

In this technical review, members of the International Society for Cell Therapy (ISCT) provide guidance in developing commercializable autologous and patient-specific manufacturing strategies from the perspective of process development. Guidance is provided to help small academic or biotech researchers determine what questions can be addressed at the bench level in order to make their cell therapy products more feasible for commercial-scale production.
Keywords: Cellular therapy, Stem cells, Stem cell culture, Clinical translation
Abstract
Cell therapy is poised to play an enormous role in regenerative medicine. However, little guidance is being made available to academic and industrial entities in the start-up phase. In this technical review, members of the International Society for Cell Therapy provide guidance in developing commercializable autologous and patient-specific manufacturing strategies from the perspective of process development. Special emphasis is placed on providing guidance to small academic or biotech researchers as to what simple questions can be addressed or answered at the bench in order to make their cell therapy products more feasible for commercial-scale production. We discuss the processes that are required for scale-out at the manufacturing level, and how many questions can be addressed at the bench level. The goal of this review is to provide guidance in the form of topics that can be addressed early in the process of development to better the chances of the product being successful for future commercialization.
Introduction
This review focuses on providing guidance to the small academic or biotech researcher in an effort to aid in cell therapy product development. Items to be addressed include typical good manufacturing practice (GMP) processes, technology transfer, cell sources, isolation procedures, bio- and cryopreservation, media, cytokines, sera, serum-free media, scalable platforms, matrices, cell densities, harvesting, genetic alterations, characterization/phenotypic assays, and safety assays (Fig. 1). Although each process is not standard for all cell types, we compare multiple cell types and propose alternate methods where available. Although cell therapy manufacturing has relied heavily on biologic manufacturing/bioprocess, we compare and contrast how shared processes might be beneficial. For example, adherent cells are commonly used for biologic production; however, the cells are normally not recovered. In the case of cell therapy, manufacturing adherent mesenchymal stem cells (MSCs) becomes a serious scalability issue. Alternative adherent scale-up/scale-out systems are available. Alternatively, some groups have successfully adapted MSCs to suspension cultures.
Figure 1.

This flowchart represents a typical cell therapy product process and production layout. Each step has multiple steps within it and can be variable depending on the cell type.
GMP Processes, Definition, and Cell Therapy-Specific Processes
Overview of GMP
What is GMP and how does one implement it for manufacturing autologous cell therapies? GMP is defined by Medicines and Healthcare Products Regulatory Agency (MHRA) in the United Kingdom as “that part of quality assurance which ensures that medicinal products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorization or product specification. GMP is concerned with both production and quality control.” Both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have similar definitions.
As defined, GMP guidelines cover not only the actual physical process of making the drug but also the quality assurance that the drug is produced under conditions that are consistent, safe, and effective for their intended use. With this intent, GMP guidelines include nearly all aspects of drug manufacturing, including but not limited to the quality control and assurance system, manufacturing facilities, equipment and devices used in the process, raw materials, media and medium supplements, storage, and shipping. In the United States, guidelines for cell-based therapeutics are regulated by the FDA (http://www.fda.gov) and are encompassed in the drug manufacturing regulations as described in Title 21 of the Code of Federal Regulations (CFR) in several sections (21CFR210, 211, 610, and 820), including the use of human tissue and cell products (21CFR1271). The EMA (http://www.ema.europa.eu/ema) for the European Union and the MHRA (http://www.mhra.gov.uk) publishes similar guidelines. Both the EMA and MHRA consider cell therapy products to be advanced-therapy medicinal products and reviewed by the Committee for Advanced Therapies. Additional guidance for cell and gene therapies may be found in Regulation (EC) No. 1394/2007. It is important to understand these regulations early in the product development phase in order to ensure that compliance can be achieved. If issues arise, they can be addressed prior to production. The intent of this review is not to provide specific guidance on how to navigate through the regulatory approval process but rather to point readers to sources of information so that they may become familiar with regulations and guidance specific to their products as they develop their cell therapies.
The overall process for preparing to initiate a phase I safety clinical trial in the United States is depicted in Figure 2. There are three major stages of activities in the process to apply for approval to conduct a phase I clinical trial: Research, Technology Transfer and Development, and the Investigational New Drug (IND) application. The Research stage is where the initial characterization, isolation, and production of the cell therapeutic product are identified and generated. Most, if not all, of these activities occur in the research laboratory of the inventor, and the cell therapy product generated in this setting is used in proof-of-concept studies in animal models of disease showing the potential clinical application of the cell therapy. If the proof-of-concept studies show promise and testing in human disease is indicated, the program moves into the Technology Transfer and Development stage, where formal preclinical studies in appropriate animal models to characterize the safety of the product are performed. This stage also includes process scale-up or scale-out, development of initial process and product specifications, and generation of documentation to support manufacturing and product quality testing. The activities in these areas are the basis of the Chemistry, Manufacturing, and Controls (CMC) section of the IND application. The clinical protocol is also developed in this stage. Lastly, these three major components (preclinical, CMC, and clinical protocol) are assembled into the IND application for regulatory agency review and approval. Both the EMA and the MHRA have similar processes to apply for a phase I trial, but developers should refer to specific regulatory documents for their countries and regions for guidance.
Figure 2.
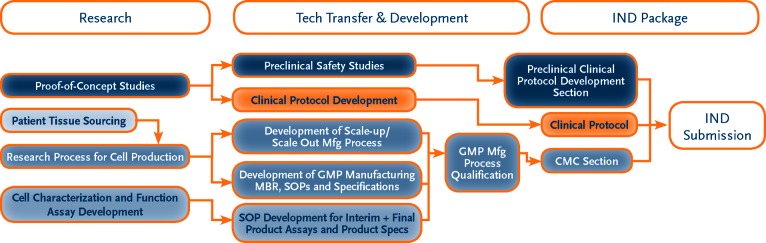
This flowchart represents the steps required for IND submission. Each developer should refer to the regulatory documents specific to its country and region for guidance prior to developing IND strategies. Abbreviations: CMC, Chemistry, Manufacturing, and Controls; GMP, Good Manufacturing Practice; IND, Investigational New Drug; MBR, Manufacturing Batch Record; Mfg, manufacturing; SOP, Standard Operating Procedure.
Considerations in Designing a GMP-Compliant Process
To implement GMP guidelines for the production of autologous cell therapy products, the approach begins at the manufacturing process design stage. It is necessary to perform a gap analysis for GMP compliance from incoming raw material quality and source to the validation of final product shipping containers. For each process step, whether it involves cell isolation or enrichment, in vitro culture, genetic modification, or final product fill and finish, the overall approach should be to reduce risk of contamination of the product, establish documentation to verify that the entire process is correctly performed, and minimize variability in the process while maintaining the salient characteristics and function of the cells of interest. Where practical, the use of single-use, disposable materials and closed systems for manipulating cells is preferred. Reagents, media and supplements, and cytokines should be manufactured under GMP, United States Pharmacopeia, or European guidelines. If not available in these forms, then additional testing may be needed to ensure the appropriate level of purity and lot-to-lot consistency. Additional considerations for developing a GMP-compliant process include qualification of any specialized equipment or instrument used in the process, qualification of the final product container, and shipping protocols. To demonstrate process control and monitor variability, assays should be developed to determine cell phenotype, genotype, and/or function at critical process steps. In addition, the final product should be tested for identity, safety (viral), purity, potency, sterility, endotoxin, and mycoplasma. This may be a challenge since there will be inherent patient-to-patient variability in the starting product for autologous products.
Scalability
Choosing production platforms that are scalable is a critical step in developing a cell-based product. Depending on the dose required and the possible number of patients to be treated during trials or once commercialized, the proper scalable platforms (e.g., bioreactors, cell harvest methods) need to be determined in the early stages of product development. Another consideration is whether production sites, along with scalable platforms, can be numerous throughout the country of interest, or whether all production must take place within one facility.
Discussion With Institutional Business Development or Technology Transfer Office
The internal institutional business development or technology transfer group should be identified as early as possible. Most if not all universities with substantial research activities have a group or department that coordinates and manages activities related to inventions produced by university employees. These activities include patent applications, prosecutions, and maintenance of intellectual property portfolios. These departments may solicit and develop agreements and contacts for the out-licensing of these assets to biotech and pharmaceutical companies interested in commercializing these inventions. This connection is important for many reasons, but primarily to secure any patent protection, agreements with external service providers, and out-license agreements for technology that might be needed as a therapy is developed.
Upon completion of the preclinical studies, the next step is to develop a scaled-up process for the production of a cell therapeutic product for clinical manufacturing. If an institution does not have access to a GMP facility or resources for developing and producing clinical-grade materials, one option is to find a contract manufacturing organization (CMO) that can do this. Selection of a CMO that has the right scientific and technical expertise, experience, and capacity to manufacture at the scale needed for the intended target patient population is a crucial step in the commercialization process of a cell therapy product. At the organizational level, the developer should evaluate the CMO's history and breadth of experience in cell therapy, including but not limited to the types of cell therapy products manufactured; the length and scale of manufacturing campaigns (e.g., phase I, II, III); the number of products manufactured, stored, and shipped; and the track record of interactions with the regulatory agencies. The developer should also assess the CMO's management and staff for the depth of technical and scientific understanding of the cell therapeutic process and product. A CMO that understands the science behind the cell therapy product of interest may become a valuable partner in identifying, evaluating, and implementing process improvements to drive manufacturing costs down without affecting final product specifications and activity.
For autologous cell therapy products, technology transfer from the developer's laboratory to the CMO is the first step toward developing a commercial manufacturing process. Generally, the technology transfer process encompasses the following steps:
Scientific and technical information transfer, including but not limited to the following: scientific background publications, cell isolation and manipulation protocols, assay protocols, bill of materials for production, raw material specifications, and process parameter data, if available. The purpose of this step is to educate the CMO scientists and technical staff on what the cell product is and how the cells are isolated or manipulated to produce the final product. An in-depth understanding of the cell product concept and manufacturing process will help the CMO staff perform a gap analysis for GMP and identify critical process steps and develop the appropriate quality control (QC) assays and specifications for process control, from procurement of raw materials and supplies to the final product container for patient administration. A process that lacks appropriate process controls will introduce manufacturing variability to an autologous cell product that will likely begin with cells that may already have inherent patient-to-patient variability. An educated CMO staff will enable better troubleshooting if interim product or in-process testing falls outside of expected specifications.
Training of the process development (PD) staff. If the manufacturing process has already been used in clinical-scale manufacturing and has final Standard Operating Procedures (SOPs), Manufacturing Batch Records (MBRs), raw materials acceptance criteria and specifications, and other documentation, then this step may be skipped and the technology transfer may proceed directly to training the CMO QC and manufacturing staff. This transfer will be smooth and relatively seamless if the manufacturing and quality documents have the details and rigor needed for commercial industrial manufacturing. But for many developers moving from preclinical phase to a phase I clinical trial, the protocols used to generate the cell product may have been performed only at the research scale for proof-of-concept or toxicity studies in animal models and may not be sufficient for producing clinical doses. In this step, the role of the PD staff is to learn the process from the developer through direct observation and hands-on training of the laboratory process so that they can then design a GMP-compliant process at the appropriate scale needed for clinical doses. The PD staff may also need to develop QC assays for process control and convert research protocols into a format that facilitates GMP compliance (e.g., SOPs and MBRs) for proper documentation during clinical manufacturing. A collaborative effort between the developer and PD staff during this step will help ensure that the process is faithfully transferred to generate the cell product with the desired identity, purity, and activity.
Training of the QC and operations manufacturing staff. The final step in the technology transfer process is the training of the technical staff that will perform the clinical manufacturing process and testing of interim and final products. If the CMO performed development work to scale up and modify the process for GMP compliance, then the trainers are normally the CMO's PD staff, although the developer may also be involved in the training process. As with the previous steps, classroom education of the operations staff on the nature of the cell product and process is recommended before hands-on training so that the staff understands the critical process parameters in production. The training should include engineering runs performed in a clean-room environment. Since these sessions are typically performed by the full complement of manufacturing operators, process verifiers, and QC test personnel, these training runs often help identify potential materials and personnel workflow issues that may introduce risk of product contamination, other process variability not evident during development, or areas where information flow is confusing or incomplete between QC and operations. The MBR should incorporate any process changes or improvements found in the engineering runs prior to process qualification runs. Lastly, documentation of successful training demonstrating that the staff is proficient and competent to manufacture the product should be included in the technology transfer process.
In summary, the technology transfer process involves the transfer of both knowledge and methodology from the developer to the CMO or other interested parties (investors). Technology transfer may include classroom education and hands-on training but should be designed to ensure faithful replication of the process used to produce the therapeutic cell product. It is also important to identify local translational centers, either industry- or government-derived. Translational centers help facilitate research and innovative science in order to reach clinical diagnostic and therapeutic use as quickly and as productively as possible. These centers can help with funding opportunities and advise on business initiative directions, and some offer in-house research facilities. These include, for example, the National Center for Advancing Translational Sciences in the United States, the Medical Research Council in the United Kingdom, and Medicen in France.
Processes
Cell Sources and Isolation Processes
Cell sources and isolation processes vary greatly, and it is best to address any concerns early in development. As an example, we will discuss the isolation and sourcing for MSCs. Despite the vast variety of source tissues such as bone marrow, adipose, periodontal, and others, MSCs show some common characteristics that include fibroblast-like shape in culture, multipotent differentiation, extensive proliferation capacity, plastic adherence, and a common surface marker profile (e.g., CD34−, CD45−, CD31−, CD44+, CD90+, CD166+, and CD105+). However, there is no single surface marker that uniquely defines MSCs. The common characteristics of MSCs are the basis for the isolation techniques. The techniques can be gathered in to three main groups: size/weight separation, plastic adhesion, and CD marker selection.
It is important to note that, if available, control material or cells (not primary patient-derived) should be considered for all process development work. For instance, K562 cells (an immortalized human erythroleukemic cell line) are ideal for T-cell process optimization.
The first and most commonly used isolation technology is Ficoll-Paque gradients. Ficoll (GE Healthcare, Little Chalfont, U.K., http://www.gehealthcare.com) is a neutral, highly branched, high-mass, hydrophilic polysaccharide that dissolves readily in aqueous solutions. It is prepared by reaction of the polysaccharide with epichlorohydrin. Ficoll is part of Ficoll-Paque, which is used in laboratories to separate blood or bone marrow to its components (erythrocytes, leukocytes, etc.). Ficoll-Paque is normally placed at the bottom of a conical tube, and the aspirate is then slowly layered above Ficoll-Paque. After centrifugation, the following layers are visible: upper layer of plasma and other constituents, a layer of mononuclear cells called buffy coat (peripheral blood mononuclear cells [PBMCs]/mononuclear cells [MNCs]/MSCs), Ficoll-Paque, and erythrocytes and granulocytes, which should be present in pellet form. Once separating the layer, the cell mixture can be further processed using selective adherence or CD marker selection. Many devices (such as the Cobe 2991 [Terumo BCT, Lakewood, CO, http://www.terumobct.com], Cell Saver [Haemonetics, Braintree, MA, http://www.haemonetics.com], and Sepax [BioSafe, Eysins, Switzerland, http://www.biosafe.ch] devices) have separate protocols for closed-system use with Ficoll. Several developments based on Ficoll-Paque have evolved over the years, from prepacked tubes with a polymer layer that separates the Ficoll-Paque and the buffy coat layer, all the way to prepacked columns.
One such product is the SepMate-50 (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com), a specialized tube containing a porous insert that forms a physical barrier between the Ficoll-Paque and sample. Although the product is labeled for research use only, this barrier allows the sample to be rapidly pipetted onto the insert, avoiding the need for overlaying it directly onto Ficoll-Paque. The SepMate insert also reduces the duration of the centrifugation step, and after centrifugation, the top layer containing plasma and PBMCs can be poured into a new tube. Other devices include a column containing a porous, high-density polyethylene barrier. These products allow blood to be layered on much more quickly without mixing polysaccharide and blood.
An alternate method of isolating MSCs and other progenitor cells on the basis of physical properties involves the use of filtration-based systems. One such product is the Purecell Select System (Pall Corporation, Port Washington, NY, http://www.pall.com), which can be used with blood, bone marrow, and umbilical cord blood to prepare MSCs based on their entrapment in and adsorption to a fibrous polyester filter matrix [1, 2]. Following the filtration/collection step, cells are then recovered by retrograde rinsing of the filter with an appropriate harvest solution, such as PlasmaLyte-A (Baxter Healthcare, Deerfield, IL, http://www.baxter.com) or saline/dextran solutions. This method can be completed in a short period of time (15–30 minutes) and provides levels of MNCs and CD34+ cells that are comparable to or higher than those of Ficoll-based methods, albeit with somewhat higher levels of granulocytes. Filter-based separations have the advantage of being rapid and easy to use without the need for a centrifuge, and they can be done at the point of care within a disposable closed system to minimize contamination risk.
Another device for point-of-care extraction of adipose-derived MSC is the Celution System (Cytori, San Diego, CA, http://www.cytori.com). The Celution System consists of an electromechanical software-driven device, a presterilized single-use consumable set, and a proprietary processing reagent. The adipose-derived regenerative cells are isolated from the adipose tissue matrix in a highly optimized automated process using enzymatic digestion, gravity-based separation, and centrifugal concentration.
Phenotypic isolation can be achieved by selection of plastic adhesion in which cells after gradient separation are placed on coated cell culture dishes. MSCs adhere to plastic; therefore, once the MSCs have been seeded for 4–24 hours, the media can be replaced and adherent cells will colonize. Some laboratories skip the separation steps and seed the aspirate directly, relying only on the adherence for isolation of the MSCs. Additionally, cell sorting technologies that can sort cells by size or shape can be used. Such technologies include fluorescence-activated cell sorting (FACS) using selective marker labeling or size exclusion. This technique is limited by sample size, cost, and aseptic handling.
An additional option for MSC isolation is based on selection using antibodies against the common MSC CD markers by cell sorters. One example of magnetic sorting technology is the MACS (magnetic-activated cell sorter; Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com). The principle behind this technology is the use of CD antibodies that are attached to magnetic beads. The aspirate or cells are incubated with the beads, allowing specific binding between the antibody and CD marker on the cell. Once bound, the mixture is placed on a magnetic column, and the beads with the bound cells stick to the column because of the magnetic nature of the bead. The retained, antibody-bound cells can then be washed out by removing the magnet and washing out the cells. It should be determined whether the cell source will be obtained fresh or frozen. Issues regarding cryopreservation, logistics, and so forth must be thought of prior to product development.
Cryopreservation for Cellular Therapies
Cryopreservation of cells and long-term storage options permit completion of safety and quality control testing, along with transport from the collection site to the processing facility and subsequently to the clinical site [3–5]. Although the basic concepts surrounding cell cryopreservation are the same, no single method is universally applied for a variety of reasons—the basic reason is that no cell therapy product is essentially the same. The general processes are typically established on the basis of a number of standard parameters, including the following:
Harvesting and processing of cells
Addition of the cryopreservation media
Cooling protocol
Storage temperature and container
Warming/thawing
Post-thaw assessment
The current understanding and practice of cryopreservation for cells and more specifically cell therapy have been well documented, and suboptimal cryopreservation techniques can severely impact cell/product stability [6]. Modifying any of these steps can impact cell utility and cell therapy development. Here we highlight some of the critical aspects and provide additional support when considering cryopreservation protocol development for cell therapies. Raw material and finished product stability, or shelf life, is very important when working with cells [6]. To maximize cell stability, recovery, and function, each of the steps should be considered.
Cell Harvesting/Processing
Following cell/tissue harvest, cells are processed and pelleted (centrifugation) to concentrate and remove any excess growth media. Further processing may be performed to remove growth factors, serum, and so forth. Once concentrated, cells should be resuspended in an appropriate medium as soon as possible to limit any undue stress. Several assessment methods are available, and the best method will depend in part on the cell model, but oftentimes the use of multiple methods will provide a better overall assessment of the population. Standard cell counts—using trypan blue or live/dead assays, for example—can provide an overall cell number, but additional functional assays should be included [3, 7, 8]. Once prefreeze counts are determined, cells can be prepared for cryopreservation.
Cryopreservation Media
The cryopreservation media used for freezing cells have numerous variations, but typically include culture media or balanced salt solution, cryoprotective agent (CPA), and proteins. Many studies have been performed to determine the optimal CPA and concentration for preserving cells, and standard protocols include a 5%–10% dimethyl sulfoxide (DMSO) solution [3–5]. Although certain perceived standard protocols exist, it is important that each cell product be evaluated for the optimal base medium solution and DMSO concentration used. Although culture media have typically been used as the base solution, more optimized balanced salt solutions are available and may provide enhanced buffering and stability thereby reducing cell stress [7, 9]. Since some cells are highly sensitive to DMSO, studies should be performed to determine the optimal concentration [9]. Twenty-four-hour post-thaw viability is a more reliable parameter than the viability at time of thaw, which can differ quite dramatically depending on the solution and the freezing protocol.
Cooling and Storage
Although the influence of cooling rate on post-thaw survival has been documented for various cell types, standard protocol for most therapeutic cell models is performed with a controlled rate freezing (CRF) device having a rate of 1°C–2°C per minute [3–5]. Manual techniques, such as using the Nalgene Mr. Frosty (Nalgene, Rochester, NY, http://www.nalgene.com), are effective alternatives if a CRF device is not available. One drawback to manual protocols is that these methods are suitable only for cryovial containers and are not conducive to larger scale production. Cells are then transferred to storage in either the liquid (−196°C) or vapor (−156°C) phase of nitrogen. Although storage at −80°C can be used, long-term storage is typically performed at temperatures below −150°C—the temperature at which all enzymatic activity is suppressed [3, 5]. Cells and products can essentially be preserved indefinitely if these temperatures are maintained.
Thawing and Post-Thaw Assessment
Thawing of cryopreserved cells is typically performed as rapidly as possible, and standard practice uses a water bath at 37°C. Rapid warming rates are commonly achieved by agitating the sample in a water bath until all visible ice crystals have melted. Once thawed, samples should be immediately removed from the water bath and slowly diluted with media and washed [3–5]. DMSO can be toxic to cells, so limiting their exposure to higher concentrations and temperatures above 2°C–8°C is recommended. Dilution and washing requirements will be dependent upon the intended use of the cells or product and should be determined. Following the freeze/thaw process, cells can be fragile, and care should be taken when handling cells to minimize potential cell loss.
Post-thaw viability and recovery testing is critical to assessing the health of the cell population and developing cryopreservation protocols. Using the wrong assays or testing at the wrong time can significantly impact efficacy and subsequent utility of the cells. Assessment is often performed immediately post-thaw, but the literature has shown that this can lead to inaccurate results [3, 8]. Ideally, assessment studies should be performed immediately, as well as over a 24–48-hour post-thaw recovery period to determine the true recovery profile. Just as important are the assays used to assess the recovery and viability. Viability and recovery assays can be divided into multiple categories consisting of physical/membrane integrity, metabolic activity, mechanical activity, proliferation, and functional/engraftment potential. Typically, more than one measure is used to determine the post-thaw viability of cells. The functional activity is often very critical to overall efficacy.
Cryopreservation Containers
Although overlooked at times, especially earlier in the development process, the freezing/storage container used is important. The use of cryovials for cryopreservation storage is standard practice, especially for research and development uses. These products are not ideal where clinical and larger scale applications are intended. Cryovials often cannot support large volumes (greater than 5 ml) and rely on open processing. If clinical applications are anticipated, disposable closed-system cell freezing bags should be considered. These specialized containers allow for aseptic filling and removal and reliable sterile containment. Other considerations should include information on the cell contact material of the container, potential leachables and extractables, and particulate levels.
Maintaining the quality of the starting material, or cell source material, will impact the cryopreservation and post-thaw results—essentially, what you put in is what you get out. Each of the aspects discussed above is critical when developing an effective cell therapy cryopreservation strategy and protocol. Although general processes exist, minor modifications can and should be performed to create an optimal protocol.
Media, Cytokines, Sera, and Serum-Free Media
There are many important factors in a cell culture process, but the cell culture medium is arguably the most critical. In the most basic sense, cell culture media supply critical nutrients to the cells and allow them to grow and remain viable. Generally, cell culture media contain energy sources, amino acids, vitamins, lipids, inorganic metals, salts, and buffering agents. These components are designed and balanced to present a physiological environment for the specific cell of interest [10]. The quality of the components used and the consistency of manufacture of these factors are critical to the performance of the medium.
Serum is commonly added to cell culture media. It is often derived from bovine, human, or horse sources and is processed and treated in various ways (irradiated, heat inactivated, etc.). Serum is a relatively undefined material that varies by source and lot. It contains a rich mixture of lipids, growth factors, proteins, metabolites, inorganic minerals, and other nutrients that can induce cell growth in various culture systems. Serum also contains extracellular matrix proteins that promote the attachment of cells to tissue culture plates. Although there are benefits to using serum, there are also some drawbacks. Serum presents lot-to-lot variability. This is a problem when the consistency of manufacturing is a goal. Often lots of serum must be qualified to ensure that performance meets specifications. This involves a considerable amount of labor and validation. Serum can also present a regulatory risk since it may be a source of risk for adventitious agent contamination. Finally, cost and limited lot sizes are additional negative aspects of using serum, as well as demand on a limited supply of serum. In addition, human platelet lysate can be used to replace fetal bovine serum (FBS), possibly in autologous applications.
For these reasons, the use of serum-free medium (SFM) has been increasingly preferred [11]. SFM presents a greater degree of definition, reproducibility, and manufacturability than traditional serum-supplemented formulations. To replace serum, SFM often include proteins, lipids, trace metals, and growth factors at empirically determined concentrations for the specific cell type and application of interest. The components chosen may be synthetic, recombinant, or from native tissue extracts (serum albumin, transferrin, etc.). Recombinant growth factors and cytokines are available and may be included in media to promote cell growth or to drive cellular responses according to a specific signaling pathway. Similar to serum, the incorporation of recombinant proteins adds a degree of cost and complexity to the formulation.
Regardless of the components used, one challenge in the design of cell culture media for cell therapy applications is to produce a formulation that delivers consistent results for multiple donors. In a patient-specific autologous therapy, the medium ideally will provide predictable results for each patient. It is important during the development of a cell culture medium that multiple donor pools are evaluated.
Adherence Versus Suspension
If the cell product requires ex vivo culture for expansion, activation, differentiation, or genetic or other manipulation, the developer must consider the type of device in which to culture the cells. Two basic cell culture systems exist: vessels for adherent cells and vessels for cells that grow in suspension. Each type of cell type presents different technical issues for scaling up and scaling out. For adherent cells, one must provide a solid substrate or surface on which the cells must attach to grow. This may be accomplished by simply providing a flat surface as found with traditional cell culture flasks or through the use of beads suspended in media. Traditional flat-surface vessels allow the cells to be visualized during culture to assess the relative health, morphology, and density of the cells. Scaling up or scaling out, however, may become logistically challenging if large numbers of cells are required per dose or per lot. Several manufacturers have developed large and small flasks with multiple layers that help reduce the number of flasks needed per lot. Additionally, manipulating adherent cells in multiple flasks may increase the risk of contamination. Device manufacturers have designed and developed robotic systems to automate the handling of flasks and closed devices with large surface areas that allow easier scale-up and scale-out processes.
Another solution to the scaling of an adherent cell product is the use of beads in suspension cultures. Growing adherent cells on beads may also reduce manufacturing logistics by allowing one to manipulate the cells in vessels as if they were nonadherent cells. Technical issues with scaling up and scaling out are reduced, but visualization of the cells during culture may be difficult or impossible. Because many adherent cell products use percentage of confluence as an important parameter for cell health and function, bead-based systems may not be suitable for all adherent cell types.
There are many devices and vessels for culturing cells that grow in suspension already available to cell therapy developers from the pharmaceutical and biotech fields. Systems for growing cells to produce recombinant proteins and antibodies may be adapted for growing stem cells, T cells and other lymphocyte populations, dendritic cells, and many other types of cells.
Scalable Platforms: Scale-Up Versus Scale-Out
To provide for the increased cell numbers or patient doses required for clinical trials and commercialization of cell-based therapies, the need to expand or increase the manufacturing capacity becomes “mission critical” for a successful new therapeutic product.
The term “scale-up” typically refers to increasing the manufacturing output or capacity, which is achieved by increasing the number of cells or volume processed for each manufacturing lot performed. The term “scale-out” refers to the alternate approach of keeping the manufacturing lot size the same but increasing or expanding out the number of manufacturing lines or unit operations that can be run concurrently. Considerations for whether to scale up or scale out a manufacturing process have several aspects, including the type of cells, the total number of cells or patient doses needed per manufactured lot, and the practicality of manufacturing increased cell numbers or volume in a GMP-compliant process, preferably with a closed manufacturing process to minimize contamination risk.
For allogeneic products, it is most often a scaling up of the process that is used to provide between 5 billion and 50 billion cells per manufactured lot. For such scale-ups, the current approach is to use either more cell culture flasks, cell factories/hyperstacks, or larger scale single-use bioreactors for adherent or suspension cells.
For autologous products, the goal is to be able to produce a suitable number of cells for one to five patient doses of cells, typically in the 1–5 billion range per manufactured run, and to then be able to scale out the number of manufactured runs that can be performed on a regular (daily or weekly) basis. In these scale-out cases, the choices are again to use conventional cell culture flasks, cell factories or static-bags, and bioreactors.
Bioreactors
There are several different types of bioreactors that can be used to generate large quantities of cells for the production of therapeutic proteins, viral vaccines, tissue engineering, or cell therapy applications. The advantage of using bioreactor technology is that variables such as temperature, pH, dissolved gasses, and agitation can be monitored and closely controlled at a large scale in a closed environment. This can help maintain a process that is consistent and reproducible at various scales. Bioreactor technology can be generally separated by size, mode of agitation, and cell type (suspension, adherent, etc.). The most common types of vessel designs include stirred-tank, packed-bed, hollow-fiber, and cellbag bioreactors.
The simplest form of a stirred-tank bioreactor is the spinner flask. These vessels have a limited operating volume and are mostly used for process optimization. On the other end of the spectrum, stirred-tank bioreactors for the processing of mammalian cells can reach up to 20,000 liters in scale.
Stirred-tank bioreactors are defined by an internal impeller that provides the mixing of the medium and gases. In addition, the impeller provides the needed uplift to keep the cells in suspension. Thus, cells are floating freely in the reactor. A major advantage of these bioreactors is that they allow for the close monitoring of critical process parameters, such as oxygen, pH, and temperature. In addition, sampling can be performed at any stage of the process without major interruption (e.g., via sterile sampling ports such as Millipore's NovaSeptum system [Billerica, MA, http://www.millipore.com]). This allows for a close in-line or off-line monitoring of the cells (e.g., via FACS analysis). Although stirred-tank systems have been used routinely for protein/monoclonal antibody manufacturing for decades, their use for the production of cell therapy products is in the early stages. For one thing, traditional stainless steel tanks require an intensive cleaning procedure between runs and in addition have to be validated for each new run. Since many cell therapy applications, such as expansion of patient-specific T cells, do not require large quantities of cells, the cost and labor associated with the cleaning procedure and validation are not justified for these traditional systems. However, in recent years various single-use bioreactor systems have been developed and introduced to the marketplace. These systems are commercially available at different sizes ranging from 2 to 250 liters and allow for convenient cost-effective manufacturing of cell therapy products.
A special case in stirred-tank reactors is the growth of anchorage-dependent cells. Since these bioreactors do not provide a surface on which cells can grow, the addition of microcarriers to the bioreactor is needed. These particles range in size from 100 to 300 μm and provide the surface area needed for cell adherence and growth. Several microcarriers are available in a range of sizes, core materials, and surface structures, and the appropriate carrier has to be determined for each cell type independently.
Independent of how the cells are grown, either in suspension or on microcarriers, one attribute of stirred-tank bioreactors that needs to be considered are the shear forces that are introduced into the system through the agitation. Thus, in the process development phase, the impact of shear forces on the cells needs to be analyzed. This is especially critical for processes involving stem cells as various studies have shown that a high level of shear forces could influence the cell potency.
Although stirred vessels rely on an internal impeller to drive agitation of the culture medium, cellbag bioreactors instead use a rocking motion to agitate the fluid [12, 13]. This provides a gentle mixing that greatly reduces shear stress compared with other agitated systems. Because of this, wave motion bioreactors are popular cell types where the minimization of shear stress is necessary. The most well-known system is the WAVE Bioreactor (GE Healthcare), which has been used in many different applications and to generate material for many clinical trials. Cellbag reactors are single-use systems and eliminate the need for tank sterilization and complex setup schemes, which makes the process less laborious and reduces the need for cleaning validation. Wave motion bioreactors are popular options for expansion of patient-specific T cells and anchorage-dependent cells used in cell therapy applications (grown on microcarriers), and they have been used for the production of monoclonal antibodies for clinical-scale productions. These systems offer control of mixing rate, temperature, and oxygenation at a scale-up to 500 liters.
Packed-bed bioreactors are systems designed to expand cells that are immobilized on a growth surface that may include beads, porous matrices, or mesh structures [14]. The growth surfaces are packed into the bioreactor vessel in a tight configuration that allows for a very high cell density (more than 1 × 108 cells per milliliter). Cell culture media can be perfused through the system at a controlled rate to provide sufficient nutritional and growth requirements for the cells. These systems are amenable to the production of expressed proteins because the spent medium containing the molecule of interest can be removed without disturbing the cells. Recently, packed-bed systems have been used more commonly for the production of cell products such as adherent adult stem cells and human embryonic stem cells. The advantages of this system include the potential for very high density cultures, very low shear stress, and extensive process parameter control. Although the potential for growing large numbers of cells is high, the efficient and effective harvest of the cells presents a substantial challenge. Cell harvest in a packed-bed process will require optimization of dissociation and washing steps to reach a high yield. Currently, there are available packed-bed systems on the market such as the Quantum (Beckman Coulter, Fullerton, CA, http://www.beckmancoulter.com), iCELLis (ATMI, Danbury, CT, http://www.atmi.com), and the Celligen PBR system (New Brunswick, Enfield, CT, http://newbrunswick.eppendorf.com).
Monitoring of bioreactor runs, regardless of the type of system used, is a critical activity to ensure run consistency and optimal performance. Parameters such as dissolved oxygen, pH, temperature, mixing rate, and gassing rate are typically measured in real time using in-line sensors placed within the reactor vessel. Additional variables such as lactate, glutamine, glutamate, glucose, cell number, and cell viability can be measured through off-line devices (such as the BioProfile Analyzer; Nova Biomedical, Waltham, MA, http://www.novabiomedical.com). Taken together these variables can be tracked and used to model specific processes. These data can be used to optimize cell output and process yield. Real-time monitoring can also be used to quickly identify problems and allow the scientist to adjust controls to avoid scrapping an expensive run [15].
Although most in-line parameters are measured by traditional sensor technology that is either chemical- or optical-based, there is notable research being conducted on next generation monitoring technology [15]. Raman spectroscopy has been applied to generate sensors that are capable of analyzing a culture to predict glutamine, glutamate, glucose, lactate, ammonium, and viable cell density simultaneously. This would represent a significant advancement by reducing the need for extensive offline analysis and the subsequent delayed response in parameter control.
Batch, Fed-Batch, and Perfusion
Primarily driven by bioprocess applications, there are three different modes of operation for primary cell culture process: batch, fed-batch, and perfusion. Batch refers to inoculating a cell culture system with a predetermined quantity of medium and cell concentration and allowing the culture to run with no feeds or medium exchange until harvest. Fed-batch refers to inoculating a cell culture system and adding media, cytokines/growth factors, nutrients, or a pH-controlling buffer to increase the cell density and maintain high viability for the majority of the culture time. Perfusion refers to inoculating a cell culture system and continuously feeding and harvesting to maintain health, viability, and productivity as long as possible (significantly longer than a batch or fed-batch process). Perfusion removes and replaces media within a container, leaving the cells in the original cell culture system, without compromising the sterility of the cell culture system [16]. The process is performed through controlled cycles of harvesting spent media out of the cell culture system (the cells remaining in the container or bag), while feeding in fresh media. Perfusion is typically done using either a stand-alone bioreactor (for adherent cells) or a cellbag-based bioreactor (for suspension cells). Harvesting of spent media is performed using either a hollow fiber or membrane sheet type filters. It allows for high cell densities to be achieved and is scalable for a wide variety of volumes. Perfusion is a highly efficient way of culturing cells with higher cell viability than batch or fed-batch systems, and it can produce much greater cell numbers from the same size container. This is ideal for autologous therapies where high cell densities are needed while reducing the overall of cost of the media and supplements.
Antibiotics
In general, most cell therapy laboratories and production facilities do not use antibiotics. The introduction of antibiotics adds additional nonhuman components to the product. Carryover of antibiotics into the final cell product may also have serious consequences for patient safety, such as an allergic reaction to penicillin. It also increases the possibility of false-negative results during sterility/bacterial testing prior to patient treatment.
Matrices/Coatings
In vivo, many cell types depend on the surrounding extracellular matrix (ECM) and associated cell signaling components for growth and proper function. In cell culture, adherent cell lines or primary cells require ECM for attachment to cell growth surfaces such as flasks, plates, microcarriers, and scaffolds. ECM consists of components such as proteins (such as laminin, collagen, fibronectin, and elastin) and proteoglycans. Certain cell types that are grown in culture are not able to produce sufficient ECM for proper cell attachment. Several strategies have been developed to overcome this. Serum-supplemented media are widely used for many cell types because of their growth-promoting properties, as well as the broad complement of cellular attachment substrates that are contained in serum. For serum-free systems, ECM molecules can be used to treat cultureware to promote cell attachment. Additionally, recombinant molecules or synthetic chemical processes can be incorporated into the manufacture of culture surfaces to facilitate cell attachment without any pretreatment. These precoated products are commercially available from many vendors and present a convenient option. For many tissue engineering applications, the use of a three-dimensional lattice (such as collagen scaffolds) for cell growth may provide a more physiologically relevant environment to encourage tissue-forming properties [17].
Seed Densities
Optimization of an efficient expansion protocol is a crucial part of manufacturing and process development. Researchers have reported the lack of standardization of expansion protocols, including cell seeding density for cell therapy [18]. The initial plating of cells in cultureware containing a specific medium formulation marks the beginning of cell expansion for the scale-up process. Seeding density can significantly affect cell quality and cell yield and thus requires optimization at an early stage of the process. Determining optimal seeding density is, however, highly dependent on parameters such as medium composition (e.g., serum-free formulation, FBS-containing formulation, cytokine composition, glucose level), tissue source, quality of donor (age, medical history), culture method (low oxygen, two-dimensional [2D] culture, bioreactor, scaffolds, enzymes for dissociation), and culture flasks [19–22]. Seeding density can be optimized once medium composition, tissue source, and culture method have been determined. Both initial cell seeding density (from primary tissue) and passaging cell density will determine the output of a scale-up procedure in terms of cell yield and cost (time of culture, labor and materials). In some practices, cells are purchased from a supplier where cells are isolated and culture-expanded in one particular medium formulation and then cryopreserved. Following freeze-thaw, cells could be culture-expanded in a different formulation with a modified protocol. Changing of culture medium and protocol require further optimization of seeding density post-thaw for long-term cell expansion.
Researchers have reported in tissue engineering applications, certain biomechanical properties of hyaluronic acid hydrogels showed improvement with higher MSC seeding densities [23], although others have reported that lower seeding densities of MSCs favor a higher proliferation rate and maintenance of stemness properties in a 2D culture system [21, 24]. However, an increased cell proliferation rate does not necessarily translate to optimal cell yield. Optimization of seeding density strategies need to be established to find a balance between time, labor, and materials cost and optimal cell yield. Every scale-up system is unique, and therefore seeding density needs to be optimized according to each system.
Harvesting
The harvesting of expanded cells is a critical step in the manufacturing of cell therapy products, as this is often the final processing step prior to patient infusion or cryopreservation. Cell harvesting can be a laborious and time-intensive process, and as the volume of cells being processed increases, this step can quickly become a process bottleneck [14]. The objective in the harvesting process is to achieve a reduction in volume or an increase in cell concentration with high yield and recovery while maintaining the integrity and functionality of the cells.
It is important early on in the development of a cell therapy product to estimate what scale of commercial production will be needed, so that in initial preclinical and process development and phase I efforts one works with a method that can be scaled up or scaled out to meet the product requirements for later stage trials and commercialization. For most autologous cell products that are grown in suspension culture, such as antigen-specific T cells, dendritic cells, and natural killer (NK) cells, a typical production volume is in the 2–10-liter range, whereas CD34+ hematopoietic stems cells and tumor-infiltrating lymphocytes are often in the 10–15-liter range, and in some cases as high as 50–70 liters. For adherent cells, such as bone marrow-derived MSCs the volume of cells harvested from cell culture vessels are typically produced in 5–10 liters of culture fluid.
Several technology choices exist for cell harvesting depending on the volume of cells and the available infrastructure and personnel resources. The most commonly or widely used method is centrifugation with 250- or 500-ml centrifuge bottles, although with harvest volumes greater than 5 liters, manual centrifugation becomes increasingly time-consuming and labor-intensive and can be a significant driver of the cost of goods. Another drawback of centrifugation is that it involves multiple open manipulations in the decanting and resuspending of the cells, which introduces contamination risk and therefore requires a full clean-room facility. For these reasons, other “closed” technologies are also being adapted for use with cellular therapies, such as blood processing equipment/cell processors and tangential flow filtration devices available from several suppliers.
Gas Conditions
Although most mammalian cell culture uses 5% CO2 as the desired agent, it is important to note that variations in the concentrations of both CO2 and O2 might provide positive growth conditions and must be explored. For example, hypoxic conditions could possibly provide better pH and overall medium stabilization, as well as increased growth.
Feeder Cells and Antigen-Presenting Cells
Many cell therapy products require feeder cells or antigen-presenting cells to aid in cell production. Although each cellular product is different, we discuss this application for T-cell production.
T cells are optimally activated/stimulated by professional antigen-presenting cells (APCs), or dendritic cells (DCs). These APCs provide the antigen-specific primary signal and the secondary costimulatory signal to the T cells. However, the manufacturing of personalized DCs is expensive, labor-intensive, and highly variable. In addition, DCs from disease-state patients may be impaired in their ability to properly activate T cells. Bulk T cells can be activated using irradiated allogeneic PBMCs and OKT3 antibody (a monoclonal antibody that blocks the function of CD3 on T cells, rapid expansion protocol). This process can also be costly and have inconsistent/irregular results because of the donor-to-donor variability of the PBMCs, as well as limited supply. Artificial activation modalities with defined molecular profiles are a highly desired alternative to the DCs and allo-PBMC feeder cell schema. The CD3/CD28 DynaBeads (Life Technologies, Rockville, MD, http://www.lifetech.com) are an alternative to DCs and allo-PBMC activation/stimulation. The paramagnetic beads are coated with anti-CD3 and anti-CD28 antibodies and deliver robust signals capable of activating and expanding bulk T cells through the T cell receptor (TcR) complex and the CD28 costimulatory molecule. However, these beads are capable of expanding only a subset of T cells, which may not necessarily represent the complete population of desired cells. A more consistent and comprehensive tool is the K562-based artificial APC system. The K562 erythroleukemic cell line provides an excellent platform because it does not express human leukocyte antigen (HLA) molecules, thereby mitigating allogeneic responses. The cell line also expresses adhesion molecules that facilitate APC:T-cell interactions. In addition, the cell can be genetically modified to express HLA molecules, costimulatory molecules, and cytokines. Expression of the high affinity Fc receptor on K562 cells also allows the presentation of antibodies such as anti-CD3 and anti-CD28 to the T cells, making it an attractive off-the-shelf product that can be used for expansion of any clinical sample. HLA-expressing K562 cells can also be used to selectively expand antigen-specific T cells by pulsing with specific peptides. The ability to manufacture a robust/consistent APC product for the activation and expansion of a T-cell sample is critical for the commercialization of an autologous T-cell product.
Genetic Alterations and Vector Design
Although the frequency of any given subset of cells in the body or an isolated procedure can be extremely low, advances in genetic engineering have allowed the production of well-defined phenotypic cell subpopulations from a bulk starting material. Cells can be genetically modified in a transient manner (via RNA electroporation) or a durable permanent fashion (viral, plasmid). The genetic modifications made to these cells may also be coupled to an ex vivo expansion phase, with the transient alterations occurring at the end of the amplification whereas the permanent changes typically occur near the onset of the culture. Although T cells and DCs are the two most common types of cells poised for genetic modification, hematopoietic stem cells (HSCs) and NK cells can also be a potential source. The cell type and amount of starting material will have a dramatic impact on the culture time and cell divisions required to achieve the desired output.
The cell product can be transiently modified using a large-scale GMP electroporation system, such as a MaxCyte system (MaxCyte, Gaithersburg, MD, http://www.maxcyte.com), and RNA or plasmid. Although the expression is temporary, modifications to enhance stability of the genetic material can maintain expression for extended periods of time. Permanently modified cells typically involve viral vectors (lentivirus, retrovirus, adenovirus) or plasmid DNA maintained by selection or episomal replication. T cells can be modified to express a variety of genes that impact their function and phenotype, including cytokines, cell surface molecules for redirected specificity (TcRs and chimeric antigen receptors), intracellular modulators, and DNA-modifying enzymes (zinc-finger nucleases). DCs can be altered to express target antigens (Provenge; Dendreon, Seattle, WA, http://www.dendreon.com), costimulatory molecules, and maturation molecules.
The choice and design of vector for the genetic modifications is also a critical component for the cell product. The virus encoding the gene of interest must be able to efficiently transduce target cells with minimal toxicity; if long-term engraftment of cells is required, minimal immunogenicity is also imperative. Expression of the gene is driven by an internal promoter that should maintain stable expression in the cell. Typically, the promoters are chosen to be ubiquitously active (elongation factor-1α, cyclic GMP-dependent protein kinase [PKG]) in all cell types; however, promoters may also be cell type-specific or expression may be regulated by environmental cues, such as hypoxia, or drug-induced (e.g., tet-on and tet-off). The potential for in vivo toxicity is of great concern, and safety mechanisms to address the potential need also be considered for the vector design. Suicide switches/genes can also be incorporated into the vector allowing for the immediate removal of the cells. Insertional activation of oncogenes is a concern for safety, as is the potential for generating recombinant replicating virus. The safety requirements are key components that need to be factored into the release of the final product.
Stability and Reproducibility Assays
A critical step is to determine how stable each product is throughout the production process. It is advised that during each step, characterization be performed initially. This will determine how each process step could potentially alter a cell's phenotype. It is also suggested that as the process or materials within it change, cell characterization assays be performed to determine how or whether the change affected the product. In addition, if components of the assay itself change (by the end user or manufacturer), product changes must also be determined.
Variability in Processes and Cellular Raw Material
Although determining variations in the overall process, including evaluation of raw materials and supply management, is the specialty of the CMO, it is important to understand how critical this process is. Variations, such as cell potency and patient-specific properties, should be addressed. Factors that can minimize such variations can be identified as alternatives, allowing the downstream process individuals to be aware prior to determining the overall risk assessment road map. Are there lot-to-lot variations in any materials, including sera? Items to be assessed at the bench scale are starting/source materials (cell lines, viral/bacterial stocks, media, chemicals, serum, water) and in-process materials (resins, buffers, filters, column housings, tubing, reagents) [25]. Packaging and device/delivery components will be addressed by the CMO.
Phenotypic Assays
A defined characterization profile ensures that the manufacturing process reproducibly yields the intended product. It is an evolving process from laboratory research and across clinical development. Among the arsenal of assays that are available to characterize cell therapy products, cell phenotyping by multiparametric flow cytometry has been the premier tool to identify and enumerate cell subsets [26]. Over the last decade, progress at the level of hardware technology (lasers, fluidics, optics, electronics), fluorochrome chemistry (greater variety of antibody-fluorochrome conjugates), and software interface has led to the advent of polychromatic flow cytometry where more than 5 and up to 20 parameters can be analyzed simultaneously at the single-cell level [27]. This remarkable technology has made its mark in the diagnostic world, where it has been used to dissect the complexities of the hematopoietic and immune system. This platform is ideally suited to interrogating and defining the inherent heterogeneity of cell therapy products, through the development of a product-specific phenotype assay.
The fundamentals of phenotyping assays are reagent choice, panel design, and instrument calibration. Starting early in product and process development with a broad screening of cell surface markers is a good way to gain in-depth knowledge about the product phenotype and choose appropriate markers. The next step is to distinguish the critical phenotypic attributes from less relevant markers. The diversity of cell therapy products is such that there are no commercially available optimized panels (with a few exceptions), so validating the selected antibody cocktail(s) has to be done for each type of product. It is a labor-intensive iterative process that involves antibody titration (to optimize the signal-to-noise ratio), antibody pairing with fluorochromes (dim markers with high stain index fluorochrome and vice versa), and making sure that single stain performances are not compromised by spectral overlap [28, 29]. At this stage, proper controls, compensation matrix, and acquisition templates can be defined. At the instrument level, a comprehensive quality assurance and quality control program must be in place to calibrate, optimize, and maintain cytometer performance [30]. The development of a phenotype assay has to be completed by putting in place a data analysis plan to manage the high-content information and extract the interrogated product profile.
The product's ultimate signature profile delivers information about its identity (what the product is, which can include positive and negative markers), purity (what are the cellular contaminants, if any), and release criteria (expressed as percentage of cells as well as mean fluorescence intensity). In some cases, surface markers with inducible or regulated expression can be part of a potency assay as well. Part or all of the profile can also be used in process testing and in comparability studies. Polychromatic flow cytometry can bring a wealth of information about a product and fits in as part of a matrix of analytical assays for cell therapy product characterization.
Potency, Safety Assays, Toxicity, Immunodeficient Mice, and Animal Imaging
The ability to generate large numbers of phenotypically and functionally defined cells provides a promising strategy for the treatment of numerous diseases. Given the recent success of autologous cell therapy in the treatment of cancer, it is poised to become a standard care for many malignancies. Each manufactured lot is patient-specific and will need to meet numerous criteria for release. This section describes the assays to be implemented in order to address identity, safety, and potency. It also discusses the use of animal models to assess in vivo efficacy and different imaging systems to monitor trafficking and engraftment of the infused cells. The International Society for Cellular Therapy (ISCT) has recently published a position paper on potency assays for the cell therapy industry [31].
Large-scale manufacturing of patient-specific lots will require a robust identity testing to ensure that each lot is matched with the intended patient. The most accurate method for this testing will be polymerase chain reaction (PCR)-based. The production/expansion of the cells will require significant culture periods and assays that can address the purity and phenotype need to also be implemented. In the case of T cells, the expanded cells will need to be assessed for a “rested” phenotype, thereby minimizing potential toxicities. In the case of dendritic cells, they would need to be examined for the presence of immature DCs, which could pose hurdles for vaccine efficacy. Assays to address toxicities and off-target effects of transgene expressing T cells can be examined in multiparameter tests. The final T-cell product can be tested against target-negative and target-positive controls or artificial target cells (K562). This system can be used to measure toxicity and off-target effects against the negative controls and efficacy/potency against the positive controls. For the case of genetically modified T cells (viral vectors), two major concerns are the insertional transformation of the T cells and the production of replication-competent virus. For the oncogenic transformation component, long-term or chronic stimulation of the modified T cells should not result in the outgrowth of a clonal subpopulation of cells. The assay for recombinant virus can be assessed by a p24-enzyme-linked immunosorbent assay or either a PCR-based or cell-based amplification assay of the T-cell supernatant.
These assays provide valuable information regarding in vitro function; however, they do not necessarily correlate with in vivo function. Immunodeficient mice models can help answer some questions regarding in vivo functions such as trafficking, biodistribution, and organ-specific nesting. The NSG mouse (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) is a severely immunocompromised mouse strain that supports the engraftment of a wide variety of primary human cells. Xenografted cells can include primary tumor cells, T cells, DCs, hematopoietic stem cells, and skin cells and can be transferred into the mice by multiple routes (intravascular, intraperitoneal, subcutaneous). The biodistribution of the cells is an important component to assess since it may directly influence both efficacy and toxicity in nondisease organs. Historically, complete organ/tissue harvest of engrafted mice can be performed at various time points, and a full necropsy of all tissues and organs can be performed. Critical organs and tissues include blood, spleen, lymph nodes, and bone marrow; other important tissues include the liver, lung, kidney, brain, gonads, heart, and skin. Engrafted cells can be detected by flow cytometry, PCR, and immunohistochemistry. Both the presence and absolute amounts of engrafted cells can be determined with time, providing a detailed kinetic profile of their distribution. Long-term persistence in these organs may be an indicator of toxicities and off-target effects.
Noninvasive imaging of xenografted mice allows for longitudinal tracking of the cells without terminating the study. Intravenous infusion of cells is usually followed by a temporary residence in the lung tissue for ∼24 hours, after which the cells distribute throughout the body. If an immediate placement of the cells is required, then alternate routes of entry may be required, such as intraperitoneal or direct intraorgan placement. Intraperitoneal infusion is also an option for delivery; however, trafficking may favor the lymphatic system rather than a more systemic distribution, and imaging studies can address the distribution as well as the magnitude of the cell dose. Several modes of imaging can be used to track and follow the xenografted cells. Luciferase-tagged cells can be located and quantitated with the addition of luciferin substrate using a charge-coupled device camera (Xenogen system; STTARR, Toronto, ON, Canada, http://www.sttarr.com). Cells can also be tracked using fluorescent proteins. For cells located deep in the mouse, proteins such as TurboFP635 or tdTomato may be more applicable for in vivo use because of their ability to emit through tissue. However, fluorescence is diminished in a hypoxic environment such as a tumor, which may limit its use in certain models. Cells can also be loaded with nanoparticles and tracked in mouse models; however, dividing cells will dilute the signal and may provide tracking/traceability only for several population doublings.
The development of assays that address safety and toxicity as well as in vivo trafficking and propagation will establish an important basis for the cellular product. It may also provide important details that may be correlated to clinical outcome.
More traditional tumorigenicity assessment includes subcutaneous or intramuscular administration of cells, followed by assessment of growth of the injection area relative to tumor growth after injection of known tumorigenic cell types, such as HT-1080 cells. However, intravenous injection is a common route of administration for cell therapy products, and the objective of this section is to describe safety study designs to evaluate tumorigenicity after intravenous administration of a cell therapy product in a nude mouse model. This type of assessment has been used in FDA-approved INDs and may therefore serve as a general example. Still, for each product, agreement with regulatory agencies should be sought for the experimental design based on the specifics of the product driving the controls, time frames, and specific endpoints that should be used, in the intended mode of administration in the clinic [32].
Conclusion
Determining the factors that can enhance the success of a cell therapy product early in development is critical to the overall success of the product. Even if all of the processes described above are not achievable, addressing them in the development stage will benefit downstream interested parties. Making data available, whether positive or negative, is a critical component to success. Processes for each cell product vary greatly, but the field will benefit greatly from the establishment of a road map for cell therapy process variations and options.
Acknowledgments
We thank the ISCT, the Alliance for Regenerative Medicine, the California Institute for Regenerative Medicine, and the NIH Center for Regenerative Medicine for their input and advice.
Author Contributions
S.E., M.A., H.B., S.B., A.C., C.C., D.C., T.F., O.K., K.N., W.V.H., and R.W.: manuscript writing.
Disclosure of Potential Conflicts of Interest
A.C. has compensated employment with Life Technologies. T.F. has compensated employment and compensated stock options with PCT Cell Therapy Services. K.N. has compensated employment with Novartis. W.V.H. has compensated employment, is a patent holder, and holds stock options with Athersys, Inc. R.W. has compensated employment with StemCell Technologies, Inc.
References
- 1.Hibino N, Nalbandian A, Devine L, et al. Comparison of human bone marrow mononuclear cell isolation methods for creating tissue-engineered vascular grafts: Novel filter system versus traditional density centrifugation method. Tissue Eng Part C Methods. 2011;17:993–998. doi: 10.1089/ten.tec.2011.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sowemimo-Coker SO, Andrade F, Kim A, et al. A simple filtration system for red blood cell depletion and volume reduction in routine processing of human umbilical cord blood. Vox Sang. 2009;96:138–145. doi: 10.1111/j.1423-0410.2008.01124.x. [DOI] [PubMed] [Google Scholar]
- 3.Hubel A. Advancing the preservation of cellular therapy products. Transfusion. 2011;51(suppl 4):82S–86S. doi: 10.1111/j.1537-2995.2011.03370.x. [DOI] [PubMed] [Google Scholar]
- 4.Hunt CJ. Cryopreservation of human stem cells for clinical application: A review. Transfus Med Hemother. 2011;38:107–123. doi: 10.1159/000326623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thirumala S, Goebel WS, Woods EJ. Clinical grade adult stem cell banking. Organogenesis. 2009;5:143–154. doi: 10.4161/org.5.3.9811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger S. Biopreservation stability considerations for cell therapy development and commercialization. BioPreservation Today. 2009;1:4–5. [Google Scholar]
- 7.Clarke DM. Optimizing biopreservation yield: Steps to improving cryopreservation outcomes. BioPreservation Today. 2009;1:6. [Google Scholar]
- 8.Mathew AJ. What is your viability assay really saying? BioPreservation Today. 2009;1:8. [Google Scholar]
- 9.Clarke DM, Yadock DJ, Nicoud IB, et al. Improved post-thaw recovery of peripheral blood stem/progenitor cells using a novel intracellular-like cryopreservation solution. Cytotherapy. 2009;11:472–479. doi: 10.1080/14653240902887242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorfien SF, Campbell A, Vemuri MC. Design of culture media. In: Murray M-Y, editor. Comprehensive Biotechnology. 2nd ed. Burlington, VT: Academic Press; 2011. pp. 205–215. [Google Scholar]
- 11.Jayme DW. Serum-free and protein-free media. In: Stacey GN, Davis J, editors. Biological Medicines from Cell Culture. London, U.K.: Wiley; 2007. pp. 29–44. [Google Scholar]
- 12.Eibl R, Werner S, Eibl D. Bag bioreactor based on wave-induced motion: Characteristics and applications. Adv Biochem Eng Biotechnol. 2010;115:55–87. doi: 10.1007/10_2008_15. [DOI] [PubMed] [Google Scholar]
- 13.Singh V. Disposable bioreactor for cell culture using wave-induced agitation. Cytotechnology. 1999;30:149–158. doi: 10.1023/A:1008025016272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rowley J, Abraham E, Campbell A, et al. Meeting lot-size challenges of manufacturing adherent cells for therapy. BioProcess Int. 2012;10:16–22. [Google Scholar]
- 15.Abu-Absi NR, Kenty BM, Cuellar ME, et al. Real time monitoring of multiple parameters in mammalian cell culture bioreactors using an in-line Raman spectroscopy probe. Biotechnol Bioeng. 2011;108:1215–1221. doi: 10.1002/bit.23023. [DOI] [PubMed] [Google Scholar]
- 16.Tran CA, Burton L, Russom D, et al. Manufacturing of large numbers of patient-specific T cells for adoptive immunotherapy: An approach to improving product safety, composition, and production capacity. J Immunother. 2007;30:644–654. doi: 10.1097/CJI.0b013e318052e1f4. [DOI] [PubMed] [Google Scholar]
- 17.Artym VV, Matsumoto K. Imaging cells in three-dimensional collagen matrix. Curr Protoc Cell Biol. 2010 doi: 10.1002/0471143030.cb1018s48. Chapter 10:Unit 10.18.1–18.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fekete N, Rojewski MT, Furst D, et al. GMP-compliant isolation and large-scale expansion of bone marrow-derived MSC. PLoS One. 2012;7:e43255. doi: 10.1371/journal.pone.0043255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fossett E, Khan WS. Optimising human mesenchymal stem cell numbers for clinical application: A literature review. Stem Cells Int. 2012;2012:465259. doi: 10.1155/2012/465259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fossett E, Khan WS, Longo UG, et al. Effect of age and gender on cell proliferation and cell surface characterization of synovial fat pad derived mesenchymal stem cells. J Orthop Res. 2012;30:1013–1018. doi: 10.1002/jor.22057. [DOI] [PubMed] [Google Scholar]
- 21.Sekiya I, Larson BL, Smith JR, et al. Expansion of human adult stem cells from bone marrow stroma: Conditions that maximize the yields of early progenitors and evaluate their quality. Stem Cells. 2002;20:530–541. doi: 10.1634/stemcells.20-6-530. [DOI] [PubMed] [Google Scholar]
- 22.Sotiropoulou PA, Perez SA, Salagianni M, et al. Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells. 2006;24:462–471. doi: 10.1634/stemcells.2004-0331. [DOI] [PubMed] [Google Scholar]
- 23.Erickson IE, Kestle SR, Zellars KH, et al. High mesenchymal stem cell seeding densities in hyaluronic acid hydrogels produce engineered cartilage with native tissue properties. Acta Biomater. 2012;8:3027–3034. doi: 10.1016/j.actbio.2012.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colter DC, Sekiya I, Prockop DJ. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc Natl Acad Sci USA. 2001;98:7841–7845. doi: 10.1073/pnas.141221698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beck G, Schenerman M, Dougherty J, et al. Raw material control strategies for bioprocesses. BioProcess Int. 2009;7:18–33. [Google Scholar]
- 26.Shapiro HM. Practical Flow Cytometry. 4th ed. Hoboken, NJ: Wiley-Liss; 2003. [Google Scholar]
- 27.Chattopadhyay PK, Hogerkorp CM, Roederer M. A chromatic explosion: The development and future of multiparameter flow cytometry. Immunology. 2008;125:441–449. doi: 10.1111/j.1365-2567.2008.02989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maecker H, Trotter J. Becton Dickinson Application Note 23-9538-01. Franklin Lakes, NJ: Becton Dickinson; 2008. Selecting reagents for multicolor flow cytometry. [Google Scholar]
- 29.Mahnke YD, Roederer M. Optimizing a multicolor immunophenotyping assay. Clin Lab Med. 2007;27:469–485. v. doi: 10.1016/j.cll.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perfetto SP, Ambrozak D, Nguyen R, et al. Quality assurance for polychromatic flow cytometry. Nat Protoc. 2006;1:1522–1530. doi: 10.1038/nprot.2006.250. [DOI] [PubMed] [Google Scholar]
- 31.Bravery CA, Carmen J, Fong T, et al. Potency assay development for cellular therapy products: An ISCT review of the requirements and experiences in the industry. Cytotherapy. 2013;15:9–19. e19. doi: 10.1016/j.jcyt.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 32.Bailey AM. Balancing tissue and tumor formation in regenerative medicine. Sci Transl Med. 2012;4:147fs28. doi: 10.1126/scitranslmed.3003685. [DOI] [PubMed] [Google Scholar]