

Abstract
Human association studies of common genetic polymorphisms have identified many loci that are associated with risk of complex diseases, although individual loci typically have small effects. However, by envisaging genetic associations in terms of cellular pathways, rather than any specific polymorphism, combined effects of many biologically-relevant alleles can be detected. The effects are likely to be most apparent in investigations of phenotypically-homogenous subtypes of complex diseases. We report findings from a case-control, genetic association study of relationships between 2,925 SNPs and two subtypes of a commonly occurring chronic facial pain condition, temporomandibular disorder (TMD): 1) localized TMD; and 2) TMD with widespread pain. When compared to healthy controls, cases with localized TMD differed in allelic frequency of SNPs that mapped to a serotonergic receptor pathway (P=0.0012), while cases of TMD with widespread pain differed in allelic frequency of SNPs that mapped to a T-cell receptor pathway (P=0.0014). A risk index representing combined effects of six SNPs from the serotonergic pathway was associated with greater odds of localized TMD (odds ratio = 2.7, P=1.3×10−9), and the result was reproduced in a replication case-control cohort study of 639 people (odds ratio = 1.6, P=0.014). A risk index representing combined effects of eight SNPs from the T-cell receptor pathway was associated with greater odds of TMD with widespread pain (P=1.9×10−8), although the result was not significant in the replication cohort. These findings illustrate potential for clinical classification of chronic pain based on distinct molecular profiles and genetic background.
Keywords: Temporomandibular disorder, human genetics, serotonergic receptor, case-control study
Introduction
Temporomandibular disorder (TMD) is a commonly-occurring clinical condition characterized by pain in the temporomandibular joints and the masticatory muscles. In the US population, TMD symptoms affect 3% of men and 6% of women[10] and incidence of 3.5% per annum has been reported in women.[37]. Diverse treatments, including surgical and behavioral interventions, are used to treat TMD, although evidence of efficacy is equivocal for some of them[4] and no medicines have been approved specifically for TMD. Despite its burden in the population, the pathophysiology of TMD remains largely unknown.[5] More fundamentally, it is not clear whether TMD should be regarded as a facial-regional pain condition, or whether TMD is a manifestation of other widespread pain syndromes, such as fibromyalgia and chronic widespread pain.[47] Evidence for the latter comes from the clinical observation that TMD patients often present with other chronic pain conditions. Furthermore, clinical studies show that TMD patients are sensitive to a wide range of experimental pain modalities outside of the facial area. [5; 17; 18] This enhanced sensitivity has been attributed to pathological changes in central pain processing such as sensitization of spinal nociceptive neurons and disturbances of descending noxious control systems. [9; 26]
The distinction between localized and generalized pain in TMD is important both for patient diagnosis and for proper understanding of the etiology and pathophysiology of chronic pain. For example, if there are specific biological mechanisms, such as central sensitization, contributing to subtypes of TMD that manifest as widespread pain, it is likely that therapies targeting those mechanisms would produce greater benefit than localized treatments, such as dental-orthopedic and physical therapy treatments that target the temporomandibular joints and related muscles.
Here we investigated heterogeneity in clinical subgroups of TMD, seeking genetic evidence that different biological mechanisms might contribute to localized- versus-widespread pain. We expected that the analysis of genetic variants associated selectively with clinical subgroups of TMD might improve the power to identify candidate genes associated with each specific subgroup. We further anticipated that quantitatively-small etiologic influences of each single nucleotide polymorphism (SNP) would become larger in aggregate when genes were grouped according to their known contribution to cellular pathways known to affect pain processing. Additionally, we expected that known cellular pathways involved in pain processing would unmask genetic associations that conventionally are dismissed because they fail to exceed a Bonferroni-corrected P-value when evaluated in conventional genetic association analysis.
The specific aims of the study were to characterize subgroups of people with TMD, classified according to the presence or absence of widespread palpation-evoked pain, comparing each subgroup to healthy controls in order to identify genotypes that are associated differentially with the occurrence of widespread and localized pain. To do so, we used a case-control study design, evaluating over 300 pain-related genes to identify putative etiologic contributions of underlying molecular signals and pain mechanisms.
Material and Methods
Study participants and data collection
This paper investigates genetic associations with two subtypes of TMD using data from discovery and replication cohorts. The discovery cohort was a case-control study of TMD in women aged 18–60. Details of the study have been described in previous papers that investigated contributions of cigarette smoking[30] and circulating cytokines[36] to TMD. In summary, 398 self-identified white women aged 18–60 years were enrolled into the study between 2005–09. As described below, 385 of them with complete data were analyzed for this paper. All study participants were volunteers recruited from the communities surrounding UNC, Chapel Hill, using mass emails and flyers. The replication dataset came from a case-control study of TMD conducted at four US study sites. Details of study methods are described elsewhere,[35] and other papers have reported associations between TMD and psychological characteristics, pain sensitivity, autonomic function, clinical characteristics and common genetic variants (for overviews, see Maixner et al[16] and Fillingim et al[8]). The replication cohort enrolled both males and females aged 18–44 years of any racial/ethnic group between 2006–8, although this the analysis for this paper was limited to self-declared whites. In all other respects, the two studies used equivalent methods and criteria for subject selection, case-classification, phenotypic measurement, and genotyping.
Selection criteria
The discovery cohort comprised 18–60 year-old white females, while the replication cohort comprised 18–44 year-old whites of both genders. Exclusion criteria when enrolling participants in discovery and replication cohorts were self-reported history of one or more of 14 health-related conditions: diabetes; drug or alcohol abuse; hypertension not controlled by medication; hyperthyroidism; lupus erythematosis; psychiatric illness requiring hospitalization; respiratory disease (other than asthma) not controlled by medication; rheumatoid arthritis; kidney failure or dialysis; epilepsy; current chemotherapy or radiation treatment; pregnant or nursing mothers; facial injury or surgery; current orthodontic treatment.
Case-classifications and other phenotypic measurements
During a two hour visit to the research clinics at study sties, study examiners determined case-classification using a standardized protocol based on the Research Diagnostics Criteria for TMD.[6] The examination protocol and diagnostic criteria were equivalent in the discovery and replication cohorts. To be classified with TMD, study participants required presence of both: a) reported facial pain for at least five days during the previous two weeks, and b) examination-evoked pain in ≥3 temporomandibular muscles and/or ≥1 TM joints during digital palpation and jaw maneuver. Examiners also palpated eighteen body sites and widespread pain was classified as present when palpation elicited pain at diagonally-opposite quadrants of the body (i.e., above and below the waist, both on the left and right sides). Self-reported history of bodily pain did not contribute to the classification of widespread pain. In the discovery cohort, there were 104 females who had TMD with widespread pain, 93 who had TMD without widespread pain (i.e., localized TMD), and 188 women had neither TMD nor widespread pain (i.e., controls). In the OPPERA case-control study of 1,818 people,[16] 725 met the same criteria for race, and case-classification: 71 females and 15 males had TMD with widespread pain; 32 females and 9 males had localized TMD; and 320 females and 278 males were controls.
Following the clinical examination, study participants underwent quantitative sensory testing to determine responsiveness to thermal and pressure-pain stimuli. This paper reports descriptive findings only from the discovery cohort in order to characterize pain-sensitivity phenotypes associated with the two subgroups of TMD. Measurement procedures have been described previously.[36] In summary, threshold and tolerance to thermal pain was assessed on the forearm by applying a thermode that increased in temperature at a rate of 0.5°C/sec. Separate ratings of thermal pain “windup” were made in response to 10 repeated thermal stimuli of 50°C applied to the right hand were evaluated.[27] Stimuli of 0.5 sec duration were repeated once every three seconds and pain was rated on a 0–100 numerical rating scale (NRS). Finally, pressure pain thresholds in response to a hand-held pressure algometer [11] were assessed bilaterally at five locations: temporalis, masseter, TM joints, trapezius and lateral epicondyle.
Study participants also completed standardized psychosocial questionnaires, and descriptive findings from the discovery cohort are reported here to provide a more substantial characterization of the subgroups. A body pain symptom questionnaire was added to the study after approximately one half of the participants had been enrolled. It included a body manikin that was shaded to indicate location of pain symptoms that had lasted for at least one day in the preceding three months. Widespread pain symptoms were classified when both the axial skeleton and at least diagonally-opposite quadrants were shaded. The percentage of participants reporting symptoms was compared between controls and the two types of cases to characterize their body pain symptomatology. Finally, a sample of venous blood was collected for DNA extraction and genotyping. All study participants provided informed verbal and written consent. The studies and consent procedures were approved by the institutional review boards at each enrollment site
Genotyping
For both the discovery and replication cohorts, DNA extracted from whole blood was genotyped by Cogenics Inc. (Morrisville, NC, now Beckman-Coulter Genomics) using the Algynomics Pain Research Panel. This platform is a dedicated array for the targeted assessment of genes involved in acute and chronic pain conditions, utilizing the Affymetrix MegAllele platform. The panel genotypes 3295 SNPs corresponding to over 350 genes that represent three overlapping domains: (i) genes that mediate the transmission of pain signals by sensory nerve fibers and by neural pathways of the central nervous system that mediate the perception of pain; (ii) genes that mediate peripheral and central inflammatory responses to tissue injury or psychological stress; (iii) genes that influence mood and affective states associated with chronic pain conditions. Genotyping calls were checked for quality and filtered using utilities implemented in PLINK version 1.06[28]. Subjects with genotypically identified non-white ancestry or evidence of cryptic relatedness were eliminated from analyses. Samples were removed after applying a filter for overall genotype call rate <95%; SNPs were also subjected to a call rate >95% threshold, and were also filtered for minor allele frequency <1% and agreement across repeated samples >99%. The final cleaned dataset included 2720 SNPs in the discovery cohort (Supplementary Table 1) and 2924 SNPs in the replication cohort. [38]
Phenotypic characterization of TMD subgroups and controls in the discovery cohort
Eleven summary measures of experimental pain were computed. Thermal pain threshold was defined as the temperature (°C) at which pain was first reported, while tolerance was defined as the highest temperature that could be tolerated (with an upper limit of 50°C). NRS responses to repeated thermal stimuli were plotted against time to derive two summary measures of temporal “windup” for each subject: a) highest NRS response minus lowest NRS response, and b) slope of a linear regression line fitted for the first five NRS responses. Also, the subject’s first NRS was used as a measure of initial thermal sensitivity, and the sum of all 10 NRS was computed as a measure of overall thermal sensitivity. Mechanical pressure threshold at each of five anatomical sites was expressed as the algometer loading (kg) at which pain was first detected.
Standard scoring algorithms were used to compute summary measures from psychosocial questionnaires. Responses to the McGill Pain Questionnaire[19] were summed to produce three scores: a) the sensory components of pain (sum of 11 items); b) the affective component of pain (sum of 5 items), and c) pain intensity (1 item). The 54-item Pennebaker Index of Limbic Languidness[25] asked about somatic symptoms. Responses were summed to produce a single score. Responses to the 10-item Perceived Stress Scale[2] were recoded, as required, and summed to produce a single score. Six measures of mood were assessed using the 72-item Profile of Mood States questionnaire.[15] Scales represent a continuum of six bi-directional scales, with higher values representing more positive moods: a) agreeable-hostile, b) elated-depressed, c) confident-unsure, d) energetic-tired, e) clearheaded-confused, and f) composed-anxious. Measures of state and trait anxiety were computed from responses to the State-Trait Anxiety Inventory. [39]
Descriptive statistics for each measure of psychological status and experimental pain sensitivity were generated to provide profiles describing pain sensitivity of controls and the two subgroups of TMD in the discovery cohort. Analysis of variance was used with post-hoc contrasts between controls and each of the TMD subgroups, and the results were plotted.
Genetic association analyses
For the discovery cohort, case-control analysis for genetic associations was performed using logistic regression as implemented in PLINK. Case status was considered as a binary dependent variable, necessitating two logistic regression models for each SNP: a) localized TMD versus controls, and b) TMD with widespread pain versus controls. For each model, the number of minor alleles was the independent variable in a test of an additive inheritance model, and person’s age was a covariate. As a simple Bonferroni correction is overly conservative, we used spectral decomposition [22] to estimate the number of effectively independent SNPs tested by the Algynomics Pain Research Panel after accounting for the linkage disequilibrium between neighboring SNPs. This yielded a critical threshold of P<3.0×10−5, accounting for 1720 effectively independent SNPs, as the effective Bonferroni correction for multiple genetic associations.
Pathway analysis
We employed a statistical method that combines association signals from a set of several genes that constitute a predefined pathway. As population variation in quantitative traits and common genetic disorders are believed to arise from the combined effects of multiple inherited genetic variants, it is possible for several weak signals to reinforce one another when the signals are considered together.
For the discovery cohort, biologic pathways were identified using a variation of the Gene Set Enrichment Analysis method [41] adapted to use P-values from logistic regression models of the preceding step.[24] [48]. There are three major challenges faced by the pathway analysis of association signals. First, the variable number of SNPs for different genes needs to be taken into account: the magnitude of the strongest SNP signal is expected to be larger for a gene with more SNPs. Second, linkage disequilibrium between SNPs within a gene lowers the expected magnitude of the strongest SNP signal in a gene. Third, only a small percentage of SNPs may show association in a gene that harbors genuine causative variation; that is, whatever method is used to aggregate signals into a per-gene statistic faces a very unfavorable signal-to-noise ratio. To compute gene-specific P-values, we used the Simes test [34] which is robust in the presence of positive correlations between P-values and which has good power under heterogeneity of effects.[31; 49] As a way of obtaining a single per-gene P-value, this method provides advantage over common combination methods when only a few SNPs within a gene may be associated.[50] Ranks of per-gene P-values for a given pathway were compared against the background by the one-sided Mann-Whitney test. This test gives a single pathway-specific P-value. Because various pathways have overlapping genes, Mann-Whitney pathway P-values are not independent, and a simple Bonferroni multiple-testing correction for the number of tested pathways is overly conservative. Therefore, significance levels for the minimum of all pathway P-values were obtained via permutation analysis.
Each step of the permutation algorithm randomly shuffles SNP-specific P-values among all SNPs genes and repeats the pathway P-value calculation. This allows to build an empirical distribution for the pathway P-values under the hypothesis of no pathway effect and to determine the critical values.
Multivariable genetic risk prediction model
SNPs from two significant pathways identified in the preceding step were evaluated in two multivariable binary logistic regression models: one model evaluated contributions of SNPs from the serotonergic pathway to the odds of localized TMD; the other model evaluated contributions of SNPs from the T-cell pathway to the odds of TMD with widespread pain. For each model, SNPs were coded to represent the number of copies of the minor allele. When SNPs within a pathway had high pairwise correlations due to linkage disequilibrium, one of the pair was excluded and all remaining SNPs were used as predictor variables. Parameter estimates from the model were multiplied by the number of copies of the relevant SNP for each person to produce a “risk index” for each individual. Then, a univariate binary logistic regression model was created using only the risk index as the predictor variable. In the discovery study, the parameter estimate for the risk index necessarily had a value of 1.0 because the index represents the linear predictor which, in logistic regression, is calibrated to yield an estimate of 1.0 when it is used as the only covariate.[40]
The same algorithm using parameter estimates from the discovery cohort was then applied to the replication cohort to compute a risk index for each person in the replication cohort. The effect of the risk index was tested in a binary logistic regression model in which the risk index was the main predictor variable, together with dummy variables for study site and for gender. The alternate to the null hypothesis in the replication cohort was that the parameter estimate for the risk index was greater than zero (i.e., odds ratio greater than 1.0), and the null hypothesis was rejected if the one-tailed P-value was less than 0.05. The replication test was then repeated with a logistic regression model using only females from the replication cohort. Replication with both genders was conducted to be inclusive of all people enrolled in the OPPERA study, while replication among females only was conducted to provide strict comparability with the discovery cohort.
Results
Phenotypes characteristics of TMD subtypes and controls
The 385 white females in the discovery cohort ranged in age from 18 to 60 years (median=29 years, interquartile range = 23, 43). In the replication cohort, there were 423 females ranging in age from 18 to 44 years (median=24 years, interquartile range=21, 30) and 302 males ranging in age from 18 to 44 years (median=23 years, interquartile range=20, 27).
TMD cases with widespread pain had palpation tenderness at a median of 11 sites from among the 18 body sites palpated (lower and upper quartiles were 6 and 14 sites, respectively). For cases with localized TMD, corresponding statistics were 1 (0, 3), while controls had no body palpation tenderness. For the 240 participants who completed the body pain symptom questionnaire, widespread pain symptoms were reported by 46% of cases with widespread pain, 14% of cases with localized TMD and none of the controls.
In the discovery cohort, TMD subtypes differed from controls in most phenotypic characteristics, although not for measures of thermal pain sensitivity (Fig 1). In general, phenotypic contrasts between cases and controls were more pronounced for the TMD with widespread pain subtype than for the localized TMD subtype. The magnitude of the difference varied significantly (P<0.002) according to TMD subtype for three types of measures: sensitivity to pressure pain, pain perceptions, and somatic awareness.
Figure 1.
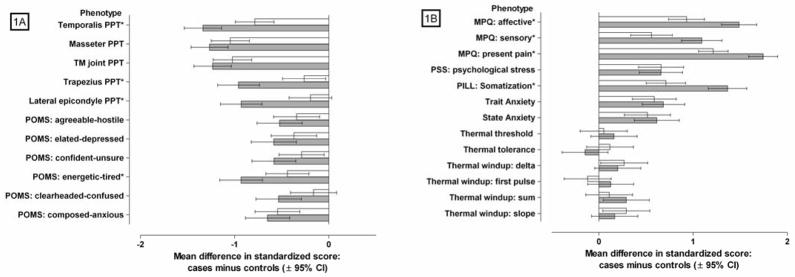
Case-control differences in intermediate phenotypes for localized TMD and TMD with widespread pain
Mean (±95% confidence interval) differences in quantitative phenotypes between n=192 TMD controls and two TMD subtypes ▭ localized TMD (n=94), and
 TMD with widespread pain (n=105)]. Phenotypes in Fig 1A are measures of pressure pain thresholds (PPT) and mood, where lower means in TMD subtypes relative to controls signify greater pain sensitivity and more negative mood (respectively). Phenotypes in Fig 1B are measures of pain perceptions, psychological states, and numerical ratings of thermal pain, where higher means in TMD subtypes relative to controls signify more severe pain, greater psychological distress, and greater pain sensitivity (respectively). Mood was measured using the Profile of Mood States (POMS) questionnaire. Pain perceptions were measured using the McGill Pain Questionnaire (MPQ). Psychological stress was measured using the Perceived Stress Scale (PSS). Somatization was measure using the Pennebaker Index of Limbic Languidness (PILL). All phenotypic measures were transformed to standardized z-scores with a mean of zero and standard deviation of 1.0 for the full cohort of TMD cases and controls.
TMD with widespread pain (n=105)]. Phenotypes in Fig 1A are measures of pressure pain thresholds (PPT) and mood, where lower means in TMD subtypes relative to controls signify greater pain sensitivity and more negative mood (respectively). Phenotypes in Fig 1B are measures of pain perceptions, psychological states, and numerical ratings of thermal pain, where higher means in TMD subtypes relative to controls signify more severe pain, greater psychological distress, and greater pain sensitivity (respectively). Mood was measured using the Profile of Mood States (POMS) questionnaire. Pain perceptions were measured using the McGill Pain Questionnaire (MPQ). Psychological stress was measured using the Perceived Stress Scale (PSS). Somatization was measure using the Pennebaker Index of Limbic Languidness (PILL). All phenotypic measures were transformed to standardized z-scores with a mean of zero and standard deviation of 1.0 for the full cohort of TMD cases and controls.
* indicates phenotypes where the case-control difference varies significantly (P<0.002) according to TMD subtype.
Genetic associations with TMD and its subtypes
The overall genotyping rate for the discovery cohort using the Pain Research Panel was 99.7% (Supplementary Table 1), indicating a very low probability that differential missingness rates in cases and controls might influence group allele frequencies. All of the subjects in the discovery cohort were Caucasian by self-report and by genetic clustering (see Supplementary Figure 1), but the potential remains for bias due to subtle differences in ethnic makeup between case and control populations. Examination of the distribution of test statistics, by Q-Q plot (Supplementary Figure 2) and by the genomic inflation factor statistic lambda (λ<1.0), suggested no systematic deviation from their expected distribution under the null that would indicate population stratification. The quality control metrics of all SNPs with p-values <0.01, including Hardy-Weinberg equilibrium p-values and missingness rates, showed no indication that these associations are due to technical errors in sample quality or genotype calling accuracy (Supplementary Table 1).
Results for the regression test contrasting all TMD cases against controls are presented in the Supplementary Table 2. The threshold for significance, adjusted for the number of SNPs tested and linkage disequilibrium between them, was set to p<3.0×10−5. We observed no significantly associated SNPs, either in contrasts between all TMD cases and controls, or in the contrasts of TMD subtypes and controls.
Pathway analysis
To explore cellular mechanisms that underlie TMD subtypes, we applied bioinformatics tools to identify signaling networks implicated by the association results in the discovery cohort. The multiple-testing-adjusted significance threshold was determined to be 0.002 for the 5% level, as determined through permutation analysis. Using association results from all TMD cases compared to controls, no single cellular pathway was statistically significant, although the serotonin receptor R2 (HTR2)-> ELK-SRF/GATA4 signaling pathway approached statistical significance (P=0.0026, Table 1A). However, when SNP association results from the analysis of localized TMD were used, the HTR2-> ELK-SRF/GATA4 signaling pathway was statistically significant (Table 1B, P=0.0013). Conversely, this same pathway was not significant (P=0.3579, Table 1B) based on SNP association results for TMD with widespread pain, suggesting that association with this signaling pathway is driven by the localized TMD subtype. However, association results from analysis of TMD with widespread pain revealed a statistically significant effect of the T-cell receptor -> CREBBP signaling pathway (Table 1C, P=0.0017). Conversely, the same pathway was not significant (P=0.9840) based on SNP association results for the localized TMD subtype.
Table 1.
P-values of Cellular Pathways Associated with TMD Rank Ordered according to TMD case-classification: (A)all TMD cases; (B) loclized TMD or (C)TMD with widespread paip.
| A | Top pathways for all TMD cases | All TMD Cases: P-value* | Localized TMD: P-value* | TMD with widespread pain P-value* |
|---|---|---|---|---|
| HTR2 -> ELK-SRF/GATA4 signaling | 0.003 | 0.001 | 0.358 | |
| HTR2 -> membrane transport | 0.004 | 0.015 | 0.426 | |
| ADORA2A/B -> vasodilation | 0.008 | 0.069 | 0.113 | |
| ADORA1/2A -> exocytosis | 0.031 | 0.011 | 0.587 | |
| B | Top pathways for localized TMD | |||
| HTR2-> ELK-SRF/GATA4 signaling pathways | 0.003 | 0.001 | 0.358 | |
| ADORA1/2A -> exocytosis | 0.031 | 0.011 | 0.587 | |
| LeptinR -> ELK-SRF signaling | 0.285 | 0.014 | 0.913 | |
| PECAM -> SP1 signaling | 0.285 | 0.014 | 0.913 | |
| PTPRC -> BCL6 signaling | 0.285 | 0.014 | 0.913 | |
| TNFRSF6 -> HSF1 signaling | 0.285 | 0.014 | 0.913 | |
| HTR2 -> membrane transport | 0.004 | 0.015 | 0.426 | |
| PTGER1/4 -> vascular motility | 0.650 | 0.015 | 0.954 | |
| ADRB1 -> prostaglandin generation | 0.329 | 0.017 | 0.864 | |
| IGF1R -> ELK-SRF/HIF1A/MYC/SREBF | 0.459 | 0.020 | 0.875 | |
| signaling | ||||
| ADRA1 -> vasoconstriction | 0.313 | 0.022 | 0.963 | |
| ADRA1 -> prostaglandin generation | 0.328 | 0.031 | 0.839 | |
| CholinergicRn -> CREB signaling | 0.339 | 0.032 | 0.542 | |
| CHRM1/2/3 -> vascular motility | 0.299 | 0.033 | 0.768 | |
| AGER -> CREB/SP1 signaling | 0.238 | 0.036 | 0.720 | |
| P2RY2/12/13/14 -> N-type calcium channel | 0.262 | 0.036 | 0.750 | |
| NMDA receptors -> Ca2+/CREB | 0.425 | 0.039 | 0.656 | |
| activation/PGE2 synthesis | ||||
| CHRNA7 -> NOS1 production | 0.465 | 0.040 | 0.972 | |
| BDKRB1/2 -> prostaglandin generation | 0.127 | 0.042 | 0.501 | |
| AdrenergicRa -> ELK-SRF signaling | 0.315 | 0.043 | 0.592 | |
| AdrenergicRa -> STAT3 signaling | 0.315 | 0.043 | 0.592 | |
| HRH2/4 -> IL6/10 production | 0.405 | 0.045 | 0.907 | |
| Notch -> TCF3 signaling | 0.375 | 0.046 | 0.707 | |
| GALR1/2/3 -> POMC/NPY production | 0.539 | 0.046 | 0.805 | |
| ADRA2C/ADRB2 -> vasoconstriction | 0.068 | 0.047 | 0.096 | |
| C | Top pathways for TMD with widespread pain | |||
| T-cell receptor -> CREBBP signaling pathway | 0.110 | 0.984 | 0.002 | |
| VasopressinR1/R2 -> | 0.708 | 0.943 | 0.043 | |
| MEF/MYOD/NFATC/MYOG signaling | ||||
| NeuropeptideYR -> ATF/CREB signaling | 0.546 | 0.634 | 0.045 | |
| EphrinR -> actin signaling | 0.127 | 0.808 | 0.047 |
P-values were obtained via permutation analysis. The multiple-testing-adjusted significance threshold was 0.002 for 5% type I error.
Further investigation of the HTR2-> ELK-SRF/GATA4 signaling pathway in localized TMD revealed contributions from SNPs situated within the following genes: HTR2A and HTR2C serotonin receptor 2; mitogen-activated protein kinase 1 (MAPK1, ERK2); and mitogen-activated protein kinase 1 (MAP2K1, MAPKK1, MEK1) (Figure 2A, Supplementary Table 2). Meanwhile, for the T-cell receptor -> CREBBP signaling pathway in TMD with widespread pain, contributions were found for two genes: calcium/calmodulin-dependent protein kinase IV (CAMK4) and calmodulin 2 (CALM2) (Figure 2B, Supplemental Table 2).
Figure 2.

Cellular pathways associated with (A) localized TMD and (B) TMD with widespread pain.
Genes shown in blue are significantly associated with case status with P<0.05. (A) HTR2-> ELK-SRF/GATA4 pathway. HTR2A – 5-hydroxytryptamine receptor 2A (OMIM 182135), HTR2C - 5-hydroxytryptamine receptor 2C (OMIM 312861), MAPK1 – mitogen-activated protein kinase 1 (OMIM 176948), MAP2K1 - mitogen-activated protein kinase 1 (OMIM 176872). (B) T-cell receptor -> CREBBP pathway. CAMK4 - calcium/calmodulin-dependent protein kinase IV (OMIM 114080), CALM2 - calmodulin 2 (OMIM 114182).
Multivariable genetic risk prediction model
Of the10 SNPs involved in the serotonergic signaling pathway for localized TMD (rs9316233, rs4776783, rs12439516, rs12440176, rs2276008, rs6928, rs8136867, rs4821402, rs3813928 and rs3813929), four were excluded from multivariable modeling in the discovery cohort due to linkage disequilibrium that created very high pairwise correlations. Specifically rs12440176 was excluded because of correlation (r=1.0) with rs12439516; rs4821402 was excluded because of correlation (r=1.0) with rs6928; rs8136867 was excluded because of correlation (r=0.98 with rs6928); and rs3813929 was excluded because of correlation (r=−1.0) with rs3813928. In the discovery cohort, the remaining six SNPs in combination were strongly associated with odds of localized TMD when evaluated in a multivariable binary logistic regression model (P=2.0 × 10−6 for the full model - Table 2A). The risk index that combined all six SNPs were likewise was a significant (P=1.3 × 10−9) predictor of localized TMD. By virtue of the method used to calculate the index, a one-unit increase in the index was associated with 2.72-fold greater odds of localized TMD (95%CI = 1.93, 3.95). When the same algorithm was used to create an identical risk index in the replication study, the association with localized TMD was in the same direction and statistically significant (odds ratio=1.58, lower limit of 1-tailed 95% CI = 1.16, 1-tailed P-value = 0.007). The result was virtually identical when the replication cohort was restricted to females (odds ratio=1.53, lower 1-tailed 95%CI = 1.07, 1-tailed P=0.024).
Table 2.
Logistic regression models of associations between serotonergic pathway SNPs and localized TMD
| 2a: Discovery phase model of six serotonergic pathway SNPs
| |||
|---|---|---|---|
| SNP (gene) | Maximum likelihood estimates | ||
| odds ratio | 95% confidence limits | P-value | |
| rs9316233 (HTR2A) | 0.39 | 0.22, 0.67 | 0.001 |
| rs4776783 (MAP2K1) | 2.04 | 1.19, 3.55 | 0.010 |
| RS12439516 (MAP2K1) | 0.64 | 0.31, 1.25 | 0.207 |
| rs2276008 (MAPK1) | 2.99 | 1.12, 8.38 | 0.031 |
| RS6928 (MAPK1) | 0.74 | 0.50, 1.10 | 0.138 |
| rs3813928 (HTR2C) | 1.79 | 1.10, 2.91 | 0.018 |
| 2b: Discovery phase model of serotonergic pathway risk index
| |||
|---|---|---|---|
| Maximum likelihood estimates | |||
| odds ratio | 95% confidence limits | P-value | |
| Risk index from 6 SNPs | 2.72 | 1.93, 3.95 | 1.3 × 10−9 |
| 2c: Male and female replication phase model of six serotonergic pathway SNPs
| |||
|---|---|---|---|
| SNP (gene) | Maximum likelihood estimates | ||
| 1-tailed | 1-tailed | ||
| odds ratio | 95% confidence limit* | P-value | |
| rs9316233 (HTR2A) | 0.66 | n/a, 1.16 | 0.122 |
| rs4776783 (MAP2K1) | 1.86 | 1.09, na | 0.025 |
| RS12439516 (MAP2K1) | 0.79 | n/a, 1.66 | 0.314 |
| rs2276008 (MAPK1) | 1.50 | 0.58, na | 0.224 |
| RS6928 (MAPK1) | 0.89 | n/a, 1.34 | 0.317 |
| rs3813928 (HTR2C) | 1.07 | 0.66, n/a | 0.410 |
| 2d: Male and female replication phase model of serotinergic pathway risk index
| |||
|---|---|---|---|
| Maximum likelihood estimates | |||
| 1-tailed | 1-tailed | ||
| odds ratio | 95% confidence limit* | P-value | |
| Risk index from 6 SNPs | 1.58 | 1.16, n/a | 0.007 |
| 2e: Female replication phase model of serotonergic pathway risk index | |||
|---|---|---|---|
| Maximum likelihood estimates | |||
| 1-tailed | 1-tailed | ||
| odds ratio | 95% confidence limit* | P-value | |
| Risk index from 6 SNPs | 1.53 | 1.07, n/a | 0.024 |
Discovery phase models 2a and 2b are based on 91 cases of localized TMD and 185 conrols in the COMT cohort. Replication phase models 2c and 2d are based on 38 female and male cases of localized TMD and 596 female and male controls in the OPPERA study. Models 2c and 2d additionally control for OPPERA study site and gender. Replication phase model 2e is based on 30 female cases of localized TMD and 315 female controls in the OPPERA study. Model 2e additionally controls for OPPERA study site. In models 2a and 2c, genotypes are modeled as number of copies of minor allele (CMA) assuming codominant effects. In models 2b, 2d and 2e, the risk index is CMA[rs9316233] × −0.941 + CMA[rs4776783] × 0.712 + CMA[rs12439516] × −0.444 + CMA[rs2276008] × 1.094 + CMA[rs6928] × −0.295 + CMA[rs3813928] × 0.580
n/a = limit of confidence interval is not applicable to the one-tailed hypothesis
Eight SNPs involved in the T-cell receptor pathway could be used in multivariable analysis of TMD with widespread pain (Supplementary Table 3), where a risk index based on those SNPs was significantly (P=1.9E-08) associated with case status. Again, the method used to calculate the index meant that a one-unit increase in the index was associated with 2.72-fold greater odds of TMD with widespread pain (95%CI =1.93, 3.95). While the index had the same direction of effect in the replication cohort, it was not statistically significant (odds ratio=1.08, lower 1-tailed 95%CI = 0.88, P=0.31) (Supplementary Table 3).
Discussion
In revealing a novel association of the serotonergic signaling pathway with localized orofacial pain, this study illustrates the benefits of bioinformatics tools and careful phenotyping when studying effects of multiple genes in an association study. While case-control studies are used widely to investigate effects on disease of large numbers of genetic markers, interpretation of the findings has been hampered by the problem of false-negative associations when each SNP is evaluated independently. False negatives occur when SNPs in genes that truly alter disease susceptibility are declared to be unrelated to the disease under study. The problem is only partly addressed by conducting studies of large cohorts in which a priori calculations show sufficient power to detect nominated thresholds of quantitative effects (i.e., odds ratios) for individual SNPs. In practice, single SNPs yield very small odds ratios in association studies of complex diseases. In many instances odds ratios with magnitude close to the null value of 1.0 are observed, even for genes found to be true positives when tested in replication-cohorts. Part of the reason for small quantitative effects is due to statistical noise in studies of complex diseases, where genetic heterogeneity of the condition itself hampers the ability to detect association. The consequence of false negatives is that disease susceptibility genes remain unrecognized and they are not selected for follow-up analysis. Opportunities for biological and clinical discoveries therefore are lost.
In this study, we used two analytic strategies to deal with the problems noted above. First, we reduced clinical heterogeneity in the classification of TMD by creating two clinical subtypes: TMD cases where pain was localized to the facial area, and TMD cases with widespread bodily pain. The sub-classification was based on evidence that people with widespread pain often exhibit pathological changes in central pain processing mechanisms, where proposed mechanisms include sensitization of spinal nociceptive neurons and disturbances of descending noxious control systems [9] [26]. We found evidence consistent with such mechanisms in a separate investigation of quantitative sensory testing among participants in the current discovery cohort.[1] The method used to sub-classify TMD was based on assessments of tenderness to digital palpation at multiple body sites. Digital palpation is a quick and non-invasive procedure that is feasible to perform in everyday clinical practice. Heterogeneity among these TMD subtypes was confirmed when we found that TMD cases with widespread pain had greater sensitivity to pressure pain, higher levels of somatization and more severe perceptions of their clinical pain than TMD cases with localized pain (Figure 1). These findings show that this simple clinical procedure provides an effective way to distinguish between clinically-meaningful subtypes of TMD.
When we undertook conventional genetic association analysis using approximately 3,000 SNPs from genes known to regulate pain processing, we found that different sets of SNPs were differentially associated with the two subtypes of TMD. When analyzed using conventional methods for genetic association studies, the associations approached statistical significance although none met the Bonferroni-adjusted threshold required to account for multiple comparison. We therefore applied a second analytic strategy to identify potentially small effects of individual SNPs that, in combination, had larger influences on cellular pathways that are biologically-relevant to pain mechanisms. This was achieved using pathway analysis that combines gene-disease association signals from several sets of genes that constitute a predefined cellular pathway.[7; 45] The method makes it possible for several weak signals to reinforce one another when the signals are considered together. The signals may be combined directly into an overall test statistic, or ranked together with the “background” values for all of the genes in a study. In the later approach, a statistical test is used to ascertain whether the ranks for a given pathway appear to be different from those for the background genes.
In the pathway analysis of association results for the combined TMD case-classification, none of the 248 cellular pathways reached the threshold for statistical significance. However, when the method was used for TMD subtypes, two cellular pathways emerged: the HTR2-> ELK-SRF/GATA4 signaling pathway was significant in the analysis of localized TMD; and the T-cell receptor -> CREBBP signaling pathway was significant in the analysis of TMD with widespread pain. Risk indices, representing multivariable combinations of SNPs from each pathway, were associated with the respective subtypes in the discovery cohort. In the replication cohort, the direction of association was the same for both pathways, and for the HTR2-> ELK-SRF/GATA4 signaling pathway, the risk index was replicated as a significant predictor of localized TMD. Although we had available only a limited number of cases in the replication cohort, the association with the serotonergic pathway was replicated with convincing protection against type I error (i.e., P<0.025). The association of TMD with widespread pain and the T-cell receptor pathway was not statistically significant, although to rule out type II error, the relationship should be replicated in a larger cohort.
The replicated association between serotonergic signaling and localized TMD represents a novel finding regarding etiology of chronic pain. While commonly associated with antinociception in animal experimental studies [29; 32], serotonin (5-hydroxytryptamine, or 5-HT) is also known to produce a hyperalgesic response when injected subcutaneously or into rodents’ deep tissues.[43] [42] Different subtypes of 5-HT receptors are associated with hypersensitive responses to various noxious stimuli. For example, mechanical hypersensitivity (allodynia) induced by serotonin has been linked to HT1A, HT2B, and HT2C receptors [43] [14], whereas the HT2A receptor appears to be responsible for HT-induced thermal hypersensitivity [44]. Furthermore, recent studies indicate that the descending serotonin (5-HT) system from the rostral ventromedial medulla (RVM) in the brainstem and the 5-HT(3) receptor subtype in the spinal dorsal horn are involved in enhanced descending pain facilitation after tissue and nerve injury[46] [13].
A plausible interpretation of the current findings is that localized facial pain is influenced by mechanisms operating through the stimulation of peripheral HT2 receptors that are modified by an individual’s genetic landscape. Importantly, the localized TMD subgroup had less depressive mood than the TMD subgroup with widespread pain (Fig. 1A), consistent with activation of a central serotonin receptor pathway. Taken together, these observations suggest that people with localized TMD may have an up-regulated serotonergic pathway. This up-regulation leads to local hyperalgesia through peripheral HTR2s but also an adequate central serotonergic response mechanism, limiting the potential for more widespread pain. It is important, however, to keep in mind that pathway analysis does not provide the directionality of the association, and alternative hypotheses should be investigated.
The observed association between T-cell receptor pathway and TMD with widespread pain corroborates recent studies of the role of T-cell-mediated mechanisms in pain. T-cell infiltration of injured sciatic nerves was demonstrated in the chronic constriction injury model of peripheral neuropathic pain in rats, while the absence of functional T-cells in animals resulted in reduced hyperalgesia.[12; 21] Compared to young animals, adults had a greater T-cell infiltration and activation in the dorsal horn of the spinal cord after peripheral nerve injury, showing that T-cells contribute in evolution of pain response.[3] In multiple sclerosis patients who often suffer from pain, T-cells localize to sites of demyelination within the central nervous system.[23] Our finding of T-cell pathway involvement in TMD with widespread pain indicates the importance of immune cells in the development and maintenance of persistent widespread pain.[20]
Effect sizes and P-values observed initially in the discovery cohort were weaker upon re-testing in the replication cohort. This is a consequence of the winner’s curse phenomenon that may introduce substantial bias among top-ranking results of discovery studies. The extent of the bias is difficult to quantify as it depends not only on the number of tests, but also on the number of true signals and their effect sizes, which are generally unknown.[33] This phenomenon underlines importance of replication studies that provide unbiased estimates of association effects.
In summary, common genetic variants usually contribute to molecular pathophysiological processes in ways that produce weak associations between individuals SNPs and clinical disease when assessed in human association studies. In this study we distinguished phenotypically between biologically relevant subtypes of chronic TMD and applied a new analytic approach that searches for combinations of usually-small genetic influences on known cellular signaling pathways. We found that anatomically localized and generalized types of pain represent clinically meaningful subgroups of TMD and that they have distinct molecular profiles with correspondingly-distinct genetic backgrounds. The results reveal a distinct role for a serotonergic pathway in pathophysiology of TMD, although further work is needed to uncover the direction of this contribution. Potentially, this major cellular pathway contributing to development of localized TMD might be effectively treated using serotonin receptor 2 selective ligands. Furthermore, our strategy of genetic modeling using pathway analysis could hold promise for studies of other pain conditions where widespread pain is a significant feature, such as headache, back pain and fibromyalgia.
Supplementary Material
Summary.
Cellular pathway analysis from a human genetic association study identified a serotonergic receptor 2 pathway that contributed to localized manifestations of facial pain.
Acknowledgments
This work was supported by National Institutes of Health, National Institutes of Dental and Cranial Research [grant numbers RO1-DE16558 and UO1-DE017018 to LD, GS, WM]; D.V.Z was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences. We are grateful to investigators at OPPERA study sites for their contribution in directing data collection: Dr. Joel Greenspan, University of Maryland, MD; Mr. Charles Knott, Battelle Memorial Inc, Durham, NC.
Footnotes
Conflict of Interest Statement
Drs. Slade, Diatchenko, Fillingim and Maixner are consultants with equity interests in Algynomics Inc., a company providing research services in personalized pain medication and diagnostics. All other authors declare no financial relationships that might represent a possible conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen H, Slade G, Lim PF, Miller V, Maixner W, Diatchenko L. Relationship between temporomandibular disorders, widespread palpation tenderness, and multiple pain conditions: a case-control study. The journal of pain : official journal of the American Pain Society. 2012;13(10):1016–1027. doi: 10.1016/j.jpain.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen S, Kamarck T, Mermelstein R. A global measure of perceived stress. J Health Soc Behav. 1983;24(4):385–396. [PubMed] [Google Scholar]
- 3.Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, Barrett L, Brenner GJ, Vardeh D, Woolf CJ, Fitzgerald M. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. 2009;29(46):14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dao TT, Lavigne GJ, Charbonneau A, Feine JS, Lund JP. The efficacy of oral splints in the treatment of myofascial pain of the jaw muscles: a controlled clinical trial. Pain. 1994;56(1):85–94. doi: 10.1016/0304-3959(94)90153-8. [DOI] [PubMed] [Google Scholar]
- 5.Diatchenko L, Nackley AG, Slade GD, Fillingim RB, Maixner W. Idiopathic pain disorders--pathways of vulnerability. Pain. 2006;123(3):226–230. doi: 10.1016/j.pain.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 6.Dworkin SF, LeResche L. Research diagnostic criteria for temporomandibular disorders: review, criteria, examinations and specifications, critique. J Craniomandib Disord. 1992;6(4):301–355. [PubMed] [Google Scholar]
- 7.Elbers CC, van Eijk KR, Franke L, Mulder F, van der Schouw YT, Wijmenga C, Onland-Moret NC. Using genome-wide pathway analysis to unravel the etiology of complex diseases. Genet Epidemiol. 2009;33(5):419–431. doi: 10.1002/gepi.20395. [DOI] [PubMed] [Google Scholar]
- 8.Fillingim RB, Slade GD, Diatchenko L, Dubner R, Greenspan JD, Knott C, Ohrbach R, Maixner W. Summary of findings from the OPPERA baseline case-control study: implications and future directions. J Pain. 2011;12(11 Suppl):T102–107. doi: 10.1016/j.jpain.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gracely RH, Grant MA, Giesecke T. Evoked pain measures in fibromyalgia. Best Pract Res Clin Rheumatol. 2003;17(4):593–609. doi: 10.1016/s1521-6942(03)00036-6. [DOI] [PubMed] [Google Scholar]
- 10.Isong U, Gansky S, Plesh O. Temporomandibular joint and muscle disorder-type pain in U.S. adults: the National Health Interview Survey. J Orofac Pain. 2008;22(4):317–322. [PMC free article] [PubMed] [Google Scholar]
- 11.Jaeger B, Reeves JL. Quantification of changes in myofascial trigger point sensitivity with the pressure algometer following passive stretch. Pain. 1986;27(2):203–210. doi: 10.1016/0304-3959(86)90211-3. [DOI] [PubMed] [Google Scholar]
- 12.Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp Neurol. 2006;200(2):480–485. doi: 10.1016/j.expneurol.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 13.Lagraize SC, Guo W, Yang K, Wei F, Ren K, Dubner R. Spinal cord mechanisms mediating behavioral hyperalgesia induced by neurokinin-1 tachykinin receptor activation in the rostral ventromedial medulla. Neuroscience. 2010;171(4):1341–1356. doi: 10.1016/j.neuroscience.2010.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin SY, Chang WJ, Lin CS, Huang CY, Wang HF, Sun WH. Serotonin receptor 5-HT2B mediates serotonin-induced mechanical hyperalgesia. J Neurosci. 2011;31(4):1410–1418. doi: 10.1523/JNEUROSCI.4682-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lorr M, McNair DM. Profile of Mood States-Bipolar form (POMS-BI) San Diego, CA: Educational and Industrial Testing Service; 1988. [Google Scholar]
- 16.Maixner W, Diatchenko L, Dubner R, Fillingim RB, Greenspan JD, Knott C, Ohrbach R, Weir B, Slade GD. Orofacial Pain Prospective Evaluation and Risk Assessment study--the OPPERA study. J Pain. 2011;12(11 Suppl):T4–11. e11–12. doi: 10.1016/j.jpain.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maixner W, Fillingim R, Booker D, Sigurdsson A. Sensitivity of patients with painful temporomandibular disorders to experimentally evoked pain. Pain. 1995;63(3):341–351. doi: 10.1016/0304-3959(95)00068-2. [DOI] [PubMed] [Google Scholar]
- 18.Maixner W, Fillingim R, Sigurdsson A, Kincaid S, Silva S. Sensitivity of patients with painful temporomandibular disorders to experimentally evoked pain: evidence for altered temporal summation of pain. Pain. 1998;76(1–2):71–81. doi: 10.1016/s0304-3959(98)00028-1. [DOI] [PubMed] [Google Scholar]
- 19.Melzack R. The short-form McGill Pain Questionnaire. Pain. 1987;30(2):191–197. doi: 10.1016/0304-3959(87)91074-8. [DOI] [PubMed] [Google Scholar]
- 20.Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. 2006;51(2):240–264. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. 2004;129(3):767–777. doi: 10.1016/j.neuroscience.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 22.Mulder EJ, Van Baal C, Gaist D, Kallela M, Kaprio J, Svensson DA, Nyholt DR, Martin NG, MacGregor AJ, Cherkas LF, Boomsma DI, Palotie A. Genetic and environmental influences on migraine: a twin study across six countries. Twin Res. 2003;6(5):422–431. doi: 10.1375/136905203770326420. [DOI] [PubMed] [Google Scholar]
- 23.O’Connor AB, Schwid SR, Herrmann DN, Markman JD, Dworkin RH. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain. 2008;137(1):96–111. doi: 10.1016/j.pain.2007.08.024. [DOI] [PubMed] [Google Scholar]
- 24.Peng G, Luo L, Siu H, Zhu Y, Hu P, Hong S, Zhao J, Zhou X, Reveille JD, Jin L, Amos CI, Xiong M. Gene and pathway-based second-wave analysis of genome-wide association studies. Eur J Hum Genet. 2010;18(1):111–117. doi: 10.1038/ejhg.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pennebaker JW. The psychology of physical symptoms. New York: Springer-Verlag; 1982. [Google Scholar]
- 26.Pfau DB, Rolke R, Nickel R, Treede RD, Daublaender M. Somatosensory profiles in subgroups of patients with myogenic temporomandibular disorders and Fibromyalgia Syndrome. Pain. 2009;147(1–3):72–83. doi: 10.1016/j.pain.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Price DD, Hu JW, Dubner R, Gracely RH. Peripheral suppression of first pain and central summation of second pain evoked by noxious heat pulses. Pain. 1977;3(1):57–68. doi: 10.1016/0304-3959(77)90035-5. [DOI] [PubMed] [Google Scholar]
- 28.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts MH. 5-Hydroxytryptamine and antinociception. Neuropharmacology. 1984;23(12B):1529–1536. doi: 10.1016/0028-3908(84)90097-2. [DOI] [PubMed] [Google Scholar]
- 30.Sanders AE, Maixner W, Nackley AG, Diatchenko L, By K, Miller VE, Slade GD. Excess risk of temporomandibular disorder associated with cigarette smoking in young adults. J Pain. 2012;13(1):21–31. doi: 10.1016/j.jpain.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkar SK, Chang C-K. The Simes Method for Multiple Hypothesis Testing With Positively Dependent Test Statistics. Journal of the American Statistical Association. 1997;92(440):1601–1608. [Google Scholar]
- 32.Schul R, Frenk H. The role of serotonin in analgesia elicited by morphine in the periaqueductal gray matter (PAG) Brain Res. 1991;553(2):353–357. doi: 10.1016/0006-8993(91)90849-q. [DOI] [PubMed] [Google Scholar]
- 33.Senn S. A Note Concerning a Selection “Paradox” of Dawid’s. The American Statistician. 2008;62(3):206–210. [Google Scholar]
- 34.Simes RJ. An improved Bonferroni procedure for multiple tests of significance. Biometrika. 1986;73(3):751–754. [Google Scholar]
- 35.Slade GD, Bair E, By K, Mulkey F, Baraian C, Rothwell R, Reynolds M, Miller V, Gonzalez Y, Gordon S, Ribeiro-Dasilva M, Lim PF, Greenspan JD, Dubner R, Fillingim RB, Diatchenko L, Maixner W, Dampier D, Knott C, Ohrbach R. Study methods, recruitment, sociodemographic findings, and demographic representativeness in the OPPERA study. J Pain. 2011;12(11 Suppl):T12–26. doi: 10.1016/j.jpain.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slade GD, Conrad MS, Diatchenko L, Rashid NU, Zhong S, Smith S, Rhodes J, Medvedev A, Makarov S, Maixner W, Nackley AG. Cytokine biomarkers and chronic pain: association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain. 2011;152(12):2802–2812. doi: 10.1016/j.pain.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slade GD, Diatchenko L, Bhalang K, Sigurdsson A, Fillingim RB, Belfer I, Max MB, Goldman D, Maixner W. Influence of psychological factors on risk of temporomandibular disorders. J Dent Res. 2007;86(11):1120–1125. doi: 10.1177/154405910708601119. [DOI] [PubMed] [Google Scholar]
- 38.Smith SB, Maixner DW, Greenspan JD, Dubner R, Fillingim RB, Ohrbach R, Knott C, Slade GD, Bair E, Gibson DG, Zaykin DV, Weir BS, Maixner W, Diatchenko L. Potential genetic risk factors for chronic TMD: genetic associations from the OPPERA case control study. J Pain. 2011;12(11 Suppl):T92–101. doi: 10.1016/j.jpain.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spielberger C, Gorusch RL, Lushene R, Vagg PR, Jacobs GA. Manual for the State-Trait Anxiety Inventory (Form Y1) Palo Alto, CA: Consulting Psychologists Press; 1983. [Google Scholar]
- 40.Steyerberg EW. Book Clinical prediction models a practical approach to development, validation, and updating. City: Springer; 2009. Clinical prediction models a practical approach to development, validation, and updating; p. xxviii.p. 497. [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sufka KJ, Schomburg FM, Giordano J. Receptor mediation of 5-HT-induced inflammation and nociception in rats. Pharmacol Biochem Behav. 1992;41(1):53–56. doi: 10.1016/0091-3057(92)90058-n. [DOI] [PubMed] [Google Scholar]
- 43.Taiwo YO, Levine JD. Serotonin is a directly-acting hyperalgesic agent in the rat. Neuroscience. 1992;48(2):485–490. doi: 10.1016/0306-4522(92)90508-y. [DOI] [PubMed] [Google Scholar]
- 44.Tokunaga A, Saika M, Senba E. 5-HT2A receptor subtype is involved in the thermal hyperalgesic mechanism of serotonin in the periphery. Pain. 1998;76(3):349–355. doi: 10.1016/S0304-3959(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 45.Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet. 2010;11(12):843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- 46.Wei F, Dubner R, Zou S, Ren K, Bai G, Wei D, Guo W. Molecular depletion of descending serotonin unmasks its novel facilitatory role in the development of persistent pain. J Neurosci. 2010;30(25):8624–8636. doi: 10.1523/JNEUROSCI.5389-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wright EF, Des Rosier KF, Clark MK, Bifano SL. Identifying undiagnosed rheumatic disorders among patients with TMD. J Am Dent Assoc. 1997;128(6):738–744. doi: 10.14219/jada.archive.1997.0297. [DOI] [PubMed] [Google Scholar]
- 48.Yu K, Li Q, Bergen AW, Pfeiffer RM, Rosenberg PS, Caporaso N, Kraft P, Chatterjee N. Pathway analysis by adaptive combination of P-values. Genet Epidemiol. 2009;33(8):700–709. doi: 10.1002/gepi.20422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zaykin DV, Zhivotovsky LA, Czika W, Shao S, Wolfinger RD. Combining p-values in large-scale genomics experiments. Pharm Stat. 2007;6(3):217–226. doi: 10.1002/pst.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaykin DV, Zhivotovsky LA, Westfall PH, Weir BS. Truncated product method for combining P-values. Genet Epidemiol. 2002;22(2):170–185. doi: 10.1002/gepi.0042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.