

Abstract
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) is an established method for the interrogation of protein conformation and dynamics. While the data analysis challenge of HDX-MS has been addressed by a number of software packages, new computational tools are needed to keep pace with the improved methods and throughput of this technique. To address these needs, we report an integrated desktop program titled HDX Workbench, which facilitates automation, management, visualization, and statistical cross-comparison of large HDX data sets. Using the software, validated data analysis can be achieved at the rate of generation. The application is available at the project home page http://hdx.florida.scripps.edu.
Keywords: Mass spectrometry, HDX, Software, H/D exchange, Protein dynamics, Automation
Introduction
Differential hydrogen/deuterium exchange (HDX) mass spectrometry is an effective method for characterizing protein dynamics, protein–ligand interactions, and protein–protein interactions [1]. Historically, the analysis, statistical validation, and display of data have been the most time consuming facets of HDX experiments in the common peptide-based workflow. The informatics problem starts where the acquisition ends, in which each peptide is associated with an isotope cluster and is assigned a level of deuterium content (%D). The essential challenge remains the confident determination of the level of deuterium content for every peptide and the subsequent rendering of these data. The large number of data points in a conventional HDX experiment makes this task cumbersome and time-consuming. In addition, many HDX projects require the statistical cross-comparison of many data sets. Here we describe an integrated software solution that provides a comprehensive solution to these problems.
Algorithmic Detection
The HDX data analysis bottleneck has been mitigated to a large degree by several recently developed computational methods and tools [2–17]. Algorithms are available to compare the theoretical and experimental isotopic distributions using least-squares regression. The least-squares score can then be used to identify the presence of each peptide isotope cluster, determine its retention time (RT) range, and estimate the level of percent deuterium incorporation [4, 6, 8, 10, 12, 15, 18]. Methods to digest the protein sequence in silico and search for the expected peptides based on least-squares fit score have been described in an effort to increase sequence coverage [17]. Data consolidation approaches have been used with overlapping peptides to extract deuterium content at a higher resolution than that of the peptide fragments [9, 19]. Other solutions use maximum entropy or Fourier deconvolution methods [4, 8, 20] to determine the level of deuterium content. As a result of these efforts, with good quality data, much of the peptide detection and extraction of deuterium content in both the H2O control and on-exchange samples can be accomplished with reasonable accuracy and minimal human intervention.
Statistical Approaches and Data Visualization
Statistical methods have been described that evaluate the significance of differences for ligand screening using Tukey multiple comparison [21]. Other methods have been integrated into software workflows, ranging from the calculation of the aggregate %D for a sample to the usage of t-tests to determine the differences between the states of two proteins [11]. Examples include interactive displays and tools allowing for rapid adjustment and validation of peptide ion centroid m/z values. Automated visual representations of HDX data have been facilitated by new tools and scripts, which generate sequence coverage heat maps, perturbation tables, deuterium uptake plots, and 3D structure overlays of publication quality [9, 12, 16]. Alternate visualization approaches such as the mirror plot have been developed to aid in the interpretation of data from two samples [22].
Limitations of Current Software
Despite the list of software features given above, many existing solutions (including our own HD Desktop application) have limitations from a design and usability standpoint. Remote uploading requirements may make it difficult to manage large data sets and may compromise data confidentiality [6, 9]. Other applications are only available commercially or have commercial software dependencies [18]. Client server solutions usually employ a centralized backend database, which can lead to greater maintenance, archiving issues, and reduced data portability when tracking experimental HDX data sets over long periods of time. Other barriers to users are difficult installation requirements and the need for command line input. Lastly, some projects have been abandoned or provide little documentation or support.
Justification of Our Work
The need for HDX analysis continues to grow. The rate of data generation continues to increase attributable to improved methods, more sensitive instruments, higher throughput robotics, and more efficient liquid chromatography systems. Whereas previous HDX studies have usually been limited to two sample conditions, some laboratories are now able to conduct experiments involving the comparison of as many as 100 ligands in a single study. Our automation has recently been updated to include dual column, parallel high-performance liquid chromatography (HPLC), effectively doubling the rate of data acquisition. Furthermore, the proteins studied with HDX are becoming larger and more complex, contributing to increased data volume. Mass spectrometers require less material and are more efficient, allowing for the interrogation of proteins that were previously not possible. For example, a recent project from our laboratory presents multiple HDX data sets from the protein AMP kinase, which has a molecular weight of 146 kDa. A Mascot search found that AMP kinase generated 492 peptides confirmed by MS/MS (manuscript in preparation). HDX analysis on a protein of this magnitude would have been prohibitively difficult and time-consuming just a few years ago. Lastly, reviews on publications in the HDX space in 2001 [23] and 2009 [24] indicate that the number of laboratories conducting HDX experiments continues to grow.
There is a strong need for software that pushes the boundaries of HDX data analysis. Although existing software does address many needs of the HDX community, no single application to date integrates all of the essential features required for the rapid and robust processing, display, management, and statistical analysis of large HDX data sets. The difficulties to be overcome are integration of essential features in a user-friendly manner, addressing the issues associated with large volumes of data, and increasing the analysis speed for an individual experiment. Another point that has become clear is the extreme difficulty in fully automating the extraction of %D values from HDX data. Despite recent advancements, no algorithm is completely accurate, especially in complex situations and the possible presence of mass conflicts makes it necessary to have robust validation tools to go along with improved isotopic detection approaches. The lessons learned developing our previous two solutions, “The Deuterator” and “HD Desktop” (HDD), and a survey of the now mature HDX analysis landscape led to the development of a new application, named HDX Workbench (HDX-WB). The HDX-WB software manages target protein information, automates the detection of peptides, the extraction of %D values, and the display of %D versus time plots that contain intrinsic exchange rate information. In addition, HDX-WB provides interactive interfaces allowing for rapid data validation, adjustment, and presentation. New tools support current statistical methods and allow for the cross comparison of HDX data from hundreds of experiments and is accomplished on a multi-project scale. The software is available from our website http://hdx.florida.scripps.edu.
Methods
HDX Workbench was developed using Java/Swing technologies, allowing the core code base to be modular and the front end to provide a rich, interactive user experience. The open source projects BioJava [25] and JFreechart [26] were integrated to provide plots, spectral views, and sequence coverage heat maps. The software installation and data processing is accomplished locally, which is not a performance issue, as computer performance has been increasing to a point where server side processing is less of a necessity. This is also an improvement from the previous client-server backend database models, as users are now able to maintain data confidentiality and prevent large file uploads. The backend database has been replaced with an XML data model, which can be stored locally or onto a common network share. This facilitates data sharing from multiple users, archiving, data management, and addresses scalability concerns.
A common recurring task of HDX-WB is to average, or “co-add,” spectral data within a defined range of scans. Conducting this task using the standardized MS data formats, such as mzML and mzXML [27] has been computationally expensive because of inherent inefficiencies of XML, particularly in the area of read speed. As a result, HDX-WB now makes use of the Thermo Scientific MSFileReader library, which is a freely available COM based interface allowing direct programmatic access to raw file data using predefined methods. Using Java Native Interface (JNI) technology, we have developed a Java-COM bridge to access all functions within the MSFileReader library (e.g., co-add and extracted ion chromatogram [XIC]), therefore enabling invocation of local system calls and direct integration with the software. Our previous software parsed the open file format mzXML for this task, as this library was not available at the time of development. While this does limit the software to Thermo Scientific instrument data, in the future we anticipate that we will be able to integrate other native instrument libraries into HDX-WB, as well as the new mz5 data format [28]. By accomplishing this, much of the inefficiency issues associated with the XML based formats will be resolved and will allow the software to become MS platform independent.
Building on our previous efforts in processing bottom up HDX data and our experience with the Deuterator and HD Desktop, we have standardized a common workflow for HDX data analysis (Figure 1). The workflow begins at the data acquisition phase and ends with finalizing and archiving the data.
Figure 1.

The standardized workflow for HDX analysis outlines the common steps for data analysis of an HDX experiment. This workflow has been established over several years and has driven the design of the software
Protein Definition
Once experimental data is acquired, the first task is to define a protein using the Protein Editor interface (Figure 2). A list of candidate peptides and a backbone protein sequence are submitted with inputs of sequence, charge, and optionally retention time, database search score, and maximally H/D exchanged control (Dmax) %D measurements. The list of target peptides is usually obtained from searches with database software such as SEQUEST or Mascot. The results may either be converted to CSV format and imported, or entered directly through the software interface. The input retention time will be used if a peptide isotopic envelope is not detected algorithmically within the user-defined limits of ppm error, minimum signal intensity, and maximum theoretical fit score. The input peptide retention times will, however, be retained in the user interface to allow for manual curating of the data.
Figure 2.
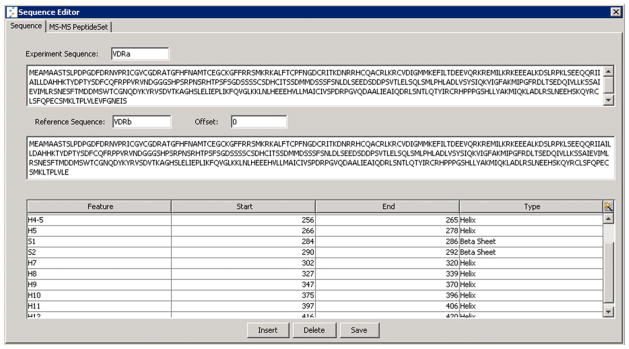
The protein editor allows for the creation and editing of all aspects of a protein used in HDX analysis. Sequence, peptide set, and secondary structure features can be managed using this interface. The protein configuration can be saved for reuse or shared with other users
It is common to have protein sequences that are different from the canonical sequence. To accommodate these situations, an optional reference sequence may be entered to allow sequence alignment between experimental versions of the protein sequence such as a sub-domain, and the intact protein sequence. The interface allows secondary structure features, such as helixes and beta sheets to be defined and propagated throughout the software. All of the protein and peptide set-related information can be saved for subsequent reuse or shared with other users.
Experiment Wizard
Upon completion of the protein definition, the next series of inputs includes information related to the experiment setup. This is accomplished via a wizard-based tool. The user provides the experiment input parameters and the software builds the template for the experiment. At this stage, instrument *.raw files are associated with each time point replicate using a drag and drop interface. Once this is completed, the user enters parameters for initial detection of peptides within the H2O control data files. The list of parameters for both the detection and experiment wizard is shown in Table 1.
Table 1.
Input Variables for the Experiment Wizard Up and Detection Algorithm
| Parameter | Description | Interface |
|---|---|---|
| Job name | Name of the detection job | Detection job |
| Job description | Description of the detection job | Detection job |
| Minimum peak intensity | Minimum peak intensity cutoff of the highest peak within a peptide isotopic cluster (absolute units from *.raw file) | Detection job |
| Minimum scan time minutes | Minimum retention time the algorithm will look for peptide isotope clusters | Detection job |
| Maximum scan time minutes | Maximum retention time the algorithm will look for peptide isotope clusters | Detection job |
| PPM Error | Mass accuracy error in parts per million | Detection job |
| Maximum signal score | Algorithmic score threshold related to χ2 sore (recommended settings = 10 for t=0 detect, and 20 for HDX detect) | Detection job |
| Retention time window | Expected retention time width over which the peptide is expected to elute (min) | Detection job |
| Instrument resolution – mz | Resolving power of the instrument – used for defining sub-range width | Detection job |
| Parent Project | Predefined project folder within which the experiment will be created | Experiment Wizard |
| Experiment name | Name of the experiment | Experiment Wizard |
| Number of proteins | Number of proteins in the experiment | Experiment Wizard |
| Number of samples | Number of samples | Experiment Wizard |
| Number of time points | Number of on-exchange time points, including the t=0 files | Experiment Wizard |
| Number of replicates | Number of replicates for each sample | Experiment Wizard |
| Temperature | On-exchange solution temperature | Experiment Wizard |
| pH | On-exchange solution pH | Experiment Wizard |
| Deuterium solution concentration | Decimal percent concentration of deuterium in solution used for the experiment (range = 0.00 to 1.00) | Experiment Wizard |
| Recovery estimate | Decimal percentage estimate for back exchange (range = 0.00 to −1.00); note that this value is ignored if the peptide set contains a Dmax value | Experiment Wizard |
| Description | Description of the experiment and placeholder for additional experiment attributes | Experiment Wizard |
Peptide Detection Algorithm
As in previous solutions [6, 8–10, 18], the software attempts to determine the correct RT range for each peptide automatically. The input retention times are provided in the peptide set, presented in the downstream interface, and used to define RT range in the event that the peptide is not found within the specified limits of the detection algorithm (e.g., signal intensity, ppm, and fit score). The HDX-WB peptide isotope detection algorithm follows a similar approach to HD Desktop, in which the theoretical distribution for the peptide is initially calculated with Qmass [29, 30], and is then compared with the experimental spectra with a least squares regression. For this version of software, we precalculate and save all possible theoretical distributions for a given peptide, and then compare them with the experimental data from individual scans. Because we now exclusively acquire data with high resolution FT-MS instruments (Orbitrap), we no longer require a co-add moving window approach as described in HD Desktop. Filters are then applied such as mass accuracy, m/z range, retention time range, and intensity to define the best matched %D value. This approach of indexing all possible theoretical distributions for each scan increases the speed of peptide detection without compromising accuracy.
In cases where MS/MS-based peptide identification is unavailable or limited in sequence coverage, HDX-WB provides the ability to extract all possible peptides from the protein sequence in place of a predetermined input peptide list. This operates in a manner similar to Hexicon [5]. To account for low enzymatic specificity, the software determines all possible combinations of peptide sequences between user defined residues in length and runs them through the detect algorithm however cleavage after H, K, P, and R may be eliminated from consideration based upon the Hamuro rules of pepsin specificity [31]. This has been shown to be a reasonable approach with novel or common enzymes used in HDX, such as pepsin or Fungal XIII, albeit somewhat more computationally expensive. The input list of peptides is not a requirement if this option is used and the approach has been shown to provide increased sequence coverage [5]. However, care should be taken when using this approach, as no product ion information is considered in the peptide identification.
An important consideration when searching MS1 data from predefined peptide sets is the detection of mass conflicts, in which a putative peptide can share the same or nearly the same mass with one, or many, other peptides within the peptide set. The software defines a mass conflict as two or more peptides within the peptide set whose theoretical monoisotopic mass is less than or equal to the error tolerance designated in the experiment set up. A mass conflict will indicate potential false positives from the detection process, as peptides with the same elemental composition will result in the same isotopic distribution and mass. HDX-WB provides the ability to automatically detect and flag peptides with mass conflicts within a user’s dataset, and allow the user to validate them manually.
HDX-WB is able to detect the potential presence of modifications from raw data; however, site localization is not possible because it is MS1 raw data being interrogated. For example, a search for one serine phosphorylation site on the peptide LULSSTVK would need to consider the forms LULpSSTVK and LULSpSTVK. Both of these are comprised of the same elemental composition and, as a result, the MS1 spectra are identical. The configuration of the available modifications within HDX-WB is made available in an external file allowing users to customize them as needed. These installed modifications are subsequently made available via the detection interface. Modifications may also be added directly into the peptide set if the site is well characterized. The software additionally provides support for detection of point mutations.
Calculation of %D
Calculation of the percent level of deuterium content is attained with either a theoretical fit or intensity weighted centroid m/z method. The former is accomplished by iterating from 0 to 100 % in theoretical deuterium incorporation using the software Qmass [29, 30] to retain the best theoretical match to the observed spectrum using a least squares approach. The second ‘centroid’ method is achieved by calculating the weighted average of all spectral data within defined m/z limits, and determining %D by comparing the result to defined minimum and maximum m/z values [32]. To automate the centroiding process, HDX-WB automatically defines the m/z limits by examination of the beginning (head) and end (tail) of the peptide isotopic cluster for consecutive peaks above 2 % of the most abundant peak within the isotope cluster.
HDX Workbench further disregards isotopes from interfering peptide ions or chemical noise by exploiting a “subranges” approach, which is based on three variables: (1) the equidistant nature of peptide isotopes based on the charge, (2) the resolving power of the instrument, and (3) the expected mass accuracy of the instrument. Using this information, an m/z sub-range width is defined and peptide isotope data can be expected to reside exclusively within these ranges. With higher resolution instruments, this approach has been shown to be effective at disregarding non-peak data in complex situations [9, 10]. As in HDD, HDX-WB allows for an estimate of the linear m/z-dependent resolving power exhibited by Fourier transform-based instruments. We have empirically determined that m/z ÷ 25,000 is appropriate for FT-MS instruments with a resolving power of 60,000 at m/z 400. When set to m/z ÷ 25,000, the subrange width is 0.05 at 1250 Th and 0.02 at 500 Th. Extraction of %D for a single sample is determined by simply calculating the mean of the individual time point replicate averages. The differential %D is determined by calculating the difference between the individual sample %D values [9].
Results
Data Review Interface
The primary graphical user interface (GUI) for data review and adjustment is shown in Figure 3. Most of the panels can be dynamically collapsed and resized to customize the workspace as needed. The tree view (Figure 3a) provides the primary means to review and manage experiment data. The underlying data for each experiment is stored in a consistent folder structure that is represented in this tree view. The folders are organized hierarchically, allowing users to navigate from the project to the experiment data, launch detection jobs, and view results. Completed HDX detection jobs are loaded into the data review interface in the right pane (Figure 3b–h), which presents a central point for the analysis of HDX experiment data. This pane is comprised of several individual components and tables with the goal of providing a comprehensive feature set to enable users to rapidly validate, curate, and visualize HDX data. The following series of paragraphs will describe the features of these tools.
Figure 3.

The Perturbation View provides a central feature set for integrating several graphic tools for editing and review of HDX data. (a) Tree pane is the primary launch point of the software which allows users to manage project and experiment data. A wide range of functions are available contextually from each node such as launching a detect job, editing experiment information, editing protein information, and loading result data. Only projects associated with the specific user are shown. (b) Peptide summary table provides experiment level information for each peptide. Peptide selection will launch results in the subsequent tools. (c) The extracted ion chromatogram (XIC) from selected replicates are color coded and displayed in a single view, facilitating validation of the peptide assignments and providing feedback regarding chromatographic consistency over several runs. (d) Deuterium uptake plots display the perturbation data for all time point replicates in one view, and are updated automatically. The results of t-tests between samples are shown above each time point to illustrate statistical significance. Also shown above are the intrinsic exchange rate plot (black) for the selected peptide calculated based on the work from Walter Englander. (e) Spectra pane displays the co-added raw spectral data for one or many replicates. Sub-range bars are colored grey. Shown above are the spectra for Apo replicate 1 at 0 and 3600 s rendered concurrently. (f) Information toolbar allows recalculation of input values for centroiding approach and displays additional information. Tables (g) and (h) allow users to load one to many replicates in the spectral and XIC panes for review. The replicate table (h) displays %D results for each time point replicate determined by both centroid and theoretical methods. Right clicking a cell will discard the replicate from the data set, automatically updating all views. (c), (d) (e) Support zoom in/out functionality
The peptide summary grid (Figure 3b) is the initial launching point for accessing all data within the perturbation view. The extracted ion chromatogram (XIC) is displayed in (Figure 3c), the %D versus time plot is shown in (Figure 3d), and the mass spectrum is displayed in (Figure 3e). Each row of the peptide summary grid (Figure 3b) is representative of one peptide along with corresponding information. Selection of a peptide will populate the saved percent deuterium results for this peptide into the other two tables (Figure 3g–h) as well as the deuterium uptake plots (Figure 3d). The “aggregate grid” (Figure 3g) displays the mean percent deuterium incorporation for each individual sample and time point. Selection of a cell will load all the replicate spectra and XICs into a single view. The “data point grid” shows all the individual replicate %D values used to calculate the mean values presented in the aggregate grid (Figure 3g). In addition, (Figure 3h) allows users to load or discard individual peptide replicate data. Both the aggregate and replicate grids support loading of multiple cells, allowing the visualization of the XIC and mass spectrum for many replicates within a single view.
When the spectra and XIC windows are populated, the information toolbar (Figure 3f) provides the means to adjust the retention and m/z ranges for all spectra currently showing in the pane. The sub-range width can be adjusted here at the individual peptide replicate level to include/exclude peak data. If a single replicate is loaded in the spectral pane, associated information will be displayed on the information portion of the toolbar, which includes centroid, centroid %D, scan range, and the theoretical %D. The textboxes are prepopulated with the m/z and retention time ranges that were considered to generate the spectra. When multiple replicates are selected, the range textboxes will be blank. Recalculation of multiple replicates will update the values for the textboxes that are manually populated; otherwise the saved values will be used. This, for example, would allow the user to update the retention times without updating the m/z ranges or sub-range width values for multiple replicates. The ability to select and curate multiple replicates simultaneously reduces the time it takes to curate a dataset dramatically. A conventional experiment processed in a few days using HD Desktop now can be processed and validated in a few hours.
The spectral pane (Figure 3e) displays the co-added spectrum for the selected peptide replicates. Sub-ranges are displayed in grey from which peak data are extracted to calculate the centroid. When multiple replicates are viewed at the same time, they are color coded to allow for differentiation of the spectra. The extracted ion chromatogram pane (Figure 3c) displays the selected ion chromatographic data across the entire gradient for the selected peptide replicates. The midpoint m/z value of the sub-range bar immediately preceding the centroid is determined and this value, ±0.01, is then extracted over the entire gradient. This permits users to investigate all possible scan ranges over which the selected peptide may elute. The retention time range used to co-add the spectra is displayed with vertical bars and shares the same color as the peptide XIC. Multi-XICs can also be loaded and are colored in the same manner, which can provide visual feedback related to chromatographic consistency. The input retention times from the peptide set are displayed in black in this pane for reference purposes. Deuterium uptake plots are presented in a smaller form directly in the main view (Figure 3d) and in a larger form in a tabbed pane, and are updated concurrently with curation. The software additionally can display the intrinsic uptake plots, based on the previous work of Walter Englander [33]. The P values from t-tests between the two samples at each time point are calculated on the fly and are displayed above every time point.
Sequence coverage view (Figure 4) is available via a tabbed pane and provides several types of coverage views across the backbone protein sequence. This tool renders coverage heat maps for individual samples (Figure 4a), perturbation (Figure 4b), or consolidated views. In the perturbation coverage view (Figure 4b), each peptide representation is accompanied with the percent deuterium incorporation and standard deviation. Users can select from various options, including rendering perturbation data, or heat map coverage for individual samples. Secondary structure features defined in the Protein Editor are rendered automatically according to their positional coordinates.
Figure 4.

Sequence coverage view. Panel (a) shows heat map coverage plots for a single sample. The level of deuterium incorporation %D for each time point is rendered in colored horizontal layers for each peptide, allowing for visualization of the rate of exchange in a single block. Panel (b) displays colored differential HDX data rendered onto the protein sequence. The difference level of deuterium incorporation measured as a percentage (delta %D), and it associated standard deviation are displayed on each peptide to facilitate the validation process. The bar at the beginning of each peptide represents the first two residues in which the exchange rate occurs too quickly to be measured. The secondary structure elements are rendered automatically from the features defined in the protein editor and provide additional context to the peptide coverage
Experiment Comparison
A central feature of HDX Workbench is the ability to manage and organize many large experimental data sets over long periods of time. While much of this task has been addressed by the data architecture and organization, custom tools are needed to interrogate, cross-compare, and implement statistical analysis of these data. The Experiment Comparison Tool (Figure 5) addresses these needs by automating the production of the common tabular format for perturbation data across several experiments. Users initially select the experiments of interest and the perturbation data from each experiment is extracted, collated, and sorted in an interactive tabular format. Perturbation data from like peptides are then consolidated, loaded into grid format, and colored according to the heat map legend. Individual peptides can be removed if they are not common to all samples or are not desired in the final peptide list. The results can be exported in graphical format or copied into a spreadsheet.
Figure 5.

Experiment comparison. This tool allows cross comparison of result data from multiple experiments. Shown above is perturbation data for Vitamin D receptor in the presence of 31 ligands displayed in a single view. Cells are colored according to a heat map legend, facilitating visual identification of regions of stability or exposure. Column selection, sorting, and formatting can be adjusted or ordered according to similarity. Peptides can be removed if not present in all experiments. Results can be exported from this view in text or graphical format. Right clicking a peptide will launch statistical analysis features in which the user may select a representative time point, and the results from all experiments for the selected peptide/ time point combination will be compared for statistical significance using one-way ANOVA and Tukey’s multiple comparison
Additional analysis is needed to determine if the differences between experiment results are statistically significant. In multi-ligand studies it is often important to define peptide/ ligand distinctions when the difference in %D values is small. While it may be straightforward to define disparity between ligands for a particular peptide if the difference between their perturbation values is large (exceeding 5 %D), it is much less obvious if the differences are smaller. In the plots view, as described previously, t-tests are conducted using the data from the two samples to generate a P value for each time point. Users of HDX-WB are able to conduct comparisons of data from more than two groups of experiments, thus the appropriate approach to determine statistical significance between these data is by using Tukey’s multiple comparison test. The goal of this analysis is to compare the perturbation data from multiple experiments and determine if there is a statistically significant difference. The first step in this process is the manual selection of a representative time point, and the replicates data points from both samples at this time point are used to conduct a one-way analysis of variance (ANOVA) to evaluate whether there is a divergence between the means of the experiments. If there is a difference, the Tukey method is then used to determine statistical significance if the resulting P value is less than 0.05. In the case where there is a comparison between two experiments, a t-test is used. This workflow has been described previously [21] and has been integrated into the Experiment Comparison Tool.
It is noteworthy to reiterate that this tool allows the researcher to extract information from large amounts of data. Each perturbation %D cell in the grid represents the results from deuterium uptake plots from two samples. For a typical experiment with seven time points in triplicate, each cell would represent 42 individual data points.
Once the review process has been completed for an entire experimental dataset, it can be marked as finalized and the comprehensive data can be exported as a single file in comma separated format (CSV). This is an important step in the process, as it completes the analysis workflow by providing a finalized data set archive for future reference that can be accessed independent from the software.
Conclusions
The development of HDX Workbench was motivated by the need for software to accommodate the ever-changing HDX landscape. Great emphasis was placed on achieving the three main goals of increased analysis speed, user-friendly graphic interface, and strong support for large HDX data sets. These developments have allowed us to process and refine data to a level acceptable for publication at the same rate as data are generated. The improvements brought by the software in terms of reduced time spent analyzing large volumes of HDX data, as well as on the cross comparison of many experiments have been presented. Data analysis for HDX experiments has been an active area of research. It is our belief that finally we have now reached the point with this software where the data analysis problem is not a bottleneck. This system has facilitated access to information-rich datasets from which we have gained important insights into the characterization of perturbations from many ligand bound proteins. We believe that software of this nature can lead to greater adoption of HDX technique. Future iterations of the software will include support for additional instrument data types, HDX sub-localization of deuteration data from MS/MS spectra, electron-transfer dissociation (ETD) and electron-capture dissociation (ECD) support, and improvements to algorithmic accuracy. HDX Workbench is available as a desktop application with no commercial software dependencies at http://hdx.florida.scripps.edu.
Acknowledgments
The authors acknowledge support for this work by US National Institutes of Health (NIH) National Institute of General Medical Sciences R01-GM084041.
References
- 1.Englander SW. Hydrogen Exchange Mass Spectrometry: A Historical Perspective. J Am Soc Mass Spectrom. 2006;17(11):1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abzalimov RR, Kaltashov IA. Extraction of Local Hydrogen Exchange Data from HDX CAD MS Measurements by Deconvolution of Isotopic Distributions of Fragment Ions. J Am Soc Mass Spectrom. 2006;17 (11):1543–1551. doi: 10.1016/j.jasms.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 3.Chik JK, Vande Graaf JL, Schriemer DC. Quantitating the statistical distribution of deuterium incorporation to extend the utility of H/D exchange MS data. Anal Chem. 2006;78(1):207–214. doi: 10.1021/ac050988l. [DOI] [PubMed] [Google Scholar]
- 4.Hotchko M, Anand GS, Komives EA, Ten Eyck LF. Automated extraction of backbone deuteration levels from amide H/2H mass spectrometry experiments. Protein Sci. 2006;15(3):583–601. doi: 10.1110/ps.051774906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lou XH, Kirchner M, Renard BY, Kothe U, Boppel S, Graf C, Lee CT, Steen JAJ, Steen H, Mayer MP, Hamprecht FA. Deuteration Distribution Estimation with Improved Sequence Coverage for HX/MS Experiments. Bioinformatics. 2010;26:1535–1541. doi: 10.1093/bioinformatics/btq165. [DOI] [PubMed] [Google Scholar]
- 6.Miller DE, Prasannan CB, Villar MT, Fenton AW, Artigues A. HDXFinder: automated analysis and data reporting of deuterium/ hydrogen exchange mass spectrometry. J Am Soc Mass Spectrom. 2012;23 (2):425–429. doi: 10.1007/s13361-011-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikamanon P, Pun E, Chou W, Koter MD, Gershon PD. “TOF2H”: a precision toolbox for rapid, high density/high coverage hydrogen-deuterium exchange mass spectrometry via an LC-MALDI approach, covering the data pipeline from spectral acquisition to HDX rate analysis. BMC Bioinforma. 2008;9:387. doi: 10.1186/1471-2105-9-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmblad M, Buijs J, Hakansson P. Automatic Analysis of Hydrogen/Deuterium Exchange Mass Spectra of Peptides and Proteins Using Calculations of Isotopic Distributions. J Am Soc Mass Spectrom. 2001;12:1153–1162. doi: 10.1016/S1044-0305(01)00301-4. [DOI] [PubMed] [Google Scholar]
- 9.Pascal BD, Chalmers MJ, Busby SA, Griffin PR. HD desktop: An integrated platform for the analysis and visualization of H/D exchange data. J Am Soc Mass Spectrom. 2009;20:601–610. doi: 10.1016/j.jasms.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pascal BD, Chalmers MJ, Busby SA, Mader CC, Southern MR, Tsinoremas NF, Griffin PR. The Deuterator: software for the determination of backbone amide deuterium levels from H/D exchange MS data. BMC Bioinforma. 2007;8:156. doi: 10.1186/1471-2105-8-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu S, Liu L, Uzuner U, Zhou X, Gu M, Shi W, Zhang Y, Dai SY, Yuan JS. HDX-analyzer: a novel package for statistical analysis of protein structure dynamics. BMC Bioinforma. 2011;12(Suppl 1):S43. doi: 10.1186/1471-2105-12-S1-S43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slysz GW, Baker CA, Bozsa BM, Dang A, Percy AJ, Bennett M, Schriemer DC. Hydra: Software for tailored processing of H/D exchange data from MS or tandem MS analyses. BMC Bioinforma. 2009;10:162. doi: 10.1186/1471-2105-10-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weis DD, Engen JR, Kass IJ. Semi-Automated Data Processing of Hydrogen Exchange Mass Spectra Using HX-Express. J Am Soc Mass Spectrom. 2006;17(12):1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 14.Weis DD, Hotchko M, Wales TE, Ten Eyck LF, Engen JR. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J Am Soc Mass Spectrom. 2006;17 (11):1498–1509. doi: 10.1016/j.jasms.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 15.Woods VLJ, Hamuro Y. High resolution, high-throughput amide deuterium exchange-mass spectrometry (DXMS) determination of protein binding site structure and dynamics: utility in pharmaceutical design. J Cell Biochem. 2001;(Suppl 37):89–98. doi: 10.1002/jcb.10069. [DOI] [PubMed] [Google Scholar]
- 16.Kaven D, Man P. MSTools—Web based application for visualization and presentation of HXMS data. Int J Mass Spectrom. 2010;302:53–58. [Google Scholar]
- 17.Kreshuk A, Stankiewicz M, Lou X, Kirchner M, Hamprecht FA, Mayer MP. Automated detection and analysis of bimodal isotope peak distributions in H/D exchange mass spectrometry using HEXICON. J Am Soc Mass Spectrom. 2011;302:125–131. [Google Scholar]
- 18.Kan ZY, Mayne L, Chetty PS, Englander SW. ExMS: Data Analysis for HX-MS Experiments. J Am Soc Mass Spectrom. 2011;22:1906–1915. doi: 10.1007/s13361-011-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burns-Hamuro LL, Hamuro Y, Kim JS, Sigala P, Fayos R, Stranz DD, Jennings PA, Taylor SS, Woods VL., Jr Distinct interaction modes of an AKAP bound to two regulatory subunit isoforms of protein kinase A revealed by amide hydrogen/deuterium exchange. Protein Sci. 2005;14(12):2982–2992. doi: 10.1110/ps.051687305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Marshall AG. A Universal Algorithm for Fast and Automated Charge State Deconvolution of Electrospray Mass-to-Charge Ratio Spectra. J Am Soc Mass Spectrom. 1998;9(3):225–233. doi: 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- 21.Chalmers MJ, Pascal BD, Willis S, Zhang J, Iturria SJ, Dodge JA, Griffin PR. Methods for the Analysis of High Precision Differential Hydrogen Deuterium Exchange Data. Int J Mass Spectrom. 2011;302(1–3):59–68. doi: 10.1016/j.ijms.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houde D, Berkowitz SA, Engen JR. The Utility of Hydrogen/ Deuterium Exchange Mass Spectrometry in Biopharmaceutical Comparability Studies. J Pharm Sci. 2011;100(6):2071–2086. doi: 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Englander SW. Hydrogen exchange. Nat Struct Biol. 2001;8(9):1–2. doi: 10.1038/nsb0901-741. [DOI] [PubMed] [Google Scholar]
- 24.Engen JR. Analysis of Protein Conformation and Dynamics by Hydrogen/Deuterium Exchange MS. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holland RCG, Down T, Pocock M, Prlić A, Huen D, James K, Foisy S, Dräger A, Yates A, Heuer M, Schreiber MJ. BioJava: an Open-Source Framework for Bioinformatics. Bioinformatics. 2008;24(18):2096–2097. doi: 10.1093/bioinformatics/btn397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilbert D, Morgner T. JFreeChart, a free java class library for generating charts. 2007 Available at http://www.jfree.org/jfreechart/
- 27.Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, Aebersold R. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 28.Wilhelm M, Kirchner M, Steen JA, Steen H. mz5: Space- and Time-efficient Storage of Mass Spectrometry Data Sets. Mol Cell Proteom. 2012;11(1):1–5. doi: 10.1074/mcp.O111.011379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rockwood AL, Haimi P. Efficient calculation of accurate masses of isotopic peaks. J Am Soc Mass Spectrom. 2006;17(3):415–419. doi: 10.1016/j.jasms.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Rockwood AL, Van Orman JR, Dearden DV. Isotopic compositions and accurate masses of single isotopic peaks. J Am Soc Mass Spectrom. 2004;15(1):12–21. doi: 10.1016/j.jasms.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 31.Hamuro Y, Coales SJ, Molnar KS, Tuske SJ, Morrow JA. Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun Mass Spectrom. 2008;22:1041–1046. doi: 10.1002/rcm.3467. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2(4):522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai Y, Milne JS, Englander SW. Primary Structure Effects on Peptide Group Hydrogen Exchange. Protein Struct Funct Genet. 1993;17 (1):75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]