

Using PIG-A gene targeting and an inducible PIG-A expression system, this study established a conditional PIG-A knockout model in human induced pluripotent stem cells that allowed for the production of glycosylphosphatidylinositol-anchored protein (GPI-AP)-deficient blood cells. This conditional PIG-A knockout model should be a valuable tool for studying the importance of GPI-APs in hematopoiesis and human development.
Keywords: Human iPSCs, GPI-anchor protein, Hematopoiesis, PIG-A gene knockout
Abstract
PIG-A is an X-linked gene required for the biosynthesis of glycosylphosphatidylinositol (GPI) anchors; thus, PIG-A mutant cells have a deficiency or absence of all GPI-anchored proteins (GPI-APs). Acquired mutations in hematopoietic stem cells result in the disease paroxysmal nocturnal hemoglobinuria, and hypomorphic germline PIG-A mutations lead to severe developmental abnormalities, seizures, and early death. Human induced pluripotent stem cells (iPSCs) can differentiate into cell types derived from all three germ layers, providing a novel developmental system for modeling human diseases. Using PIG-A gene targeting and an inducible PIG-A expression system, we have established, for the first time, a conditional PIG-A knockout model in human iPSCs that allows for the production of GPI-AP-deficient blood cells. PIG-A-null iPSCs were unable to generate hematopoietic cells or any cells expressing the CD34 marker and were defective in generating mesodermal cells expressing KDR/VEGFR2 (kinase insert domain receptor) and CD56 markers. In addition, PIG-A-null iPSCs had a block in embryonic development prior to mesoderm differentiation that appears to be due to defective signaling through bone morphogenetic protein 4. However, early inducible PIG-A transgene expression allowed for the generation of GPI-AP-deficient blood cells. This conditional PIG-A knockout model should be a valuable tool for studying the importance of GPI-APs in hematopoiesis and human development.
Introduction
Covalent linkage to glycosylphosphatidylinositol (GPI) is an important means of anchoring many cell-surface glycoproteins to the cell membrane [1–3]. GPI-anchored proteins (GPI-APs) function as complement regulatory proteins, enzymes, blood group antigens, receptors, and adhesion molecules. GPI-APs also serve as receptors for proaerolysin, a pore-forming bacterial toxin secreted by Aeromonas hydrophila [4, 5]. The GPI anchor is synthesized in the endoplasmic reticulum membrane and involves at least 10 reactions and more than 20 different gene products [3]. The first step in this pathway is the transfer of N-acetylglucosamine (GlcNAc) from UDP-GlcNAc to phosphatidylinositol (PI) to yield GlcNAc-PI. This step is catalyzed by GlcNAc:PI a1-6 GlcNAc transferase, an enzyme whose subunits are encoded by seven different genes: PIG-A [6], PIG-C [7], PIG-H [8], GPI1 [9, 10], PIG-Y [11], PIG-P, and DPM2 [12]. GPI anchor assembly continues in the endoplasmic reticulum with acylation of the inositol and stepwise addition of mannosyl and phosphoethanolamine residues. The preassembled GPI is linked to proteins that contain a C-terminal GPI-attachment signal peptide, displacing it in a transamidase reaction [13]. The mature GPI-APs then transit the secretory pathway to reach its final destination at the plasma membrane.
Somatic PIG-A mutations arise in hematopoietic stem cells in patients with the human disease paroxysmal nocturnal hemoglobinuria (PNH) [14, 15]; however, the impact of germline PIG-A mutations has led to seemingly contradictory results. Pig-a knockout mice result in low levels of chimerism in early embryos and are embryonic lethal, suggesting an important role for GPI-APs in early development [16, 17]. Consistent with this, a Pig-a-null mouse embryonic stem (ES) cell clone was unable form embryoid bodies (EBs) efficiently or generate hematopoietic cells [18]. Conversely, a similar mouse ES cell model, which tested multiple clones, reported that Pig-a-null ES cells are competent for hematopoiesis, albeit at a severely reduced efficiency [17]. It was previously shown that PIG-A mutant human ES cells made by proaerolysin selection form EBs and hematopoietic cells but do not form trophoblasts [19]. Thus, the precise role of PIG-A in development remains unclear. Interestingly, we recently described a male-specific lethal X-linked disorder caused by a hypomorphic germline PIG-A mutation, suggesting a dose-dependent phenotype of PIG-A mutants [20]. All three males in this pedigree presented with intractable seizures and severe developmental abnormalities and died within 3 months after birth. Hypomorphic mutations in other genes involved in GPI anchor biosynthesis that lead to partial GPI-AP deficiency have been reported to have a similar phenotype of severe developmental abnormalities, seizures, and early death [21–23].
In order to better understand the consequence of PIG-A mutations in human development and in hematopoiesis, we have further analyzed previously described human ES and induced pluripotent stem cell (iPSC) lines that lack PIG-A expression [19, 24]. Cell lines generated from male H1 human ES cells by proaerolysin selection lack PIG-A gene expression and any GPI-APs; however, no genetic mutations in all six exons and exon/intron junctions of the PIG-A gene were detected after extensive sequencing of the transcribed region [19]. We now show that the lack of PIG-A expression in these cells is due to transcriptional silencing after enforced and brief proaerolysin selection, and a low level of GPI-AP expression is observed after extended cell cultures. To generate human cells that stably and completely lack the PIG-A gene, zinc finger nucleases were engineered to create a PIG-A-null allele via homologous recombination in hFib2-iPS5 [24]. We found that PIG-A-null iPSCs have a block in embryonic development prior to mesoderm differentiation. However, using an inducible promoter to transiently express a PIG-A transgene and restore GPI-APs in the first 10–14 days, we were able to produce GPI-AP-deficient blood cells. These findings demonstrate that GPI-APs are critical for the establishment of embryonic hematopoiesis.
Materials and Methods
Cell Culture of Human ES Cells and iPSCs
The human iPSC lines hFib2-iPS5, derived from adult male dermal fibroblasts, and FPHR (GPI-AP−), which was previously established by PIG-A gene knockout in the hFib2-iPS5 cell line using zinc finger nuclease-mediated homologous recombination, were used in this study [24]. Human ES cell lines such as H1 and H9 and an H1-derived AR-c1 clone were also used as previously described [19]. All iPSC as well as ES cell lines were maintained on irradiated mouse embryonic fibroblasts (MEFs; passage 3) in 2 ml of human ES cell medium containing Dulbecco's Modified Eagle's Medium: Nutrient Mixture F-12 (DMEM/F12) medium (Life Technologies, Grand Island, NY, http://www.lifetechnologies.com) supplemented with 20% knockout serum replacement (Life Technologies), 5 ng/ml basic fibroblast growth factor (R&D Systems Inc., Minneapolis, MN, http://www.rndsystems.com), 0.1 mM (Minimum Essential Media) nonessential amino acids (Life Technologies), and 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com). The cells were fed daily and were passaged every 4–7 days. For passaging, cells were incubated with 1 mg/ml collagenase IV (Life Technologies) in DMEM/F12 for 5 minutes and replated in a six-well MEF plate precoated with 0.1% gelatin.
PIG-A Overexpression in FPHR (PIG-Anull, GPI-AP−) iPSCs
FPHR cells were transduced with the lentiviral (LV) supernatant of pLV-PIG-A vector containing the full-length human PIG-A transgene (iDuet101a/PIG-A) and 8 μg/ml polybrene (Sigma-Aldrich) as previously described [25]. The transduced cells were enzymatically passaged and expanded. After 1 week, the cells were stained with CD59 antibody, and CD59-positive cells were isolated by cell sorting (BD Biosciences, San Diego, CA, http://www.bdbiosciences.com) and expanded. The level of expression of CD59 for restoration of the PIG-A gene from FPHR cells was confirmed by fluorescence-activated cell sorting (FACS), thus establishing the FPHR-PIG-A (GPI-AP+) cell line.
Inducible PIG-A Gene Expression System in FPHR (PIG-Anull, GPI-AP−) Cells
The inducible PIG-A cell line was prepared as previously described [25]. FPHR cells were cotransduced by two LV vectors: the regulator of pLV-tTR-KRAB (tTS/RFP) containing the tetracycline (tet)-SUPER inducible transgene expression system and the other of pLV-PIG-A (iDuet101/PIG-A) expressing human PIG-A cDNA controlled by the human EF1α promoter. Once doxycycline (Dox) was added, PIG-A gene expression was turned on because Dox was binding to a tet transcriptional suppressor (tTS) to release the blocking of EF1a promoter-driven PIG-A expression. When Dox was absent, PIG-A gene expression was suppressed, thus establishing an inducible PIG-A cell line, FPHR-Krab-PIG-A (FPHR-KP). FPHR-KP cells were cultured with and without 50 ng/ml of Dox in human embryonic stem (hES) medium and were collected at a various time points and stained with an allophycocyanin (APC)-conjugated anti-CD59 (Novus Biologicals, Littleton, CO, http://www.novusbio.com) for FACS analysis. After 3 days following Dox treatment, cell-surface CD59 expression was restored.
EB Generation by Force Aggregation
To induce hematopoietic mesoderm differentiation, EBs were generated via aggregation as previously described [26, 27]. Briefly, 70%–90% confluent human iPSCs (hFib2-iPS5, passages 70–78; both FPHR and FPHR-PIG-A, passages 66–72) were enzymatically treated with TrypLE (Life Technologies) or Accutase (Sigma-Aldrich) and incubated at 37°C for 5 minutes to dissociate the colonies into a single cell suspension. The cells were washed and resuspended in serum-free medium (SFM) and then added to 96-well plates supplemented with 10 ng/ml basic fibroblast growth factor, 10 ng/ml bone morphogenetic protein 4 (BMP-4), 10 ng/ml vascular endothelial growth factor (VEGF), and 50 ng/ml stem cell factor (SCF) at a density of 3,000–5,000 cells per well. Next, the cells were aggregated at the bottom of the 96-well plates by centrifugation at 3,000 rpm for 5 minutes and placed in a humidified incubator at 37°C with 5% CO2. Every 3 days, the cell medium was changed with 50 μl of fresh SFM containing cytokines.
Hematopoietic Expansion and Differentiation From EBs
After 10 days of mesoderm induction, EBs were surrounded by blood cells. At days 10–14, all the cells were collected, and EBs were dissociated by TrypLE or Accutase into single cells. EB-derived single cells were stained with anti-CD34 and anti-CD45 monoclonal antibodies. The level of CD34 and CD45 expression was analyzed by FACS. Next, EB-derived single cells were further differentiated into myeloid and erythroid cells.
Myeloid Progenitor Induction
EB-derived blood cells were cultured in SFM supplemented with 10 ng/ml Flt3 ligand, 50 mg/ml granulocyte-macrophage colony-stimulating factor, 10 ng/ml interleukin-3 (IL-3), 50 ng/ml SCF, 3 U/ml erythropoietin (EPO), and 10 ng/ml thrombopoietin for 5 days. Following differentiation and expansion, the cells were harvested, washed, and stained with anti-human CD45-phycoerythrin (PE), CD15-fluorescein isothiocyanate (FITC), and CD33-PE for FACS analysis.
Erythroid Induction
EB-derived blood cells were cultured in SFM with 10 ng/ml IL-3, 50 ng/ml SCF, and 3 U/ml EPO for 8 days, followed by SFM with SCF (50 ng/ml) and EPO (3 U/ml) for another 3 days and SFM with EPO (3 U/ml) only until days 11–20 as previously described [28]. The cultures were maintained at 37°C with 5% CO2. Cells were harvested and stained with anti-CD45, anti-CD71, and anti-GlyA (CD235a) antibody for FACS analysis.
Colony-Forming Unit Assay
Blood cells dissociated from day 12–15 EBs were added to 3 ml of MethoCult (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com) at a density of 50,000 cells per tube and mixed well by vortexing. The cell mixture was dispensed into 36-mm Petri dishes in duplicate and incubated at 37°C with 5% CO2 humidity for 14 days. Counting and characterization of colony-forming units (CFUs) was performed using an inverted microscope.
FACS Analysis
Cells were washed in phosphate-buffered saline (PBS) and resuspended in 100 μl of FACS buffer at a concentration of 106 cells per ml. Before staining, cells were treated with either 5% fetal bovine serum or 0.5% bovine serum albumin in PBS (blocking buffer) for 10 minutes. The cells were stained with the appropriate dilution of the antibody and incubated for 30 minutes at 4°C in the dark. The antibodies used were CD33-PE, CD34-FITC, CD34-PE, CD45-PE, CD56-PE, CD71-FITC, CD235a (GlyA)-FITC, CD235a (GlyA)-APC, and CD309-PE (BD Biosciences); CD59 APC (Novus Biologicals); annexin V-FITC Kit (MASC; Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com); and SSEA-4-PE (R&D Systems). The isotype controls were IgG1-FITC, IgG2a-APC, and IgG1-PE (BD Biosciences). After staining, cells were washed in PBS and resuspended in FACS buffer. A minimum of 104 events was acquired for each sample using a FACSCalibur (BD Biosciences). FACS results were analyzed with FlowJo software (Tree Star, Ashland, OR, http://www.treestar.com).
Quantitative Polymerase Chain Reaction
Total RNA was isolated from EBs at day 0, 3, or 7 of differentiation using the RNeasy kit (Qiagen, Hilden, Germany, http://www.qiagen.com), followed by cDNA synthesis with SuperScript III reverse transcriptase following manufacturer's protocol. Quantitative polymerase chain reaction (PCR) and data analysis were performed using SYBR Select reagent (Life Technologies) and the StepOne Plus Real-Time PCR system (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com). Glyceraldehyde-3-phosphate dehydrogenase was used an endogenous control. The primers were Brachyury (forward, GGATGAAGGCTCCCGTCTC; reverse, GCTGTGATCTCCTCGTTCTGATA), MIXL1 (forward, CGGCGTCAGAGTGGGAAAT; reverse, GTTCGGGCAGGCAGTTCA), SOX17 (forward, GCTTTCATGGTGTGGGCTAA; reverse, CGCCTTCCACGACTTGC), FOXA2 (forward, TACAGGCGCAGCTACACGCACGCAAAG; reverse, GCGGGGCACCTTCAGGAAACAGTCGT), PAX6 (forward, CATCTTTGCTTGGGAAATCCG; reverse, GGCAGCCATCTTGCGTAGGTT), and Sox1 (forward, GGTCAAACGGCCCATGAACGC; reverse, TCCTTCTTGAGCAGCGTCTTGGTCTT).
Western Blot
hFib2-iPS5 and FPHR cells were plated on a six-well plate coated by Matrigel (BD Biosciences). When cells were at 80%–90% confluence, they were treated with or without 50 ng/ml of BMP-4 for 4 hours. Four hours postinduction, the cells were lysed using RIPA buffer (Sigma-Aldrich) and centrifuged to collect the supernatants. Fifteen micrograms of protein from each sample was loaded in 4%–12% Bis-Tris gel (Invitrogen). A monoclonal antibody (Cell Signaling Technology, Beverly, MA, http://www.cellsignal.com) that specifically recognizes phospho-Smad1 (Ser463/465), Smad5 (Ser463/428), or Smad8 (Ser426/428) was used to detect the level of phosphorylated sites of Smad1/5/8. A mouse antibody recognizing the β-actin (Cell Signaling Technology) was used for normalization.
Aerolysin Assay
Apoptosis Induction
Cells were incubated with 1 nM aerolysin for 60 minutes at 37°C as previously described [29]. After incubation, each sample was washed, centrifuged, and stained with CD59-APC antibody and annexin V-FITC and propidium iodide for FACS analysis.
Aerolysin Cell Viability Assay
hFib2-iPS5 and FPHR-KP without Dox were exposed to 1 nM aerolysin at 37°C, diluted (1:1) with 0.4% trypan blue. Viable cells were counted using an inverted microscope in triplicate at 5-minute intervals.
Further details are given in the supplemental online data.
Results
Epigenetic Silencing of PIG-A Expression in Proaerolysin-Selected ES Cells
We previously used proaerolysin selection to establish multiple PIG-A-deficient human ES cell lines such as AR1-c1 [19]. Although this clone lacks GPI-AP and PIG-A gene expression, we did not observe a genetic mutation in the PIG-A locus. In addition, a fraction of cells gradually regained PIG-A expression and GPI-APs, especially after long-term culture and during the process of differentiation. This pitfall compromised studies aiming to elucidate the importance of GPI-APs in differentiated cells such as those in the blood system, which require several weeks in culture to proceed through multiple developmental processes. We hypothesized that this observation was due to inefficient epigenetic silencing of PIG-A expression over time. Thus, we analyzed DNA methylation and its relationship to PIG-A/GPI-AP expression in AR1-c1 cells. After weeks of continuous culture without selection, we typically observed that 2%–3% of AR1-c1 cells (genetically labeled with GFP) expressed GPI-APs, such as CD55, CD59, and CD90 (supplemental online Fig. 1A). We next treated the cells for 2 days with two types of small-molecule epigenetic modulators, 5′-aza-cytosine or trichonstain A. Both agents are commonly used to activate gene expression from silenced genes. Three days after treatment (day 5), we observed that 28%–36% of AR1-c1 hES cells expressed GPI-APs (supplemental online Fig. 1A; supplemental online data). To obtain direct evidence that the silencing of PIG-A expression is due to DNA methylation that blocks PIG-A gene transcription, we examined the DNA methylation status at the PIG-A promoter, which is a typical TATA-less promoter containing 54 CpG sites within the 586-base pair DNA segment (a CpG island) spanning the transcription start site (supplemental online Figs. 1B, 2). The status of DNA methylation in the PIG-A promoter in AR1-c1 and control cells was examined after genomic DNA was treated with bisulfite. The results showed that in the AR1-c1 clone, the CpG island region in the PIG-A promoter was extensively hypermethylated. However, the CpG island region was unmethylated in the PIG-A promoter of parent G-GFP hES cells. In H9 human ES cells (XX), only one allele was methylated, as expected, because of the X-chromosome inactivation (supplemental online Figs. 1C, 3). These data indicate that deficiency of PIG-A and GPI-APs can result from epigenetic silencing of the PIG-A gene.
FPHR iPSCs Are Unable to Generate Hematopoietic Cells
In order to continue to study the role of GPI-APs in hematopoietic development, an ES or iPSC model in which the PIG-A gene has been stably inactivated was required. We focused our efforts on the FPHR iPSCs, which were derived from the male hFib2-iPS5 iPSC line, in which the only PIG-A allele was genetically inactivated by gene targeting [24]. Although FPHR iPSCs lacking PIG-A and GPI-APs grew normally as undifferentiated cells, they failed to form teratomas in vivo and performed poorly when differentiating into trophoblasts in vitro [24].
To determine whether the observed effects were due to PIG-A deficiency, we established an iPSC line reconstituted with PIG-A transgene expression by transduction with a lentiviral vector containing the full-length PIG-A cDNA (FPHR-PIG-A; Fig. 1A). As expected, cell-surface expression of GPI-APs such as CD59 was present in the parental hFib2-iPS5 cells but absent in FPHR and then was restored in FPHR-PIG-A cells (Fig. 1B). We next examined the potential of hFib2-iPS5, FPHR, and FPHR-PIG-A iPSC lines to form EBs and generate hematopoietic cells. We found that hFib2-iPS5 and FPHR-PIG-A cells, but not FPHR cells, formed morphologically normal EBs with evidence of blood cells surrounding the EBs after 12–15 days in culture (Fig. 2A). Blood cell formation was confirmed in hFib2-iPS5 and FPHR-PIG-A cells by analysis of cells derived from day 15 EBs for the expression of hematopoietic cell-surface proteins (CD34 and CD45; Fig. 2B) and for the ability to generate hematopoietic colonies (Fig. 2C). FPHR cells did not express CD59, CD34, or CD45 (Fig. 2B) and were unable to generate hematopoietic colonies (Fig. 2C). Taken together, these data suggest that GPI-APs are critical for the formation of normal EBs and for primitive hematopoiesis.
Figure 1.
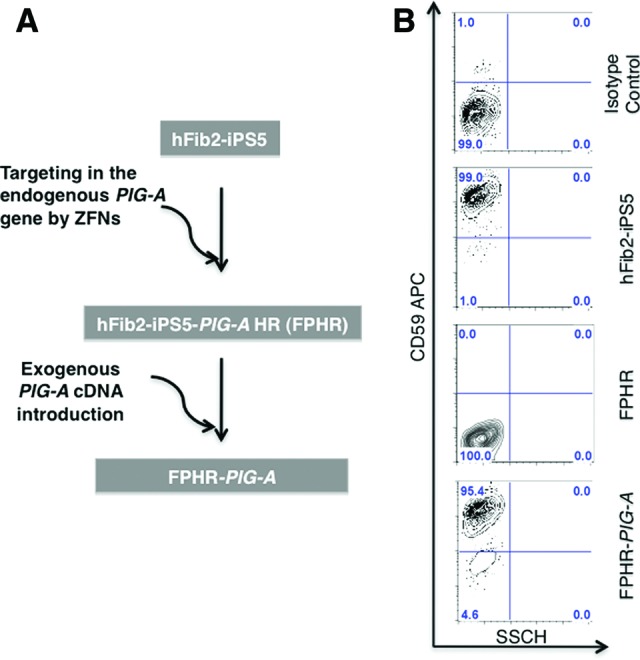
Nomenclature and CD59 surface expression of iPSC lines. (A): PIG-A was targeted with zinc finger nucleases in parental hFib2-iPS5 cells to establish the PIG-A-null induced pluripotent stem cell line hFib2-iPS5-PIG-A HR (FPHR). FPHR cells were then transduced with a lentiviral vector containing the full-length PIG-A cDNA to establish the FPHR-PIG-A cell line. (B): Flow cytometric analysis for CD59 expression in hFib2-iPS5, FPHR, and FPHR-PIG-A cells (hFib2-iPS5 cells were used for the isotype control). Abbreviations: APC, allophycocyanin; iPS, induced pluripotent stem cell; ZFN, zinc finger nuclease.
Figure 2.
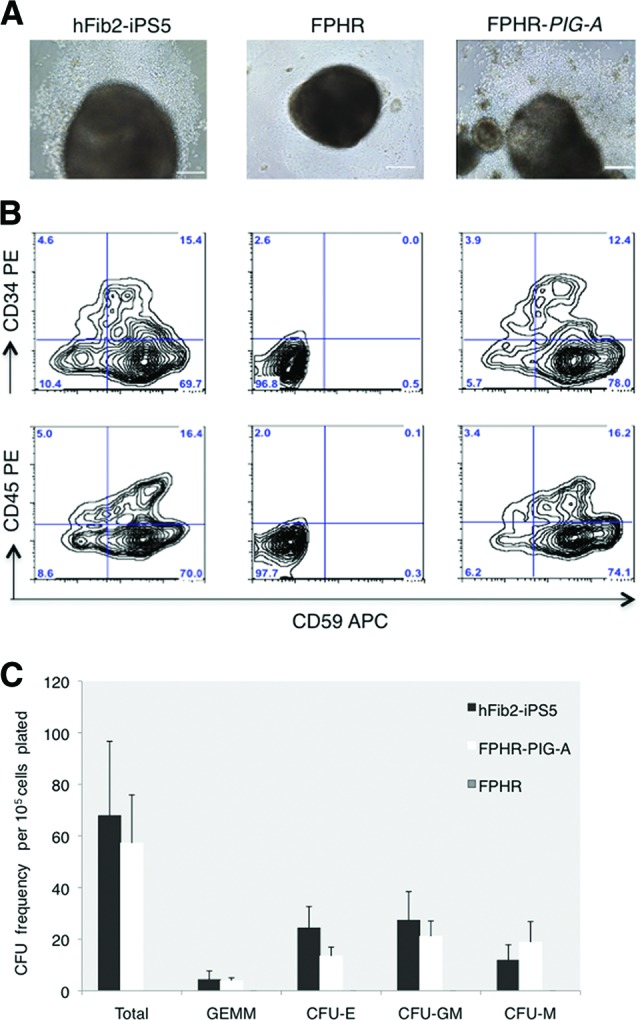
FPHR cells are unable to differentiate to hematopoietic stem cell/progenitors. Embryoid body (EB) generation was performed in hFib2-iPS5, FPHR, and FPHR-PIG-A cells using forced aggregation. (A): Morphology of the EBs from hFib2-iPS5, FPHR, and FPHR-PIG-A cells. Blood cells were observed surrounding hFib2-iPS5 (left) and FPHR-PIG-A (right) EBs but not FPHR EBs (middle). Morphology of the EBs from hFib2-iPS5, FPHR, and FPHR-PIG-A. Scale bars = 200 μm. (B): The expression of hematopoietic markers CD34 and CD45 is shown for hFib2-iPS5 cells (left), FPHR cells (middle), and FPHR-PIG-A cells (right). (C): Enumeration of CFUs from hFib2-iPS5, FPHR, and FPHR-PIG-A cells. Error bars represent the SD based on three experiments. No colonies formed from the FPHR cells; there was no significant difference in total CFU colonies between hFib2-iPS5 and FPHR-PIG-A (p > .05) cell types. Abbreviations: APC, allophycocyanin; CFU, colony-forming unit; CFU-E, colony-forming unit-erythroid; CFU-GM, colony-forming unit-granulocyte-macrophage; CFU-M, colony-forming unit-macrophage; iPS, induced pluripotent stem cell; PE, phycoerythrin.
GPI-APs Are Required for the Development of Mesoderm
Hematopoiesis is believed to be derived from mesodermal progenitors known as hemangioblasts that express KDR/VEGFR2 (also known as VEGF receptor 2 or Flk1 in mice) [30]. A recent paper suggested that CD56+ cells represent the earliest multipotent mesodermal progenitors during the early stages of differentiation from human ES cells predating the KDR/VEGFR2+/CD34+ hemangioblast [31]. Therefore, we hypothesized that FPHR cells may be unable to express KDR/VEGFR2 and/or CD56, in addition to CD34 markers. To test this hypothesis, we induced mesodermal differentiation of the hFib2-iPS5, FPHR, and FPHR-PIG-A cells and assessed cell-surface expression of CD56, KDR/VEGFR2, and CD34 on days 0, 3, 7, and 10 during the process of EB formation and differentiation (Fig. 3; supplemental online Fig. 4). Surface expression of CD56 and KDR/VEGFR2 was virtually absent from the day 3 EBs derived from the FPHR cells compared with hFib2-iPS5 and FPHR-PIG-A cells, although by day 7 some expression was detected (Fig. 3; supplemental online Fig. 4). CD34 expression remained absent in FPHR cells throughout mesoderm differentiation compared with hFib2-iPS5 and FPHR-PIG-A cells.
Figure 3.

Time course of early mesodermal markers and hematopoietic stem cell markers during EB differentiation on days 3, 7, and 10. The expression of CD56, KDR/VEGFR2, and CD34 was analyzed in hFib2-iPS5 (○, solid line), FPHR (▴, solid line), and FPHR-PIG-A (♦, dashed line). Error bars represent the SD based on six experiments. (A): CD56 expression was significantly increased in hFib2-iPS5 and FPHR-PIG-A but not in FPHR cells during EB differentiation at days 3 and 7. (B): KDR/VEGFR2 expression was significantly elevated in hFib2-iPS5 and FPHR-PIG-A compared with FPHR cells in day 3 and 7 EBs. (C): CD34 expression was significantly increased at day 7 and highly significantly increased for day 10 EBs in hFib2-iPS5 and FPHR-PIG-A cells compared with FPHR cells. *, p < .05; **, p < .01. Abbreviations: EB, embryoid body; iPS, induced pluripotent stem cell.
To better understand the influence of the loss of PIG-A in early development, we differentiated the iPSC lines via EBs and performed quantitative reverse transcription (RT)-PCR to measure lineage markers of mesoderm (BRACHYURY and MIXL1), endoderm (FOXA2 and SOX17), and ectoderm (PAX6 and SOX1) on days 0, 3, and 7 of differentiation. Expression of mesoderm and endoderm markers failed to elevate in the FPHR cells during EB differentiation; however, ectodermal markers were increased in the FPHR cells (Fig. 4A). Furthermore, the number of cells per EB derived from FPHR cells was 2.9 ± 0.8-fold less at day 7 than the number of cells per EB derived from the hFib2-iPS5 cells. Thus, the increase in ectoderm differentiation appears to result from a loss of mesoendodermal differentiation and subsequent cell death, rather than an increase in ectodermal cells. The defect appears to be specific, since failure to express markers of early mesoderm/endoderm and the acceleration of ectodermal markers in the FPHR cells was fully restored in FPHR-PIG-A cells.
Figure 4.

Influence of PIG-A mutations in early development. (A): Relative expression of germ layer markers by quantitative reverse transcription-polymerase chain reaction. By day 3 of differentiation, embryoid bodies (EBs) of both hFib2-iPS5 and FPHR-PIG-A cells showed upregulation of early mesodermal markers BRACHYURY and MIXL1 compared with FPHR cells. Endodermal markers FOXA2 and SOX17 were similarly upregulated at days 3 and 7 in both hFib2-iPS5 and FPHR-PIG-A compared with FPHR cells. Conversely, ectodermal markers PAX6 and SOX1 remained repressed throughout differentiation in both hFib2-iPS5 and FPHR-PIG-A cells but were upregulated in FPHR cells. (B): Western blot analysis for BMP signaling activation of hFib2-iPS5 and FPHR cells. All cells were treated with/without BMP-4. After 6 hours of BMP-4 induction, phosphorylated Smad 1/5/8 (60 kDa) was detected using specific phosphorylated Smad 1/5/8 antibody, and phosphorylated Smad 1/5/8 was significantly induced in hFib2-iPS5 cells but not FPHR cells. β-Actin was used as endogenous control. (C): Quantitative analysis of the intensity of fold change in hFib2-iPS5 and FPHR cells. The expression of phosphorylated Smad 1/5/8 was not detected in FPHR cells without BMP-4 and slightly increased in FPHR but significantly lower than hFib2-iPS5 cells after BMP-4 induction. *, p < .05. Abbreviations: BMP, bone morphogenetic protein; iPS, induced pluripotent stem cell.
The BMP and activin signaling pathways are required for mesoderm commitment of ES cells and subsequent hematopoiesis [32–34]. Moreover, GPI-APs such as repulsive guidance molecules (RGMs), specifically RGMa and RGMb (DRAGON), are known coreceptors (type III, in addition to essential type I and II receptors) that are important for BMP/activin signaling [35, 36]. Thus, we hypothesized that BMP-4 signaling may be attenuated in the FPHR cells lacking all the GPI-APs. We measured the level of phosphorylated Smads (forms 1/5/8) specific to BMP signaling as an indicator of activation by BMP-4 treatment. Indeed, the phosphorylated forms of Smad 1/5/8 were not detected in FPHR in the absence of BMP-4; however, BMP-4 treatment markedly increased the level of phosphorylated forms of Smad 1/5/8 in hFib2-iPS5 compared with FPHR cells (Fig. 4B, 4C). These data suggest that a defect in BMP-4 signaling in the PIG-A-null FPHR cells may explain the defect in early development including mesodermal and subsequent hematopoietic differentiation.
Generation of GPI-AP-Deficient Blood From FPHR-KP iPSCs
One objective in generating PIG-A knockout iPSCs is to establish a novel disease model of PNH that will provide a renewable source of GPI-AP-deficient blood cells. In order to overcome hurdles of generating mesodermal progenitor cells prior to hematopoiesis, we made use of a previously described inducible PIG-A expression system [20, 32]. We hypothesized that transient or conditional expression of PIG-A during early embryogenesis would allow for the formation of GPI-APs and healthy blood-forming EBs. To test this hypothesis, we transduced the FPHR cells with our previously described Dox-inducible PIG-A expression system to establish the FPHR-KP cell line (supplemental online Fig. 5A). FPHR-KP cells express the red fluorescent probe (RFP) and only in the presence of Dox express PIG-A transgene. FPHR-KP cells in Dox-containing medium begin to show cell-surface expression of the GPI-AP CD59 within 3 days (data not shown). We next used FACS sorting to collect RFP+/CD59− and RFP+/CD59+ undifferentiated FPHR-KP cells, cultured them in differentiation-inducing medium without or with Dox for up to 14 days, and counted the percentage of EBs with blood cells in 96-well plates (Fig. 5A, 5B). The RFP+/CD59− without Dox formed abnormal EBs and did not generate blood cells. Less than 5% of the RFP+/CD59+ cells formed EBs with blood cells in the absence of Dox; however, in the presence of Dox for 14 days, 50% of FPHR-KP cells made morphologically normal EBs with blood cells. These data suggest that GPI-AP expression for up to 14 days is sufficient for the formation of normal EBs.
Figure 5.
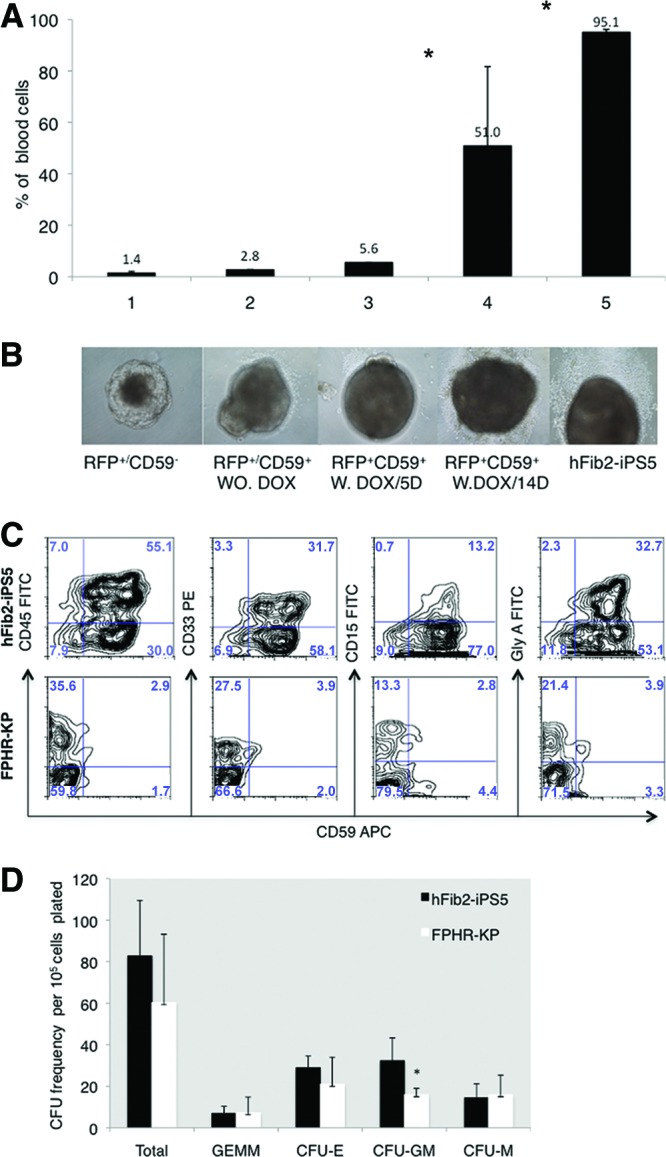
Glycosylphosphatidylinositol-anchored protein (GPI-AP) deficiency beyond the embryoid body (EB) stage does not interfere with hematopoietic differentiation. The FPHR-KP cell line was established by cotransduction with both the PIG-A expression vector and the tetracycline transcriptional suppressor/RFP (regulator). For doxycycline induction, FPHR-KP cells were cultured in human embryonic stem cell medium and exposed to doxycycline for 6 days. Following 6 days of doxycycline treatment, both positive cells for CD59+ and RFP+ were sorted for hematopoietic lineage differentiation markers, and CD59− and RFP+ cells were used as negative and positive controls, respectively. During EB generation, the cells sorted were cultured either with or without doxycycline. (A): After 14 days of EB formation, blood cells appeared, and the percentages of EB-containing blood cells were counted and normalized to a standard number (100 EBs) for each cell line based on three experiments. The percentage of EBs with blood cells in FPHR-KP was higher after 14 days of coculture with doxycycline compared with 5 days of doxycycline treatment and lower than hFib2-iPS5 cells. Significance (*, p < .05) should be between doxycycline 5 days (bar 3) and 14 days (bar 4); significance (*, p < .05) should be between doxycycline 14 days (bar 4) and hFib2-iPS5 (bar 5). (B): Photomicrographs of representative day 14 EBs are shown below each cell subset. Magnification, ×10. (C): Phenotype analysis of the cells derived from hFib2-iPS5 and FPHR-KP EBs. Plots are shown for CD45-FITC/CD59APC, CD33PE/CD59APC, CD15-FITC/CD59APC, and GlyA-FITC/CD59APC in hFib2-iPS5 (top) and FPHR-KP (bottom). (D): Quantitative CFU frequency from hFib2-iPS5 and FPHR-KP cells. Error bars represent SD based on three experiments. There was no significant difference in total CFU colonies between hFib2-iPS5 and FPHR-KP (p > .05), but the number of CFU-GM colonies in hFib2-iPS5 was significantly higher than in FPHR-KP (p < .05). Abbreviations: APC, allophycocyanin; CFU, colony-forming unit; CFU-E, colony-forming unit-erythroid; CFU-GM, colony-forming unit-granulocyte-macrophage; CFU-M, colony-forming unit-macrophage; DOX, doxycycline; FITC, fluorescein isothiocyanate; iPS, induced pluripotent stem cell; PE, phycoerythrin; RFP, red fluorescent probe; W, with; WO, without.
We next removed Dox from the culture medium of blood cells derived from the day 15 FPHR-KP EBs and cultured them in medium supplemented with either myeloid or erythroid inducing cytokines in order to generate or expand more mature GPI anchor-deficient blood cells. After 5 days of culture, we stained the differentiated hematopoietic cells with monoclonal antibodies against CD59, CD45, CD33, CD15, and glycophorin-A. FPHR-KP-derived hematopoietic cells were able to differentiate into both myeloid and erythroid lineages that were CD59-deficient (Fig. 5C). Next, blood cells derived from the FPHR-KP cells were cultured in methylcellulose supplemented with colony-stimulating factors for 14 days and scored for CFU. The FPHR-KP-derived cells were able to generate comparable numbers of CFU as blood cells derived from hFib2-iPS5 cell line (Fig. 5D).
GPI-AP-Negative Blood Cells Derived From FPHR-KP iPSCs Are Resistant to Proaerolysin
A hallmark of PNH blood cells is that they are resistant to the bacterial channel-forming toxin proaerolysin [5, 29, 37]. Proaerolysin uses GPI-AP as its receptor. After binding, the toxin oligomerizes and disrupts the membrane by forming channels that lead to either apoptosis or lysis of the cell depending upon the cell type and the concentration of proaerolysin. As anticipated, GPI-AP-deficient blood cells derived from FPHR-KP cells were resistant to proaerolysin (Fig. 6A, 6B). In addition, no CFU formed when hFib2-iPS5-derived blood cells were plated in methylcellulose supplemented with growth factors and 1 nM proaerolysin (Fig. 6C). In contrast, there was less than a 50% reduction in the number of CFU that form after 14 days when FPHR-KP-derived blood cells were plated in similar conditions. These data confirm that the majority of the colony-forming cells from the FPHR-KP-derived blood (in the absence of Dox) were GPI-AP-deficient. The fact that 100% of the colony-forming cells derived from the FPHR-KP-derived blood cells were not proaerolysin-resistant demonstrates that a minority of blood cells retain GPI-AP expression following Dox withdrawal from the culture medium.
Figure 6.

Glycosylphosphatidylinositol-anchored protein (GPI-AP)-deficient blood cells derived from FPHR-KP cells are resistant to proaerolysin. (A): Blood cells derived from parental hFib2-iPS5 or FPHR-KP embryoid bodies (EBs) following withdrawal of doxycycline (GPI-AP−) were exposed to 1 nM AE for up to 50 minutes, and viable cells were counted in triplicate using an inverted microscope. After AE treatment, cell viability in hFib2-iPS5 cells (▴) was significantly decreased with time compared with FPHR-KP (○) cells. (B): Following AE treatment, the cells were treated with annexin V-fluorescein isothiocyanate/phosphatidylinositol and analyzed by flow cytometry, which revealed CD59+ cells from hFib2-iPS5 undergoing apoptosis (top right) and CD59− cells of FPHR-KP, which were resistant to apoptosis (bottom right). (C): Representative CFU colonies derived from hFib2-iPS5 and FPHR-KP cells. The cells derived from hFib2-iPS5 EBs and FPHR-KP EBs were cultured in MethoCult methylcellulose medium (StemCell Technologies) with or without proaerolysin. After 14 days, CFUs were counted, and there were no proaerolysin-resistant CFUs in hFib2-iPS5 cells. However, proaerolysin-resistant CFU colonies were observed from FPHR-KP cells. Furthermore, there was no significant difference with or without AE in FPHR-KP cell-derived CFU colonies (p > .05). *, p < .05; **, p < .01. Abbreviations: AE, proaerolysin; APC, allophycocyanin; CFU, colony-forming unit; iPS, induced pluripotent stem cell.
Discussion
Using PIG-A gene targeting in human iPSCs and an inducible PIG-A expression system, we have established a conditional PIG-A knockout model that allows for the production of GPI-AP-deficient human blood cells. These studies show that PIG-A-null human iPSCs are unable to form hematopoietic cells. The initial block appears to be at the generation of mesodermal progenitors that express CD56, predating the deficiency of precursor cells expressing KDR/VEGFR2 and CD34 (Fig. 7). As result, hematopoiesis did not occur, consistent with results in the murine system using mouse ES cells defective in Flk1 (the mouse equivalent of KDR/VEGFR2) [38]. However, transient expression of GPI-APs during hematopoietic differentiation allowed us to generate GPI-AP-deficient blood cells in multiple hematopoietic lineages.
Figure 7.
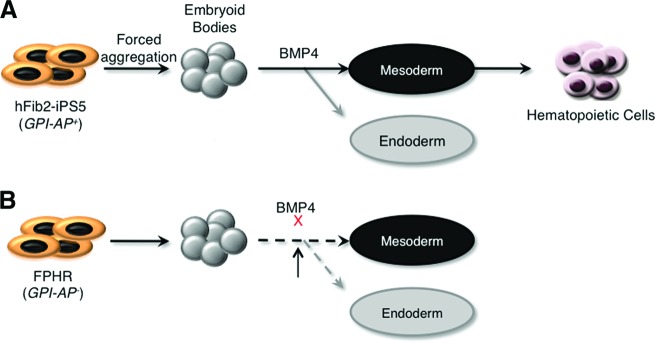
Model of GPI-AP function during embryonic stem (ES)/iPS cell differentiation. (A): In the presence of GPI-APs, ES/iPS cells can form embryoid bodies and then be induced to differentiate into the three germ layers (mesoderm, endoderm, and ectoderm). In the presence of BMP-4, cells are induced toward the mesoderm and endoderm lineages, whereas induction toward the ectoderm lineage is blocked. In the presence of additional cytokines, mesodermal cells can be further differentiated into hematopoietic cells. (B): In the absence of GPI-APs, ES/iPS cells can form embryoid bodies (though abnormal). However, they are unable to be induced to differentiate into either mesoderm or endoderm, likely because of a decrease in BMP-4 signaling activation. Abbreviations: BMP, bone morphogenetic protein; GPI-AP, glycosylphosphatidylinositol-anchored protein; iPS, induced pluripotent stem cell.
Complete GPI-AP deficiency in Pig-a−/− mice leads to neurodevelopmental defects and embryonic lethality before the 9th day of gestation [39]. Female embryos in which one Pig-a allele was disrupted by Cre such that only half of the cells in the embryo proper did not express GPI-APs because of random X-inactivation developed until 19 days post coitum but showed abnormal phenotypes such as insufficient closure of neural tube and cleft palate [39]. The PIG-A knockout human iPSCs used in our study also demonstrated complete GPI-AP deficiency, formed EBs poorly by a standard suspension culture method, and were unable to generate hematopoietic cells. Interestingly, we previously reported that PIG-A-deficient human ES cells that were generated based on resistance to proaerolysin killing are able to generate normal EBs that can differentiate into the three embryonic germ layers without overt defects at early stages of ES cell differentiation [19]. The AR1-c1 hES cells failed to form trophoblasts after induction by EB formation or by adding exogenous BMP-4 onto monolayer cultures [19]. These seemingly discordant results are likely a consequence of different methodologies used to generate PIG-A-deficient cells and incomplete activation of PIG-A in the AR1-c1 hES cells we previously used [19]. In that study, we used proaerolysin selection to establish the PIG-A-deficient ES cells. Although the proaerolysin-resistant human ES cells lacked GPI-AP and PIG-A expression, we did not observe a genetic mutation in the PIG-A locus. In the current study, we provide further direct evidence that silencing of PIG-A expression and lack of GPI-APs are due to promoter DNA methylation. These data raise the possibility that PNH could arise from epigenetic silencing of PIG-A, since the diagnosis is based on phenotype rather than genotype in most instances. Epigenetic silencing caused by mutations in the promoter region of PIG-M leading to GPI-AP deficiency is well described; furthermore, re-expression of the GPI-APs occurs in these patients following treatment with sodium butyrate [40]. Epigenetic silencing of genes involved in GPI anchor biosynthesis has also been reported in Burkitt lymphoma cells [41].
Although the PIG-A-deficient AR1-c1 human ES cells were the best available at the time for us to investigate the role of GPI-APs in early human development, the epigenetic silencing of the PIG-A gene is a drawback since a fraction of cells gradually regained PIG-A expression and GPI-APs, especially after long-term culture required for expansion or differentiation. Therefore, we generated the PIG-A-null FPHR iPSCs by homologous recombination, which is similar to the Pig-a-null mouse ES cells established by Dunn et al. [18], using gene targeting to introduce null mutations. Similar to mouse Pig-a-null ES cells, the FPHR iPSCs that have a defective PIG-A gene could not readily form EBs via forced aggregation. Although the cells aggregated and formed cell spheres, they were abnormal and did not form hematopoietic cells surrounding the main EBs. Further analysis indicated that CD34 cells were completely lacking, as were precursor cells expressing KDR/VEGFR2 and CD56 at earlier stages. RT-PCR analysis revealed that markers of mesoderm and endoderm failed to elevate in the FPHR cells during EB differentiation; however, expression of ectodermal markers was increased. Since the relative number of cells in the FPHR EBs decreased by day 7, our data suggest that the increase in ectodermal markers is secondary to lack of mesoendodermal differentiation. Moreover, the PIG-A-null cells (FPHR) demonstrated reduced signaling through BMP-4. Taken together, these data suggest that one or more GPI-APs, likely RGMa and/or RGMb, are required for early development leading to blood formation and that temporary restoration of PIG-A expression (and subsequent GPI-APs) is sufficient to overcome this defect. BMP-4 signaling is required for mesoderm formation, and BMP-4 knockout in transgenic mice leads to embryonic lethality between days 6.5 and 9.5, which corresponds to the early developmental block we observe in the FPHR cells [32]. The observed ectodermal differentiation in the PIG-A-null iPSC is likely a consequence of reduced or delayed differentiation toward other germline lineages. It is believed that differentiation into neuronal cells is a default fate of ES cells under the inhibitory control (or in the absence) of mesoderm- and/or endoderm-inducing factors such as BMP-4 [42]. Our results also show that hematopoietic growth and differentiation beyond the level of the EB stage is not dependent upon GPI-APs. Transient expression of GPI-APs for just 14 days enabled the cells to form EBs and to terminally differentiate into GPI-AP-deficient myeloid and erythroid cells.
Conclusion
The iPSC lines established here should serve as a valuable model for studying inherited and acquired GPI-AP deficiency. The human disease PNH is caused by somatic PIG-A mutations occurring in hematopoietic stem cells. PNH is a clonal hematopoietic disorder, and the mechanism(s) whereby the PNH cells achieve clonal dominance are still unclear. It has been hypothesized that immune selection of the PNH clone and/or secondary mutations are required to achieve clonal dominance [43–46]. We did not find that the PIG-A-null blood cells generated from human iPSCs (hFib2-iPS5) had an intrinsic growth advantage over the wild-type PIG-A iPSCs. These findings are in agreement with conditional Pig-a knockout studies performed in mice [47, 48]. In the future, PNH patient-specific iPSCs transduced with an inducible PIG-A gene could be used to more directly test these and other hypotheses.
Human PIG-A-null iPSCs may also be useful for studying the developmental pathways that are perturbed in inherited GPI-AP deficiency. Germline hypomorphic mutations leading to reduced GPI-AP expression have been described for PIG-A and five other genes involved in GPI anchor biosynthesis: PIG-M [21], PIG-V [22], PIG-O [49], PIG-L [50], and PIG-N [23]. These patients all have similar phenotypes characterized by multiple developmental abnormalities, facial dysmorphism, severe neurologic disease, seizures, and early death. Interestingly, none of these patients had significant hematopoietic defects, suggesting that partial GPI-AP deficiency is sufficient for blood formation but not sufficient normal neurologic development. Modeling these mutations in PIG-A mutant human iPSCs should provide insight into the developmental abnormalities and signaling pathways that are affected by GPI anchor deficiency.
Supplementary Material
Acknowledgments
We thank Dr. Saul Sharkis for helpful discussions and critical review and Dr. Zack Wang for providing primers and for helpful discussions. This study was supported in part by several grants from the NIH (P01-CA70970, R01-HL073781, and T32-HL007525).
Author Contributions
X.Y.: conception and design, performance of experiments, collection and assembly of data, data analysis and interpretation, manuscript writing; E.M.B.: collection and assembly of data, data analysis and interpretation; Z.Y.: conception and design, data analysis and interpretation, provision of study material; C.F.L.: assembly of data, provision of study material, performance of experiments; G.C.: provision of study material, performance of experiments; J.Z.: provision of study material; L.C.: conception and design, data analysis and interpretation, provision of study material, manuscript writing, final approval of manuscript; R.A.B.: conception and design, financial support, administrative support, data analysis, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Low MG, Saltiel AR. Structural and functional roles of glycosyl-phosphatidylinositol in membranes. Science. 1988;239:268–275. doi: 10.1126/science.3276003. [DOI] [PubMed] [Google Scholar]
- 2.Udenfriend S, Kodukula K. How glycosylphosphatidylinositol-anchored membrane proteins are made. Annu Rev Biochem. 1995;64:563–591. doi: 10.1146/annurev.bi.64.070195.003023. [DOI] [PubMed] [Google Scholar]
- 3.Kinoshita T, Inoue N. Dissecting and manipulating the pathway for glycosylphosphatidylinositol-anchor biosynthesis. Curr Opin Chem Biol. 2000;4:632–638. doi: 10.1016/s1367-5931(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 4.Diep DB, Nelson KL, Raja SM, et al. Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J Biol Chem. 1998;273:2355–2360. doi: 10.1074/jbc.273.4.2355. [DOI] [PubMed] [Google Scholar]
- 5.Brodsky RA, Mukhina GL, Nelson KL, et al. Resistance of paroxysmal nocturnal hemoglobinuria cells to the glycosylphosphatidylinositol-binding toxin aerolysin. Blood. 1999;93:1749–1756. [PubMed] [Google Scholar]
- 6.Miyata T, Takeda J, Iida Y, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. 1993;259:1318–1320. doi: 10.1126/science.7680492. [DOI] [PubMed] [Google Scholar]
- 7.Inoue N, Watanabe R, Takeda J, et al. PIG-C, one of the three human genes involved in the first step of glycosylphosphatidylinositol biosynthesis is a homologue of Saccharomyces cerevisiae GPI2. Biochem Biophys Res Commun. 1996;226:193–199. doi: 10.1006/bbrc.1996.1332. [DOI] [PubMed] [Google Scholar]
- 8.Kamitani T, Chang HM, Rollins C, et al. Correction of the class H defect in glycosylphosphatidylinositol anchor biosynthesis in Ltk− cells by a human cDNA clone. J Biol Chem. 1993;268:20733–20736. [PubMed] [Google Scholar]
- 9.Tiede A, Schubert J, Nischan C, et al. Human and mouse Gpi1p homologues restore glycosylphosphatidylinositol membrane anchor biosynthesis in yeast mutants. Biochem J. 1998;334:609–616. doi: 10.1042/bj3340609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe R, Inoue N, Westfall B, et al. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG-A, PIG-H, PIG-C and GPI1. EMBO J. 1998;17:877–885. doi: 10.1093/emboj/17.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murakami Y, Siripanyaphinyo U, Hong Y, et al. The initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-Y, a seventh component. Mol Biol Cell. 2005;16:5236–5246. doi: 10.1091/mbc.E05-08-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe R, Murakami Y, Marmor MD, et al. Initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-P and is regulated by DPM2. EMBO J. 2000;19:4402–4411. doi: 10.1093/emboj/19.16.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma DK, Vidugiriene J, Bangs JD, et al. A cell-free assay for glycosylphosphatidylinositol anchoring in African trypanosomes. Demonstration of a transamidation reaction mechanism. J Biol Chem. 1999;274:16479–16486. doi: 10.1074/jbc.274.23.16479. [DOI] [PubMed] [Google Scholar]
- 14.Takeda J, Miyata T, Kawagoe K, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 15.Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13:110–117. doi: 10.1002/j.1460-2075.1994.tb06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawagoe K, Kitamura D, Okabe M, et al. Glycosylphosphatidylinositol-anchor-deficient mice: Implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:3600–3606. [PubMed] [Google Scholar]
- 17.Rosti V, Tremml G, Soares V, et al. Murine embryonic stem cells without pig-a gene activity are competent for hematopoiesis with the PNH phenotype but not for clonal expansion. J Clin Invest. 1997;100:1028–1036. doi: 10.1172/JCI119613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunn DE, Yu J, Nagarajan S, et al. A knock-out model of paroxysmal nocturnal hemoglobinuria: Pig-a− hematopoiesis is reconstituted following intercellular transfer of GPI-anchored proteins. Proc Natl Acad Sci USA. 1996;93:7938–7943. doi: 10.1073/pnas.93.15.7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen G, Ye Z, Yu X, et al. Trophoblast differentiation defect in human embryonic stem cells lacking PIG-A and GPI-anchored cell-surface proteins. Cell Stem Cell. 2008;2:345–355. doi: 10.1016/j.stem.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston JJ, Gropman AL, Sapp JC, et al. The phenotype of a germline mutation in PIGA: The gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet. 2012;90:295–300. doi: 10.1016/j.ajhg.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Almeida AM, Murakami Y, Layton DM, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med. 2006;12:846–851. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 22.Krawitz PM, Schweiger MR, Rodelsperger C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42:827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 23.Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet. 2011;48:383–389. doi: 10.1136/jmg.2010.087114. [DOI] [PubMed] [Google Scholar]
- 24.Zou J, Maeder ML, Mali P, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou BY, Ye Z, Chen G, et al. Inducible and reversible transgene expression in human stem cells after efficient and stable gene transfer. Stem Cells. 2007;25:779–789. doi: 10.1634/stemcells.2006-0128. [DOI] [PubMed] [Google Scholar]
- 26.Ng ES, Davis RP, Hatzistavrou T, et al. Directed differentiation of human embryonic stem cells as spin embryoid bodies and a description of the hematopoietic blast colony forming assay. Curr Protoc Stem Cell Biol. 2008 doi: 10.1002/9780470151808.sc01d03s4. Chapter 1:Unit 1D.3. [DOI] [PubMed] [Google Scholar]
- 27.Ye Z, Zhan H, Mali P, et al. Human induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lapillonne H, Kobari L, Mazurier C, et al. Red blood cell generation from human induced pluripotent stem cells: Perspectives for transfusion medicine. Haematologica. 2010;95:1651–1659. doi: 10.3324/haematol.2010.023556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson KL, Brodsky RA, Buckley JT. Channels formed by subnanomolar concentrations of the toxin aerolysin trigger apoptosis of T lymphomas. Cell Microbiol. 1999;1:69–74. doi: 10.1046/j.1462-5822.1999.00009.x. [DOI] [PubMed] [Google Scholar]
- 30.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 31.Evseenko D, Zhu Y, Schenke-Layland K, et al. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc Natl Acad Sci USA. 2010;107:13742–13747. doi: 10.1073/pnas.1002077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winnier G, Blessing M, Labosky PA, et al. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- 33.Johansson BM, Wiles MV. Evidence for involvement of activin A and bone morphogenetic protein 4 in mammalian mesoderm and hematopoietic development. Mol Cell Biol. 1995;15:141–151. doi: 10.1128/mcb.15.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maeno M, Mead PE, Kelley C, et al. The role of BMP-4 and GATA-2 in the induction and differentiation of hematopoietic mesoderm in Xenopus laevis. Blood. 1996;88:1965–1972. [PubMed] [Google Scholar]
- 35.Babitt JL, Zhang Y, Samad TA, et al. Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co-receptor. J Biol Chem. 2005;280:29820–29827. doi: 10.1074/jbc.M503511200. [DOI] [PubMed] [Google Scholar]
- 36.Samad TA, Rebbapragada A, Bell E, et al. DRAGON, a bone morphogenetic protein co-receptor. J Biol Chem. 2005;280:14122–14129. doi: 10.1074/jbc.M410034200. [DOI] [PubMed] [Google Scholar]
- 37.Brodsky RA, Mukhina GL, Li S, et al. Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol. 2000;114:459–466. doi: 10.1093/ajcp/114.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lugus JJ, Park C, Ma YD, et al. Both primitive and definitive blood cells are derived from Flk-1+ mesoderm. Blood. 2009;113:563–566. doi: 10.1182/blood-2008-06-162750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nozaki M, Ohishi K, Yamada N, et al. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab Invest. 1999;79:293–299. [PubMed] [Google Scholar]
- 40.Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med. 2007;356:1641–1647. doi: 10.1056/NEJMoa063369. [DOI] [PubMed] [Google Scholar]
- 41.Hu R, Mukhina GL, Lee SH, et al. Silencing of genes required for glycosylphosphatidylinositol anchor biosynthesis in Burkitt lymphoma. Exp Hematol. 2009;37:423–434. doi: 10.1016/j.exphem.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hemmati-Brivanlou A, Melton D. Vertebrate embryonic cells will become nerve cells unless told otherwise. Cell. 1997;88:13–17. doi: 10.1016/s0092-8674(00)81853-x. [DOI] [PubMed] [Google Scholar]
- 43.Luzzatto L, Bessler M, Rotoli B. Somatic mutations in paroxysmal nocturnal hemoglobinuria: A blessing in disguise? Cell. 1997;88:1–4. doi: 10.1016/s0092-8674(00)81850-4. [DOI] [PubMed] [Google Scholar]
- 44.Young NS. The problem of clonality in aplastic anemia: Dr. Dameshek's riddle, restated. Blood. 1992;79:1385–1392. [PubMed] [Google Scholar]
- 45.Hanaoka N, Kawaguchi T, Horikawa K, et al. Immunoselection by natural killer cells of PIGA mutant cells missing stress-inducible ULBP. Blood. 2006;107:1184–1191. doi: 10.1182/blood-2005-03-1337. [DOI] [PubMed] [Google Scholar]
- 46.Inoue N, Izui-Sarumaru T, Murakami Y, et al. Molecular basis of clonal expansion of hematopoiesis in two patients with paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2006;108:4232–4236. doi: 10.1182/blood-2006-05-025148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murakami Y, Kinoshita T, Maeda Y, et al. Different roles of glycosylphosphatidylinositol in various hematopoietic cells as revealed by a mouse model of paroxysmal nocturnal hemoglobinuria. Blood. 1999;94:2963–2970. [PubMed] [Google Scholar]
- 48.Tremml G, Dominguez C, Rosti V, et al. Increased sensitivity to complement and a decreased red blood cell life span in mice mosaic for a nonfunctional Piga gene. Blood. 1999;94:2945–2954. [PubMed] [Google Scholar]
- 49.Krawitz PM, Murakami Y, Hecht J, et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet. 2012;91:146–151. doi: 10.1016/j.ajhg.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ng BG, Hackmann K, Jones MA, et al. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet. 2012;90:685–688. doi: 10.1016/j.ajhg.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.