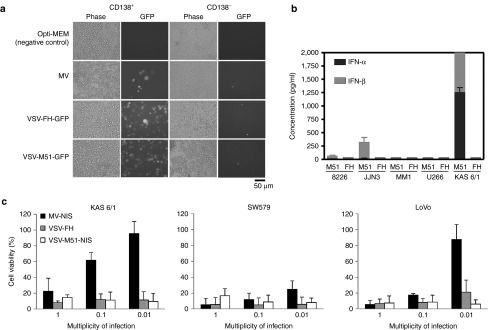
VSV-FH infection of human cells. (a) VSV-FH specificity on CD138+ plasma cells (myeloma) and CD138− nonplasma cells derived from the bone marrow of patients with multiple myeloma. 5 × 106 cells were infected with the indicated viruses at MOI = 0.1. Pictures were taken at 24 hours after infection. Representative example from two patients is shown. (b) Induction of IFN-α and -β in myeloma cells. 5 × 105 cells were infected with either VSV-FH or VSV-M51-NIS. Secreted IFN-α or -β (at 48 hpi) for the indicated cell lines was quantified and plotted (8226 = RPMI 8226; M51 = VSV-M51-NIS; FH = VSV-FH) (c) Viability of infected cancer cell lines. Human cancer cells were infected with the indicated viruses at three different multiplicity of infections. Three days after infection, cell viability was measured by MTS assay. Bars represent average of three experiments (mean ± SD). VSV, vesicular stomatitis virus.
As we are also interested to test VSV-FH against solid tumors, we included SW579 (thyroid cancer) and LoVo (colorectal cancer) cell lines in the cytotoxicity assays. VSV-FH was found to have varying degrees of potency (Figure 4c). MV-NIS, measles expressing the human sodium iodide symporter (NIS), previously shown to be effective against multiple myeloma, was used as the therapeutic virus in this comparative study.26,27 At an MOI of 1.0, both viruses were potent in all cell lines tested. At a low MOI of 0.01, MV-NIS was generally not as potent as VSV-FH. In vitro, VSV-M51 was able to kill all the tested cell lines very efficiently.
Superior activity of VSV-FH is due to rapid fusogenic viral spread between cells
To understand why VSV-FH is more active than MV in the plasmacytoma model, VSV-FH virus was compared with MV in various infection assays. First, to assess the tropism of VSV-FH, Chinese hamster ovary (CHO) cells expressing MV receptors, CD46, SLAM, or Nectin-4,29 were infected by the viruses (Figure 5a). Fusion triggering of VSV-G glycoprotein is activated by low pH but measles fusion is pH independent and is initiated upon binding of H to one of the three MV receptors (CD46, SLAM, or Nectin-4).29,30,31,32,33 MV and VSV-FH share a similar tropism (Figure 5a). They are nonpermissive on CHO cells that lack MV receptors, but are infectious on CD46, SLAM, or Nectin-4 positive CHO cell lines. In contrast, VSV was able to infect all of the four cell lines, including parental CHO. These data confirmed that the VSV-FH tropism is now dictated exclusively by the measles H and F proteins incorporated on the viral coat.
Figure 5.

Viral replication and cytopathic effects of VSV-FH and the parental viruses. (a) Specificity of receptor usage by VSV-FH. A panel of CHO cells (2.5 × 105 cells), expressing specific MV receptors, was infected with MV (MOI = 0.1), VSV (MOI = 0.01), or VSV-FH (MOI = 0.01) and stained with crystal violet at 24 hours (VSV and VSV-FH) or 48 hours (MV). (b) Viral progeny production over time in Vero cells. Cells were infected with a MOI of 0.1 (MV) or 0.00001 (VSV or VSV-FH), cells and supernatant were harvested at the indicated times and the amount of infectious particles was determined by TCID50 titrations (mean ± SD, n = 3). (c) Cytopathic effect at 72 hours in cells previously infected with MV, VSV-FH, or VSV at the indicated multiplicities of infection. Cells were infected at different MOIs with the indicated viruses. At 3 days after infection, cells were fixed and stained with crystal violet. MOI, multiplicity of infection; MV, measles virus; VSV, vesicular stomatitis virus.
Next, the production of infectious particles was determined in Vero producer cells. These cells were infected with MV, VSV-FH, and VSV and the amount of infectious particles in the cells or released into the supernatant was quantified by TCID50 titration (Figure 5b). MV is a cell-associated virus and most of the MV progeny is in the infected cells, with little released into the supernatant. In contrast, VSV releases most of its virions into the supernatant. VSV-FH has an intermediate phenotype; at 24 hours after infection, most of the virions were found in the cells, but at later time points, most virions were released into the supernatant. Of note here is that Vero cells were infected by MV at MOI of 0.1 whereas MOI of 0.00001 was used for VSV-FH and VSV. A lower MOI was used for VSV-FH and VSV as otherwise their rapid replication would have resulted in complete destruction of the cell monolayer before 72 hours.
Cell-infection assays show that VSV-FH induces rapid intercellular fusion in the monolayer. This CPE of syncytial formation is characteristic of MV infection, but not of VSV which instead causes cells to round up and lyse. Intercellular fusion of VSV-FH infected cells can be detected at 12 hours after infection (MOI 0.001, data not shown) and the numbers of infectious foci and syncytia numbers continued to grow rapidly. To compare the CPE of the viruses, Vero cells were infected with VSV-FH, MV, and VSV with MOIs of 1.0 to 0.00001. Cells were stained with 2% crystal violet 72 hours later. Total destruction of the monolayer occurred at MOI of 0.1 for MV. In contrast, the VSV-FH and VSV viruses caused total destruction of the cells at 4-log lower MOI of 0.00001(Figure 5c). Together, these data show a more potent cell killing activity of VSV-FH compared with parental MV.
Discussion
In this study, we amalgamated VSV and MV to generate VSV-FH and shown its promise as a systemic therapy for multiple myeloma. Incorporation of MV envelope glycoproteins onto a VSV core yielded a stable fully replicative virus without the neurovirulence properties associated with VSV. VSV-FH, unlike the parental VSV, did not cause adverse clinical signs or weight loss when given intravenously to MV (CD46) receptor positive mice. The virus amalgam is now fusogenic, and has acquired the tropism of MV which includes a preference for tumors expressing high levels of CD46, a complement regulatory protein that is overexpressed in different types of cancers whereas expressed at relatively low levels in normal cells.17 CD138+ malignant plasma cells express 7–10 times higher CD46 on their cell surface compared with normal bone marrow stromal cells.34 Indeed, we showed here that VSV-FH is able to efficiently infect primary CD138+ myeloma cells and not CD138− cells derived from normal bone marrow. Compared with MV, VSV-FH replicates and spreads faster with large syncytia. The faster replication of VSV-FH is evident in infection assays on Vero cells as well as in human cancer cell lines.
A single dose of VSV-FH was able to induce rapid regression of subcutaneous KAS 6/1 plasmacytomas very early after treatment, an effect that was not observed in MV-NIS treated tumors despite mice given a tenfold higher dose of MV-NIS.
It is worth noting that in the KAS6/1 human myeloma (systemic disease or plasmacytoma) models, VSV-M51 did not demonstrate antitumor efficacy. This is likely due to the antiviral mechanisms of these cancer cells. VSV-M51, due to the mutation in the matrix protein, is designed to induce IFN production in cells with functional IFN pathways. Indeed, we determined that KAS 6/1 cells were able to produce IFN-α/-β as a response against VSV-M51 infection (Figure 4b). Data here underscore the importance of developing methods to optimally identify the patient population suitable for virotherapy as cancer cell lines that retain functional IFN responsiveness and sensitivity should not be treated by viruses that induce IFN production.
We were able to successfully rescue the fully replicative VSV-FH with titers around 107 TCID50/ml. Interestingly, in a previous work done by Schnell et al.,35 although trying to engineer VSV to express either MV-H or MV-F, it was observed that a VSV expressing MV-F had a limited replication, and that MV-F gene was lost after a few passages. It was thought that the F gene was not compatible with VSV and somehow, toxic to the producer cells or poor virus budding. Here, in contrast to the previous study, we have inserted both H and F into the VSV genome, thus significantly extending its size by 2.1 kb. Interestingly, the average size of the VSV-FH virions was 204 × 80 nm (Figure 1c), compared with the reported 180 × 70 nm of parental VSV.36 As VSV-FH infection depends on the expression of both MV-H and MV-F, it is possible that there is a selective pressure for the presence of both of these genes, preventing the accumulation of negative mutations in these genes. Interestingly, a series of small structures are present on the surface of VSV-FH (magnified area of Figure 1c), these structures are similar to those on MV virions but are absent in VSV, suggesting that they might be the incorporated MV-F and MV-H glycoproteins on the virions of VSV-FH. We are currently collaborating with other laboratories to characterize the structures of MV glycoproteins on VSV-FH.
We have passaged VSV-FH in Vero cells (using a MOI of 0.00001 and collecting the supernatant after 72 hpi) for >10 continuous passages and have not seen nonfusogenic viruses or loss of viability of the viral stocks. Moreover, the specificity of this virus is conserved after these passages and it is still able to infect CHO cells expressing CD46, SLAM, or Nectin-4, but not MV-receptor negative CHO cells. It is noteworthy that maximum titer VSV-FH is around 2–3 logs less than parental VSV, perhaps due to the fact that VSV-FH genome size is ~2,000 nucleotides longer than VSV. Also, the incorporation of MV glycoproteins into VSV might be less efficient than VSV-G incorporation. Indeed, as shown in previous reports, VSV pseudotyped with MV glycoproteins have titers around ten times less than VSV pseudotyped with VSV-G.21 One advantage of VSV-FH that was not studied in this report is that its infection can be retargeted by engineering the H protein to bear a scFV or ligand specific for different cellular receptors. This strategy has been used to retarget MV and VSV pseudotypes to cells expressing PSMA, folate receptor, and epidermal growth factor receptor.21,37,38,39 Thus, VSV-FH oncolytic activity can potentially be retargeted via these cancer associated receptors for specific cancer types.
Another method to restrict VSV tropism was developed by Bergman et al.,40 in which the VSV-G was replaced with a modified gene coding for Sindbis virus glycoprotein; this Sindbis glycoprotein contained a single-chain antibody directed to Her2/neu. The virus was found to infect preferentially cells expressing Her2/neu. Our approach to restrict VSV tropism is based also in the expression of foreign glycoproteins and the resulting viruses grow at similar titers. However, one of the advantages of using MV proteins to restrict VSV infection is that the expression of MV-F and MV-H results in the formation of syncytia, helping therefore to the intracellular spreading of the virus.
One of the main drawbacks of using MV as an oncolytic agent is the presence of neutralizing antibodies found in measles vaccinated individuals. As VSV-FH glycoproteins are the same as MV, it is likely that antibodies raised against MV are able to neutralize VSV-FH. Although patients with myeloma have very low levels of MV-specific antibodies,41 the application of VSV-FH for systemic therapy in other cancer types can be problematic. Fortunately, there are multiple technologies that can be used to bypass this issue, such as PEGylation,42 the use of cell carriers for delivering virus to the tumor site although evading the immune response,43 or the use of immunosuppressant drugs, like cyclophosphamide (which is also used as an anticancer drug).27,44,45 These strategies had been used to increase the antitumor activities of MV and VSV in tumor models.
In summary, VSV-FH is a new promising oncolytic agent that combines VSV fast replication machinery with the MV safety profile and flexible retargeting platform. This results in a faster replication, bigger syncytia, release of more infectious particles and higher titers which has been a major limiting factor in manufacture of clinical grade MV. Future experiments are necessary to assess multiple parameters of VSV-FH as an oncolytic agent such as the minimum effective dose, highest tolerated dose against multiple myeloma, and to explore its use against other malignancies.
Materials and Methods
Cell culture. All the cells were cultured at 37 °C in 5% CO2 atmosphere. Vero (ATCC No.CCL-81), baby hamster kidney (ATCC No.CRL-12071), SW579 (squamous cell carcinoma from the thyroid; ATCC No.HTB-107), and LoVo (colorectal adenocarcinoma; ATCC No. CCL-229) cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA). Human Multiple Myeloma cell line KAS 6/1 was a gift from Diane Jelinek (Mayo Clinic, Rochester, MN), KAS 6/1 F/G-Luc cells were generated by transduction using lentiviral vectors expressing Gaussia and Firefly Luciferase proteins as previously described;46 RPMI 8226 were a gift from John Lust (Mayo Clinic); MM1 and JJN3 were a gift from Rafael Fonseca (Mayo Clinic). U266 cells were purchased from ATCC (No. TIB-196). CHO cells and CHO cells expressing CD46 (CHO-CD46) or SLAM (CHO-SLAM) have been described previously,47 CHO cells expressing Nectin-4 (CHO-Nectin-4) were a gift from Christopher D. Richardson (Dalhousie University, Halifax, Nova Scotia, Canada).29
Cloning and rescue of VSV-FH. MV-F was subcloned into Zero Blunt Topo vector (Invitrogen, Carlsbad, CA), using the plasmid pCGF as template for PCR, primers are shown in Supplementary Table S1. Then MV-F was digested with NotI and cloned into a plasmid containing VSV-mIFN full-genome sequence (a gift from Glen Barber, University of Miami School of Medicine, Miami, FL. Plasmid is based on pVSV-XN2, Indiana serotype,48 and further modified by Obuchi et al.13 and Naik et al.2). MV-F gene contains the untranslated regions (UTR) of MV-F at the 5′ and 3′ ends, and it was cloned immediately after the VSV Intergenic region corresponding to the VSV-G gene. To remove VSV-G from this construct, the plasmid was digested with NotI and religated. To remove mIFN gene, plasmid was digested with NheI and XhoI, the ends were blunted by using the Quick Blunting Kit (New England Biolabs, Ipswich, MA) and the plasmid was religated; the resulting plasmid is VSVDG-F. MV-H was subcloned into Zero Blunt Topo Vector by PCR amplification (primers shown in Supplementary Table S1, FWD primer contains VSV Intergenic region) of the MV-H gene present in the pCGH plasmid. This gene, containing the 5′ and 3′ UTR of MV-H, was then excised using SphI and cloned into a SphI site present at the end of the MV-F sequence of pVSVDG-F. GFP gene was cloned into the XhoI site immediately after the VSV Intergenic region present at the end of MV-H. Fully replication-competent VSV-FH and VSV-FH-GFP were obtained using plasmids pVSVDG-FH or pVSVDG-FH-GFPwith pMD.G following the VSV rescue system previously described.48
To produce large amounts of VSV-FH, 2 × 107 Vero cells in 150 mm2 dishes were infected with VSV-FH at a MOI of 0.00001 in 13 ml of Opti-MEM (Invitrogen). Supernatant was harvested at 3 days after infection, and cell debris were spun down at 3,000 rpm. To concentrate the virus, supernatants were concentrated using Amicon Ultra-15 Centrifugal Filters (Millipore, Billerica, MA).
Western blots. 1.5 × 105 TCID50 particles were loaded per lane and fractionated PAGE in 10% Tris–HCl Criterion precast gel (Bio-Rad, Hercules, CA) and transferred to a polyvinylidene difluoride membrane (Bio-Rad). Membranes were blocked (5% nonfat milk in Tris-buffered saline–Tween) and incubated with primary antibodies (monoclonal mouse αMV-N (Abcam, Cambridge, MA), polyclonal rabbit αMV-H and αMV-F, and polyclonal αVSV structural proteins).49,50 After five washes with Tris-buffered saline–Tween, membranes were incubated with peroxidase-conjugated secondary antibody, and washed five times with Tris-buffered saline–Tween. Signal was then developed using Pierce ECL western blotting substrate kit (Thermo Scientific, Waltham, MA).
Infectious viral particles production. 1 × 106 Vero cells per well of a six-well plate were infected with MV (MOI = 0.1), or VSV-FH (MOI = 0.00001) for 3 hours in 1 ml of Opti-MEM. Then inoculum was removed and replaced with 2 ml of DMEM 5% FBS (v/v). At the indicated times after infection, supernatant was recovered, cell debris were removed by centrifugation (3,000 rpm for 5 minutes) and stored at −80 °C. Cells were washed once with opti-MEM, resuspended in 2 ml of media, scraped from the plate and stored at −80 °C. Frozen cells and supernatant were freeze-thawed once and the amount of infectious particles per ml was titered in Vero cells as previously described.49
VSV-FH specificity in CHO cells. 5 × 105 CHO, CHO-CD46, CHO-SLAM, CHO-Nectin-4, or Vero cells were plated per well of a six-well plate. Next day cells were infected with MV (MOI = 0.1), VSV-FH (MOI = 0.001), or VSV (MOI = 0.001). After 3 hours, inoculum was removed and replaced with fresh growing media. Cells were incubated for 24 hours (for VSV-FH and VSV) or 48 hours (for MV) at 37 °C. After this incubation time, cells were fixed with 5% glutaraldehyde and stained with 0.1% crystal violet.
Cell viability assays. SW579 and LoVo cells (14,000 cells per well) were seeded in a 96-well plate and infected next day with the indicated viruses at MOI of 1, 0.1, and 0.01 diluted in 50 µl of opti-MEM. KAS 6/1 (5 × 105 cells per well) were infected for 3 hours with the indicated viruses at MOIs of 1, 0.1, and 0.01, then media was removed and replaced with 100 µl of growing media. At 3 days after infection, cell viability was measured using the CellTiter 96 AQueous Assay (Promega, Fitchburg, WI), following the manufacturer recommendations.
Induction of IFN-α and -β in multiple myeloma cells by VSV-M51 or VSV-FH. 5 × 105 cells were infected with either VSV-FH or VSV-M51-NIS at a MOI of 1. Forty-eight hours after infection, the supernatant was harvested. Secreted IFN-α or -β was quantified using Human IFN ELISA kit (R&D Systems, Minneapolis, MN) following manufacturer instructions.
In vivo experiments. All the experiments were approved by Institutional Animal Care and Use Committee (IACUC).
VSV-FH safety. C57bl/6 IFN/CD46+ 4- to 5-week-old mice23 were injected with 1 × 107 TCID50 units of the VSV-FH (n = 7), VSV-GFP (n = 6), or 100 µl of opti-MEM (n = 4). Body weight was measured every day for the first 12 days after injection. Mice were euthanized when neurotoxicity symptoms were observed (e.g., limb paralysis, tremors, lethargic behavior, low-weight, etc.). At day 30 after injection, blood was extracted from surviving mice and assayed for the presence of αMV and αVSV antibodies by ELISA and the presence of neutralizing antibodies against MV or VSV by plaque-reduction neutralization as previously described.21 Ifnartm-CD46Ge mice25 4–5 weeks old were injected with 1 × 107 TCID50 units of VSV-FH (n = 3) or MV-NIS (n = 3), 1 × 106 TCID50 units of VSV-GFP (n = 3), or 100 µl of opti-MEM (n = 3). Mice were euthanized when neurotoxicity symptoms were observed (e.g., limb paralysis, tremors, lethargic behavior, low-weight, etc.).
VSV-FH efficacy against subcutaneous plasmacytomas. ICR SCID mice of 4–6 weeks old were purchased from Taconic (Germantown, NY). One day before implantation of xenografts, mice were whole body irradiated (2 Gy). Next day, 2 × 106 KAS 6/1 cells were implanted subcutaneously in the right flank of the mice. When tumors reached a volume of 50 mm3, 1 × 107 TCID50 units of MV-NIS (n = 6), 1 × 106 TCID50 units of VSV-FH (n = 7), 1 × 107 TCID50 units of VSV-M51-NIS (n = 8), or 100 µl of saline solution (n = 8) were injected through tail vein injection. Tumor volume was measured three times per week, and mice euthanized when tumor reached a volume equal or larger than 2,000 mm3 or mice presented paralysis, head drop, lethargy, or weight loss >20%.
VSV-FH efficacy against KAS 6/1 disseminated model. ICR SCID mice (Taconic) of 4–6 weeks old were injected with 1 × 107 lentivirus-transduced KAS 6/1 cells expressing Firefly and Gaussia Luciferase.46 Tumor burden was monitored by quantifying the presence of Gaussia Luciferase in blood using a Top Count NXT Scintillation and Luminescence Counter (Perkin Elmer, Waltham, MA) in a black 96-well plate at wavelength of 470 nm and the Biolux Gaussia luciferase assay kit (New England Biolabs) following manufacturer instructions. Mice were treated when most of the animals presented relative lights units around 30,000/5 µl of blood. Groups were intravenously injected with three doses of 1 × 106 TCID50 units in 100 µl of Opti-MEM of the indicated viruses or vehicle only (n = 10 per group) at days 31, 38, and 41 after implantation. Mice were monitored daily and euthanized when presented paralysis, head drop, lethargy, or weight loss >20%.
SUPPLEMENTARY MATERIAL Table S1. Primers used to flank F and H for PCR cloning of the genes into VSV-FH genome.
Acknowledgments
We thank Suzanne Greiner for expert technical assistance with the animal experiments and Christopher D Richardson (Dalhousie University, Halifax, Nova Scotia, Canada) for the CHO-Nectin-4 cells. This work is funded by grants from the NIH/NCI (CA129193, CA129966). The authors declared no conflict of interest.
Supplementary Material
References
- Blackham AU, Northrup SA, Willingham M, D'Agostino RB, Jr, Lyles DS, Stewart JH., 4th Variation in susceptibility of human malignant melanomas to oncolytic vesicular stomatitis virus. Surgery. 2013;153:333–343. doi: 10.1016/j.surg.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Nace R, Barber GN, Russell SJ. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-ß. Cancer Gene Ther. 2012;19:443–450. doi: 10.1038/cgt.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AM, Besmer DM, Moerdyk-Schauwecker M, Moestl N, Ornelles DA, Mukherjee P, et al. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J Virol. 2012;86:3073–3087. doi: 10.1128/JVI.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiber JF, Barber GN. Vesicular stomatitis virus expressing tumor suppressor p53 is a highly attenuated, potent oncolytic agent. J Virol. 2011;85:10440–10450. doi: 10.1128/JVI.05408-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Boeuf F, Niknejad N, Wang J, Auer R, Weberpals JI, Bell JC, et al. Sensitivity of cervical carcinoma cells to vesicular stomatitis virus-induced oncolysis: potential role of human papilloma virus infection. Int J Cancer. 2012;131:E204–E215. doi: 10.1002/ijc.27404. [DOI] [PubMed] [Google Scholar]
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 2004;17:516–527. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- Bi Z, Barna M, Komatsu T, Reiss CS. Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J Virol. 1995;69:6466–6472. doi: 10.1128/jvi.69.10.6466-6472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BL, Rhodes RB, McKenzie M, Lyles DS. The role of vesicular stomatitis virus matrix protein in inhibition of host-directed gene expression is genetically separable from its function in virus assembly. J Virol. 1993;67:4814–4821. doi: 10.1128/jvi.67.8.4814-4821.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol. 2003;77:4646–4657. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. 2004;330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Obuchi M, Fernandez M, Barber GN. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol. 2003;77:8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou J, Soriano R, Hayward SW, Cunha GR, Williams PM, Gao WQ. Expression profiling of a human cell line model of prostatic cancer reveals a direct involvement of interferon signaling in prostate tumor progression. Proc Natl Acad Sci USA. 2002;99:2830–2835. doi: 10.1073/pnas.052705299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloura V, Wang LC, Fridlender ZG, Sun J, Cheng G, Kapoor V, et al. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum Gene Ther. 2010;21:51–64. doi: 10.1089/hum.2009.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matin SF, Rackley RR, Sadhukhan PC, Kim MS, Novick AC, Bandyopadhyay SK. Impaired alpha-interferon signaling in transitional cell carcinoma: lack of p48 expression in 5637 cells. Cancer Res. 2001;61:2261–2266. [PubMed] [Google Scholar]
- Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. 2003;40:109–123. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64:4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- Galanis E. Therapeutic potential of oncolytic measles virus: promises and challenges. Clin Pharmacol Ther. 2010;88:620–625. doi: 10.1038/clpt.2010.211. [DOI] [PubMed] [Google Scholar]
- Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala-Breton C, Barber GN, Russell SJ, Peng KW. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum Gene Ther. 2012;23:484–491. doi: 10.1089/hum.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin AB, Olitsky PK. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus: IV. Variations in neuroinvasiveness in difierent species. J Exp Med. 1938;67:229–249. doi: 10.1084/jem.67.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper C, Leung M, Stephensen CB, Pinkert CA, Liszewski MK, Cattaneo R, et al. Membrane cofactor protein (MCP; CD46) expression in transgenic mice. Clin Exp Immunol. 2001;124:180–189. doi: 10.1046/j.1365-2249.2001.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz CJ, Gerlier D, Hu A, Cathomen T, Liszewski MK, Atkinson JP, et al. Selective expression of a subset of measles virus receptor-competent CD46 isoforms in human brain. Virology. 1996;217:349–355. doi: 10.1006/viro.1996.0122. [DOI] [PubMed] [Google Scholar]
- Mrkic B, Pavlovic J, Rülicke T, Volpe P, Buchholz CJ, Hourcade D, et al. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72:7420–7427. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingli D, Peng KW, Harvey ME, Greipp PR, O'Connor MK, Cattaneo R, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103:1641–1646. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, et al. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther. 2007;82:700–710. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Carlson SK, Classic KL, Greiner S, Naik S, Power AT, et al. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood. 2007;110:2342–2350. doi: 10.1182/blood-2007-01-065573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, et al. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011;7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, et al. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlebach MD, Mateo M, Sinn PL, Prüfer S, Uhlig KM, Leonard VH, et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature. 2011;480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuo H, Ono N, Tanaka K, Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- Ong HT, Timm MM, Greipp PR, Witzig TE, Dispenzieri A, Russell SJ, et al. Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Exp Hematol. 2006;34:713–720. doi: 10.1016/j.exphem.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Schnell MJ, Buonocore L, Kretzschmar E, Johnson E, Rose JK. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc Natl Acad Sci USA. 1996;93:11359–11365. doi: 10.1073/pnas.93.21.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SP. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009;5:e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Hasegawa K, Russell SJ, Sadelain M, Peng KW. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate. 2009;69:1128–1141. doi: 10.1002/pros.20962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Nakamura T, Harvey M, Ikeda Y, Oberg A, Figini M, et al. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res. 2006;12 20 Pt 1:6170–6178. doi: 10.1158/1078-0432.CCR-06-0992. [DOI] [PubMed] [Google Scholar]
- Paraskevakou G, Allen C, Nakamura T, Zollman P, James CD, Peng KW, et al. Epidermal growth factor receptor (EGFR)-retargeted measles virus strains effectively target EGFR- or EGFRvIII expressing gliomas. Mol Ther. 2007;15:677–686. doi: 10.1038/sj.mt.6300105. [DOI] [PubMed] [Google Scholar]
- Bergman I, Whitaker-Dowling P, Gao Y, Griffin JA. Preferential targeting of vesicular stomatitis virus to breast cancer cells. Virology. 2004;330:24–33. doi: 10.1016/j.virol.2004.06.048. [DOI] [PubMed] [Google Scholar]
- Dingli D, Peng KW, Harvey ME, Vongpunsawad S, Bergert ER, Kyle RA, et al. Interaction of measles virus vectors with Auger electron emitting radioisotopes. Biochem Biophys Res Commun. 2005;337:22–29. doi: 10.1016/j.bbrc.2005.08.261. [DOI] [PubMed] [Google Scholar]
- Tesfay MZ, Kirk AC, Hadac EM, Griesmann GE, Federspiel MJ, Barber GN, et al. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J Virol. 2013;87:3752–3759. doi: 10.1128/JVI.02832-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmon C, Harrington K, Kottke T, Prestwich R, Melcher A, Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009;17:1667–1676. doi: 10.1038/mt.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci USA. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browder T, Butterfield CE, Kräling BM, Shi B, Marshall B, O'Reilly MS, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]
- Liu C, Russell SJ, Peng KW. Systemic therapy of disseminated myeloma in passively immunized mice using measles virus-infected cell carriers. Mol Ther. 2010;18:1155–1164. doi: 10.1038/mt.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Peng KW, Vongpunsawad S, Harvey M, Mizuguchi H, Hayakawa T, et al. Antibody-targeted cell fusion. Nat Biotechnol. 2004;22:331–336. doi: 10.1038/nbt942. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Stillman EA, Whitt MA, Rose JK. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadac EM, Peng KW, Nakamura T, Russell SJ. Reengineering paramyxovirus tropism. Virology. 2004;329:217–225. doi: 10.1016/j.virol.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Jenks N, Myers R, Greiner SM, Thompson J, Mader EK, Greenslade A, et al. Safety studies on intrahepatic or intratumoral injection of oncolytic vesicular stomatitis virus expressing interferon-beta in rodents and nonhuman primates. Hum Gene Ther. 2010;21:451–462. doi: 10.1089/hum.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.