

This study examined nestin-positive stem cells in myocardium of dystrophin/utrophin-deficient mice, a model for Duchenne muscular dystrophy. Findings indicate that new cardiomyocytes form in dystrophic heart, and nestin-positive interstitial cells may generate them in addition to other cells of the cardiac lineage.
Keywords: Cardiac, Muscular dystrophy, Nestin, Sca-1, Tissue regeneration, Muscle stem cells
Abstract
Nestin+ cardiac stem cells differentiate into striated cells following myocardial infarct. Transplantation of exogenous stem cells into myocardium of a murine model for Duchenne muscular dystrophy (DMD) increased proliferation of endogenous nestin+ stem cells and resulted in the appearance of nestin+ striated cells. This correlated with, and may be responsible for, prevention of dilated cardiomyopathy. We examined nestin+ stem cells in the myocardium of dystrophin/utrophin-deficient (mdx/utrn−/−) mice, a model for DMD. We found that 92% of nestin+ interstitial cells expressed Flk-1, a marker present on cardiac progenitor cells that differentiate into the cardiac lineage, and that a subset expressed Sca-1, present on adult cardiac cells that become cardiomyocytes. Nestin+ interstitial cells maintained expression of Flk-1 but lost Sca-1 expression with age and were present in lower numbers in dystrophin-deficient heart than in wild-type heart. Unexpectedly, large clusters of nestin+ striated cells ranging in size from 20 to 250 cells and extending up to 500 μm were present in mdx/utrn−/− heart near the end stage of disease. These cells were also present in dystrophin-deficient mdx/utrn+/− and mdx heart but not wild-type heart. Nestin+ striated cells expressed cardiac troponin I, desmin, and Connexin 43 and correlated with proinflammatory CD68+ macrophages. Elongated nestin+ interstitial cells with striations were observed that did not express Flk-1 or the late cardiac marker cardiac troponin I but strongly expressed the early cardiac marker desmin. Nestin was also detected in endothelial and smooth muscle cells. These data indicate that new cardiomyocytes form in dystrophic heart, and nestin+ interstitial cells may generate them in addition to other cells of the cardiac lineage.
Introduction
Nestin is an intermediate filament protein involved in cytoskeletal remodeling, and it is expressed transiently during development in the central nervous system, heart, and skeletal muscle [1–4]. Nestin is specifically upregulated during differentiation of stem cells, as expression of lineage-specific marker proteins is occurring [5]. Nestin expression is downregulated following differentiation, as expression of tissue-specific intermediate filaments begins to occur. Nestin is not present in adult tissues with the exception of expression in stem and progenitor cells and transient expression in regenerating tissues. Nestin is expressed by neural progenitors, hair follicle stem cells, bone marrow stem cells, mesenchymal stem cells, and skeletal and cardiac muscle stem cells [5–9], which may contribute to tissue homeostasis and repair following injury. In agreement with this, nestin has also been observed transiently in adults during regeneration of skeletal muscle, following injury to the central nervous system, and in heart following myocardial infarction [10–17].
Endogenous nestin+ stem cells in the adult heart have neural potential, differentiating into neural cells in vitro [18], directly innervating damaged, infarcted heart tissue in vivo [19], and facilitating sympathetic sprouting by other neural stem cells in infarcted heart [14, 19]. However, they also have the potential to differentiate into cardiac muscle cells. A subset of nestin+ stem cells in the heart express cardiac transcription factors, including Nkx2.5 in the cytosol, and nestin+ cells isolated from cardiospheres or by Sca-1 expression from adult heart differentiate into cardiomyocytes in culture [15, 18, 20]. Some studies have reported that these cells generate nestin+ striated cardiomyocytes in the adult rat heart following ischemic damage in rats and in adult human heart following myocardial infarct or during end-stage heart failure [14–17]. Lineage tracing studies support this possibility: it was found that endogenous cardiac stem cells expressing nestin originate from the neural crest, and although most remain in a dormant state in adult heart, some become cardiomyocytes [14, 18].
In a recent study by our group we reported a significant increase in the division of endogenous nestin+ cardiac stem cells following transplantation of mesoangioblasts into the heart of dystrophin- and utrophin-deficient (mdx/utrn−/−) mice, a phenotypic model for Duchenne muscular dystrophy (DMD) [21]. This was accompanied by an overall decline in the number of nestin+ cardiac stem cells as well as the appearance of clusters of nestin+ striated cells, originating from host cells and expressing cardiac markers. These events coincided with a delay in the onset of dilated cardiomyopathy resulting from dystrophin deficiency, despite the lack of dystrophin restoration by the exogenous cells. Because nestin expression in adult tissues is associated with stem cells and tissue regeneration, the presence of nestin+ striated cells suggested that regeneration was occurring in the heart of mice injected with exogenous mesoangioblasts. Additionally, the simultaneous activation and depletion of endogenous nestin+ stem cells indicated that they may have been generating the nestin+ cardiomyocytes observed in the heart.
The generation of new cardiomyocytes in dystrophin-deficient cardiac muscle from an endogenous population of cardiac stem cells suggests a potential mechanism that may be exploited to delay or prevent dilated cardiomyopathy (DCM) in DMD. Mutations in the dystrophin gene cause Becker muscular dystrophy (BMD) and DMD. BMD has an incidence of 1 in 8,000–10,000 males and results in decreased life expectancy, variable muscle weakness, and onset of DCM between 13 and 21 years of age (reviewed in [22]). DMD occurs in 1 of every 3,500 boys born [23]. Nearly 60% of DMD patients develop dilated cardiomyopathy by 10 years of age, and all patients have DCM by 18 years of age, resulting in the death of between 10%–40% of patients [24–30]. Beta blockers, angiotensin-converting enzyme inhibitors, and steroids have been used to treat the symptoms of cardiomyopathy in DMD [31–44], but these agents cannot replace lost or damaged cardiomyocytes that result from lack of fully functional dystrophin.
Stem cell therapy has the potential to generate new cardiomyocytes in damaged heart, and may therefore prevent, or in combination with the aforementioned agents further delay, the onset of dilated cardiomyopathy. Three different populations of cells have been tested for their efficacy in restoring dystrophin and/or generating new cardiomyocytes in the heart of animal models for DMD. First, fetal cardiomyocytes with a functional copy of the dystrophin gene were transplanted into the heart of dystrophin-deficient mice and dogs and restored dystrophin expression in the heart. The donor cells integrated into host tissue and formed gap junctions with surrounding cardiomyocytes [45]. Although these studies are promising, the availability of donor fetal cardiomyocytes in sufficient numbers for transplantation into DMD patients may not be feasible and may also present an ethical dilemma. A second study used skeletal muscle-derived stem cells (MDSCs), transplanting the undifferentiated cells into the heart of a mouse model for DMD. MDSCs expressed dystrophin but became skeletal muscle cells in the heart rather than cardiac myocytes [46]. Previous studies in human patients with skeletal muscle-derived stem cells known as myoblasts have demonstrated that differentiation of stem cells into skeletal muscle cells in the heart may result in the development of arrhythmias because the skeletal muscle cells do not form gap junctions necessary for electrical coupling with host cardiomyocytes [47, 48]. In a third study, we transplanted undifferentiated aorta-derived mesoangioblasts (ADMs) with a functional copy of the dystrophin gene into the heart of mouse models for DMD, which prevented the onset of DCM [21]. However, donor cell differentiation into dystrophin-positive cardiomyocytes was rare. Instead, there was a significant increase in the division of nestin+ endogenous cardiac stem cells, and nestin+ striated cells expressing cardiac troponin I were detected specifically in mesoangioblast-transplanted heart.
These data suggested that nestin+ cardiac stem cells might be capable of generating new cardiac myocytes in dystrophin-deficient heart. We therefore examined nestin+ stem cells in dystrophin-deficient heart to determine whether these cells declined in number with disease progression, as do other endogenous stem cells from dystrophin-deficient skeletal and cardiac muscle [49–54]. The decline in stem cell number and differentiation potential in dystrophin-deficient muscle has been attributed to the continuous cycles of regeneration required to counter the progressive damage and degeneration resulting from the absence of dystrophin [55, 56] and has important implications for the feasibility of autologous cell therapy in DMD patients. Although we did find a significantly lower number of nestin+ stem cells in the heart of mouse models for DMD, we also report a surprising finding indicating that dystrophin-deficient cardiac muscle is capable of regeneration at the end stages of muscle disease.
Materials and Methods
Mice
All mice were handled according to a protocol approved by the University of Illinois Institutional Animal Care and Use Committee. Mdx/utrn−/− dystrophin- and utrophin-deficient mice are a phenotypic model for Duchenne muscular dystrophy; they develop progressive pathology in skeletal and cardiac muscle, ventricular dilation, and a resulting decrease in heart function [57, 58]. Mdx, Mdx/utrn+/− and Mdx/utrn−/− mice were generated by interbreeding mdx/utrn+/− mice [57], and the progeny were genotyped as previously described [59]. Because the mdx and utrn−/− mice used to generate mdx/utrn+/− mice were maintained on a C57Bl background, we used C57Bl/10 mice for age-matched wild-type controls. Cardiac ultrasound data from the mdx/utrn−/− and wild-type mice used in this study have been published previously [58].
Tissue Collection, Immunocytochemistry, and Data Quantitation
For immunohistochemistry of cardiac tissue, hearts were collected, weighed, and frozen in liquid nitrogen-cooled 2-methylbutane and stored at −80°C. Skeletal muscle was also frozen in liquid nitrogen-cooled 2-methylbutane and stored at −80°C. Consecutive serial sections were cut in 10-μm-thick slices and collected on numbered slides. Sections were fixed in 3.7% formaldehyde for 10 minutes and incubated with blocking solution (1× phosphate-buffered saline [PBS]/2% horse serum/5% bovine serum albumin, or 10% normal goat serum [Invitrogen, Frederick, MD, http://www.invitrogen.com] for CD163, Flk-1, desmin, and Sca-1) for 1 hour at room temperature (RT), followed by incubation with primary antibodies for 1 hour at RT. An exception is the immunostaining protocol for Flk-1 and nestin, in which tissue was fixed with ice-cold acetone for 2 minutes, followed by incubation with blocking solution for 1 hour at RT and primary antibody overnight at 4°C. Primary antibodies used were as follows: chicken antibody to Nestin (CH23001, 1:100; Neuromics, Edina, MN, http://www.neuromics.com), mouse monoclonal anti-cardiac troponin I (MAB1691, 1:300; Millipore, Billerica, MA, http://www.millipore.com), mouse antibody to desmin (DE-R-11, 1:50; Santa Cruz Biotechnology Inc., Santa Cruz, CA, http://www.scbt.com), rat antibody to CD68 (1:50; BioLegend, San Diego, CA, http://www.biolegend.com), rabbit polyclonal antibody to CD163 (1:50, M-96; Santa Cruz Biotechnology), rat antibody to Sca-1 (1:100; BD Biosciences, San Jose, CA, http://www.bdbiosciences.com), rabbit antibody to Nkx2.5 (1:50, sc14033; Santa Cruz Biotechnology), rat antibody to Flk-1 (1:10, AVAS 12α1; BD Biosciences), mouse monoclonal antibody to α-smooth muscle actin (1:100; Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com), rabbit polyclonal antibody to CD31 (1:100, ab28364; Abcam, Cambridge, MA, http://www.abcam.com), and rabbit polyclonal anti-Connexin 43 (Cxn43) (1:100, C6219; Sigma-Aldrich). The mouse monoclonal antibody to fetal myosin heavy chain (feMHC) (clone 47A, 1:10) was kindly provided by Dr. Stephen Kaufman (University of Illinois). Next, slides were washed with 1× PBS and incubated with secondary antibodies (Jackson Immunoresearch Laboratories, West Grove, PA, http://www.jacksonimmuno.com) for 1 hour at RT, and then coverslips were mounted using Vectashield mounting medium including 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, http://www.vectorlabs.com).
Images were captured with a Retiga 2000R digital camera (QImaging, Surrey, BC, Canada, http://www.qimaging.com) mounted on a Leica inverted DMI 4000B microscope (Leica, Heerbrugg, Switzerland, http://www.leica.com). Images were assigned color and merged using Image-Pro Plus software (MediaCybernetics, Bethesda, MD, http://www.mediacy.com).
Nestin+ interstitial stem cells were quantified from 10 randomly captured images containing Nestin fluorescein isothiocyanate and DAPI-stained nuclei from each mouse in each of the following groups, with four mice in each group: groups 1 and 2, 5-week-old wild-type and mdx/utrn−/− mice; groups 3 and 4, 10-week-old wild-type and mdx/utrn−/− mice; groups 5 and 6, 15-week-old wild-type and mdx/utrn−/− mice. Nestin+ striated cells expressing cardiac troponin I were quantified in tissue from the hearts of three different 15-week-old mdx/utrn−/− mice, from multiple tissue sections from each mouse, were examined for a total of 500 nestin+ cells. The frequency of nestin+ striated cells adjacent to CD68+ macrophages was determined in six tissue sections from each of three 15-week-old mdx/utrn−/− mice. Macrophage clusters consisted of ≥8 macrophages, and 2–5 clusters per section were typically observed. To quantify nestin+ interstitial cells expressing Sca-1, four randomly chosen fields were captured with a ×20 objective, and single-stained and merged images were used to calculate the number of nestin+ cells and the percentage that expressed Sca-1. Numbers of cells per field varied, with a minimum of 491 cells counted for one animal and a maximum of 992 cells counted for another animal. Five-, 10-, and 15-week-old mdx/utrn−/− mice were examined, four mice per group. To quantify nestin+ interstitial cells expressing Flk-1, four randomly chosen fields of view were captured with a ×40 objective. Images of individual fluorophores for nestin+ and Flk1+ and merged images with both fluorophores were used to quantify the number of nestin+ cells and the percentage coexpressing Flk-1. Five-, 10-, and 15-week-old mdx/utrn−/− and 65-week-old mdx/utrn+/− mice, with two mice per group. A minimum of 200 cells were examined for each mouse.
Statistical significance was determined by the two-tailed Student's t test, using the GraphPad QuickCalc free online software (http://graphpad.com/quickcalcs/ttest2.cfm) from GraphPad Software, Inc. (La Jolla, CA, http://www.graphpad.com).
Results
Nestin+ Striated Cells Are Present in the Heart of mdx/utrn−/− Mice at 15 Weeks of Age
We have previously reported that mdx/utrn−/− mice develop regions of extensive fibrosis, ventricular dilation, thinning of the left ventricular wall, and a decline in heart function measured by cardiac ultrasound between 10 and 15 weeks of age [58]. Mdx/utrn−/− mice develop tachycardia by 5 weeks of age, which also precedes dilated cardiomyopathy in DMD patients. Thinning of the left ventricular wall and septum follow at 10 weeks of age. By 15 weeks of age pathology is evident in the form of damaged cardiomyocyte membranes, increased interstitial collagen, and large regions of fibrosis and inflammatory cell infiltration throughout the myocardium of the ventricles. Ventricular remodeling and declining function is significant by 15 weeks of age as well, with dilation of the left ventricle and a decrease in fractional shortening, ejection fraction, and end diastolic volume [58]. Using tissue collected from the ventricles of these mdx/utrn−/− and wild-type mice, we examined nestin+ cells in hearts collected at 5, 10, and 15 weeks of age. Unexpectedly, we detected clusters of nestin+ striated cells in the heart of mdx/utrn−/− mice at 15 weeks of age (Fig. 1A–1C, four of four mice). Nestin+ striated cells were detected in all mdx/utrn−/− hearts at 15 weeks of age and in one heart at 10 weeks of age, but not in the heart of 5-week-old mice or wild-type mice (Fig. 1A–1C, 1L–1O). The expression of nestin was more intense in smaller striated cells, with a gradual decrease in intensity with increasing size of the cells (Fig. 1I). This is similar to the transient expression of nestin reported in regenerating skeletal muscle of DMD patients [60]. We observed the same trend in nestin expression in small caliber, feMHC-positive regenerating skeletal muscle fibers in mdx/utrn−/− mice, with the most intense expression in the smallest fibers and decreasing intensity with increasing fiber size (Fig. 1E–1G). Nestin+ striated cells were not present in nondamaged, wild-type hearts at 5, 10, or 15 weeks of age (Fig. 1L, 1O, zero of four mice per group).
Figure 1.

Nestin+ striated cells are present in mdx/utrn−/− dystrophic myocardium. (A–C): Intense nestin immunofluorescence in striated cells (green, white arrows; *, ventricle) and interstitial stem cells (white arrowheads) in mdx/utrn−/− heart. (D): Negative control, no nestin primary antibody, fluorescein isothiocyanate secondary antibody only. (E–G): Nestin expression (red, white arrows in [E] and [G]) in small-caliber skeletal muscle fibers in mdx/utrn−/− that express the fetal form of myosin heavy chain (feMHC) (green, white arrows in [E] and [F]), with greater intensity of both markers in smaller fibers. *, damaged fibers; #, mature muscle fibers. (H): Negative control, no feMHC primary antibody, tetramethylrhodamine isothiocyanate secondary antibody only. (I–K): Nestin expression in striated cells in the mdx/utrn−/− heart was more intense in smaller striated cells (white arrowheads, [I]) than in large striated cells (white arrows, [I–K]). (L–N): Nestin+ interstitial stem cells (white arrowheads), but not striated cells, were observed in the heart of 15 week wt (L), 5 week (M), and 10 week (N) mdx/utrn−/− mice. (O): Quantitation of nestin+ striated cells in mdx/utrn−/− heart. Abbreviations: D, day; DAPI, 4′,6-diamidino-2-phenylindole; dKO, double knockout; LV, left ventricle; MHC, myosin heavy chain; RV, right ventricle; wks, weeks; Wt, wild-type.
We examined myocardium of the mdx and mdx/utrn+/− mice, also deficient in dystrophin, for the presence of nestin+ striated cells. The mdx mouse is the genotypic model for Duchenne muscular dystrophy and develops pathology in the heart and dilated cardiomyopathy over a period between 12 and 21 months of age [61–63]. Mdx/utrn+/− mice contain the same mutation in dystrophin as the mdx mouse but also lack one allele of utrophin, a gene coding for a protein that compensates functionally for dystrophin in mice. Heart function, measured by stroke volume and ejection fraction, declines in the mdx/utrn+/− mouse, and fibrosis is detectable by 10 months of age [64]. Nestin+ striated cells were present in the myocardium of both mdx (eight of eight mice) and mdx/utrn+/− (two of two mice) mice 14–16 months old (Fig. 2A–2F). The majority of nestin+ striated cells detected in mdx heart were isolated, single cells (Fig. 2A, 2B), rather than clusters similar to those present in the 15-week mdx/utrn−/− heart. We did, however, observe clusters of nestin+ striated cells in mdx/utrn+/− heart, with more intense expression in smaller striated cells (Fig. 2C–2F), similar to mdx/utrn−/− heart.
Figure 2.
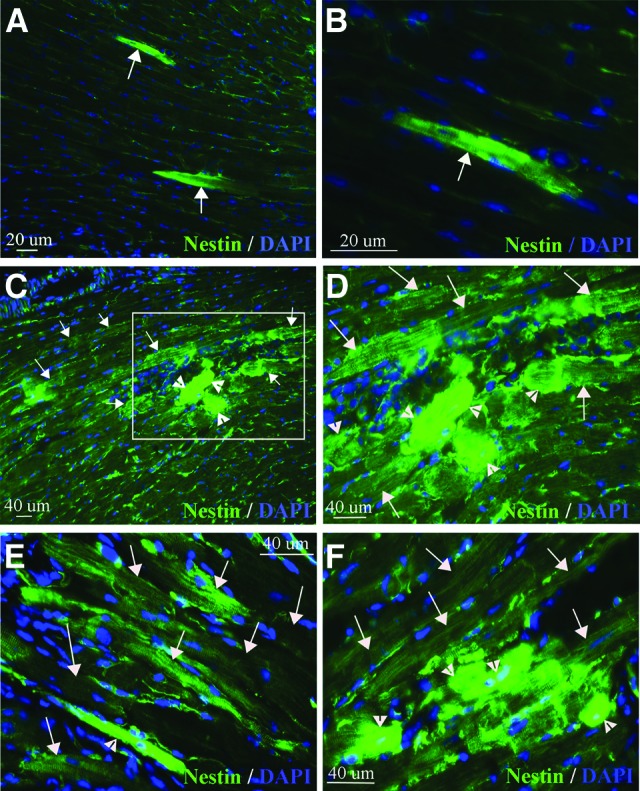
Nestin+ striated cells are present in mdx and mdx/utrn+/− dystrophin-deficient heart. (A, B): Nestin+ striated cells (green, white arrows) in the heart of 16-month-old mdx mice. (C–F): Large clusters of nestin+ striated cells (green, white arrows and arrowheads) were present in the heart of 16-month-old mdx/utrn+/− mice, with greatest intensity in the smaller cells (small striated cells indicated by white arrowheads, larger striated cells indicated by white arrows). (D) is an enlargement of the boxed area in (C). Abbreviation: DAPI, 4′,6-diamidino-2-phenylindole.
Many of the nestin+ striated cells in mdx/utrn−/− and mdx/utrn+/− heart were present in regions of damaged and disorganized tissue, with inflammatory cell infiltration evident. In such regions, the staining of nestin+ striated cells varied in intensity and the cells were frequently disordered and of different sizes (Fig. 1A–1C, 1I). However, we also observed large regions of well-ordered nestin+ striated cells with more homogeneous, less intense staining, in which the striated cells were similar in size (Fig. 1J, 1K). In mdx, mdx/utrn−/− and mdx/utrn+/− mice nestin+ striated cells were present in the left or right ventricular wall or the septum, and in the mdx/utrn−/− and mdx/utrn+/− hearts there was frequently more than one cluster present in the same tissue section (Fig. 1O). Nestin+ striated cells were not present in every section of the heart. When present, clusters of nestin+ striated cells in the mdx/utrn−/− heart ranged in size from 20 to more than 200 cells per cluster, and the same clusters were frequently present throughout consecutive serial sections on multiple slides, at distances ranging from 70 μm to 500 μm (Fig. 1O).
Nestin+ Striated Cells Express Markers of Cardiac Myocytes
We examined nestin+ striated cells for expression of proteins present in cardiomyocytes. We observed that 98.18 ± 2.10% of nestin+ striated cells, but not the surrounding nestin+ interstitial stem cells, expressed cardiac troponin I (Fig. 3A–3C). We also analyzed cells for expression of the gap junction protein Cxn43, which is essential for electrical coupling between cardiomyocytes. In a previous report, nestin+ striated cells in postinfarcted heart expressed the gap junction protein Cxn43 in an aberrant pattern [15]. We detected Cxn43 expression in an aberrant pattern in some nestin+ striated cells in mdx/utrn−/− heart (Fig. 3F, 3G), but many large nestin+ striated cells, particularly those with lower expression of nestin as judged by fluorescence intensity, expressed Cxn43 at the junction with adjacent cardiomyocytes, as expected (Fig. 3E–3G). Nkx2.5, a transcription factor transiently expressed during cardiac development, was also present in the nuclei of a few nestin+ striated cells (Fig. 3I–3K, three cells in 10 tissue sections). The low number of nestin+ striated cells expressing this developmental transcription factor may be the result of the transient nature of Nkx2.5 expression during cardiac differentiation. It was found that 99.60% of nestin+ striated cells also expressed desmin (Fig. 3M–3O), one of the earliest markers of both cardiac and skeletal muscle that is also present in adult cardiomyocytes [65–68]. Desmin expression was more intense in the nestin+ striated cells in the heart than in surrounding, striated cells that did not express nestin (Fig. 3M–3O). This is similar to desmin expression in regenerating skeletal myofibers in mdx/utrn−/− mice (Fig. 3Q–3S) and is consistent with reports indicating that desmin expression increases and peaks during terminal differentiation of skeletal muscle [65] and during cardiogenesis [66–68].
Figure 3.

Nestin+ striated cells express cardiac markers. (A–P): nestin+ striated (green in [A, C, E, G, I, K, N, O] and red in [J, L], indicated by white arrows) but not interstitial (# in [A–C, E–G, M–O]) cells expressed cTnI (red [B, C]), Cxn43 (red [F, G], gap junctions indicated by white arrowheads), Nkx2.5 (red [I–K]), and desmin (green [N, O]; greater intensity in nestin+ cells). (D, H, L, P): Myocardium incubated with secondary antibody only as a control for background, nonspecific immunofluorescence. (F, G): Cxn43 was observed in the cytosol in some nestin+ striated cells (*). (O): Enlargement of region shown in (M) and (N), indicated by the white box in (M). (Q–S): Intense desmin expression (green [R, S]), in small-caliber nestin+ myofibers (red [Q, S]) in mdx/utrn−/− skeletal muscle. (T): Skeletal muscle incubated with secondary antibody only as a control for background, nonspecific immunofluorescence. White asterisks in (M–O) represent myofibers not undergoing regeneration. Blue, DAPI-labeled nuclei. Abbreviations: cTpnI, cardiac troponin I; Cxn43, Connexin 43; DAPI, 4′,6-diamidino-2-phenylindole.
Nestin+ Cardiomyocyte Cells Are Localized to Regions of Inflammatory Cell Infiltration Containing M1 Macrophages
Nestin+ striated cells were frequently detected in regions of the heart where tissue was disorganized (Fig. 4A). Intraperitoneal (i.p.) injections of Evans Blue Dye (EBD) in 15-week-old mdx/utrn−/− mice were used to detect cardiomyocytes with damaged membranes. Upon examination of myocardium from EBD-injected mice (n = 2), it was apparent that nestin+ striated cells surrounded regions of damaged cardiomyocytes (Fig. 4B, 4C). Inflammatory cell infiltration was also present in many of these regions (Fig. 4A). Macrophage infiltration in damaged skeletal muscle is necessary for myofiber regeneration [69]; therefore, we examined whether the inflammatory cells near nestin+ striated cells were macrophages. Large clusters of CD68+ macrophages were detected in mdx/utrn−/− but not wild-type heart. Nestin+ striated cells were adjacent to or surrounding the macrophages in 82.06 ± 1.21% of clusters of ≥8 CD68+ macrophages (Fig. 4D, 4E, n = 3 mice).
Figure 4.
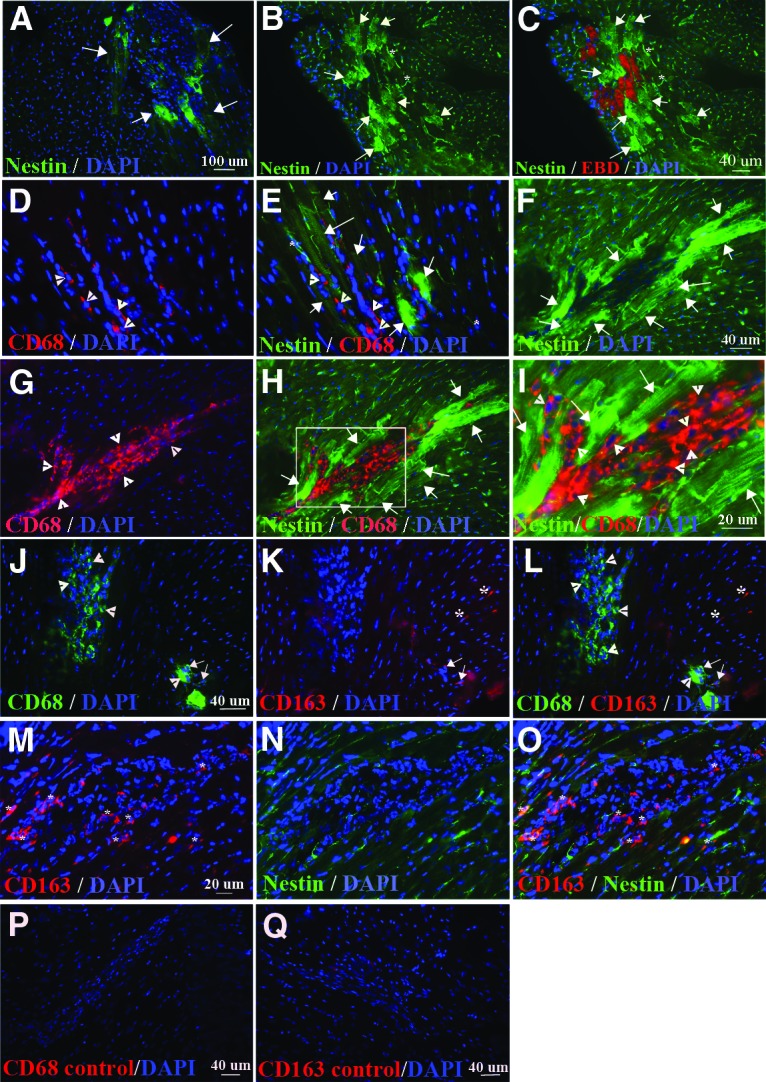
Nestin+ striated cells are adjacent to clusters of M1 macrophages. (A–I): Nestin+ striated cells (green, white arrows [A–C, E, F, H, I]) intermingle with nestin+ interstitial cells (green, white asterisks [B, C, E]) and clusters of CD68+ M1 macrophages (red, yellow arrows [D, E, G–I]) in the heart of 15-week-old (A–E) (n = 4) and 10-week-old (G–I) (n = 1/4) mdx/utrn−/− mice. (C): Nestin+ striated cells (green, white arrows) were present surrounding areas of cardiomyocytes with membrane damage, detected by EBD (red) binding to serum proteins inside the damaged cells. (I): Enlargement of a region from (H), indicated by the white box. (J–L): CD68+ M1 (green, indicated by white arrowheads [J, L]) and CD163+ M2 (red, indicated by white asterisks and white arrows [J–L]) macrophages in dystrophin-deficient heart. M2 macrophages expressing both CD163 and CD68 are indicated by white arrows (J–L), and M2 macrophages expressing only CD163 are indicated by white asterisks (K, L). (M–O): Nestin+ striated cells were not present surrounding CD163+ macrophages (red, indicated by white asterisks in [M, O]). (P, Q): Myocardial tissue incubated with secondary antibody, but not CD68 (P) or CD163 (Q) primary antibody, as a control for background, nonspecific immunofluorescence. Blue, DAPI-labeled nuclei. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; EBD, Evans Blue Dye.
We had not initially observed nestin+ striated cells in the hearts of 5- and 10-week-old mdx/utrn−/− mice. To further establish whether there was a link between the presence of macrophages in the dystrophin-deficient heart and nestin+ striated cells, we examined additional heart tissue from the same 5- and 10-week-old mice for the presence of macrophages. We did not observe macrophages in the hearts of 5-week-old mice (n = 4) or three out of four 10-week-old mice. A single cluster of CD68+ macrophages was present in one 10-week-old mdx/utrn−/− heart (Fig. 4F–4I). Immunostaining for nestin revealed a large cluster of nestin+ striated cells surrounding the macrophage cluster (Fig. 4F–I).
Anti-inflammatory type II (M2) macrophages stimulate myogenesis and myofiber growth following injury in skeletal muscle [69]. To determine whether the macrophages adjacent to, or in the midst of, nestin+ striated cells were inflammatory (M1) or M2 macrophages, we stained for markers of both cells. CD163 or the presence of both CD163 and CD68 was used to identify M2 cells, whereas expression of CD68 alone indicated M1 macrophages [70–72]. Very few CD163+ macrophages were present in dystrophin-deficient heart (Fig. 4J–4L), and only one cluster of these cells was observed (Fig. 4M–4O). Costaining for both markers revealed that CD68+ rarely colocalized with CD163 (Fig. 4J–4L). We did not observe nestin+ striated cells surrounding CD163+ M2 macrophages (Fig. 4M–4O).
Nestin+ “Transition” Cells Express Early Markers of Muscle but Not Stem Cell Markers or Late Cardiac Markers
Nestin+ cells that appeared to be in transition from interstitial stem cells to striated cells were occasionally detected, were frequently adjacent to macrophages, and exhibited elongated processes reminiscent of nestin+ stem cells as well as striations (Fig. 5A–5C). In contrast to the large nestin+ striated cells with cardiomyocyte morphology (Fig. 1B, 1C, 1G–1I, Fig. 2A–2D), these cells did not express cardiac troponin I (Fig. 5D–5F). They also did not express Sca-1 (Fig. 5G–5I) or Flk-1 (Fig. 5J–5L), stem cell markers present on nestin+ interstitial cells. However, the elongated nestin+ cells did express desmin (Fig. 5M–5O), one of the earliest muscle markers detected in the heart during development [66].
Figure 5.

Nestin+ transitional cells express early, but not late, markers of muscle cells and do not express stem cell markers. (A–O): Nestin+ elongated cells with striations (green in [A–D, F, G, I], red in [J, L, M, O], indicated by white arrowheads) were frequently observed in close proximity to CD68+ macrophages (red, indicated by white asterisks in [A]). They did not express cTpnI (red [E, F]) (a marker of mature cardiomyocytes), Sca-1 (red [H, I]), or Flk-1 (green [K, L]) stem cell markers on nestin+ interstitial cells, but they did express desmin (green [K, L]), an early marker of striated muscle. White arrows indicate nestin+ interstitial cells (A, G–I, J–L). White asterisks indicate nestin+ interstitial cells expressing Sca-1 (G–I), Flk-1 (J–L), or desmin (M–O). Blue, DAPI-labeled nuclei. Abbreviations: cTpnI, cardiac troponin I; DAPI, 4′,6-diamidino-2-phenylindole.
Nestin+ Interstitial Cells Express Markers of Cardiac Stem Cells and Are Present in Lower Numbers in Dystrophin-Deficient Cardiac Muscle
Previous studies have reported expression of nestin in vitro on stem cells isolated from cardiac muscle by the presence of the marker Sca-1 [20] or by dye exclusion (side population cells) [18]. We sought to establish whether the nestin+ interstitial cells we observed in vivo expressed markers known to be present on cardiac stem cells. Nestin+ interstitial cells had small-cell bodies and processes and were present throughout the myocardium, as reported previously in rat, mouse, and human heart (Figs. 6A–6C, 1A, 1J) [15, 17, 73]. We observed that 92%–95% of nestin+ interstitial cells expressed Flk-1 (Fig. 6D–6F, 6H), a surface marker present on cardiac stem cells with potential to differentiate into cardiomyocytes, smooth muscle cells, and endothelial cells [74, 75]. The percentage of nestin+ interstitial stem cells expressing Flk-1 remained constant with increasing age in the mdx/utrn−/− mice and was similar in 16-month-old mdx/utrn+/− mice (Fig. 6H). Approximately 25% of nestin+ interstitial stem cells also expressed Sca-1 (Fig. 6I–6K, 6M), a marker also present on some stem cells from adult cardiac muscle [76]. In contrast to Flk-1, however, the percentage of nestin+ cells expressing Sca-1 declined over time (Fig. 6M). Some endogenous cardiac and skeletal muscle stem cells decrease in number and exhibit a marked decline in proliferative potential in the absence of dystrophin [49–54]. To determine whether nestin+ interstitial stem cells declined in number over time with disease progression, we quantified them in the ventricles of 5-, 10-, and 15-week-old mdx/utrn−/− and wild-type mice. Although the number of nestin+ stem cells did not decline over time in the mdx/utrn−/− heart, there were fewer nestin+ cells in dystrophin-deficient heart than in wild-type heart at 5 and 15 weeks of age (Fig. 6N).
Figure 6.

Nestin+ interstitial cells express stem cell markers and are present in decreased numbers in dystrophin-deficient heart. (A–F): Nestin+ interstitial cells with processes (green [A–C], red [D, F], indicated by white arrows) in myocardium of the ventricles expressed the cardiac stem cell marker Flk-1 (green [E, F]). (G): Myocardium stained with secondary antibody only as a control for background, nonspecific immunofluorescence in (D–F). (H): Nestin+ interstitial cells maintained expression of Flk-1 with increasing age (similar to the percentage in 10-week-old wild-type heart, 95.48% ± 5.99%, n = 1). (D–F): Nestin-expressing cells in vessels did not express Flk-1 (white arrowheads). (I–K): Some nestin+ interstitial cells (green [I, K]) expressed Sca-1 (red [J, K], white arrows), although this decreased with age in dystrophic heart (M). (L): Myocardium stained with secondary antibody only as a control for background, nonspecific immunofluorescence in (I–K). (N): There were fewer nestin+ interstitial cells in dystrophin-deficient heart (mdx/utrn−/−, white bars) than in wild-type heart (black bars) at 5 and 15 weeks of age. There was not a difference in nestin+ cells at 10 weeks of age between the mdx/utrn−/− and wild-type mice (p = .3466). Blue, DAPI-labeled nuclei (B, C, E–G, I–L). Images in (A–C) are from myocardium of wild-type mice; images in (D–F) and (H–J) are from mdx/utrn−/− myocardium. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; Dko, double knockout; dKO, double knockout; wk, weeks; WT, wild-type.
Nestin Is Expressed by Smooth Muscle and Endothelial Cells in Dystrophin-Deficient Cardiac Muscle
In addition to differentiating into cardiomyocytes, nestin+ stem cells derived from cardiospheres differentiate into smooth muscle cells in vitro and following transplantation into check embryos [18] and into smooth muscle cells and endothelial cells following transplantation into the infarcted heart [14]. We examined the heart of dystrophin-deficient mice for smooth muscle cells and endothelial cells expressing nestin. We observed nestin+ cells that also expressed α-smooth muscle actin in large vessels in the heart of mdx/utrn−/− and mdx/utrn+/− mice (Fig. 7A–7F). Nestin+ endothelial cells were not observed in large vessels, but some nestin+ cells in the interstitium were observed to express the endothelial marker CD31 and may represent angiogenic endothelial cells similar to nestin+ endothelial cells reported in regenerating pancreas [77] (Fig. 7G–7L). Nestin+ cells were not observed in the walls of vessels in mdx or wild-type cardiac muscle.
Figure 7.

Nestin is present in smooth muscle and endothelial cells in dystrophin-deficient heart. (A–F): Nestin (green [A, C, D, F]) was present in smooth muscle cells expressing SMA (red [B, C, E, F], white arrows) in dystrophic heart. (D–F): Enlargements of (A–C), respectively (enlarged region indicated by the white box in [A]). White arrowheads indicate endothelial cells. (G–L): Nestin (green [G, I, J, L]) was not expressed by CD31+ endothelial cells (red, white arrowheads [G–L]) in large vessels but was expressed by endothelial cells in the interstitium (red [G–L], white asterisks). (J–L): Enlargements of (G–I), respectively (enlarged region indicated by the white box in [G]). (M, N): Myocardial tissue incubated with secondary antibody, but not SMA (M) or CD31 (N) primary antibody, as a control for background, nonspecific immunofluorescence. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; SMA, smooth muscle actin.
Discussion
Nestin is an intermediate filament expressed during development in a variety of tissues including skeletal and cardiac muscle [1–2]. Nestin is downregulated following development, and is typically absent from differentiated cells, including cardiomyocytes in adult heart [2]. Expression of nestin in adult tissues is associated either with stem and progenitor cells or regenerating tissue [5–17]. Transient expression of nestin has been observed in skeletal muscle from DMD and BMD patients, in small-caliber muscle fibers containing centrally localized nuclei [60], features of regenerating myofibers. We observed this same expression pattern of nestin in the skeletal muscle of mdx/utrn−/− mice colocalizing with feMHC in de novo myofibers (Fig. 1E–1G). We now report that nestin is also present in striated cells in adult heart of dystrophin-deficient mdx, mdx/utrn−/− and mdx/utrn+/− mice. Nestin+ striated cells in mdx/utrn−/− heart were present in regions of inflammatory cell infiltration and varied in size and intensity of staining, exhibiting more intense staining in smaller striated cells than larger striated cells (Figs. 1I, 2C–2F, 4A–4E), consistent with declining expression observed following differentiation and regeneration of other tissues [5]. In addition, the nestin+ striated cells in the mdx/utrn−/− and mdx/utrn+/− heart with the greatest intensity of nestin also had more intense immunostaining for desmin (Fig. 3M–3O), as did small caliber, nestin+ skeletal myofibers of mdx/utrn−/− mice (Fig. 3Q–3S). Desmin is one of the earliest markers detected in cardiac and skeletal muscle during development [65–66] and expression has been reported to peak as skeletal muscle cells undergo terminal differentiation in the embryo [65] and as heart development progresses [66–68]. Based on these data and previous reports linking nestin expression in adult tissues to damage and repair/regeneration, the expression of nestin in striated cells of the mdx/utrn−/− heart may also be indicative of regeneration. These data are similar to reports of new nestin+ striated cells in adult mouse, human, and rat heart following myocardial infarction, which were attributed to endogenous nestin+ cardiac stem cells [16, 17].
We previously reported nestin+ striated cells of host origin in the heart of 10-week-old mdx/utrn−/− mice following transplantation of exogenous mesoangioblast stem cells. The appearance of the nestin+ striated cells correlated with a delay in the onset of DCM [21], indicating that their presence might be beneficial to the heart. We observed nestin+ striated cells in 75% of mesoangioblast cell-injected hearts, but only one sham-injected control mouse contained a small number of nestin+ striated cells. Therefore the appearance of nestin+ striated cells in mdx/utrn−/− heart appeared to be primarily an indirect result of the injection of exogenous mesoangioblasts. It was surprising that nestin+ striated cells were present in clusters throughout the ventricles of 15-week-old mdx/utrn−/− mice that had not been injected with mesoangioblasts, as well as 16-month-old mdx and mdx/utrn+/− mice. These data demonstrate that the appearance of regenerating, nestin+ cardiomyocytes in dystrophin-deficient heart occurs in response to factors produced within the heart, as well as in response to mesoangioblast transplantation, albeit at later ages. The generation of these cells may be in response to pathologic changes in the heart, including extensive fibrosis, cardiac myocyte membrane damage, inflammatory cell infiltration, or ventricular remodeling, which develop in the mdx/utrn−/− mice between 10 and 15 weeks of age [58]. Consistent with this possibility, nestin+ striated cells are observed surrounding EBD-positive, damaged cardiomyocytes and in nearly all cases where clusters of CD68+ macrophages are present in dystrophin-deficient heart (Fig. 4). Macrophages promote differentiation of skeletal muscle stem cells into myofibers in regenerating skeletal muscle [69] (reviewed in [71]). Likewise, macrophages may stimulate cardiac progenitor cells to form nestin+ cardiomyocytes. Although we did observe a correlation between macrophages and nestin+ striated cells, in contrast to skeletal muscle, it is CD68+ proinflammatory rather than CD163+ anti-inflammatory macrophages that are observed in the midst of the nestin+ striated cells. There is no precedent for macrophage stimulation of new cardiomyocyte formation in injured heart, but macrophage depletion following cryoinjury in the heart of mice resulted in increased mortality, ventricular dilation, and wall thinning [78], indicating that macrophages are necessary for repair of damage in the heart. Selective depletion of proinflammatory macrophages following infarct resulted in increased areas of debris and necrotic tissue [79], underscoring the importance of inflammatory macrophages, specifically, in repair of damaged cardiac tissue. Macrophage stimulation of new cardiomyocytes in injured heart may also explain the appearance of nestin+ striated cells in mdx/utrn−/− muscle following mesoangioblast transplantation. Approximately 15% of transplanted ADMs are phagocytosed by CD68+ macrophages in the skeletal muscle of mdx/utrn−/− mice [80]. It is likely that transplantation of mesoangioblasts into the dystrophic heart also results in clearance of some donor cells, possibly those damaged during injection. An increase in the number of CD68+ macrophages following ADM transplantation in mdx/utrn−/− cardiac muscle might therefore induce appearance of nestin+ cardiomyocytes. Alternatively, mesoangioblasts may produce factors that stimulate formation of new cardiomyocytes.
The expression of nestin in cardiac muscle cells in dystrophin-deficient heart is indicative of regeneration by endogenous stem cells. This is of interest because many endogenous stem cells in dystrophin-deficient skeletal and cardiac muscle exhibit features of stem cell exhaustion and may not have the capacity to produce large numbers of new muscle cells near the end stages of disease. If endogenous nestin+ cardiac stem cells are capable of generating new, functional cardiomyocytes, they may be good candidates for cell therapy for the heart in DMD. Given our previous study, generation of nestin+ cardiomyocytes prior to the onset of pathology, before the damage and functional decline may be irreversible, may lead to a delay in the onset of DCM [21]. As a result, it is important to confirm the stem cell nature of nestin+ interstitial cells and whether they may be the source of the nestin+ cardiomyocytes.
We observed that the majority of nestin+ interstitial cells expressed Flk-1, a marker that is expressed in lateral mesoderm where cardiogenesis occurs and that is present on embryonic cells that differentiate into the cardiac lineage in vitro. Kattman et al. [75] reported that embryonic cardiovascular progenitors expressing Flk-1 give rise to cardiomyocytes, smooth muscle cells, and endothelial cells when exposed to factors present during cardiogenesis. Similarly, Iida et al. have reported that Flk-1+ cells derived from day 3 and 4 embryoid bodies form spontaneously contracting colonies and produce endothelial cells, cardiomyocytes, and hematopoietic cells [74], and a study by Moretti et al. demonstrated that Flk-1+/Isl1+ progenitors from the secondary heart field are also capable of generating these cell types from the cardiac lineage [81]. Although Flk-1 is present on cardiac progenitor cell populations during development, most cardiac progenitor cells present in adult tissue have not been reported to express Flk-1, indicating that the nestin+/Flk-1+ interstitial cells may be a unique population of cardiac progenitor cells. In support of this, most of the cardiac progenitor populations present in adult heart, including Isl1+, c-kit+, and Sca-1+ cells, are rare (approximately 500–600 Isl1+ progenitors per postnatal heart), whereas nestin+ interstitial cells are frequent throughout the ventricles of mouse, rat, and human heart (Fig. 6) [15, 17, 73]. A subset of the nestin+ interstitial cells also expressed Sca-1, a marker on stem cells in the adult myocardium that give rise to cardiomyocytes [76, 82]. The presence of these markers of cardiac progenitor cells supports previous reports that nestin+ interstitial cells are stem or progenitor cells with cardiogenic potential [14, 18, 73].
Also in agreement with this possibility, we observed transitional cells near macrophages and regions of mature nestin+ striated cells. The transitional cells exhibited striations; an elongated, slender morphology similar in length to cardiomyocytes; and long processes similar to those of nestin+ interstitial cells (Fig. 5). Because these cells did not express Flk-1 or Sca-1, they are distinct from the nestin+ interstitial stem cells observed throughout the ventricles. However, these transitional cells also did not express cardiac troponin I despite the presence of striations, and are therefore not mature cardiomyocytes. The only marker we have detected along with nestin in these cells is desmin, an early muscle marker in the heart [66], indicating that the transitional cells are in early stages of differentiation into muscle. Additional data that support the potential differentiation of the nestin+ interstitial stem cells into cells of the cardiac lineage include nestin+ expression in smooth muscle cells and endothelial cells in the mdx/utrn−/− and mdx/utrn+/− heart (Fig. 7). These results are similar to previous reports of the differentiation potential of nestin+ stem cells derived from the heart. Tomita et al. [18] have demonstrated that a side population of nestin+ cells isolated from rodent heart differentiates into cardiac and smooth muscle cells in vitro. In addition, El-Helou et al. demonstrated that nestin+ cells isolated from heart contribute to new vessel formation when transplanted into an animal model of infarct, giving rise to both endothelial and smooth muscle cells [14]. Taken together, the observations of nestin+ expression in cardiomyocytes, smooth muscle cells, and endothelial cells in dystrophin-deficient heart are in agreement with these earlier reports and indicate that the nestin+ stem or progenitor cells from the heart may be generating these mature cells from the cardiac lineage.
Despite a recent report of stem cell exhaustion among some cardiac progenitor cells in dystrophin-deficient dogs [54] and the lower numbers of nestin+ interstitial cells observed in the dystrophin-deficient mice (Fig. 6L), the large clusters of nestin+ cardiomyocytes that we observed in the mdx/utrn−/− heart at 15 weeks of age indicate that regeneration is occurring from endogenous cells in dystrophic heart. Although the number of nestin+ interstitial cells and the percentage expressing Sca-1 declined, the cells were still present in the heart at late stages of disease and continued to express the cardiac stem cell marker Flk-1 at all ages examined. It is therefore possible that the nestin+ interstitial cells generate the nestin+ striated, smooth muscle, and endothelial cells in dystrophic heart. Alternatively, other populations of cells present within the adult heart are capable of generating cells from the cardiac lineage. For example, Smart et al. have demonstrated that epicardial stem cells expressing Wt1 differentiate into de novo cardiomyocytes following infarct, although the cells do not produce endothelial cells or smooth muscle cells in that particular injury model [83]. Isl-1+ and ckit+ cardiac progenitor cells are also present in adult heart and differentiate into cardiomyocytes, endothelial cells, and smooth muscle cells [81, 84, 85], and Sca-1+/Flk-1− progenitor cells produce new cardiomyocytes in vivo [76]. Additionally, a recent study by Senyo et al. demonstrated that existing cardiac muscle cells divide to produce new cardiac myocytes during homeostasis in uninjured heart as well as following myocardial infarction [86].
Conclusion
Further studies are necessary to define whether nestin+ cardiomyocytes in dystrophin-deficient heart are fully functional and the mechanism by which they are produced. Although generation of new cardiomyocytes may be of functional benefit to the dystrophin-deficient heart, and their presence following mesoangioblast transplantation correlated with a delay in functional decline, there is an alternative interpretation of the data. Other effects were observed with mesoangioblast transplantation that may account for the functional benefit, such as differentiation of a small number of donor cells into cardiomyocytes, and angiogenesis. As a result, the nestin+ striated cells may not have been the cause for delayed onset of DCM. In the current study, nestin+ cells were observed at the end stages of disease, when we have established that these same mice are developing fibrosis, ventricular dilation, and wall thinning [58]. Thus, the mice have declining heart function despite the presence of the clusters of nestin+ striated cells. The presence of nestin+ striated cells in severely damaged heart is consistent with studies in human heart in which nestin+ striated cells are present near end-stage disease or following death from myocardial infarct [16, 17]. These data may also indicate that formation of nestin+ striated cells is not beneficial to the heart and in fact exacerbates functional decline, as postulated by Béguin et al. [15].
However, new nestin+ cardiomyocytes may appear in response to damaged cardiomyocytes or other events known to occur in dystrophin-deficient muscle, such as inflammatory cell infiltration or fibrosis. These events occur in the heart of mdx/utrn−/− mice between 10 and 15 weeks of age [58], when nestin+ striated cells are also observed, and would also be consistent with the report by Béguin et al., in which the authors observed an increase in nestin+ striated cells only after tissue loss [15]. It is necessary to determine whether nestin+ striated cells are beneficial or detrimental to dystrophic heart function, to establish whether nestin+ cardiac stem cells are the source of new cardiomyocytes, and to define the mechanism by which the nestin+ cardiomyocytes appear. These data will indicate whether activation or isolation of nestin+ cardiac interstitial cells from DMD patients may lead to production of new cardiomyocytes and may delay or prevent the onset of dilated cardiomyopathy, or alternatively whether blocking formation of nestin+ cardiomyocytes may offer a functional benefit.
Acknowledgments
This work was supported in part by funds from the Illinois Regenerative Medicine Institute.
Author Contributions
S.E.B., conception and design, collection and assembly of data, data analysis and interpretation, financial support, manuscript writing, final approval of manuscript; P.A., collection and assembly of data, data analysis; J.L.C., collection of data; J.H., collection and assembly of data.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Sejersen T, Lendahl U. Transient expression of the intermediate filament nestin during skeletal muscle development. J Cell Sci. 1993;106:1291–1300. doi: 10.1242/jcs.106.4.1291. [DOI] [PubMed] [Google Scholar]
- 2.Kachinsky AM, Dominov JA, Miller JB. Intermediate filaments in cardiac myogenesis: Nestin in the developing mouse heart. J Histochem Cytochem. 1995;43:843–847. doi: 10.1177/43.8.7542682. [DOI] [PubMed] [Google Scholar]
- 3.Cattaneo E, McKay R. Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature. 1990;347:762–765. doi: 10.1038/347762a0. [DOI] [PubMed] [Google Scholar]
- 4.Lendahl U, Zimmerman L, McKay R. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 5.Wiese C, Rolletschek A, Kania G, et al. Nestin expression: A property of multilineage progenitor cells? Cell Mol Life Sci. 2004;61:2510–2522. doi: 10.1007/s00018-004-4144-6. [DOI] [PubMed] [Google Scholar]
- 6.Sugiyama T, Nagasawa T. Bone marrow niches for hematopoietic stem cells and immune cells. Inflamm Allergy Drug Targets. 2012;11:201–206. doi: 10.2174/187152812800392689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wojakowski W, Ratajczak MZ, Tendera M. Mobilization of very small embryonic-like stem cells in acute coronary syndromes and stroke. Herz. 2010;35:467–472. doi: 10.1007/s00059-010-3389-0. [DOI] [PubMed] [Google Scholar]
- 8.Amoh Y, Li L, Katsuoka K, et al. Multipotent nestin-expressing hair follicle stem cells. J Dermatol. 2009;36:1–9. doi: 10.1111/j.1346-8138.2008.00578.x. [DOI] [PubMed] [Google Scholar]
- 9.Michalczyk K, Ziman M. Nestin structure and predicted function in cellular cytoskeletal organization. Histol Histopathol. 2005;20:665–671. doi: 10.14670/HH-20.665. [DOI] [PubMed] [Google Scholar]
- 10.Lendahl U. Transgenic analysis of central nervous system development and regeneration. Acta Anaesthesiol Scand Suppl. 1997;110:116–118. doi: 10.1111/j.1399-6576.1997.tb05524.x. [DOI] [PubMed] [Google Scholar]
- 11.Krum J, Rosenstein J. Transient coexpression of nestin, GFAP, and vascular endothelial growth factor in mature reactive astroglia following neural grafting or brain wounds. Exp Neurol. 1999;160:348–360. doi: 10.1006/exnr.1999.7222. [DOI] [PubMed] [Google Scholar]
- 12.Namiki J, Tator C. Cell proliferation and nestin expression in the ependymal of the adult rat spinal cord after injury. J Neuropathol Exp Neurol. 1999;58:489–498. doi: 10.1097/00005072-199905000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Vaittinen S, Lukka R, Sahlgren C, et al. The expression of intermediate filament nestin as related to vimentin and desmin in regenerating skeletal muscle. J Neuropathol Exp Neurol. 2001;60:588–597. doi: 10.1093/jnen/60.6.588. [DOI] [PubMed] [Google Scholar]
- 14.El-Helou V, Béguin PC, Assimakopoulos J, et al. The rat heart contains a neural stem cell population; Role in sympathetic sprouting and angiogenesis. J Mol Cell Cardiol. 2008;45:694–702. doi: 10.1016/j.yjmcc.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 15.Béguin P, El-Helou V, Assimakopoulos J, et al. The phenotype and potential origin of nestin+ cardiac myocyte-like cells following infarction. J Appl Physiol. 2009;107:1241–1248. doi: 10.1152/japplphysiol.00564.2009. [DOI] [PubMed] [Google Scholar]
- 16.Mokrý J, Karbanová J, Filip S, et al. Phenotypic and morphological characterization of in vitro oligodendrogliogenesis. Stem Cells Dev. 2008;17:333–341. doi: 10.1089/scd.2007.0091. [DOI] [PubMed] [Google Scholar]
- 17.Scobioala S, Klocke R, Kuhlmann M, et al. Up-regulation of nestin in the infarcted myocardium potentially indicates differentiation of resident cardiac stem cells into various lineages including cardiomyocytes. FASEB J. 2008;22:1021–1031. doi: 10.1096/fj.07-8252com. [DOI] [PubMed] [Google Scholar]
- 18.Tomita Y, Matsumura K, Wakamatsu Y, et al. Cardiac neural crest cells contribute to the dormant multipotent stem cell in the mammalian heart. J Cell Biol. 2005;170:1135–1146. doi: 10.1083/jcb.200504061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Béguin PC, El-Helou V, Gillis M, et al. Nestin+ stem cells independently contribute to neural remodeling of the ischemic heart. J Cell Physiol. 2011;226:1157–1165. doi: 10.1002/jcp.22441. [DOI] [PubMed] [Google Scholar]
- 20.Takamiya M, Haider KH, Ashraf M. Identification and characterization of a novel multipotent sub-population of Sca-1+ cardiac progenitor cells for myocardial regeneration. PLoS One. 2011;6:e25265. doi: 10.1371/journal.pone.0025265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chun J, O'Brien R, Song MH, et al. Injection of vessel-derived stem cells prevents dilated cardiomyopathy and promotes angiogenesis and endogenous cardiac stem cell proliferation in mdx/utrn−/− but not aged mdx mouse models for Duchenne muscular dystrophy. Stem Cells Translational Medicine. 2013;2:68–80. doi: 10.5966/sctm.2012-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison L. Dystrophinopathies. Handb Clin Neurol. 2011;101:11–39. doi: 10.1016/B978-0-08-045031-5.00002-5. [DOI] [PubMed] [Google Scholar]
- 23.Emery AE. Population frequencies of inherited neuromuscular diseases: A world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 24.Spurney CF, Guerron AD, Yu Q, et al. Membrane sealant poloxamer P188 protects against Isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc Disord. 2011;11:20–30. doi: 10.1186/1471-2261-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eagle M, Bourke J, Bullock R, et al. Managing Duchenne muscular dystrophy: The additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord. 2007;17:470–475. doi: 10.1016/j.nmd.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Eagle M, Baudouin SV, Chandler C, et al. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12:926–929. doi: 10.1016/s0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- 27.Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990;26:271–277. doi: 10.1016/0167-5273(90)90082-g. [DOI] [PubMed] [Google Scholar]
- 28.Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: The management of cardiac involvement in muscular dystrophy and myotonic dystrophy: 7th–9th June 2002, Naarden, the Netherlands. Neuromuscular Disord. 2003;13:166–172. doi: 10.1016/s0960-8966(02)00213-4. [DOI] [PubMed] [Google Scholar]
- 29.Baxter P. Treatment of the heart in Duchenne muscular dystrophy. Dev Med Child Neurol. 2006;48:163. doi: 10.1017/S0012162206000351. [DOI] [PubMed] [Google Scholar]
- 30.Romfh A, McNally EM. Cardiac assessment in Duchenne and Becker muscular dystrophies. Curr Heart Fail Rep. 2010;7:212–218. doi: 10.1007/s11897-010-0028-2. [DOI] [PubMed] [Google Scholar]
- 31.Duboc D, Meune C, Lerebours G, et al. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45:855–857. doi: 10.1016/j.jacc.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 32.Ramaciotti C, Heistein LC, Coursey M, et al. Left ventricular function and response to Enalapril in patients with Duchenne muscular dystrophy during the second decade of life. Am J Cardiol. 2006;98:825–827. doi: 10.1016/j.amjcard.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 33.Duboc D, Meune C, Pierre B, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years' follow-up. Am Heart J. 2007;154:596–602. doi: 10.1016/j.ahj.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Rhodes J, Margossian R, Darras BT, et al. Safety and efficacy of carvedilol therapy for patients with dilated cardiomyopathy secondary to muscular dystrophy. Pediatr Cardiol. 2008;29:343–351. doi: 10.1007/s00246-007-9113-z. [DOI] [PubMed] [Google Scholar]
- 35.Kajimoto H, Ishigaki K, Okumura K, et al. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ J. 2006;70:991–994. doi: 10.1253/circj.70.991. [DOI] [PubMed] [Google Scholar]
- 36.Viollet L, Thrush PT, Flanigan KM, et al. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am J Cardiol. 2012;110:98–102. doi: 10.1016/j.amjcard.2012.02.064. [DOI] [PubMed] [Google Scholar]
- 37.Ishikawa Y, Bach JR, Minami R. Cardioprotection for Duchenne's muscular dystrophy. Am Heart J. 1999;137:895–902. doi: 10.1016/s0002-8703(99)70414-x. [DOI] [PubMed] [Google Scholar]
- 38.Jefferies JL, Eidem BW, Belmont JW, et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112:2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- 39.Ogata H, Ishikawa U, Ishikawa Y, et al. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J Cardiol. 2009;53:72–78. doi: 10.1016/j.jjcc.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 40.Biggar WD, Harris VA, Eliasoph L, et al. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006;16:249–255. doi: 10.1016/j.nmd.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 41.Silversides CK, Webb GD, Harris, et al. Effects of Deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. Am J Cardiol. 2003;91:769–772. doi: 10.1016/s0002-9149(02)03429-x. [DOI] [PubMed] [Google Scholar]
- 42.Markham LW, Spicer RL, Khoury PR, et al. Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol. 2005;26:768–771. doi: 10.1007/s00246-005-0909-4. [DOI] [PubMed] [Google Scholar]
- 43.Markham LW, Kinnett K, Wong BL, et al. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul Disord. 2008;18:365–370. doi: 10.1016/j.nmd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Houde S, Filiatrault M, Fournier A, et al. Deflazacort use in Duchenne muscular dystrophy: An 8-year follow-up. Pediatr Neurol. 2008;38:200–206. doi: 10.1016/j.pediatrneurol.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Koh GY, Soonpaa MH, Klug MG, et al. Stable fetal cardiomyocyte grafts in the hearts of dystrophic mice and dogs. J Clin Invest. 1995;96:2034–2042. doi: 10.1172/JCI118251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Payne TR, Oshima H, Sakai R, et al. Regeneration of dystrophin-expressing myocytes in the mdx heart by skeletal muscle stem cells. Gene Ther. 2005;12:1264–1274. doi: 10.1038/sj.gt.3302521. [DOI] [PubMed] [Google Scholar]
- 47.Hagège AA, Marolleau JP, Vilquin JT, et al. Skeletal myoblast transplantation in ischemic heart failure. Long-term follow-up of the first phase I cohort of patients. Circulation. 2006;114:I108–I113. doi: 10.1161/CIRCULATIONAHA.105.000521. [DOI] [PubMed] [Google Scholar]
- 48.Menasché P. Stem cell therapy for heart failure: Are arrhythmias a real safety concern? Circulation. 2009;119:2735–2740. doi: 10.1161/CIRCULATIONAHA.108.812693. [DOI] [PubMed] [Google Scholar]
- 49.Jasmin G, Tautu C, Vanasse M, et al. Impaired muscle differentiation in explant cultures of Duchenne muscular dystrophy. Lab Invest. 1984;50:197–207. [PubMed] [Google Scholar]
- 50.Iannaccone ST, Nagy B, Samaha FJ. Decreased creatine kinase activity in cultured Duchenne dystrophic muscle cells. J Child Neurol. 1987;2:17–21. doi: 10.1177/088307388700200103. [DOI] [PubMed] [Google Scholar]
- 51.Delaporte C, Dehaupas M, Fardeau M. Comparison between the growth pattern of cell cultures from normal and Duchenne dystrophy muscle. J Neurol Sci. 1984;64:149–160. doi: 10.1016/0022-510x(84)90033-9. [DOI] [PubMed] [Google Scholar]
- 52.Webster C, Blau HM. Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: Implications for cell and gene therapy. Somat Cell Mol Genet. 1990;16:557–565. doi: 10.1007/BF01233096. [DOI] [PubMed] [Google Scholar]
- 53.Blau HM, Webster C, Pavlath GK. Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 1983;80:4856–4860. doi: 10.1073/pnas.80.15.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cassano M, Berardi E, Crippa S, et al. Alteration of cardiac progenitor cell potency in GRMD dogs. Cell Transplant. 2012;21:1945–1967. doi: 10.3727/096368912X638919. [DOI] [PubMed] [Google Scholar]
- 55.Decary S, Hamida CB, Mouly V, et al. Shorter telomeres in dystrophic muscle consistent with extensive regeneration in young children. Neuromuscul Disord. 2000;10:113–120. doi: 10.1016/s0960-8966(99)00093-0. [DOI] [PubMed] [Google Scholar]
- 56.Luz MA, Marques MJ, Santo NH. Impaired regeneration of dystrophin-deficient muscle fibers is caused by exhaustion of myogenic cells. Braz J Med Biol Res. 2002;35:691–695. doi: 10.1590/s0100-879x2002000600009. [DOI] [PubMed] [Google Scholar]
- 57.Grady RM, Teng H, Nichol MC, et al. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 58.Chun JL, O'Brien R, Berry SE. Cardiac dysfunction and pathology in the dystrophin and utrophin-deficient mouse during development of dilated cardiomyopathy. Neuromuscul Disord. 2012;22:368–379. doi: 10.1016/j.nmd.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 59.Burkin DJ, Wallace GQ, Nicol KJ, et al. Enhanced expression of the α7β1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sjöberg G, Edström L, Lendahl U, et al. Myofibers from Duchenne/Becker muscular dystrophy and myositis express the intermediate filament nestin. J Neuropathol Exp Neurol. 1994;53:416–423. doi: 10.1097/00005072-199407000-00014. [DOI] [PubMed] [Google Scholar]
- 61.Bridges LR. The association of cardiac muscle necrosis and inflammation with the degenerative and persistent myopathy of MDX mice. J Neurol Sci. 1986;72:147–157. doi: 10.1016/0022-510x(86)90003-1. [DOI] [PubMed] [Google Scholar]
- 62.Quinlan JG, Hahn HS, Wong BL, et al. Evolution of the mdx mouse cardiomyopathy: Physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 63.Bostick B, Yue Y, Long C, Duan D. Prevention of dystrophindeficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ Res. 2008;102:121–130. doi: 10.1161/CIRCRESAHA.107.162982. [DOI] [PubMed] [Google Scholar]
- 64.Verhaart IEC, van Duijn RJM, den Adel B, et al. Assessment of cardiac function in three mouse dystrophinopathies by magnetic resonance imaging. Neuromuscul Disord. 2012;22:418–426. doi: 10.1016/j.nmd.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 65.George-Weinstein M, Foster RF, Gerhart JV, et al. In vitro and in vivo expression of α7 integrin and desmin define primary and secondary myogenic lineages. Dev Biol. 1993;156:209–229. doi: 10.1006/dbio.1993.1071. [DOI] [PubMed] [Google Scholar]
- 66.Schaart G, Viebahn C, Langmann W, et al. Desmin and titin expression in early postimplantation mouse embryos. Development. 1989;107:585–596. doi: 10.1242/dev.107.3.585. [DOI] [PubMed] [Google Scholar]
- 67.Ya J, Markman MWM, Wagenaar GTM, et al. Expression of the smooth-muscle proteins α-smooth-muscle actin and calponin, and of the intermediate filament protein desmin are parameters of cardiomyocyte maturation in the prenatal rat heart. Anat Rec. 1997;249:495–505. doi: 10.1002/(SICI)1097-0185(199712)249:4<495::AID-AR9>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 68.Loh SH, Chan WT, Gong Z, et al. Characterization of a zebrafish (Danio rerio) desmin cDNA: An early molecular marker of myogenesis. Differentiation. 2000;65:247–254. doi: 10.1046/j.1432-0436.2000.6550247.x. [DOI] [PubMed] [Google Scholar]
- 69.Arnold L, Henry A, Pronod F, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into anti-inflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schaier M, Vorwalder S, Sommerer C, et al. Role of FTY720 on M1 and M2 macrophages, lymphocytes, and chemokines in nephrectomized rats. Am J Phsiol Renal Physiol. 2009;297:F769–F780. doi: 10.1152/ajprenal.90530.2008. [DOI] [PubMed] [Google Scholar]
- 71.Chazaud B, Brigitte M, Yacoub-Youssef H, et al. Dual and beneficial roles of macrophages during skeletal muscle regeneration. Exerc Sport Sci Rev. 2009;37:18–22. doi: 10.1097/JES.0b013e318190ebdb. [DOI] [PubMed] [Google Scholar]
- 72.Villalta SA, Nguyen HX, Deng B, et al. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet. 2009;18:482–496. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.El-Helou V, Dupuis J, Proulx C. Resident nestin+ neural-like cells and fibers are detected in normal and damaged rat myocardium. Hypertension. 2005;46:1219–1225. doi: 10.1161/01.HYP.0000187888.39665.d9. [DOI] [PubMed] [Google Scholar]
- 74.Iida M, Heike T, Yoshimoto M, et al. Identification of cardiac stem cells with Flk-1, CD31, and VE-cadherin expression during embryonic stem cell differentiation. FASEB J. 2005;19:371–378. doi: 10.1096/fj.04-1998com. [DOI] [PubMed] [Google Scholar]
- 75.Kattman SJ, Huber TL, Keller GM. Multipotent Flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell. 2006;11:723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 76.Oh H, Bradfute SB, Gallardo TD, et al. Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lardon J, Rooman I, Bouwens L. Nestin expression in pancreatic stellate cells and angiogenic endothelial cells. Histochem Cell Biol. 2002;117:535–540. doi: 10.1007/s00418-002-0412-4. [DOI] [PubMed] [Google Scholar]
- 78.van Amerongen MJ, Harmsen MC, van Rooijen N, et al. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Odintsov B, Chun J, Mulligan J, et al. 14.1 T whole body MRI for detection of mesoangioblast stem cells in a murine model of Duchenne muscular dystrophy. Magn Reson Med. 2011;66:1704–1714. doi: 10.1002/mrm.22942. [DOI] [PubMed] [Google Scholar]
- 81.Moretti A, Caron L, Nakano A, et al. Multipotent embryonic Isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127:1151–1165. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 82.Matsuura K, Nagai T, Nishigaki N, et al. Adult cardiac Sca-1-positive cells differentiate into beating cardiomyocytes. J Biol Chem. 2004;279:11384–11391. doi: 10.1074/jbc.M310822200. [DOI] [PubMed] [Google Scholar]
- 83.Smart N, Bollini S, Dubé KN, et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011;474:640–644. doi: 10.1038/nature10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Laugwitz KL, Moretti A, Lam J, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–653. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beltrami AP, Barluchhi L, Torella D. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 86.Senyo SE, Steinhauser ML, Pizzimenti CL, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]