

Abstract
Exogenous high-mobility group box 1 protein (HMGB1) administration to the mouse heart, during acute myocardial infarction (MI), results in cardiac regeneration via resident c-kit+ cell (CPC) activation. Aim of the present study was to identify the molecular pathways involved in HMGB1-induced heart repair. Gene expression profiling was performed to identify differentially expressed genes in the infarcted and bordering regions of untreated and HMGB1-treated mouse hearts, 3 days after MI. Functional categorization of the transcripts, accomplished using Ingenuity Pathway Analysis software (IPA), revealed that genes involved in tissue regeneration, that is, cardiogenesis, vasculogenesis and angiogenesis, were present both in the infarcted area and in the peri-infarct zone; HMGB1 treatment further increased the expression of these genes. IPA revealed the involvement of Notch signaling pathways in HMGB1-treated hearts. Importantly, HMGB1 determined a 35 and 58% increase in cardiomyocytes and CPCs expressing Notch intracellular cytoplasmic domain, respectively. Further, Notch inhibition by systemic treatment with the γ-secretase inhibitor DAPT, which blocked the proteolytic activation of Notch receptors, reduced the number of CPCs, their proliferative fraction, and cardiomyogenic differentiation in HMGB1-treated infarcted hearts. The present study gives insight into the molecular processes involved in HMGB1-mediated cardiac regeneration and indicates Notch signaling as a key player.
Introduction
Over the last decade, the development of regenerative therapeutic strategies has opened new perspectives in the field of cardiac regeneration.1
Specifically, in vivo delivery of cytokines or growth factors has been recognized as a promising and effective tool to enhance the regenerative potential of the heart. This approach overcomes some major problems associated with cell transplantation, such as cell engraftment and survival, as well as host immune rejection in the case of allogeneic cell transplantation.2
Several growth factors have been investigated as candidates for cardiac therapies.2 These factors fall into different functional categories, that is, those promoting angiogenesis, inducing growth and differentiation of stem cells, inhibiting apoptosis, inducing the migration of progenitor cells, and stimulating myocyte proliferation.
The potential benefit of growth factor therapy is still under investigation and various preclinical studies based on growth factor infusion after myocardial infarction (MI) have been initiated. Nevertheless, substantial gaps remain in our knowledge about the mechanisms by which these factors promote cardiac progenitor cell-mediated regeneration. Interestingly, Notch signaling pathway, which controls cardiovascular development and homeostasis, has been involved in adult cardiac repair following myocardial infarction.3
The Notch pathway mediates the signaling between adjacent cells expressing transmembrane ligands (Jagged1 and 2; Delta-like1, 3, and 4) and receptors (Notch 1–4). In the mammalian heart, and specifically in cardiomyocytes, Notch activity has been linked to proliferative signals and cell cycle reentry,4,5 while in cardiac stem cells (CPC) and in cardiac mesenchymal stromal cells, it controls proliferation and differentiation.6,7
High-mobility group box-1 protein (HMGB1) is a highly conserved nuclear protein that acts as a cytokine when released by necrotic and inflammatory cells, but it also functions as an extracellular signaling molecule during inflammation and regenerative processes.8 Specifically, extracellular HMGB1 signals tissue damage by stimulating the secretion of proinflammatory molecules and inducing stem cell proliferation and migration.
Our previous studies have shown that HMGB1 administration to the mouse heart few hours after MI enhanced cardiac regeneration leading to improvement of cardiac function.9 The regenerative process involved the proliferation and differentiation of endogenous CPCs. In addition, administration of this protein had also an influence on cardiac remodeling after MI: prolonged HMGB1 administration to infarcted hearts attenuated left ventricular dilation and increased infarcted wall thickness. Moreover, we found that HMGB1 injection in chronically failing hearts improved left ventricular function and attenuated adverse cardiac remodeling inducing cardiac tissue regeneration and extracellular matrix degradation. These events were mediated, at least in part, by miR-206 inhibition of TIMP-3 expression.10
Other studies have also demonstrated a role of HMGB1 in cardiac remodeling prevention after MI,11,12 in the setting of global ischemia/reperfusion (I/R) injury13 and after the occurrence of heart failure.14
To date, the molecular pathways underlying HMGB1-mediated cardiac regeneration are still poorly defined.
Recently, gene expression analysis by microarray has been successfully applied to cardiovascular research. Several studies have sought to present a comprehensive view of genetic changes taking place in the infarcted myocardium.15,16,17 These studies have shown that following a prolonged ischemic episode, the injured myocardium responds with altered expression in gene categories that are closely related to remodeling, energy deficit, and cell death.
In the present study, we performed a comprehensive analysis of mRNA and miRNA expression levels in myocardial samples from HMGB1-treated and untreated hearts 3 days following MI. By this approach, we have identified a relationship between exogenous HMGB1 administration and notch signaling.
Results
Gene expression in the infarcted murine heart
At first, we focused on the gene expression profile changes occurring during the early stages of left ventricular remodeling following MI. Three days after MI, total RNA was prepared from two separate regions of the infarcted heart: the scar (infarcted area (IA)), and the periinfarct zone (border zone (BZ)). As a control, RNA was also prepared from the same left ventricle region of sham-operated mice. Gene expression profile revealed 2160 and 6669 significantly modulated mRNA (defined as transcripts with fold change >1.5) in the BZ and in the IA compared with the sham group, respectively (Figure 1a and 1b, Supplementary Figure S1, and Supplementary Table S2 for the whole profiling). Among these transcripts, 1166 and 3011 were upregulated while 994 and 3658 were downmodulated in the BZ and IA, respectively. Among the modulated transcripts, 676 were specific of the BZ and 5185 of the IA, whereas 1484 were concordantly modulated in both regions (Figure 1b). The expression of selected transcripts was validated by qRT-PCR (Supplementary Figure S2). Functional categorization of these transcripts, performed using Ingenuity Pathway Analysis software (IPA), Supplementary Table S3), revealed many shared common physiological roles that were grouped into categories based on their biological functions and pathways (Figure 2a and 2b). Among the enriched functions, cardiovascular disease genes as well as genes involved in the inflammatory response and in cell death, although present in the BZ, were also enriched in the IA. Similarly, healing process-related functions (cellular movement, organismal functions, cellular assembly and organization) were enriched in the IA. Functions associated with tissue regeneration were represented both in the BZ and in the IA (cardiovascular system development and function and embryonic development, cellular growth proliferation and development). Notably, most subcategories in regenerating-associated functions included genes involved in cardiogenesis, vasculogenesis and angiogenesis (Figure 2 and Supplementary Table S3). It is well known that myocardial energy metabolism is altered in the postinfarction period that is characterized by a switch away from fatty acid toward carbohydrate metabolism.18 Therefore, lipid metabolism genes were present in the BZ and enriched in the IA, while the expression of genes related to energy metabolism, carbohydrate, amino acid, and nucleic acid metabolism were present only in the IA (Figure 2a).
Figure 1.

Gene epression analysis in control and infarcted hearts. (a) Hierarchical clustering of differentially expressed genes selected by significance analysis of microarrays (SAM) in the border zone (BZ) and in the infarcted area (IA) of 3 day infarcted hearts compared to sham operated hearts. Each row represents the expression of a single gene and columns 1 and 2 correspond to a sample pool of 3 hearts. Expression levels are represented by a color tag, with red representing the highest levels and green the lowest levels of expression. (b) The Venn diagrams shows the number of differentially up and downregulated transcripts, as obtained by SAM, and the number of overlapping transcripts between the bz and the IA.
Figure 2.
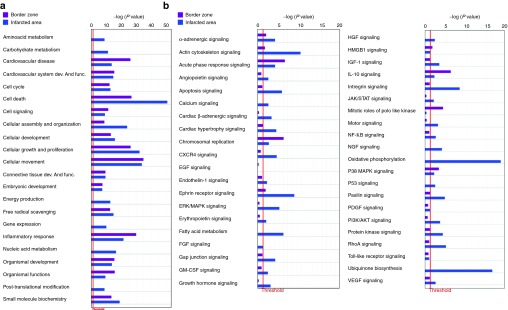
Comparison of biological functions and pathways between the border zone and the infarcted area. Ingenuity pathway analysis showing (a) selected biological functions and (b) Pathways in the border zone and in the infarcted area compared with the sham group. Bars, P values evaluated by exact Fisher test expressed in logarithmic scale. Threshold (evidenced by the red line) indicates the minimally accepted significance level of P < 0.05.
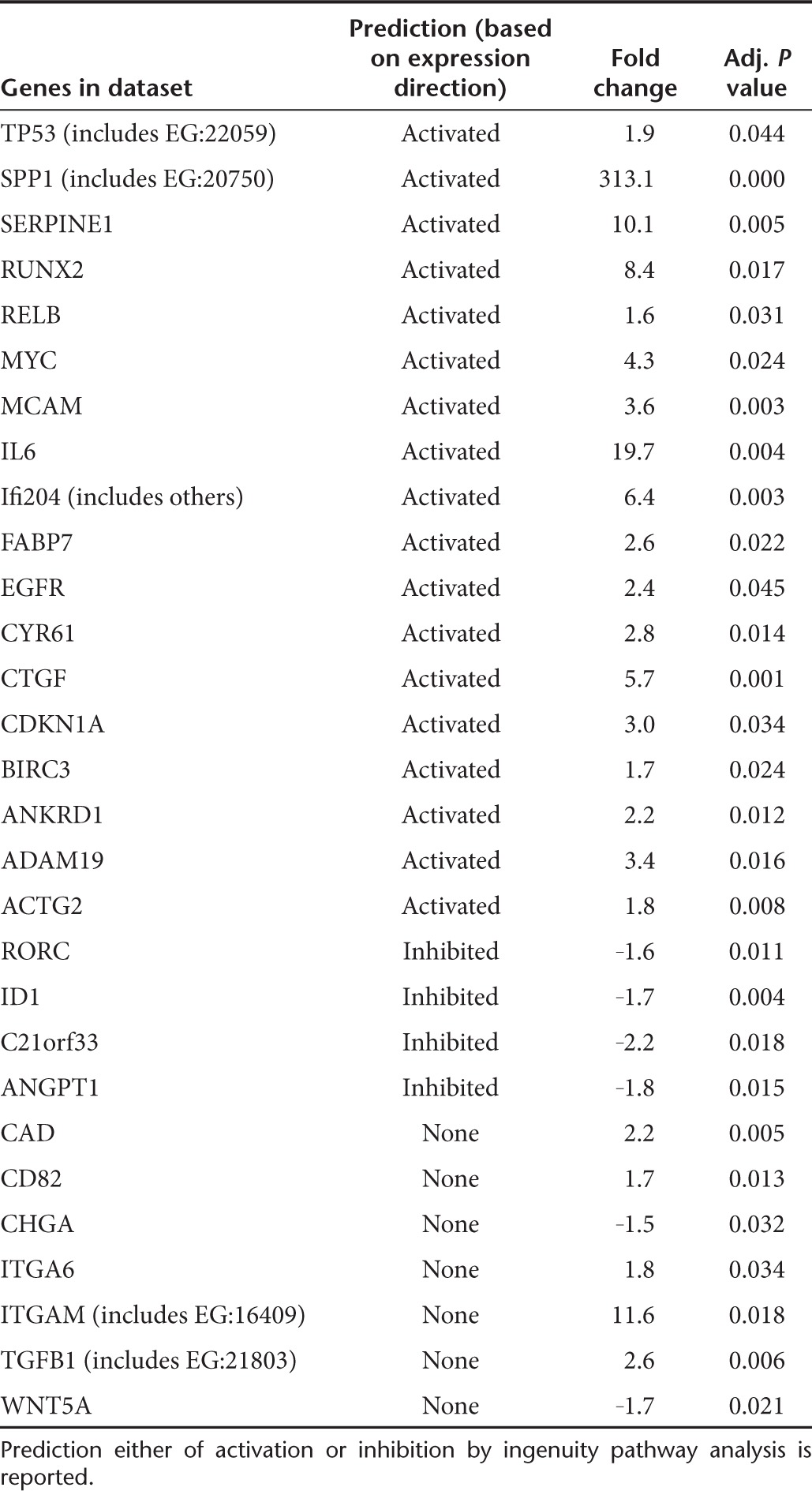
Among the top significantly enriched pathways, the acute phase responsive signaling, IL10 signaling, glucocorticoid receptor signaling, p38 MAP, HMGB1 signaling, and cell cycle control of chromosomal replication were enriched in the BZ (Figure 2b). Growth factors and receptors (IGF-1, IGF1-R, VEGF, Endothelin 1, GM-CSF, Angiopoietin, Erithropoietin, cardiac α- and β-adrenergic signaling, ephrin receptor signaling GH signaling), intracellular signaling pathways (Rho GDI, ERK5 NF-κB, RhoA PKA PI3K/AKT SAP/JNK, ERK/MAP mTOR, JAK/STAT, Calcium) and pathways involved in extracellular matrix remodeling (actin cytoskeletal signaling, paxillin, regulation of actin-based motility by Rho, gap junction signaling integrin signaling) as well as pathways associated with mitochondrial dysfunction, oxidative phosphorylation, ubiquinone biosynthesis, and fatty acid metabolism were enriched in the IA (Figure 2b and Supplementary Table S3). It is worth noting that the expression of Notch genes (Notch1, Notch2, and Notch4) increased in the IA compared with sham-operated hearts. Moreover, the “transcription factor” function of IPA analysis, aimed to predict the modulation of relevant transcription factors by looking at the genes modulated in a dataset, identified the MI-induced activation of Notch through the coherent modulation of 29 Notch downstream targets (Table 1). The activation of Notch was further validated by qRT-PCR (Supplementary Figure S2).
Table 1. Ingenuity pathway analysis of transcription factors modulated following myocardial infarction and potential Notch downstream targets.
Gene expression profiling in HMGB1-treated hearts 3 days following MI
To identify the molecular signals that initiate HMGB1-mediated regeneration, the BZ and IA were collected from 3-day HMGB1-treated hearts (Figure 3a). HMGB1 treatment specifically modulated 187 transcripts in the BZ and 124 transcripts in the IA that were not modulated in the untreated hearts (Figure 3b, and Supplementary Table S2). Among these transcripts, 94 and 81 were upregulated, while 93 and 43 were downmodulated in the BZ and IA, respectively. No common transcripts were modulated both in the BZ and in the IA (Figure 3b). The expression of selected transcripts was validated by qRT-PCR (Supplementary Figure S2).
Figure 3.

Gene expression analysis in untreated and HMGB1-treated infarcted hearts. (a) Hierarchical clustering of differentially expressed genes selected by SAM in the BZ and in the IA 3 days following MI and HMGB1 treatment. Each row represents the expression of a single gene and columns 1 and 2 correspond to a sample pool of infarcted hearts from three animals in each column. Expression levels are represented by a color tag, with red representing the highest levels and green the lowest levels of expression. (b) The Venn diagrams shows the number of differentially up and downregulated transcripts, as obtained by SAM in the BZ and in the IA. No overlapping transcripts between BZ and IA were detected.
IPA analysis, performed to explore the enriched biological functions, revealed that HMGB1 further modulated the expression of genes involved in tissue regeneration, that is, cellular movement and development, cellular growth and proliferation, cardiovascular system development and function, organ, tissue, and embryonic development (Figure 4, Supplementary Figure S3 and Supplementary Table S4 for complete IPA analysis). All these functions were enriched in the BZ and included angiogenesis and cardiogenesis subcategories. Specifically, genes involved in endothelial cell migration and proliferation (PDGFRA, FGF2, INHBA, p53, LEF1, NKX2.3) as well as in stem cell differentiation (Wnt11), mesoderm and muscle cell formation (LEF1, MYLPF, PAX3, PLAU, UTRN) were modulated. In the IA, the same functions included genes implicated in cardiomyocyte elongation and contraction (MARCKSL1, PLN, VDR) and differentiation of connective tissue cells (GDNF, LPIN1, MYD88, NR0B2, PRKG2, PTHLH, SCD, VDR). Finally, IPA analysis revealed that NFK-B and CDC42 were among the top pathways, enriched in HMGB1-treated hearts, both in the BZ and in the IA (Figure 4b). Significantly, NFK-B and CDC42 are both well-known HMGB1 downstream targets. It has been previously reported that HMGB1 may act in a paracrine manner, stimulating growth factor release from cardiac fibroblasts.19 Accordingly, growth factor signaling pathways were significantly enriched in HMGB1-treated hearts and included TGFβ, PDGF, and EGF pathways (Figure 4b and Supplementary Table S4).
Figure 4.

Comparison of biological functions and pathways between the border zone and the infarcted area of HMGB1-treated infarcted hearts. Ingenuity pathway analysis showing (a) selected biological functions and (b) pathways in BZ and in the IA 3 days following MI and HMGB1 treatment. Bars, P values evaluated by exact Fisher test expressed in logarithmic scale. Threshold indicates the minimally accepted significance level of P < 0.05.
Among transcription factors, treatment with HMGB1 in the BZ enhanced the expression of p53, myc, and LEF1, the downstream target of WNT/β catenin signaling; in the IA, HMGB1 enhanced the expression of JunB, Cebpβ and HIF1α (Supplementary Table S2). Interestingly, HIF1α has been involved in Notch-mediated regulation of vasculogenesis20 and myogenesis21 while a crosstalk between Notch and Wnt11 has been implicated in cardiogenic differentiation of both mesenchymal stem cells and endothelial progenitor cells.22,23 Although HMGB1 treatment did not appear to modulate Notch expression in Affymetrix studies, qRT-PCR revealed the upregulation of Notch2, Notch4 and Jagged2 in the IA, compared with untreated infarcted hearts (Supplementary Figure S2). These data suggest that Notch signaling pathway, already activated upon MI, is further modulated by HMGB1 treatment.
To gain further insight into the molecular mechanisms underpinning HMGB1 function, the same samples analyzed for mRNA expression were also assayed for miRNAs modulation. miRNA expression profiles were obtained and miRNAs modulated upon HMGB1 treatment in infracted hearts were identified (Supplementary Table S5). We identified and validated miR-208b and miR-301a as HMGB1-specific miRNAs downmodulated in the BZ and in the IA of treated hearts, respectively (Supplementary Figure S4).
Since miRNAs elicit their activity through the inhibition of mRNA targets, mRNAs displaying reciprocal modulation to miR-208b in BZ and to miR-301a in the IA were selected. To predict direct binding events, a miRNA target prediction analysis was then conducted using MiROnTop algorithm, allowing the identification of putative direct targets of miR-208b and miR-301a (Supplementary Table S6). In particular, TLE4 and HIF1α transcripts are upregulated upon HMGB1 treatment and putative targets of miR-208b and miR-301a, respectively. Significantly, both genes are involved in the Notch pathway, further implicating Notch in the physiologic response to HMGB1 treatment.24,25
Notch pathway is activated in HMGB1-treated infarcted hearts, in vitro and in vivo
It is already known that among Notch genes, Notch1 levels increase in response to acute MI, modulating cardiomyocyte and CPC functions.4,6,7,26 Notch1 activation is accomplished by the γ-secretase enzyme complex, resulting in the release of a Notch intracellular domain (NICD1) that translocates to the nucleus and regulates transcription.27 Indeed, 4 days after infarction, NICD1 accumulates in the nucleus of surviving cardiomyocytes.4 To verify whether HMGB1 treatment further induces Notch1 activation following infarction, we performed in vivo and in vitro studies. Specifically, in order to identify Notch1-activated cardiac cells, we performed immunohistological analysis on untreated and HMGB1-treated mouse hearts 3 days following MI. To this aim, we used an antibody directed against the cleaved portion of Notch1. Cell count revealed a 35% (1.35 fold) increase in myocytes expressing NICD1 in HMGB1-treated infarcted hearts compared with control hearts (Figure 5a). To further support HMGB1-mediated activation of Notch1, the expression levels of its targets Hes1 and Hey1 were evaluated 3 days following infarction. qRT-PCR assays showed that HMGB1 upregulated the mRNA levels of Hes1 and Hey1 in the infarcted area (Figure 5b) but not in the BZ (data not shown). It is noteworthy that Notch1 activation was present also in nonmyocyte cells (Figure 5c), and specifically in CPCs. As shown by immunofluorescence, 47 ± 4% of CPCs expressed the NICD1 fragment in untreated hearts 3 days following MI; the percentage increased up to 58 ± 4% in response to HMGB1 treatment (Figure 5c). Accordingly, Notch1 as well as Hes1 and Hey1 expression was also increased by HMGB1 (Figure 5d). Neither Notch2 nor Notch4 were modulated in similar experimental conditions (data not shown). To confirm that the CPC activation of Notch1 was HMGB1-dependent, in vitro studies were performed as well. CPCs were isolated from noninfarcted murine hearts and grown in the presence of 100 ng HMGB1 for 1 and 3 days (Figure 5e). Immunofluorescence analysis revealed a statistically significant increase (1.33- and 1.21-fold increase, respectively) in c-kit+ cells expressing NICD1 after 1 and 3 days of treatment with HMGB1 (Figure 5e). Notably, at day 1, Hes1 and NICD1 protein levels were enhanced as well (Figure 5f).
Figure 5.

HMGB1 enhances Notch expression in the border zone of HMGB1-treated infarcted hearts. (a) HMGB1 enhances Notch activation in cardiac tissue. Left panel: Average number of myocytes expressing NICD1 in the border zone of 3 day infarcted hearts treated either with HMGB1 (+) or its denatured form (-) (n = 6/each group). Right panel: representative immunohistochemical analysis showing NICD1 in myocyte nuclei (arrow). (b) qRT-PCR analysis of Hes1 and Hey1 in the IA of 3-day HMGB1-treated hearts. IA from hearts treated with denatured HMGB1 was used as control (-). (c,d) HMGB1 enhances Notch activation in CPCs in vivo. CPCs were isolated from infarcted hearts treated either with HMGB1 (+) or its denatured form (-) 3 days following infarction. (c) Average number of CPCs coexpressing c-kit and NICD1 (n = 10/each group). Lower panel: representative image of double immunofluorescence showing freshly isolated CPCs from HMGB1-treated hearts stained for c-kit (red fluorescence) and NICD1 (green fluorescence). Hoechst staining of nuclei in blue. (d) qRT-PCR analysis of Notch1, Hes1 and Hey 1 in isolated CPCs (n = 3, *P < 0.05). (e,f) HMGB1 enhances Notch activation in vitro. CPCs were isolated from non-infarcted hearts, in vitro expanded and grown for 1 and 3 days either in the absence (-) or in the presence of 100 ng HMGB1 (e) Bar graph shows the average number of cells coexpressing c-kit and NICD1 (n = 8, *P < 0.05). (f) Western blot analysis showing Hes1 and NICD1 accumulation in 1 day HMGB1-treated CPCs. The same filter was probed with anti-α-tubulin mAb to show the equal loading.
Notch pathway modulates HMGB1-mediated proliferation and differentiation of cardiac ckit+ cells in vivo
To investigate whether the Notch pathway is involved in HMGB1-induced effects on cardiac regeneration, C57/BL6 mice were injected with a γ-secretase inhibitor (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S phenylglycine t-butyl ester) (DAPT). This compound was previously shown to block both the proteolytic release of NICD and the specific activation of Notch target gene expression.28 In preliminary experiments, we showed that effective Notch1 inhibition resulted by systemic delivery of the inhibitor for 3 consecutive days (i.e., 1 day before and 2 days after MI) (Figure 6a). In these conditions, we observed in the hearts of mice receiving the inhibitor a reduction in the protein levels of NICD1 (Figure 6a) and in mRNA levels of Notch target genes Hes1, Hey1 and of its ligand Jagged1 (Figure 6b). Next, we investigated whether Notch endoproteolysis had an effect on HMGB1-mediated CPC proliferation. To verify this hypothesis, CPCs were isolated from untreated and HMGB1-treated hearts 3 days following infarction and DAPT treatment. While HMGB1 treatment enriched the population of CPCs (data not shown), the concurrent delivery of HMGB1 and DAPT significantly inhibited the HMGB1-mediated effect on CPC number (Figure 6c). Similar results were obtained for proliferating CPCs, detected as c-kit/Ki67 double positive cells (Figure 6d). Interestingly, infarcted mouse hearts treated with HMGB1 and DAPT showed a number of proliferating CPCs comparable with the number detected in untreated infarcted hearts.
Figure 6.

Notch signaling modulates in vivo CPC number and proliferation. The γ-secreatse inhibitor DAPT reduces Notch activation in infarcted hearts (a,b) and in CPCs isolated from HMGB1-treated infarcted hearts (c,d). DAPT was administered to mice 1 day before, 1 and 2 days after MI. The zone bordering the infarct was collected from six different mouse hearts and three of them were used for Western blot analysis while the others were subjected to qRT-PCR. (a) Western blot analysis was performed on heart extracts and on control brain samples (positive control), to detect NICD1. The same filter was probed with anti-Notch1 antibody. DAPT reduced NICD levels. (b) DAPT inhibited the expression of Notch-regulated genes, that is, hes, hey and jagged1 (n = 3, *P < 0.05 vs. each control). CPCs expressing c-kit (c) and c-kit/Ki67 (d) were isolated from infarcted hearts in the absence (-) and in presence (+) of DAPT, 3 days following MI and HMGB1 administration (n = 16/each group). Lower panel: representative image of double immunofluorescence showing freshly isolated CPCs from HMGB1-treated hearts stained for c-kit (red fluorescence) and Ki67 (green fluorescence). Hoechst staining of nuclei in blue.
Additional experiments were performed to establish whether the Notch pathway affected HMGB1-induced CPC cardiomyogenic differentiation and migration. Notably, Tbx5, an early transcription factor of the cardiomyocyte lineage, was increased in freshly isolated CPCs from 3 days-infarcted HMGB1-treated hearts compared with controls but this increase was significantly inhibited following DAPT delivery (Figure 7). Noteworthy, a similar trend was detected in differentiating cells coexpressing c-kit and both early markers of endothelial and smooth muscle differentiation, that is, Tie2 and smMHC (Figure 7). In contrast, HMGB1-mediated CPC migration was not affected by Notch inhibition (data not shown).
Figure 7.
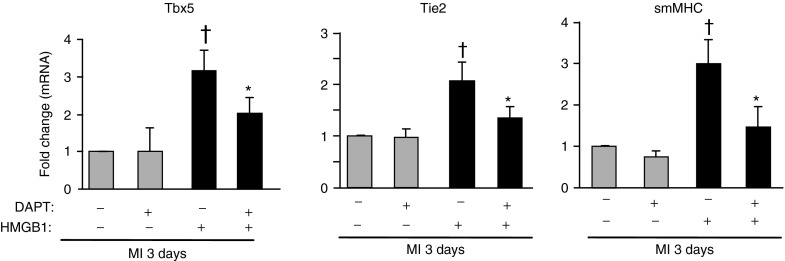
Downregulation of Notch signaling inhibits CPC cardiovascular differentiation. CPCs were isolated from infarcted hearts in the absence (-) and in the presence (+) of DAPT treatment, 3 days following MI and HMGB1 administration. qRT-PCR was performed to detect the expression of myocardial (Tbx5), endothelial (Tie2) and smooth muscle (smMHC) cell markers. n = 3, *,†P < 0.05 versus each control.
Taken together, these data indicate that Notch signaling plays a key role in HMGB1-induced CPC proliferation and differentiation in the three cardiovascular cell lineages.
Discussion
The present study utilized a transcriptomic approach to elucidate the molecular mechanisms of HMGB1-induced cardiac regeneration in a mouse model of acute MI. Our results show that HMGB1 activated a number of genes involved in cardiac protection and regeneration, and that Notch1 signaling plays a key role in HMGB1 ability to activate CPCs.
Several studies, using high-throughput screening approaches, have characterized the gene expression profile associated with acute MI. These studies, most of them performed using RNA from different regions of the infarcted left ventricular, showed that modulation of gene expression taking place in the postischemic myocardium was mainly related to cell death and cardiac remodeling.18,29,30,31 Also energy-generating processes were altered, with reduced fatty acid utilization and increased carbohydrate metabolism.18,29 Here, we determined global differences in gene expression in the BZ and in the IA of mouse hearts 3 days following MI and we identified the affected gene networks in the two regions of the infarcted left ventricular. In agreement with previous studies, we found that cell death genes as well as extracellular matrix-related genes (i.e., fibronectin and collagens) were modulated following MI, at higher extent in the IA than in the BZ. A similar result was obtained with genes associated with mitochondrial dysfunction, oxidative phosphorylation, and fatty acid metabolism. Interestingly, growth factor and angiogenic-related signaling pathways, including VEGF, NGF, angiopoietin, ephrin, IGF, HGF, Erythropoietin, GM-CSF, and GH signaling, were enriched in the IA. Genes including IGF, HGF, Erythropoietin, and GM-CSF were previously shown to be upregulated in the left ventricular acutely after MI.32 Accordingly, their administration or overexpression in the infarcted heart promotes angiogenesis, cardiomyocyte proliferation and survival in different animal models.33,34,35
It is noteworthy that IGF and HGF have been implicated in CPC activation and mobilization.36 Moreover, NGF, whose expression following MI induces angiogenesis and cardiomyocyte survival, modulates CPC function enhancing the expression of the c-kit receptor ligand stem cell factor.37 Similarly, GH administration to the infarcted heart has been shown to enhance CPC number and activate cardiac repair.38 Finally, we confirmed the activation of Notch1 signaling pathway after MI. Several lines of evidence implicated Notch1 signaling as a potent modulator of regeneration in the adult heart. Regeneration of the adult zebrafish heart after amputation is preceded by Notch reactivation.39 In murine hearts, Notch1 is activated following MI4 and plays a cardioprotective role.4,6,26 At cellular and molecular levels, Notch1 activation favors CPC differentiation into cardiomyocytes6 and cell cycle re-entry of quiescent cardiomyocytes.4,5,40 Notably, HGF, whose expression is increased in the IA, enhances AKT and CPC function and recruitment in a Notch1-dependent manner.6
We previously reported that HMGB1 administration following MI attenuates cardiac function deterioration and promotes myocardial regeneration by activating endogenous CPCs9 and inducing angiogenesis.10 In the attempt to identify the molecular mechanisms involved in this process, gene expression studies were performed in HMGB1-treated hearts 3 days following MI. We found that HMGB1 treatment further increased the expression of genes related to tissue regeneration and most of these modifications occurred in the BZ. Thus, we found increased expression of LEF1 and Nkx2.3, both involved in endothelial cell proliferation and invasion,41,42 as well as of genes implicated in stem cell differentiation and muscle cell formation, that is, Wnt11, PAX3, LEF1.43,44,45 Notably, Wnt11 induces cardiomyogenic differentiation of BM-MNC via PKC/JNK-dependent activation.46 As previously reported, HMGB1 modulates cell migration and differentiation as well as endothelial cell functions through the activation of several intracellular pathways including CD42, JNK, and NF-κB.47 These pathways were enriched in the BZ of HMGB1-treated hearts. Finally, HMGB1 enhanced the expression of TGFβ, PDGF and EGF, suggesting that it may amplify regenerating signals in a paracrine manner. Accordingly, we previously reported that HMGB1 increased cytokine, chemokine and growth factor production from cultured cardiac fibroblasts; in turn, conditioned medium of HMGB1-treated cardiac fibroblasts modulated CPC proliferation, migration, and differentiation into endothelial cells.19 It is noteworthy that in the IA of HMGB1-treated hearts, there is a predominance of pathways related to cardiomyocyte functions and cell differentiation together with signaling involved in cytoskeletal organization. HMGB1 also modulated survival-related pathways such as the sphingolipid metabolism. The activation of this pathway in the IA, reduces myocardial cell apoptosis and improves left ventricular function after MI.48 Importantly, HMGB1 modulated the Notch1 signaling pathway (Figure 8). It is well known that Notch signaling is activated in the adult heart following myocardial injury. Notch activation induces cell cycle re-entry in quiescent cardiomyocytes5,40 and regulates functions and recruitment of CPCs.6 We found that HMGB1 treatment further activated Notch1 both in cardiomyocytes and in CPCs in vivo and that the inhibition of Notch pathways by γ-secretase reduced CPC proliferation as well as their differentiation in the cardiovascular cell lineages. Accordingly, the cardiogenic gene Nkx2.5, whose expression increased in heart tissue following MI and HMGB1 treatment,9 is a target of Notch genes.6 Further, Notch1 has been described as the upstream activator of Wnt11 in the regulation of gene expression in circulating progenitor cells.23 Therefore, the HMGB1-mediated induction of Wnt11 could involve Notch1 since its expression is blunted by the γ-secretase (data not shown). We also found that HMGB1 downmodulated miR-208b and miR-301a in the BZ and in the IA of treated hearts, respectively. Putative target of these miRNAs are TLE4 and HIF1α genes that were upregulated upon HMGB1 treatment and are both involved in Notch signaling.24,25 Interestingly, HIF-1α interacts with NICD and is recruited to the Notch transcriptional complex at the Hey1 promoter,21 suggesting a possible mechanism by which HMGB1 may affect Hey1 expression. It is noteworthy that HMGB1 activates NF-κB signaling pathway through its receptors RAGE and TLR2/4/9.49 Notably, a crosstalk between Notch1 and NF-κB has been demonstrated: NF-κB increases Notch receptor expression, leading to augmented Notch signaling,50 and conversely, activated Notch upregulates the expression of NF-κB members.51
Figure 8.

Schematic representation of HMGB1-mediated activation of Notch1 signaling.
HMGB1 activates Notch1 receptor through an undefined mechanism that could involve NF-kB. Notch1 is cleaved by the γ-secretase complex and the Notch1 intracellular domain (NICD1) translocates to the nucleus, forms a transcription activation complex with RBP-jk and activates the expression of target genes Hes1, Hey1, Wnt11. Hes1/Hey1 could further modulate p53 expression which in turn may increase Notch1 expression. The activation of Nkx2.5 by Notch1 could drive commitment of CPCs to the myocyte lineage. Finally, via the modulation of miR-301a, HMGB1 could control HIF1α levels.
In conclusion, exogenous HMGB1 administration to the acutely infarcted heart activates genes and signaling pathways that play a key role in cardiac cell survival and regeneration; these effects are associated with the activation of the Notch pathway in myocardial cells and CPCs, and may account for the beneficial effect of HMGB1 treatment in acute MI.
Materials and Methods
Animal model and in vivo study. MI was induced by coronary artery ligation in 8-week-old C57 BL6 female mice (20 g body weight), as previously described.9 After 4 hours, animals were reoperated, and 200 ng of purified HMGB1 in 10 μl phosphate buffered saline (PBS) solution was injected through a 32-gauge needle. Four injections (2.5 μl per injection) were made in the ventricular wall bordering the viable myocardium. Control-infarcted mice were injected with 200 ng of denatured HMGB1. After 3 days, the infarcted area and the border zone were isolated. The infarcted area was recognized as a pale zone caused by gross necrosis of the myocardium, due to interruption of the blood supply to the area. The border zone was identified as the area between the end of necrosis and the septum.
γ secretase inhibitor (DAPT, Calbiochem) was custom synthesized as bulk preparation by Calbiochem (La Jolla, California, USA) and dissolved in DMSO at a concentration of 50 mmol/l. It was stored in aliquots as a stock solution in the dark at –20°C. DAPT (50 mmol/l) was diluted in 30 µl PBS and injected intraperitoneally in each animal (body weight: ≈ 20 g) 24 hours before MI induction and daily for additional 2 days after MI.
All experimental procedures complied with the Guidelines of the Italian National Institutes of Health, with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Academy of Sciences, Bethesda, MD) and were approved by the Institutional Animal Care and Use Committee.
miRNA and mRNA isolation and quantification. Total RNA was extracted from the infarcted and bordering region of untreated and HMGB1-treated hearts (n = 6/group), 3 days after coronary artery ligation. miRNA and mRNA isolation and quantification are described in Supplementary Material.
Cell isolation. Cardiac cells were isolated from C57BL6 female mice at 2–3 months of age by collagen digestion (280 U/ml; Worthington, Lakewood, NJ) through the coronary arteries and myocytes removed by centrifugation. In some experiments, cells were cultured in F12:DMEM 1:1 supplemented with 10% fetal bovine serum and expanded. CPCs were isolated by c-kit+ positive selection using EasySep (Stem Cell Technologies, Vancouver, Canada)9 and cultured in presence of different concentrations of HMGB1 (10 ng, 100 ng, and 200 ng). Cells were fixed in PBS with 4% paraformaldehyde and permeabilized in PBS with 0.1% Triton X-100. Coverslips were rinsed and blocked for 10 minutes in PBS with 0.2% Bovine Serum Albumine (BSA) prior to incubation with antibodies. Fixed cells were incubated with primary antibodies; mouse monoclonal c-kit antibody (R&D Systems, Minneapolis, MN), mouse polyclonal anti Ki67 (clone NCL, Novocastra Laboratories, UK) and rabbit polyclonal anti NCID (Abcam, UK), followed by incubations with antibodies coupled either with FITC or TRITC. The coverglasses were mounted and analyzed with a Zeiss microscope equipped for epifluorescence.
Chemotaxis assay. Chemotaxis was performed in 48-microwell chemotaxis chambers (Neuroprobe) using 3 μm pore-size polycarbonate filters (Costar Scientific, Cambridge, MA) coated with murine collagen type IV (Becton-Dickinson, Bedford, MA). The lower compartment of each chamber was filled with 28 μl F12K with 0.1% BSA (negative control) or 100 mg/ml of HMGB1. Each well of the upper compartment contained cardiac CPCs (4 × 105 cells/ml) (isolated as described in the section cell isolation) in 50 μl F12K with 0.1% BSA either in presence of DMSO or DAPT. Each point was run in triplicate. After 6–8 hours incubation at 37°C in a 5% CO2 humidified atmosphere, the chemotaxis assay was stopped, and cells on the filter were fixed and stained using Diff Quik (Dade AG). Cells in five random fields on the lower face of the filter were counted at 40x magnification, and migration index was calculated by dividing the number of migrated cells in the presence of chemoattractants by the cells migrated in response to F12K with 0.1% BSA.
Immunohistochemistry. In mice, the abdominal aorta was cannulated and the heart was arrested in diastole with CdCl2, perfused retrogradely with 10% (vol/vol) formalin, embedded in paraffin and sectioned (1 or 3 µm thickness). Sections were deparaffinized in xylene, hydrated and rinsed in PBS. Antigen retrieval was performed by microwaving in a citric acid-EDTA buffer (10 mmol/l, pH 7.8 UCS Diagnostic, Rome, Italy). Slides were washed in PBS and the endogenous peroxidase activity was blocked in 3% hydrogen peroxide in methyl alcohol for 20 minutes. To minimize nonspecific antibody binding, slides were preincubated with PBS containing 10% BSA overnight. The anti-NCID antibody (Abcam, UK) was diluted with 10% BSA and then incubated overnight at 4°C. Sections were then incubated with biotinylated secondary antibody for 1 hour at 37°C and subsequently in Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) for 1 hour. Finally, they were developed in Novared substrate kit (Vector Laboratories, Burlingame, CA) for 3–4 minutes, counterstained with hematoxylin to identify nuclei, dehydrated and mounted in Eukitt (Bioptica, MI, Italy).
Western blot analysis. Proteins from heart tissue (ventricles) and CPCs were homogenized and extracted with RIPA buffer (10 mmol/l Tris-HCl pH 7.4, 150 mmol/l NaCl, 1% NP40, 1% Deoxycolic acid, 0.1% SDS and 10% Glycerol) containing protease and phosphatase inhibitors (2 mmol/l phenylmethylsulfpnyl fluoride, 100 U/ml of aprotinin, 10 µg/ml of leupeptin and pepstatin, 10 mmol/l sodium fluoride, 20 mmol/l sodium vanadate). Equal amounts of total cellular proteins (100 µg/lane) were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Amersham Pharmacia Biotech, Little Chalfont, UK). Membranes were probed with rabbit polyclonal anti-NCID (0.4 µg/ml, Abcam, UK), Hes1 (Abcam, UK) and α-tubulin monoclonal antibody (0.1 µg/ml, Oncogene Science, Cambridge, MA), followed by horseradish peroxidase-coupled secondary antibodies and developed by a chemiluminescence-based detection system (ECL, Amersham Pharmacia Biotech).
Data collections and statistics. Data shown as bar graphs are means ± standard error. Statistical significance between two measurements was evaluated by unpaired Student's t-test. A probability value of P < 0.05 was considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Hierarchical clustering of differentially expressed genes Figure S2. Real time PCR validation of gene chip microarray expression analysis. Figure S3. Clustered expression pattern of genes in HMGB1-treated hearts within the indicated functional groups. Figure S4. Real time PCR validation of miR208b and miR301a expression in the indicated region of the heart after MI and HMGB1 treatment. Table S1. mRNA and miRNA probe list used in validation studies. Table S2. mRNA array results in the border zone and in the infarcted area of untreated and HMGB1 treated hearts. Table S3. Ingenuity Pathway Analyisis output of expressed genes in untreated hearts. Table S4. Ingenuity Pathway Analyisis output of expressed genes in HMGB1 treated hearts. Table S5. List of modulated miRNA in the border zone and in the infracted area of untreated and HMGB1 treated hearts. Table S6. List of putative miR208b and miR301a targets.
Acknowledgments
This research was supported by grant from Italian Ministry of Health. This work was supported by the Italian Ministry of Health. F. Limana was involved in the training programme at the PhD School of Molecular and Cellular Cardiology at the University La Cattolica, Rome, Italy. F.L., A.G. and M.C.C. are owners of the patent number WO2004004763 (Use of HMGB1 in the treatment of tissue damage and/or to promote tissue repair).
Supplementary Material
mRNA and miRNA probe list used in validation studies.
mRNA array results in the border zone and in the infarcted area of untreated and HMGB1 treated hearts.
Ingenuity Pathway Analyisis output of expressed genes in untreated hearts.
Ingenuity Pathway Analyisis output of expressed genes in HMGB1 treated hearts.
List of modulated miRNA in the border zone and in the infracted area of untreated and HMGB1 treated hearts.
List of putative miR208b and miR301a targets.
References
- Leri A, Kajstura J, Anversa P. Role of cardiac stem cells in cardiac pathophysiology: a paradigm shift in human myocardial biology. Circ Res. 2011;109:941–961. doi: 10.1161/CIRCRESAHA.111.243154. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Beohar N, Rapp J, Pandya S, Losordo DW. Rebuilding the damaged heart: the potential of cytokines and growth factors in the treatment of ischemic heart disease. J Am Coll Cardiol. 2010;56:1287–1297. doi: 10.1016/j.jacc.2010.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gude N, Sussman M. Notch signaling and cardiac repair. J Mol Cell Cardiol. 2012;52:1226–1232. doi: 10.1016/j.yjmcc.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gude NA, Emmanuel G, Wu W, Cottage CT, Fischer K, Quijada P, et al. Activation of Notch-mediated protective signaling in the myocardium. Circ Res. 2008;102:1025–1035. doi: 10.1161/CIRCRESAHA.107.164749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campa VM, Gutiérrez-Lanza R, Cerignoli F, Díaz-Trelles R, Nelson B, Tsuji T, et al. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol. 2008;183:129–141. doi: 10.1083/jcb.200806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni A, Urbanek K, Nascimbene A, Hosoda T, Zheng H, Delucchi F, et al. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci USA. 2008;105:15529–15534. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nemir M, Metrich M, Plaisance I, Lepore M, Cruchet S, Berthonneche C, et al. The Notch pathway controls fibrotic and regenerative repair in the adult heart. Eur Heart J. 2012. [DOI] [PMC free article] [PubMed]
- Germani A, Limana F, Capogrossi MC. Pivotal advances: high-mobility group box 1 protein–a cytokine with a role in cardiac repair. J Leukoc Biol. 2007;81:41–45. doi: 10.1189/jlb.0306165. [DOI] [PubMed] [Google Scholar]
- Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, et al. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res. 2005;97:e73–e83. doi: 10.1161/01.RES.0000186276.06104.04. [DOI] [PubMed] [Google Scholar]
- Limana F, Esposito G, D'Arcangelo D, Di Carlo A, Romani S, Melillo G, et al. HMGB1 attenuates cardiac remodelling in the failing heart via enhanced cardiac regeneration and miR-206-mediated inhibition of TIMP-3. PLoS ONE. 2011;6:e19845. doi: 10.1371/journal.pone.0019845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitahara T, Takeishi Y, Harada M, Niizeki T, Suzuki S, Sasaki T, et al. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc Res. 2008;80:40–46. doi: 10.1093/cvr/cvn163. [DOI] [PubMed] [Google Scholar]
- Kohno T, Anzai T, Naito K, Miyasho T, Okamoto M, Yokota H, et al. Role of high-mobility group box 1 protein in post-infarction healing process and left ventricular remodelling. Cardiovasc Res. 2009;81:565–573. doi: 10.1093/cvr/cvn291. [DOI] [PubMed] [Google Scholar]
- Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Fukushima S, Yamahara K, Yashiro K, Shintani Y, Coppen SR, et al. Modulated inflammation by injection of high-mobility group box 1 recovers post-infarction chronically failing heart. Circulation. 2008;118 14 Suppl:S106–S114. doi: 10.1161/CIRCULATIONAHA.107.757443. [DOI] [PubMed] [Google Scholar]
- Hong D, Zeng X, Xu W, Ma J, Tong Y, Chen Y. Altered profiles of gene expression in curcumin-treated rats with experimentally induced myocardial infarction. Pharmacol Res. 2010;61:142–148. doi: 10.1016/j.phrs.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Kuster DW, Merkus D, Kremer A, van Ijcken WF, de Beer VJ, Verhoeven AJ, et al. Left ventricular remodeling in swine after myocardial infarction: a transcriptional genomics approach. Basic Res Cardiol. 2011;106:1269–1281. doi: 10.1007/s00395-011-0229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahtola E, Storvik M, Louhelainen M, Merasto S, Lakkisto P, Lakkisto J, et al. Effects of levosimendan on cardiac gene expression profile and post-infarct cardiac remodelling in diabetic Goto-Kakizaki rats. Basic Clin Pharmacol Toxicol. 2011;109:387–397. doi: 10.1111/j.1742-7843.2011.00743.x. [DOI] [PubMed] [Google Scholar]
- Stanton LW, Garrard LJ, Damm D, Garrick BL, Lam A, Kapoun AM, et al. Altered patterns of gene expression in response to myocardial infarction. Circ Res. 2000;86:939–945. doi: 10.1161/01.res.86.9.939. [DOI] [PubMed] [Google Scholar]
- Rossini A, Zacheo A, Mocini D, Totta P, Facchiano A, Castoldi R, et al. HMGB1-stimulated human primary cardiac fibroblasts exert a paracrine action on human and murine cardiac stem cells. J Mol Cell Cardiol. 2008;44:683–693. doi: 10.1016/j.yjmcc.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Diez H, Fischer A, Winkler A, Hu CJ, Hatzopoulos AK, Breier G, et al. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp Cell Res. 2007;313:1–9. doi: 10.1016/j.yexcr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Boopathy AV, Pendergrass KD, Che PL, Yoon YS, Davis ME. Oxidative stress-induced Notch1 signaling promotes cardiogenic gene expression in mesenchymal stem cells. Stem Cell Res Ther. 2013;4:43. doi: 10.1186/scrt190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi M, Bushoven P, Iwasaki M, Urbich C, Zeiher AM, Dimmeler S. Notch signaling contributes to the expression of cardiac markers in human circulating progenitor cells. Circ Res. 2007;101:1139–1145. doi: 10.1161/CIRCRESAHA.107.151381. [DOI] [PubMed] [Google Scholar]
- Sharma M, Brantley JG, Vassmer D, Chaturvedi G, Baas J, Vanden Heuvel GB. The homeodomain protein Cux1 interacts with Grg4 to repress p27 kip1 expression during kidney development. Gene. 2009;439:87–94. doi: 10.1016/j.gene.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Linke S, Dias JM, Zheng X, Gradin K, Wallis TP, et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci USA. 2008;105:3368–3373. doi: 10.1073/pnas.0711591105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratsios P, Catela C, Salimova E, Huth M, Berno V, Rosenthal N, et al. Distinct roles for cell-autonomous Notch signaling in cardiomyocytes of the embryonic and adult heart. Circ Res. 2010;106:559–572. doi: 10.1161/CIRCRESAHA.109.203034. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Geling A, Steiner H, Willem M, Bally-Cuif L, Haass C. A gamma-secretase inhibitor blocks Notch signaling in vivo and causes a severe neurogenic phenotype in zebrafish. EMBO Rep. 2002;3:688–694. doi: 10.1093/embo-reports/kvf124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehl PD, Tai JT, Hillan KJ, Brown LA, Goddard A, Yang R, et al. Application of cDNA microarrays in determining molecular phenotype in cardiac growth, development, and response to injury. Circulation. 2000;101:1990–1999. doi: 10.1161/01.cir.101.16.1990. [DOI] [PubMed] [Google Scholar]
- Lyn D, Liu X, Bennett NA, Emmett NL. Gene expression profile in mouse myocardium after ischemia. Physiol Genomics. 2000;2:93–100. doi: 10.1152/physiolgenomics.2000.2.3.93. [DOI] [PubMed] [Google Scholar]
- LaFramboise WA, Bombach KL, Dhir RJ, Muha N, Cullen RF, Pogozelski AR, et al. Molecular dynamics of the compensatory response to myocardial infarct. J Mol Cell Cardiol. 2005;38:103–117. doi: 10.1016/j.yjmcc.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Vandervelde S, van Luyn MJ, Rozenbaum MH, Petersen AH, Tio RA, Harmsen MC. Stem cell-related cardiac gene expression early after murine myocardial infarction. Cardiovasc Res. 2007;73:783–793. doi: 10.1016/j.cardiores.2006.11.030. [DOI] [PubMed] [Google Scholar]
- Tao Z, Chen B, Tan X, Zhao Y, Wang L, Zhu T, et al. Coexpression of VEGF and angiopoietin-1 promotes angiogenesis and cardiomyocyte proliferation reduces apoptosis in porcine myocardial infarction (MI) heart. Proc Natl Acad Sci USA. 2011;108:2064–2069. doi: 10.1073/pnas.1018925108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG, et al. Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc Natl Acad Sci USA. 2003;100:11612–11617. doi: 10.1073/pnas.1930406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Takano H, Niitsuma Y, Hasegawa H, Uchiyama R, Oka T, et al. Sonic hedgehog is a critical mediator of erythropoietin-induced cardiac protection in mice. J Clin Invest. 2010;120:2016–2029. doi: 10.1172/JCI39896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanek K, Rota M, Cascapera S, Bearzi C, Nascimbene A, De Angelis A, et al. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res. 2005;97:663–673. doi: 10.1161/01.RES.0000183733.53101.11. [DOI] [PubMed] [Google Scholar]
- Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van Linthout S, et al. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010;106:1275–1284. doi: 10.1161/CIRCRESAHA.109.210088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanashiro-Takeuchi RM, Tziomalos K, Takeuchi LM, Treuer AV, Lamirault G, Dulce R, et al. Cardioprotective effects of growth hormone-releasing hormone agonist after myocardial infarction. Proc Natl Acad Sci USA. 2010;107:2604–2609. doi: 10.1073/pnas.0914138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raya A, Koth CM, Büscher D, Kawakami Y, Itoh T, Raya RM, et al. Activation of Notch signaling pathway precedes heart regeneration in zebrafish. Proc Natl Acad Sci USA. 2003;100 Suppl 1:11889–11895. doi: 10.1073/pnas.1834204100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collesi C, Zentilin L, Sinagra G, Giacca M. Notch1 signaling stimulates proliferation of immature cardiomyocytes. J Cell Biol. 2008;183:117–128. doi: 10.1083/jcb.200806091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holnthoner W, Pillinger M, Groger M, Wolff K, Ashton AW, Albanese C, et al. Fibroblast growth factor-2 induces Lef/Tcf-dependent transcription in human endothelial cells. J Biol Chem. 2002;277:45847–45853. doi: 10.1074/jbc.M209354200. [DOI] [PubMed] [Google Scholar]
- Planutiene M, Planutis K, Holcombe RF. Lymphoid enhancer-binding factor 1, a representative of vertebrate-specific Lef1/Tcf1 sub-family, is a Wnt-beta-catenin pathway target gene in human endothelial cells which regulates matrix metalloproteinase-2 expression and promotes endothelial cell invasion. Vasc Cell. 2011;3:28. doi: 10.1186/2045-824X-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Ghani M, Dufort D, Stiles R, De Repentigny Y, Kothary R, Megeney LA. Wnt11 promotes cardiomyocyte development by caspase-mediated suppression of canonical Wnt signals. Mol Cell Biol. 2011;31:163–178. doi: 10.1128/MCB.01539-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina G, Sirabella D, Monteverde S, Galvez BG, Tonlorenzi R, Schnapp E, et al. Skeletal muscle differentiation of embryonic mesoangioblasts requires pax3 activity. Stem Cells. 2009;27:157–164. doi: 10.1634/stemcells.2008-0503. [DOI] [PubMed] [Google Scholar]
- Abu-Elmagd M, Robson L, Sweetman D, Hadley J, Francis-West P, Münsterberg A. Wnt/Lef1 signaling acts via Pitx2 to regulate somite myogenesis. Dev Biol. 2010;337:211–219. doi: 10.1016/j.ydbio.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Flaherty MP, Abdel-Latif A, Li Q, Hunt G, Ranjan S, Ou Q, et al. Noncanonical Wnt11 signaling is sufficient to induce cardiomyogenic differentiation in unfractionated bone marrow mononuclear cells. Circulation. 2008;117:2241–2252. doi: 10.1161/CIRCULATIONAHA.107.741066. [DOI] [PubMed] [Google Scholar]
- Biscetti F, Ghirlanda G, Flex A. Therapeutic potential of high mobility group box-1 in ischemic injury and tissue regeneration. Curr Vasc Pharmacol. 2011;9:677–681. doi: 10.2174/157016111797484125. [DOI] [PubMed] [Google Scholar]
- Yeh CC, Li H, Malhotra D, Huang MC, Zhu BQ, Goetzl EJ, et al. Sphingolipid signaling and treatment during remodeling of the uninfarcted ventricular wall after myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;296:H1193–H1199. doi: 10.1152/ajpheart.01032.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad G, Siddiquei MM, Othman A, Al-Shabrawey M, Abu El-Asrar AM. High-mobility group box-1 protein activates inflammatory signaling pathway components and disrupts retinal vascular-barrier in the diabetic retina. Exp Eye Res. 2013;107:101–109. doi: 10.1016/j.exer.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Moran ST, Cariappa A, Liu H, Muir B, Sgroi D, Boboila C, et al. Synergism between NF-kappa B1/p50 and Notch2 during the development of marginal zone B lymphocytes. J Immunol. 2007;179:195–200. doi: 10.4049/jimmunol.179.1.195. [DOI] [PubMed] [Google Scholar]
- Cheng P, Zlobin A, Volgina V, Gottipati S, Osborne B, Simel EJ, et al. Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. J Immunol. 2001;167:4458–4467. doi: 10.4049/jimmunol.167.8.4458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
mRNA and miRNA probe list used in validation studies.
mRNA array results in the border zone and in the infarcted area of untreated and HMGB1 treated hearts.
Ingenuity Pathway Analyisis output of expressed genes in untreated hearts.
Ingenuity Pathway Analyisis output of expressed genes in HMGB1 treated hearts.
List of modulated miRNA in the border zone and in the infracted area of untreated and HMGB1 treated hearts.
List of putative miR208b and miR301a targets.