

Abstract
Regulatory Foxp3-lineage CD4 T cells (Tregs) were named for their ability to maintain self tolerance and suppress T cell immunity. However, resting Tregs from non-inflamed tissues exhibit little suppressor activity, and must be stimulated to acquire such function. Conversely, under certain inflammatory conditions Tregs may undergo rapid re-programming to acquire helper/effector functions. In this Brief Review, we describe recent progress in elucidating physiologic processes that control the functional status of Foxp3-lineage Tregs. Emerging evidence suggests the surprising possibility that re-programmed Tregs can be an indispensable source of helper activity in some physiologic settings, such as priming CD8+ T cell responses. This suggests a novel paradigm in which Foxp3+ Tregs intrinsically possess bifunctional potential, acting as a pre-formed pool of ‘first responder’ cells at sites of local inflammation that can provide either classical regulatory/suppressor activity, or rapidly re-program to supply helper/effector activity, contingent on signals that manifest in local physiologic settings.
Introduction
Natural Foxp3-lineage Tregs (nTregs) develop in the thymus, and induced (iTregs) Tregs differentiate from naïve CD4+ T cells in response to tolerogenic processes in peripheral tissues (Fig. 1). Tregs are essential to maintain peripheral tolerance to self and innocuous antigens. Thus, mice in which Tregs are experimentally ablated developed aggressive and lethal autoimmune syndromes (1–3). The pool of circulating Tregs is small, comprising <5% of circulating lymphocytes in human and mouse blood. Despite their critical role in maintaining peripheral tolerance, when Tregs are isolated from non-inflamed lymphoid tissues or blood of healthy individuals they are functionally quiescent (resting), and must be activated to manifest functional suppressive activity (4). This obligatory requirement for activation implies that natural microenvironmental cues must induce resting Tregs to acquire and maintain regulatory (suppressive) activity in physiologic settings. Moreover, recent studies suggest that Foxp3-lineage Tregs also have the potential to undergo functional re-programming to acquire helper/effector phenotypes in certain physiologic settings. In this Brief Review we focus on recent progress in defining settings where Tregs manifest the regulatory/suppressive phenotype, discuss factors that may promote Treg activation in such settings, and consider how certain inflammatory signals may drive Tregs to re-program into biologically important helper cells. Our focus will be on the final steps from mature, pre-formed, but resting Tregs into activated suppressor cells or re-programmed helper/effector cells (Fig. 1); we will not discuss basic mechanisms of Treg development or differentiation of naïve CD4+ T cells into inducible Tregs, as these topics have been reviewed elsewhere (5, 6).
Figure 1. How Tregs respond to local stimuli.
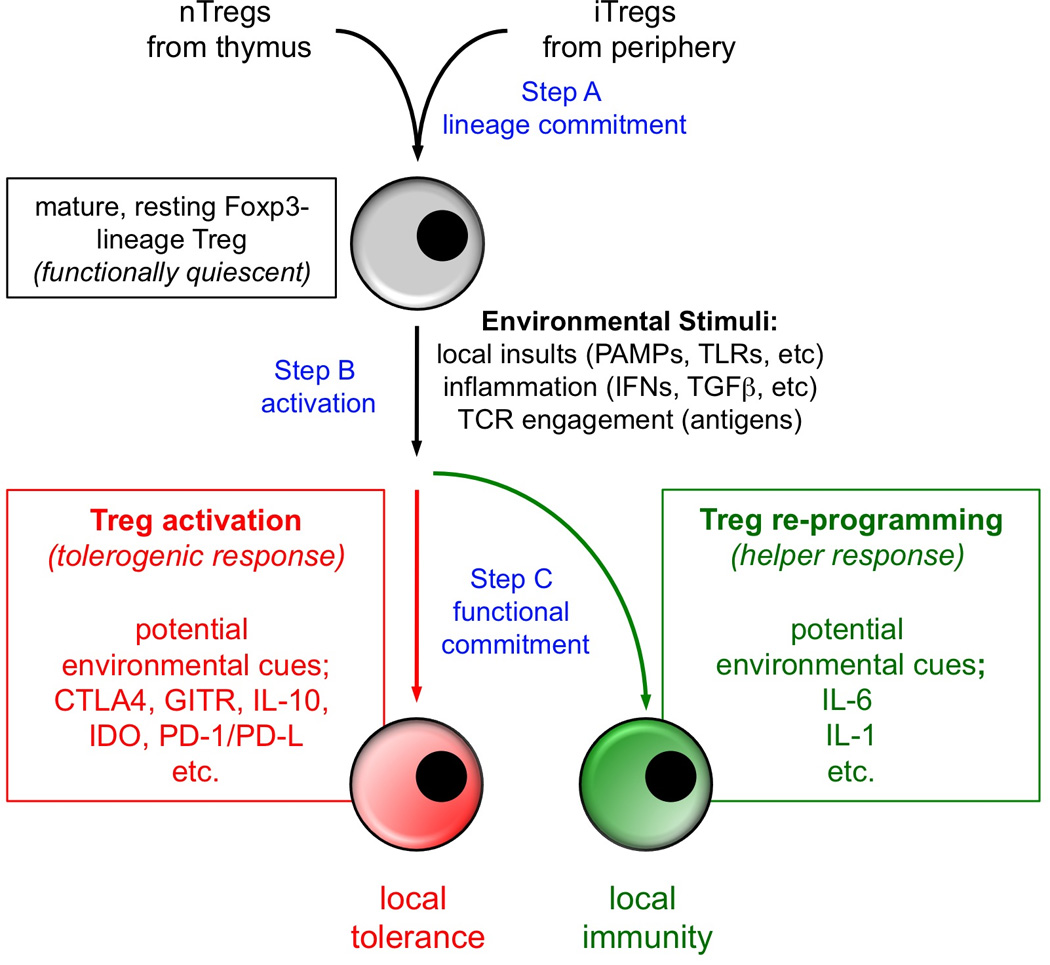
Circulating Tregs are functionally quiescent but respond rapidly to microenvironmental cues to acquire suppressive or helper/effector functions according to prevailing conditions. See text for details.
Resting Tregs must be activated to acquire functional suppressor activity
When considering Treg functional activity, it is important to emphasize that resting Tregs (isolated directly from tissues and blood, without activation or pre-treatment) are functionally quiescent (neither suppressive or pro-inflammatory). Only after some form of activation – e.g., through TCR cross-linking in vitro (4) – do Tregs acquire meaningful levels of suppressive function. Thus, simply counting the numbers of Foxp3+ Tregs in physiologic settings does not address whether these Tregs are resting or activated. Certain markers such as CCR6 and CD103 appear to correlate with an activated or “effector memory” phenotype in Tregs (7, 8). The ectonucleotidase CD39 may also mark activated Tregs in humans, although it is ubiquitously expressed by mouse Tregs (9). However, while these markers identify populations or subsets of Tregs that have experienced activation, they have not been validated as unambiguous markers of the activated functional state itself (i.e. a marker that is not expressed whenever Tregs are quiescent, and up-regulated on all functionally activated Tregs). Therefore, at present, the gold-standard for functional status still remains in vitro measurement of suppressor activity.
Given that most circulating Tregs are functionally quiescent, a key question thus becomes how local microenvironmental signals induce Tregs to express their latent functional activity. In Fig. 1, we break down this process into three sequential, conceptual steps: (A) the lineage-commitment and differentiation of progenitor cells (thymocytes or naive peripheral CD4+ T cells) that generates mature, resting Foxp3+ Tregs; (B) triggering resting Tregs to activate; and (C) inducing activated Tregs to either acquire canonical suppressive activity, or to re-program to manifest helper/effector activity. We hypothesize that distinct local environmental cues – or combinations of cues - may dictate steps (B) and (C), respectively, though this may be difficult to demonstrate experimentally. A great deal of published work already addresses the first (lineage-differentiation) step (A), and we do not deal with that here. The second (B, triggering) and third (C, functional commitment) steps have been less well studied, but there is some information available regarding the triggering of Tregs for suppressive function (depicted in the figure as the canonical or default pathway). Thus, for example, it has been shown that Tregs may become functionally activated by exposure to self antigens, or to antigens presented at mucosal surfaces, and Tregs can also be activated functionally by migrating through inflamed tissues, or by exposure to conditions created by tumors (10–13); it is generally assumed that these latter two settings also involve encounter with antigen, although the antigen may not be known. Antigen presentation appears critical for Treg activation (4), implying that APCs such as dendritic cells (DCs) may play an essential role in activating Tregs. It may be that only certain APC subsets are specialized to promote Treg activation, although this remains an open question. Also unknown is the exact role of local inflammation in triggering Treg activation, but it seems likely that inflammation plays an important role (both as an activator of local APCs, and a driver of the local cytokine milieu). Based on a variety of studies (reviewed in (14)), certain cytokines such as TGFβ, IL-10 and IL-2 may be essential for Treg activation and/or maintenance of suppressor functions, and interactions between CTLA4, GITR, PD-1 and PD-1 ligands (PD-L) on Tregs and their respective surface ligands (CD80/86, GITR-L, PD-L, PD-1) on other cell types (e.g. APCs) may also be important. In addition, we have shown that DCs expressing the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO) can directly activate mature Tregs (13); and local conditions that induce DCs to express IDO, such as tumors, TLR ligands or infections, can activate Tregs in an IDO-dependent fashion (15). Overall, the literature is inconclusive as few studies include assessments of the functional status of Tregs, and the specific triggering stimuli for resting Tregs remain incompletely understood. However, it seems clear that triggering requires antigen-driven interaction with APCs, and it is strongly influenced by the local cytokine milieu. This last point becomes relevant as we turn to the third step (C) depicted in Fig. 1, in which we entertain the possibility that alternative pathways of functional commitment exist, and that depending upon local conditions, Tregs may become activated for more functions than just classical suppression.
The concept of Treg plasticity and “re-programming”
The term “plasticity” has been used to describe the fact that Foxp3+ Tregs under certain conditions can be induced to express cytokines such as IL-17, IFNγ or IL-2, which are typically associated with helper/effector CD4+ T cell phenotypes (6, 16). This phenomenon was initially observed in vitro, either by culturing Tregs with cytokines such as IL-6 (16), or by artificially ablating Foxp3 (17). Since pro-inflammatory gene expression is normally profoundly suppressed in Foxp3+ Tregs (17), re-induction of these genes suggests that a significant change in phenotype can occur in some settings. This change has been variously described as “conversion”, “plasticity” or “re-programming” of the Tregs. In this review we continue to use these terms because they are widespread in the literature; however we offer the following modification: if it is true that Foxp3+ Tregs can rapidly adopt a pro-inflammatory phenotype in response to certain physiologic stimuli; and if this response can be shown to be biologically relevant for promoting certain immune responses; then perhaps the phenomenon should not be considered a “conversion” to a new developmental program. Rather, it may be that bifunctional potential is an intrinsic attribute of Foxp3+ Tregs – i.e., Foxp3-lineage cells are inherently capable of both suppressive and pro-inflammatory/helper activity, depending on local microenvironmental cues. This is at present a speculative hypothesis – although it has also been suggested by others (6) – and further studies are needed to elucidate the biologic role of re-programmed Tregs in vivo. However, emerging evidence suggests that local physiologic cues can control the phenotype and function of Tregs, with important consequences for the subsequent immune response.
Functional role of re-programmed Tregs
Treg plasticity is readily observed in vitro, but definitive in vivo studies are still needed to determine whether re-programmed Tregs are merely experimental artifacts, or represent fundamental participants in certain immune responses. Consistent with a possible biologic role for re-programmed Tregs, several studies identified large numbers of pro-inflammatory cells derived from former Tregs at sites of inflammation (18–20), although the biologic role of such cells was not established in these studies. In a model of lethal infection, Tregs lost Foxp3 expression and up-regulated pro-inflammatory cytokines (21); similarly, following transfer into lymphopenic hosts, many Tregs lost Foxp3 expression and acquired pathogenic effector function (22). However these are unusual biologic settings (end-stage lethal infection or profound lymphopenia) and their biologic relevance might be open to question. In studies of less extreme physiologic settings, Tsuji at al. showed that specialized helper CD4+ cells found in B cell follicles of the gut derived preferentially from former Foxp3+ Tregs (23). Wang and colleagues showed that a subset of Foxp3+ Tregs acquired the ability to produce IL-4 in vivo (via an unknown mechanism), and that IL-4 produced by converted Tregs was critical for differentiation of conventional CD4+ (TH2) helper cells (24). Consistent with the previous two studies, removal of Tregs during immunization markedly compromised the antibody response to mucosal adjuvant (25). In a transplant model, Vokaer and colleagues found that many of the effector CD4+ T cells responsible for minor-antigen disparate allograft rejection derived from re-programmed Tregs (26). And recently, we reported that the CD8+ T cell primary response to a cross-presented antigen (whole-protein ovalbumin in a CpG-based vaccine delivered to naive mice) was dependent on help from re-programmed Tregs (27). In this model, help provided by re-programmed Tregs was not mechanistically different from that provided by conventional helper T cells (e.g., it was mediated by CD40-ligand, and the requirement for re-programmed Tregs could be replaced by pre-immunizing mice with ovalbumin). Nevertheless, the key finding was that – in the absence of prior exposure to antigen – the ability of Tregs to rapidly re-program into CD40L+ helper cells was essential to support primary CD8+ T cell responses to cross-presented antigen. This model used a nominal antigen, but in light of these results it would be interesting to know whether the defect in early CD8+ T cell responses to viral infection observed when Tregs were depleted (28), might in part reflect loss of helper activity from re-programmed Tregs. Taken together, the preceding reports suggest that re-programmed Tregs may in fact play important biologic roles in certain specific physiologic settings. While suppressor activity is clearly the dominant and most critical Treg function (as shown by spontaneous autoimmunity in Treg-deficient mice), multiple studies suggest that Tregs may also play previously unappreciated roles as helper cells.
Tregs as “First-responders” model
In our vaccine model (27), the key contribution of Tregs was to provide rapid helper activity during the initial early window after vaccine administration, at a time when conventional, antigen-specific CD4+ helper cells had not started to activate and clonally expand. This rapid helper response to vaccination by Tregs that underwent functional re-programming offers a potential resolution of the long-standing paradox of how helper-dependent CD8+ T cell responses ever get started (29). Tregs have long been recognized to display attributes of constitutive activation (30). Most circulating Tregs display markers of an activated state (31), presumably due to encounter with self or other constitutively-present antigens, and resting Tregs respond more rapidly to inflammatory signals than do conventional CD4+ cells (32). Functional suppressor activity, although not constitutively present in resting Tregs, was rapidly induced to high levels in less than 24 hours, without requiring intervening proliferation or differentiation (4). Similarly, splenic Tregs acquired potent suppressor activity 12–18 hours after mice were acutely treated with high doses of CpGs administered systemically to stimulate IDO expression (15). Conversely, at local sites of low-dose CpG-induced inflammation (which does not induce IDO), Tregs quickly re-programmed to display inflammatory phenotypes and functional helper activity; these responses by Tregs far outpaced responses by naïve non-Treg CD4+ T cells, unless non-Tregs were previously primed in vivo (27). Moreover, under conditions in which IDO was blocked or ablated, high-dose systemic CpG treatment failed to activate Treg suppressor function; instead CpG-induced inflammation induced re-programming as Tregs uniformly expressed pro-inflammatory cytokines (IFNγ, IL-2, TNFα, IL-17) less than 9 hours after CpG treatment (15).
Taken together, these features suggest that Foxp3-lineage Tregs represent a highly responsive population (i.e., constitutively-primed “first-responder” cells), but the direction of the particular response elicited – either suppressive or pro-inflammatory – is exquisitely attuned to local physiologic conditions. Based on this, we propose that the fundamental, distinguishing attribute of Foxp3-lineage cells may actually be their “hair-trigger”, rapid-response capability (rather than simply their suppressor activity). Under this model, cells of the Foxp3 lineage would represent a pool of first-responder cells, capable of providing rapid suppression where appropriate; but also able – when directed by the innate immune system – to supply potent helper activity to support the early phases of immune responses (i.e., at a time when conventional primed T-helper cells are unavailable).
Loss of Foxp3 expression
One possible molecular mechanism of Treg re-programming might be loss of Foxp3 expression. In vitro, artificial ablation of Foxp3 in mature Tregs causes them to re-program into inflammatory effector cells (17). In several published models, Tregs were observed to lose Foxp3 expression when they converted into pro-inflammatory helper cells – e.g., under lymphopenic conditions (22); when Tregs converted to CD4+ follicular helper cells (23); and in “ex-Tregs” found at sites of autoimmune inflammation (20). However, several caveats apply to these observations:
Treg heterogeneity
First, peripheral Tregs are heterogeneous, and may include some cells with transient or unstable Foxp3 expression. These could be recently-converted peripherally-derived Tregs that retain the ability to revert to effector cells (33); or might reflect cells that transiently express Foxp3 at early stages during normal differentiation into TH17 cells (34). Neither of these states represent authentic, fully-committed Tregs, so their reversion to a pro-inflammatory phenotype would not constitute “re-programming”. Moreover, each study incorporated distinct experimental approaches to mark or detect Foxp3-expressing cells, which may introduce technical differences. In a recent study, Rudensky and colleagues marked Foxp3+ cells in vivo using a genetic inducible-labeling strategy intended to capture a snapshot of all cells actively expressing Foxp3 at a given time (35); this technique showed that the majority of Foxp3+ cells remained highly stable over time, even after proliferation, and in a variety of physiologic conditions (sub-lethal irradiation, Listeria infection and autoimmune diabetes). Thus, while some Tregs can lose Foxp3, it appears that most authentic Tregs retain detectable Foxp3 expression under most conditions.
Foxp3 heterogeneity
That said, however, the second caveat is that simply expressing detectable Foxp3 does not guarantee that Tregs have not undergone re-programming. In several models, we have observed continued expression of Foxp3-GFP fusion protein in Tregs exhibiting dramatic functional changes consistent with re-programming (15, 27, 36). Others have also reported continued expression of Foxp3 despite functional re-programming (16, 37). Foxp3 is known to undergo post-translational modification, and it associates with multiple regulatory factors in intracellular complexes, all of which are required for its function (38–40). Thus, if these processes were disrupted, then Foxp3 could be present in cells, but still be inactive or have abnormal function. Moreover, Foxp3 requires cooperation with other transcription factors in order to maintain the suppressive Treg phenotype: e.g., cooperation with Eos (Ikzf4) is required for Foxp3 to suppress downstream target genes; Foxp3 interacts physically with Runx1 to maintain Treg function (41), and mice lacking the Foxo1 transcription factor have markedly dysfunctional Tregs (42). Thus, there are several mechanisms by which Tregs could continue to express Foxp3, yet lose their suppressive program.
Taken together, these studies show that re-programmed Tregs may lose Foxp3 under certain circumstances, or may retain Foxp3 but still alter their function, thus emphasizing that the definition of “re-programming” needs to be functional, rather than simply loss of Foxp3. One functional test of re-programming is cytokine expression, but a more rigorous test is to ask whether sorted populations of Tregs perform a biologic helper function when adoptively transferred into new hosts (27). Another way to test for helper function by Tregs is to ask whether ablating Tregs in vivo – e.g., by using Foxp3-DTR mice (3) – unexpectedly removes a normal, biologically-important helper function.
Treg re-programming, IL-6 and innate immunity
Several years ago, Pasare and Medzhitov reported the seminal observation that signals from the innate immune system (e.g., triggered by TLR-ligands) could block the ability of Tregs to suppress immune responses (43). At the time, these studies did not address whether Tregs underwent re-programming, but Tregs clearly were no longer able to suppress effectively. In this standard model of vaccination, modulation of Treg-mediated suppression by inflammation was required in order to generate the expected (pro-inflammatory) response to the vaccine (43). Mechanistically, these early studies implicated the pro-inflammatory cytokine IL-6 as a key cytokine linking innate immune activation with abrogation of Treg-mediated suppression (44). These studies concluded that IL-6 most likely altered the effector T cells, rendering them resistant to suppression, rather than altering the Tregs. In addition to this possibility, however, subsequent studies of Treg plasticity in vitro showed that IL-6 was also a potent driver of Treg re-programming (16). We and others have confirmed that IL-6 can drive Treg re-programming in a variety of models (15, 27, 36, 45); in other models, a similar effect has been reported for another pro-inflammatory cytokine, IL-1β (46). Thus, in diverse models, cytokines of the innate immune system appear to drive Treg re-programming. Recently (using a model similar to Pasare and Medzhitov) we showed that IL-6-induced Treg re-programming was essential for successful vaccination with a TLR-ligand adjuvant (27). Importantly, this requirement for re-programmed Tregs was due not simply to a removal of suppressor activity, but to an active contribution of CD40L-mediated helper activity that was supplied exclusively by re-programmed Tregs. Thus, in this model, signals from the innate immune system (TLR-ligand, IL-6) drove Treg re-programming, and the re-programmed Tregs acted as an integral component of the downstream immunologic effects of the adjuvant. This role for Tregs was somewhat surprising, since adjuvants are classically considered strictly the province of the innate immune system. However, data from our study (27) and those of others (24, 25) suggest that re-programmed Tregs may be critical participants in the initiation of certain “innate” immune responses.
Antigen specificity of Tregs
Studies of Treg antigen specificity suggest that many Tregs recognize self antigens (47). Hence, one key concern regarding Treg re-programming is whether former Tregs would cause pathological autoimmunity. Consistent with this possibility, experimental ablation of Foxp3 in mature Tregs resulted in effector cells that appeared to cause autoimmune lesions (17); while “ex-Tregs” (former Foxp3-expressing cells) were over-represented in autoimmune diabetic lesions (20). These outcomes may arise because mature, resting Tregs attracted to sites of inflammation undergo activation (Step B), but some activated Tregs then commit to the helper/effector arm, not the suppressor arm (Step C); or activated Tregs that commit to the suppressor arm may possess attenuated suppressor functions under the influence of the prevailing inflammatory milieu in such lesions. Hence Tregs may promote pathology in autoimmune syndromes by undergoing re-programming, or manifesting less potent suppression, though – as discussed above - effector T cells in such lesions may also be more resistant to Treg-mediated suppression. On the other hand, if one of the normal physiologic functions of the Foxp3 lineage is to provide rapid helper activity for new antigens (27), then recognition of ubiquitous self antigens would be beneficial in settings where help to promote rapid development of effector T cells is desirable, because Tregs could interact with APCs at high frequency. In other words, help from Tregs would still be “antigen-driven”, but (unlike non-Treg helper cells) re-programmed Tregs would not be restricted to recognizing only the new antigens. If this is the case, then the crucial question becomes whether Treg re-programming is tightly regulated and readily reversible, or promiscuous and irreversible: if the latter, then autoimmunity would indeed be an undesirable consequence. However, if Treg re-programming is a normal physiologic function of the Treg lineage, and is tightly regulated such that it only occurs where and when it is needed, then autoimmunity is unlikely to manifest. As discussed above in the section on regulation of re-programming by the innate immune system, the available evidence suggests that Treg re-programming is highly regulated, by the same innate immune mechanisms that govern the fundamental decision to either activate or remain tolerant.
Conclusions
In this Brief Review we have summarized the emerging evidence that Tregs of the Foxp3 lineage display an unexpected degree of functional plasticity. Although the bifunctional potential of these rare cells is only beginning to be elucidated in physiologic settings, several recent studies point to significant mechanistic contributions of “re-programmed” Tregs to certain normal immune responses. Based on this, we propose that Treg plasticity may represent an intrinsic attribute of the Foxp3 lineage, and should not be considered a loss of lineage fidelity. Under most circumstances, the role of Foxp3+ Tregs is to suppress unwanted or inappropriate immune response, and certainly this is the key default attribute of Tregs (as shown by the lethal consequences of removing Tregs). However, under other circumstances Tregs appear to form a pool of rapidly-responsive helper cells, capable of supplying help to initiate T cell responses, at early times when conventional CD4+ helper cells are few in number and have not yet been primed. The same features that make Tregs effective suppressor cells – rapid response, and specificity for ubiquitous antigens – may also make them versatile “all-purpose” helper cells when required. The choice between these two consequences of Treg activation – functionally suppressive or helper states – is tightly regulated by the innate immune system, with Treg re-programming being driven by pro-inflammatory cytokines such as IL-6 and IL-1; and being inhibited by innate immunosuppressive mechanisms such as IDO.
References
- 1.Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 2.Chinen T, Volchkov PY, Chervonsky AV, Rudensky AY. A critical role for regulatory T cell-mediated control of inflammation in the absence of commensal microbiota. J Exp Med. 2010;207:2323–2330. doi: 10.1084/jem.20101235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 4.Thornton AM, Piccirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol. 2004;34:366–376. doi: 10.1002/eji.200324455. [DOI] [PubMed] [Google Scholar]
- 5.Bour-Jordan H, Bluestone JA. Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol Rev. 2009;229:41–66. doi: 10.1111/j.1600-065X.2009.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol. 2009;21:281–285. doi: 10.1016/j.coi.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleinewietfeld M, Puentes F, Borsellino G, Battistini L, Rotzschke O, Falk K. CCR6 expression defines regulatory effector/memory-like cells within the CD25(+)CD4+ T-cell subset. Blood. 2005;105:2877–2886. doi: 10.1182/blood-2004-07-2505. [DOI] [PubMed] [Google Scholar]
- 8.Stephens GL, Andersson J, Shevach EM. Distinct subsets of FoxP3+ regulatory T cells participate in the control of immune responses. J Immunol. 2007;178:6901–6911. doi: 10.4049/jimmunol.178.11.6901. [DOI] [PubMed] [Google Scholar]
- 9.Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Hopner S, Centonze D, Bernardi G, Dell'Acqua ML, Rossini PM, Battistini L, Rotzschke O, Falk K. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 10.Samy ET, Parker LA, Sharp CP, Tung KS. Continuous control of autoimmune disease by antigen-dependent polyclonal CD4+CD25+ regulatory T cells in the regional lymph node. J Exp Med. 2005;202:771–781. doi: 10.1084/jem.20041033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishikawa H, Kato T, Tawara I, Saito K, Ikeda H, Kuribayashi K, Allen PM, Schreiber RD, Sakaguchi S, Old LJ, Shiku H. Definition of target antigens for naturally occurring CD4(+) CD25(+) regulatory T cells. J Exp Med. 2005;201:681–686. doi: 10.1084/jem.20041959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, Yin N, Jessberger R, Ochando JC, Ding Y, Bromberg JS. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. 2009;30:458–469. doi: 10.1016/j.immuni.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma MD, Baban B, Chandler PR, Hou D-Y, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:1–13. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183:2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng XH, Jetten AM, Dong C. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 18.Esposito M, Ruffini F, Bergami A, Garzetti L, Borsellino G, Battistini L, Martino G, Furlan R. IL-17- and IFN-gamma-secreting Foxp3+ T cells infiltrate the target tissue in experimental autoimmunity. J Immunol. 2010;185:7467–7473. doi: 10.4049/jimmunol.1001519. [DOI] [PubMed] [Google Scholar]
- 19.O'Connor RA, Leech MD, Suffner J, Hammerling GJ, Anderton SM. Myelin-reactive, TGF-beta-induced regulatory T cells can be programmed to develop Th1-like effector function but remain less proinflammatory than myelin-reactive Th1 effectors and can suppress pathogenic T cell clonal expansion in vivo. J Immunol. 2010;185:7235–7243. doi: 10.4049/jimmunol.1001551. [DOI] [PubMed] [Google Scholar]
- 20.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O'Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duarte JH, Zelenay S, Bergman ML, Martins AC, Demengeot J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur J Immunol. 2009;39:948–955. doi: 10.1002/eji.200839196. [DOI] [PubMed] [Google Scholar]
- 23.Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science. 2009;323:1488–1492. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Souabni A, Flavell RA, Wan YY. An intrinsic mechanism predisposes Foxp3-expressing regulatory T cells to Th2 conversion in vivo. J Immunol. 2010;185:5983–5992. doi: 10.4049/jimmunol.1001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vendetti S, Davidson TS, Veglia F, Riccomi A, Negri DR, Lindstedt R, Pasquali P, Shevach EM, De Magistris MT. Polyclonal Treg cells enhance the activity of a mucosal adjuvant. Immunol Cell Biol. 2010;88:698–706. doi: 10.1038/icb.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vokaer B, Van Rompaey N, Lemaitre PH, Lhomme F, Kubjak C, Benghiat FS, Iwakura Y, Petein M, Field KA, Goldman M, Le Moine A, Charbonnier LM. Critical role of regulatory T cells in Th17-mediated minor antigen-disparate rejection. J Immunol. 2010;185:3417–3425. doi: 10.4049/jimmunol.0903961. [DOI] [PubMed] [Google Scholar]
- 27.Sharma MD, Hou DY, Baban B, Koni PA, He Y, Chandler PR, Blazar BR, Mellor AL, Munn DH. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity. 2010;33:942–954. doi: 10.1016/j.immuni.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohn M. How does the immune response get started? Cell Immunol. 2009;254:91–93. doi: 10.1016/j.cellimm.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 31.Fisson S, Darrasse-Jeze G, Litvinova E, Septier F, Klatzmann D, Liblau R, Salomon BL. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med. 2003;198:737–746. doi: 10.1084/jem.20030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Gorman WE, Dooms H, Thorne SH, Kuswanto WF, Simonds EF, Krutzik PO, Nolan GP, Abbas AK. The initial phase of an immune response functions to activate regulatory T cells. J Immunol. 2009;183:332–339. doi: 10.4049/jimmunol.0900691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A. 2009;106:1903–1908. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667–1671. doi: 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma MD, Hou D-Y, Liu Y, Koni PA, Metz R, Chandler PR, Mellor AL, He Y, Munn DH. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009;113:6102–6111. doi: 10.1182/blood-2008-12-195354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, Reis e Sousa C. DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol. 2008;38:3274–3281. doi: 10.1002/eji.200838950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li B, Samanta A, Song X, Iacono KT, Brennan P, Chatila TA, Roncador G, Banham AH, Riley JL, Wang Q, Shen Y, Saouaf SJ, Greene MI. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol. 2007;19:825–835. doi: 10.1093/intimm/dxm043. [DOI] [PubMed] [Google Scholar]
- 39.Samanta A, Li B, Song X, Bembas K, Zhang G, Katsumata M, Saouaf SJ, Wang Q, Hancock WW, Shen Y, Greene MI. TGF-beta and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc Natl Acad Sci U S A. 2008;105:14023–14027. doi: 10.1073/pnas.0806726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Zoeten EF, Lee I, Wang L, Chen C, Ge G, Wells AD, Hancock WW, Ozkaynak E. Foxp3 processing by proprotein convertases and control of regulatory T cell function. J Biol Chem. 2009;284:5709–5716. doi: 10.1074/jbc.M807322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 42.Kerdiles YM, Stone EL, Beisner DL, McGargill MA, Ch'en IL, Stockmann C, Katayama CD, Hedrick SM. Foxo transcription factors control regulatory T cell development and function. Immunity. 2010;33:890–904. doi: 10.1016/j.immuni.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 44.Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J Immunol. 2007;178:271–279. doi: 10.4049/jimmunol.178.1.271. [DOI] [PubMed] [Google Scholar]
- 45.Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, Brand D, Liu Z, Zheng SG. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol. 2010;185:2675–2679. doi: 10.4049/jimmunol.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Kim J, Boussiotis VA. IL-1beta-mediated signals preferentially drive conversion of regulatory T cells but not conventional T cells into IL-17-producing cells. J Immunol. 2010;185:4148–4153. doi: 10.4049/jimmunol.1001536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pacholczyk R, Kern J. The T-cell receptor repertoire of regulatory T cells. Immunology. 2008;125:450–458. doi: 10.1111/j.1365-2567.2008.02992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]