

Abstract
BACKGROUND
The onset of puberty is first detected as an increase in pulsatile secretion of gonadotropin-releasing hormone (GnRH). Early activation of the hypothalamic–pituitary–gonadal axis results in central precocious puberty. The timing of pubertal development is driven in part by genetic factors, but only a few, rare molecular defects associated with central precocious puberty have been identified.
METHODS
We performed whole-exome sequencing in 40 members of 15 families with central precocious puberty. Candidate variants were confirmed with Sanger sequencing. We also performed quantitative real-time polymerase-chain-reaction assays to determine levels of messenger RNA (mRNA) in the hypothalami of mice at different ages.
RESULTS
We identified four novel heterozygous mutations in MKRN3, the gene encoding makorin RING-finger protein 3, in 5 of the 15 families; both sexes were affected. The mutations included three frameshift mutations, predicted to encode truncated proteins, and one missense mutation, predicted to disrupt protein function. MKRN3 is a paternally expressed, imprinted gene located in the Prader–Willi syndrome critical region (chromosome 15q11–q13). All affected persons inherited the mutations from their fathers, a finding that indicates perfect segregation with the mode of inheritance expected for an imprinted gene. Levels of Mkrn3 mRNA were high in the arcuate nucleus of prepubertal mice, decreased immediately before puberty, and remained low after puberty.
CONCLUSIONS
Deficiency of MKRN3 causes central precocious puberty in humans. (Funded by the National Institutes of Health and others.)
The onset of puberty is first detected as an increase in the amplitude and frequency of pulses of gonadotropin-releasing hormone (GnRH) after a quiescent period during childhood. The reemergence of pulsatile GnRH secretion leads to increases in the secretion of the gonadotropins, luteinizing hormone and follicle-stimulating hormone (FSH), by the pituitary gland and the consequent activation of gonadal function.1 Early activation of the hypothalamic–pituitary–gonadal axis results in gonadotropin-dependent precocious puberty, also known as central precocious puberty, which is clinically defined by the development of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys. Pubertal timing is influenced by complex interactions among genetic, nutritional, environmental, and socioeconomic factors.2,3 The timing of puberty is associated with risks of subsequent disease; earlier age of menarche in girls is associated with increased risks of breast cancer, endometrial cancer, obesity, type 2 diabetes, and cardiovascular disease.4 Central precocious puberty has also been associated with an increased incidence of conduct and behavior disorders during adolescence.5
Compelling evidence of the influence of genetic factors on pubertal timing has been provided by population studies.6 The role of genetic factors is also illustrated by the similar age at menarche in mothers and daughters and among members of an ethnic group and by a greater concordance of pubertal timing in monozygotic than in dizygotic twins.7–9 Familial segregation analysis has shown that 27.5% of cases of central precocious puberty are familial and suggests autosomal dominant transmission with incomplete sex-dependent penetrance.10 Despite the data suggesting that age at the onset of pubertal development is primarily driven by genetic factors, the genetic determinants of the timing of human pubertal development and, in particular, central precocious puberty are largely unknown.
Extensive efforts have been made to elucidate the mechanisms that reactivate pulsatile GnRH secretion at the time of puberty. In the past decade, several genes have been identified in the complex network of inhibitory, stimulatory, and permissive neuroendocrine factors involved in the control of puberty onset. Studies in rodents and primates have shown that an enhancement of excitatory inputs and a reduction in inhibitory factors contribute to GnRH secretion and the initiation of puberty.1,11 On the basis of this knowledge, several studies in humans have used a candidate-gene approach in an attempt to detect genes associated with pubertal disorders. However, although an increasing number of genes have been implicated in congenital isolated hypogonadotropic hypogonadism and the Kallmann syndrome,12,13 only a few, rare molecular defects have been identified in patients with central precocious puberty, and no strong association has been proved.14–18 Only two mutations — one mutation in the gene encoding kiss-peptin-1 (KISS1) and one in the gene encoding its receptor (KISS1R) — have been associated with central precocious puberty, despite screening of a relatively large cohort of patients for mutations in these genes, indicating that isolated mutations in KISS1 and KISS1R genes are uncommon causes of central precocious puberty.19,20 We therefore sought to identify genetic causes of central precocious puberty by performing an exome sequence analysis in 15 families with central precocious puberty.
METHODS
PATIENTS
We selected for our study 15 probands with central precocious puberty and their affected and unaffected family members (Fig. 1; and Fig. S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org). Whole-exome sequencing was performed for 40 members of these families, 32 with central precocious puberty (27 females and 5 males) and 8 with normal pubertal timing (5 females and 3 males). Central precocious puberty was diagnosed on the basis of clinical signs of progressive pubertal development before the age of 8 years in girls and 9 years in boys; pubertal basal luteinizing hormone levels, GnRH-stimulated luteinizing hormone levels, or both; advanced bone age (determined with the use of the Greulich and Pyle method21), and normal results on magnetic resonance imaging of the central nervous system (Table 1, and Table S1 in the Supplementary Appendix). The ancestries of the families with MKRN3 defects were established by means of verbal report to clinical investigators (Table 1). The protocol was approved by the ethics committee of Sao Paulo University. Written informed consent was obtained from all participants. The last two authors vouch for the accuracy and completeness of the data and the fidelity of the study to the protocol.
Figure 1. Pedigrees of the Families with MKRN3 Mutations.

Squares indicate male family members, circles female family members, black symbols clinically affected family members, symbols with black circles asymptomatic carriers, symbols with an X deceased family members, symbols with a question mark family members whose phenotype is unknown, and arrows the proband in each family. The MKRN3 genotype is shown for family members whose DNA was available for genetic studies. A star indicates that the patient was screened by means of Sanger sequencing only. NM denotes nonmutated.
Table 1.
Clinical and Hormonal Features of 12 Patients with Central Precocious Puberty and MKRN3 Mutations in Five Families.*
| Patient No. | Sex | MKRN3 Mutation | Initial Clinical Presentation | Time of Diagnosis | LH† | FSH† | Estradiol† | Testosterone† | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DNA | Protein | Condition | Age | Age | Tanner Stage | Bone Age | Basal | After GnRH Stimulation | Basal | After GnRH Stimulation | ||||
|
|
|
|
|
|
||||||||||
| yr | yr | yr | IU/liter | IU/liter | pg/ml | ng/dl | ||||||||
| Family A | ||||||||||||||
| III-1 | Female | 637delC | Arg213Glyfs*73 | Thelarche | 5.7 | 6.5 | 2 | 7.7 | 0.8 | 13.6 | 3.8 | — | 25 | — |
| III-2 | Male | 637delC | Arg213Glyfs*73 | Testicular enlargement | 8.0 | 8.7 | 3 | 11.0 | 2.9 | 20.0 | 2.5 | — | — | 78 |
| III-3 | Female | 637delC | Arg213Glyfs*73 | Thelarche | 6.5 | 6.7 | 2 | 7.8 | 1.1 | 16.7 | 4.5 | — | <15 | — |
| Family B | ||||||||||||||
| III-1 | Female | 1171_1172insA | Tyr391* | Thelarche | 6.2 | 7.0 | 3 | 7.0 | <0.6 | 36.0 | 1.13 | 17.9 | 13.2 | — |
| III-2 | Female | 1171_1172insA | Tyr391* | Thelarche | 5.7 | 6.0 | 3 | 6.0 | <0.6 | 37.3 | 1.13 | 27.9 | 11 | — |
| Family C | ||||||||||||||
| III-1 | Female | 1095G→T | Arg365Ser | Thelarche | 6.2 | 6.4 | 2 | 9.4‡ | <0.1 | 7.3 | 7 | 7.3 | 49 | — |
| III-2 | Male | 1095G→T | Arg365Ser | Testicular enlargement and pubarche | Unknown | 9.7 | 3 | 12.0 | 2.0 | 19.5 | 4.4 | 19.5 | — | 67 |
| III-3 | Female | 1095G→T | Arg365Ser | Thelarche and pubarche | 5.4 | 5.7 | 2 | 8.5 | 0.5 | 12.3 | 3.6 | 14.5 | <15 | — |
| Family D | ||||||||||||||
| III-2 | Male | 475_476insC | Ala162Glyfs*14 | Testicular enlargement and pubarche | 5.9 | 8.1 | 3 | 10.0 | 1.18 | 6.7 | 1.53 | 2.6 | — | 116 |
| III-3 | Male | 475_476insC | Ala162Glyfs*14 | Testicular enlargement and pubarche | Unknown | 9.7 | 3 | 9.7 | 1.6 | 10.9 | 0.8 | 2.6 | — | 548 |
| Family E | ||||||||||||||
| III-1 | Male | 475_476insC | Ala162Glyfs*14 | Testicular enlargement and pubarche | 8.5 | 8.8 | 3 | 11.5 | 4.1 | — | 3.1 | — | — | 216 |
| III-2 | Female | 475_476insC | Ala162Glyfs*14 | Thelarche | 5.0 | 6.5 | 2 | 8.3 | — | 7.4 | 13 | — | 13 | — |
The available information on family ancestry is as follows: Family A reported Northwest European ancestry (from Wales, living in the United States); Family B reported living in Brazil, but their ancestry was not reported; Family C reported Northwest European ancestry (from Belgium and living in Belgium); Family D reported living in Brazil, but their ancestry was not reported; and Family E reported European ancestry (the ancestry of the proband’s father is Italian, and the mother reported European ancestry; specific details of the mother’s country of origin in Europe are not available). FSH denotes follicle-stimulating hormone, and LH luteinizing hormone. Dashes indicate that data are not available.
Normal prepubertal levels of testosterone and estradiol are less than 12.0 ng per deciliter and less than 15.0 pg per milliliter, respectively; the normal prepubertal basal level of LH is less than 0.15 IU per liter, with a peak level below 5.0 IU per liter in both girls and boys. Levels were measured at the time of diagnosis. Normal pubertal levels of FSH have not been established because the normal ranges for pubertal and prepubertal FSH overlap.
This was the patient’s bone age when she was 7.4 years old.
HORMONE ASSAYS
Serum levels of luteinizing hormone, FSH, testosterone, and estradiol were determined with the use of immunochemiluminometric assays.22 The interassay coefficient of variation was 5% or less for all assays.
During the GnRH stimulation test, serum levels of luteinizing hormone and FSH were measured 15 minutes before the intravenous administration of 100 μg of GnRH, at the time of administration, and 15, 30, 45, and 60 minutes after administration. In both sexes, basal luteinizing hormone levels higher than 0.15 U per liter were considered to be pubertal levels, and peak GnRH-stimulated luteinizing hormone levels higher than 5.0 U per liter were considered to be pubertal responses.22 Basal estradiol levels higher than 15 pg per milliliter and basal testosterone levels higher than 12 ng per deciliter were considered to be pubertal levels.
GENETIC ANALYSIS
Genomic DNA was extracted from peripheral-blood leukocytes. Whole-exome sequencing was performed for selected patients (at the Broad Institute), as previously described23 (see the Supplementary Appendix for details). We confirmed the identification of variants in the coding region of MKRN3 with the use of polymerase-chain-reaction (PCR) amplification followed by sequencing of the products with the use of the conventional Sanger method. For comparisons of the prevalence of truncating variants in the families and the exome variant server (hosted by the National Heart, Lung, and Blood Institute), we used Fisher’s exact test to compare the number of frame-shift or nonsense mutation carriers, counting each family member in the exome variant server once and counting both parents in each family to account for mutation searches across multiple offspring. For the analysis of unique variants, we included only one person (or one pair of parents) per variant in the analysis.
ASSAYS IN MICE
RNA was extracted from the arcuate nucleus of the hypothalamus of three male and three female mice at postnatal days 10, 12, 15, 18, 22, 26, 30, and 45. Pubertal development occurred between days 15 and 30 in these mice, as indicated by the increase in the expression of hypothalamic Tac2 (or neurokinin B) messenger RNA (mRNA).24 RNA was reverse-transcribed, and quantitative real-time PCR analysis was then performed to measure Mkrn3 mRNA levels, normalized to ribosomal protein L19 (see the Supplementary Appendix).
RESULTS
SEQUENCE ANALYSIS
Whole-exome sequencing performed in 40 members of 15 families with central precocious puberty identified 304,930 variants. Rigorous criteria were used to filter the variants and identify the mutations likely to be causative of the phenotype for central precocious puberty. We first analyzed exome-sequence data from a total of 15 members of the 3 largest families with pedigrees that were consistent with a dominant mode of inheritance (i.e., those families having affected members in multiple generations). Among the persons who underwent exome sequencing, we identified heterozygous nonsynonymous variants that were present in affected family members and absent in unaffected family members. Given the dominant inheritance pattern and rarity of presentation of familial precocious puberty, we excluded all variants with a minor allele frequency of more than 0.01% in either the 1000 Genomes database25 or the exome variant server.26 In addition, we excluded all putative variants that were also present in 50 of the 1000 Genomes control samples included in the variant calling process. In applying these criteria, we identified candidate genes within each family (4 in Family A, 65 in Family B, and 3 in Family F). The reason why a larger number of candidate genes were identified in Family B was that exome data were available for only 3 members of this family, as compared with 6 members each for Families A and F. One gene, MKRN3 (ENSG00000179455, gene identification number 7681), was identified in 2 families. No single gene was identified in all 3 families. Families A and B had novel frameshift mutation variants in MKRN3 (p.Arg213Glyfs*73 and p.Tyr391fs*, respectively) (Fig. 1 and 2).
Figure 2. MKRN3 Domains and the Mutations Identified in the Study Families.
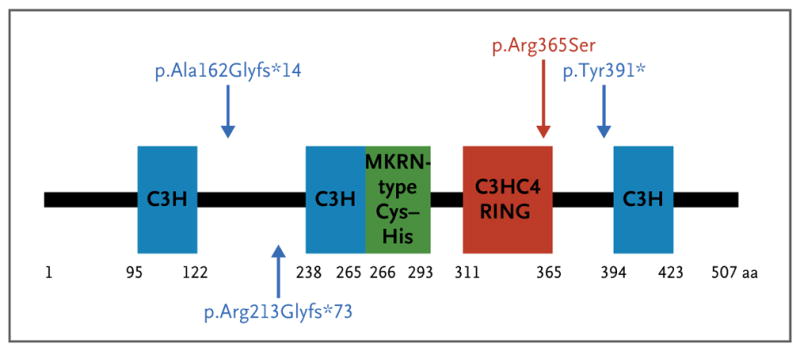
MKRN3 has four zinc-finger domains: three C3H motifs (blue) and one C3HC4 RING motif (red), which is responsible for ubiquitin ligase activity. The MKRN-specific Cys–His domain (green) is of uncertain function. The numbers correspond to the amino acid positions in the protein. Blue mutation labels and arrows indicate the location of frameshift mutations; the red mutation and arrow indicate the location of a missense mutation.
We then examined exome data from the additional 25 members of the other 12 families in the study and found another novel frameshift mutation in MKRN3 (p.Ala162Glyfs*14) in Families D and E. A novel missense variant (p.Arg365Ser) was identified in Family C (Fig. 1 and 2). This missense variant is predicted to be “probably damaging” (likely to disrupt protein function) on the basis of a Polymorphism Phenotyping, version 2 (PolyPhen2), score of 1.0 and a Protein Analysis through Evolutionary Relationship (PANTHER) score of 0.95 for the probability of being deleterious. (The range for both scores is 0 to 1.0, with 0 indicating that a change is predicted to be neutral and 1.0 indicating that it is most likely to be deleterious.) We confirmed all variants with the use of Sanger sequencing and tested for cosegregation between the variant and central precocious puberty in an additional 8 members from Families A through E who did not undergo exome sequencing. MKRN3 is an imprinted gene that is expressed only from the paternal allele.27 All affected family members inherited their mutations from their fathers, indicating perfect segregation in accordance with the imprinted mode of inheritance. The one heterozygous carrier known to have inherited his mutation from his mother (Patient II-1 in Family A) was unaffected, as expected. The remaining 10 families did not have any detectable rare coding variants in MKRN3. The prevalence of truncating variants (4 variants, 3 of which were unique, in 15 families) is much greater than that seen in population-based databases (5 variants, 4 unique, in approximately 6500 persons in the exome variant server) (P<5.0×10−8 for the prevalence of all variants, and P<2.6×10−6 for the prevalence of unique variants). Furthermore, the segregation with precocious puberty in the precise manner predicted for this imprinted gene provides additional strong and independent evidence that the MKRN3 frameshift mutations identified lead to precocious puberty in these families.
GENOTYPE–PHENOTYPE CORRELATION
In total, we identified 15 patients (8 female patients and 7 male patients) with central precocious puberty who carried mutations in MKRN3 that are predicted to be loss-of-function or damaging mutations. Each of these patients had clinical and hormonal features that are typical of premature activation of the reproductive axis, including early pubertal signs, such as breast development or testicular enlargement and pubic hair, advanced linear growth and bone age, and elevated basal luteinizing hormone levels, elevated GnRH-stimulated luteinizing hormone levels, or both. The median age at the onset of puberty in the girls was 5.75 years, ranging from 5.0 to 6.5 years (Table 1). In boys with mutations in MKRN3, the median age at the onset of puberty was 8.1 years, ranging from 5.9 to 8.5 years (Table 1). The precise time of onset of puberty was not clear in two boys, but clinical and laboratory assessment confirmed the diagnosis of central precocious puberty. The proband in Family A and her brother (Patients III-1 and III-2 in Fig. 1) have esotropia, which is a minor diagnostic criterion for the Prader–Willi syndrome.28 The boy also has a renal cyst. Neither child has any other features of the syndrome, nor do any of the other affected patients. Additional details are available in the Supplementary Appendix.
MKRN3 MRNA LEVELS IN THE MURINE ARCUATE NUCLEUS
The hypothalamic arcuate nucleus is the site of expression of several genes known to be important for puberty, including Kiss1 and Tac2.29,30 To assess Mkrn3 mRNA levels in the arcuate nucleus of mice, we performed quantitative real-time PCR (Fig. 3). In both male and female mice, Mkrn3 mRNA levels were highest on postnatal days 10 and 12, began to decline on day 15, and reached a nadir by days 18 to 22, at which time Mkrn3 expression was 10 to 20% of the levels detected at 10 days. The timing of the decline in Mkrn3 expression correlated with the ages at which arcuate Kiss1 and Tac2 expression have been shown to increase, heralding the onset of puberty.24,31 The expression of Mkrn3 remained low through day 45, the oldest age at which the mice were tested (Fig. 3).
Figure 3. Mkrn3 Messenger RNA (mRNA) Levels in the Hypothalamic Arcuate Nucleus of Male and Female Mice during Postnatal Development.
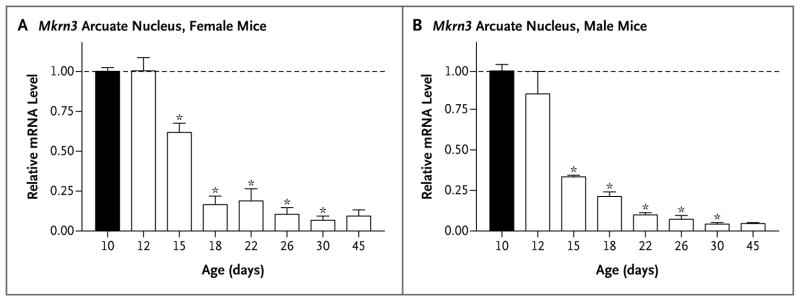
Total RNA was extracted from the hypothalamic arcuate nucleus of male and female mice at the ages indicated (number of days after birth), and Mkrn3 mRNA was quantified with the use of real-time polymerase-chain-reaction assay. The bar graphs show the relative change in mRNA levels in female mice and male mice, as compared with the level on postnatal day 10 (black bars), normalized to levels of endogenous ribosomal protein L19 mRNA. Mean (±SE) values are shown for three different mice at each age, with each measurement performed in triplicate. Significant differences (P<0.05) were measured by means of a one-way analysis of variance with a post hoc Tukey multiple-comparison test. Asterisks indicate P<0.001.
DISCUSSION
How puberty is initiated is an enigma that still captivates scientists. Many of the recent advances in our understanding of the mechanisms involved in reactivation of the hypothalamic–pituitary–gonadal, or reproductive, axis at puberty have been based on the characterization of genetic mutations associated with reproductive disorders in humans. The majority of the mutations to date were identified in patients with isolated hypogonadotropic hypogonadism, a disorder that is much less common than central precocious puberty.32,33 Genomewide association studies have identified multiple loci associated with pubertal timing,4 but aside from LIN28B (a hetero-chronic regulator of developmental timing),34 it has been difficult to implicate specific novel genes within these associated loci. Despite numerous efforts to identify genes associated with the premature activation of puberty, only two rare mutations in candidate genes have been identified in patients with central precocious puberty.19,20 To our knowledge, no strong evidence for additional causal mutations has been presented.
In our analysis of whole-exome–sequencing data in 40 members of 15 families with central precocious puberty, we identified a single gene, MKRN3, encoding the makorin RING-finger protein 3, with variants predicted to be deleterious in 5 families. MKRN3 is an intronless gene located on chromosome 15q11.2, in the Prader–Willi syndrome critical region.35 This gene is maternally imprinted, and only the paternal allele is expressed.35 The makorin protein family is distinguished by a characteristic combination of zinc-finger motifs; these proteins include two or three copies of a C3H motif in the N-terminal, followed by a novel Cys–His configuration, a C3HC4 RING zinc finger, and a final C3H motif.36 C3H zinc-finger motifs have been implicated in RNA binding, whereas the RING zinc-finger motif is found in most E3 ubiquitin ligases and is responsible for ubiquitin-ligase activity.37 The widespread species conservation of the makorin protein family suggests that it plays one or more vital roles in cells, with high levels of expression in the developing nervous system.36 MKRN3, on the other hand, is conserved only in therian mammals, and its precise function has not yet been determined.37
The deletion of chromosome 15q11-q13, which encompasses MKRN3, contributes to the Prader–Willi syndrome, but it is not yet known which specific genes in this region are related to the syndrome.38 Analyses of balanced translocations in patients with the Prader–Willi syndrome have implicated the SNURF–SNRPN locus, which is telomeric to MKRN3. One report described 2 patients with all the features of the Prader–Willi syndrome who did not have a deletion of MKRN3, suggesting that MKRN3 deletion is not necessary to cause the syndrome.38 This report also described a patient with a paternal deletion of MKRN3, MAGEL2, and NDN who had only a few features of the Prader–Willi syndrome: obesity, developmental delay, and a high pain threshold. This patient also had signs of puberty at the age of 7 years 6 months, with advanced bone age. The patient received a diagnosis of central precocious puberty, which was supported by her response to treatment with triptorelin.38 Given our data, the deletion of MKRN3 is probably the cause of early puberty in this patient.38 It is uncertain whether the obesity, developmental delay, and high pain threshold in this patient were attributable to the MKRN3 deletion. We have detailed clinical and hormonal data from 12 of the 15 persons with loss-of-function mutations in MKRN3. In this series of 12 patients, 2 had esotropia, which is a minor diagnostic criterion for the Prader–Willi syndrome.28 Other features of the syndrome were not reported. The esotropia cannot be definitively attributed to MKRN3 deletion, because esotropia can be present in up to 5% of the population.39
Three of the four mutations identified in MKRN3 in our series were frameshift mutations resulting in premature stop codons, whereas the fourth, a missense variant (p.Arg365Ser), is predicted to interfere with protein function (Fig. 2). The arginine at position 365 is located in the C3HC4 RING domain responsible for the ubiquitin ligase activity and is evolutionarily highly conserved (Fig. 2). Definitive confirmation that this missense variant causes loss of function awaits the availability of a functional assay. Although the function of MKRN3 is not well understood, and the mechanism by which MKRN3 mutations result in early activation of the central reproductive axis are not yet known, our genetic data are sufficiently compelling and statistically strong to invoke a causative role for MKRN3 in central precocious puberty.
The inheritance pattern in the affected families is consistent with the expression of MKRN3 from the paternally inherited allele only. For example, Patient II-1 in Family A inherited the mutant MKRN3 allele from his mother; because this allele was silenced, he did not have the central precocious puberty phenotype. Patients II-1 in Families C, D, and E were apparently asymptomatic heterozygous carriers of deleterious MKRN3 mutations, but since we were unable to obtain reliable pubertal histories or DNA from their parents, the parental source of their mutations is unknown. Of the 15 patients with central precocious puberty and MKRN3 mutations, 7 were male; this nearly equal sex distribution contrasts with the striking predominance of central precocious puberty in girls that has been reported previously.10 The similar incidence of central precocious puberty in association with MKRN3 mutations in the two sexes in the affected families is consistent with the autosomal pattern of inheritance. In contrast, in the 10 families without mutations in MKRN3, all affected members were female, an incidence that is similar to that reported previously10 (Fig. S1 and Table S1 in the Supplementary Appendix). Screening for mutations of MKRN3 in sporadic cases of central precocious puberty, which affects primarily girls, will add information about the role of this gene in pubertal timing. The identification of mutations in MKRN3 in families of diverse ancestry shows that the effects of MKRN3 mutations in central precocious puberty are generalizable and are not restricted to a specific ethnic group. It is possible that Families D and E are distantly related; we have neither confirmed nor excluded this possibility.
MKRN3 is associated with protein ubiquitination, in which a ubiquitin moiety is attached to a protein, thus tagging it for movement to the proteasome, where it is degraded. Ubiquitination can also be an indicator for signal transduction, cell-cycle regulation, differentiation and morphogenesis, and other nonproteolytic fates. The precise mechanism by which the deletion of MKRN3 leads to the early reactivation of pulsatile GnRH secretion remains to be elucidated. We found increased levels of Mkrn3 mRNA at young ages in the arcuate nucleus of male and female mice, with a striking reduction in levels immediately before puberty and low levels in adulthood (Fig. 3). The arcuate nucleus is considered to play a key role in puberty control in mice,29 and the pattern of Mkrn3 mRNA expression correlates with an inhibitory effect on the initiation of puberty in these animals. These data are in agreement with the identification of a loss-of-function mutation in patients with central precocious puberty, corroborating the view that the mutation has an inhibitory effect on the secretion of GnRH. The initiation of puberty is thought to result from a decrease in factors that inhibit the release of GnRH combined with an increase in stimulatory factors. Studies of hypogonadotropic hypogonadism have led to the identification of genes encoding factors that have stimulatory input.12,13 In contrast, MKRN3 seems to have an inhibitory role in humans.
Supplementary Material
Acknowledgments
Supported by grants from the National Institutes of Health (1K23HD073351, to Dr. Dauber), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (3806-11-1, to Dr. Abreu), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (1F05HD072773-01, to Dr. Abreu; U54HD28138, as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research; and R21HD066495, to Dr. Kaiser), Conselho Nacional de Desenvolvimento Científico e Tecnológico (302825/2011-8, to Dr. Latronico; and 305743/2011-2, to Dr. Mendoca), and Fundação de Amparo à Pesquisa do Estado de São Paulo (2005/04726-0, to Dr. Latronico).
Footnotes
Drs. Abreu, Dauber, Latronico, and Kaiser contributed equally to this article.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Terasawa E, Fernandez DL. Neurobio-logical mechanisms of the onset of puberty in primates. Endocr Rev. 2001;22:111–51. doi: 10.1210/edrv.22.1.0418. [DOI] [PubMed] [Google Scholar]
- 2.Gajdos ZK, Hirschhorn JN, Palmert MR. What controls the timing of puberty? An update on progress from genetic investigation. Curr Opin Endocrinol Diabetes Obes. 2009;16:16–24. doi: 10.1097/MED.0b013e328320253c. [DOI] [PubMed] [Google Scholar]
- 3.Palmert MR, Boepple PA. Variation in the timing of puberty: clinical spectrum and genetic investigation. J Clin Endocrinol Metab. 2001;86:2364–8. doi: 10.1210/jcem.86.6.7603. [DOI] [PubMed] [Google Scholar]
- 4.Elks CE, Perry JR, Sulem P, et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat Genet. 2010;42:1077–85. doi: 10.1038/ng.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golub MS, Collman GW, Foster PM, et al. Public health implications of altered puberty timing. Pediatrics. 2008;121(Suppl 3):S218–S230. doi: 10.1542/peds.2007-1813G. [DOI] [PubMed] [Google Scholar]
- 6.Palmert MR, Hirschhorn JN. Genetic approaches to stature, pubertal timing, and other complex traits. Mol Genet Metab. 2003;80:1–10. doi: 10.1016/s1096-7192(03)00107-0. [DOI] [PubMed] [Google Scholar]
- 7.Fischbein S. Intra-pair similarity in physical growth of monozygotic and of dizygotic twins during puberty. Ann Hum Biol. 1977;4:417–30. doi: 10.1080/03014467700002401. [DOI] [PubMed] [Google Scholar]
- 8.Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev. 2003;24:668–93. doi: 10.1210/er.2002-0019. [DOI] [PubMed] [Google Scholar]
- 9.Sklad M. The rate of growth and maturing of twins. Acta Genet Med Gemellol (Roma) 1977;26:221–37. doi: 10.1017/s0001566000009703. [DOI] [PubMed] [Google Scholar]
- 10.de Vries L, Kauschansky A, Shohat M, Phillip M. Familial central precocious puberty suggests autosomal dominant inheritance. J Clin Endocrinol Metab. 2004;89:1794–800. doi: 10.1210/jc.2003-030361. [DOI] [PubMed] [Google Scholar]
- 11.Plant TM, Barker-Gibb ML. Neurobio-logical mechanisms of puberty in higher primates. Hum Reprod Update. 2004;10:67–77. doi: 10.1093/humupd/dmh001. [DOI] [PubMed] [Google Scholar]
- 12.Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5:569–76. doi: 10.1038/nrendo.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semple RK, Topaloglu AK. The recent genetics of hypogonadotrophic hypogonadism — novel insights and new questions. Clin Endocrinol (Oxf ) 2010;72:427–35. doi: 10.1111/j.1365-2265.2009.03687.x. [DOI] [PubMed] [Google Scholar]
- 14.Freitas KC, Ryan G, Brito VN, et al. Molecular analysis of the neuropeptide Y1 receptor gene in human idiopathic gonadotropin-dependent precocious puberty and isolated hypogonadotropic hypogonadism. Fertil Steril. 2007;87:627–34. doi: 10.1016/j.fertnstert.2006.07.1519. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y, Chen T, Zhou Y, Li K, Xiao J. An association study between the genetic polymorphisms within GnRHI, LHβ and FSHβ genes and central precocious puberty in Chinese girls. Neurosci Lett. 2010;486:188–92. doi: 10.1016/j.neulet.2010.09.049. [DOI] [PubMed] [Google Scholar]
- 16.Parent AS, Matagne V, Westphal M, Heger S, Ojeda S, Jung H. Gene expression profiling of hypothalamic hamartomas: a search for genes associated with central precocious puberty. Horm Res. 2008;69:114–23. doi: 10.1159/000111815. [DOI] [PubMed] [Google Scholar]
- 17.Luan X, Yu H, Wei X, et al. GPR54 polymorphisms in Chinese girls with central precocious puberty. Neuroendocrinology. 2007;86:77–83. doi: 10.1159/000107511. [DOI] [PubMed] [Google Scholar]
- 18.Nakayama Y, Wondisford FE, Lash RW, et al. Analysis of gonadotropin- releasing hormone gene structure in families with familial central precocious puberty and idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1990;70:1233–8. doi: 10.1210/jcem-70-5-1233. [DOI] [PubMed] [Google Scholar]
- 19.Teles MG, Bianco SD, Brito VN, et al. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med. 2008;358:709–15. doi: 10.1056/NEJMoa073443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silveira LG, Noel SD, Silveira-Neto AP, et al. Mutations of the KISS1 gene in disorders of puberty. J Clin Endocrinol Metab. 2010;95:2276–80. doi: 10.1210/jc.2009-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greulich WW, Pyle SI. Radiographic atlas of skeletal development of the hand and wrist. 2. Stanford, CA: Stanford University Press; 1971. [Google Scholar]
- 22.Neely EK, Hintz RL, Wilson DM, et al. Normal ranges for immunochemilumino-metric gonadotropin assays. J Pediatr. 1995;127:40–6. doi: 10.1016/s0022-3476(95)70254-7. [DOI] [PubMed] [Google Scholar]
- 23.Dauber A, Stoler J, Hechter E, Safer J, Hirschhorn JN. Whole exome sequencing reveals a novel mutation in CUL7 in a patient with an undiagnosed growth disorder. J Pediatr. 2013;162(1):202.e1–204.e1. doi: 10.1016/j.jpeds.2012.07.055. [Erratum, J Pediatr 2013;162:217.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gill JC, Navarro VM, Kwong C, et al. Increased neurokinin B (Tac2) expression in the mouse arcuate nucleus is an early marker of pubertal onset with differential sensitivity to sex steroid-negative feedback than Kiss1. Endocrinology. 2012;153:4883–93. doi: 10.1210/en.2012-1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.San Lucas FA, Wang G, Scheet P, Peng B. Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bio-informatics. 2012;28:421–2. doi: 10.1093/bioinformatics/btr667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Exome variant server. Seattle: National Heart, Lung, and Blood Institute Exome Sequencing Project; 2013. ( http://evs.gs.washington.edu/EVS) [Google Scholar]
- 27.Buettner VL, Walker AM, Singer-Sam J. Novel paternally expressed intergenic transcripts at the mouse Prader-Willi/Angelman syndrome locus. Mamm Genome. 2005;16:219–27. doi: 10.1007/s00335-004-2452-7. [DOI] [PubMed] [Google Scholar]
- 28.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 29.Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA. Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/ neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci. 2009;29:11859–66. doi: 10.1523/JNEUROSCI.1569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Navarro VM, Castellano JM, McConkey SM, et al. Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat. Am J Physiol Endocrinol Metab. 2011;300:E202–10. doi: 10.1152/ajpendo.00517.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gill JC, Wang O, Kakar S, Martinelli E, Carroll RS, Kaiser UB. Reproductive hormone-dependent and -independent contributions to developmental changes in kisspeptin in GnRH-deficient hypogonadal mice. PLoS One. 2010;5(7):e11911. doi: 10.1371/journal.pone.0011911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–27. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- 33.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–6. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu H, Shah S, Shyh-Chang N, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat Genet. 2010;42:626–30. doi: 10.1038/ng.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jong MT, Gray TA, Ji Y, et al. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum Mol Genet. 1999;8:783–93. doi: 10.1093/hmg/8.5.783. [DOI] [PubMed] [Google Scholar]
- 36.Gray TA, Hernandez L, Carey AH, et al. The ancient source of a distinct gene family encoding proteins featuring RING and C(3)H zinc-finger motifs with abundant expression in developing brain and nervous system. Genomics. 2000;66:76–86. doi: 10.1006/geno.2000.6199. [DOI] [PubMed] [Google Scholar]
- 37.Böhne A, Darras A, D’Cotta H, Baroiller JF, Galiana-Arnoux D, Volff JN. The vertebrate makorin ubiquitin ligase gene family has been shaped by large-scale duplication and retroposition from an ancestral gonad-specific, maternal-effect gene. BMC Genomics. 2010;11:721. doi: 10.1186/1471-2164-11-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanber D, Giltay J, Wieczorek D, et al. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader-Willi syndrome. Eur J Hum Genet. 2009;17:582–90. doi: 10.1038/ejhg.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman DS, Repka MX, Katz J, et al. Prevalence of decreased visual acuity among preschool-aged children in an American urban population: the Baltimore Pediatric Eye Disease Study, methods, and results. Ophthalmology. 2008;115:1786–95. doi: 10.1016/j.ophtha.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.