

Abstract
Chronic intermittent hypoxia (CIH) leads to remodeling of the carotid body function manifested by augmented sensory response to hypoxia and induction of sensory long-term facilitation (LTF). It was proposed that endothelin-1 (ET-1) contributes to CIH-induced hypoxic hypersensitivity of the carotid body. The objectives of the present study were: a) to delineate the mechanisms by which CIH up regulates ET-1 expression in the carotid body, and b) to assess whether ET-1 also contributes to sensory LTF. Experiments were performed on adult, male rats exposed to alternating cycles of 5% O2 (15s) and room air (5min), 9 episodes/hr and 8hr/day for 10 days. CIH increased ET-1 levels in glomus cells without significantly altering preproendothelin-1 mRNA levels. The activity of endothelin-converting enzyme (ECE) increased with concomitant elevation of ET-1 levels in CIH exposed carotid bodies, and MnTMPyP, a membrane permeable antioxidant prevented these effects. Hypoxia facilitated ET-1 release from CIH-treated carotid body, a requisite for activation of ET receptors; however, hypoxia had no effect on ET-1 release from control carotid bodies. In CIH exposed carotid bodies, mRNAs encoding ETA receptor were up regulated and an ETA receptor specific antagonist abolished CIH-induced hypersensitivity of the hypoxic response, whereas it had no effect on the sensory LTF. These results suggest that ECE-dependent increased production of ET-1 coupled with hypoxia-evoked ET-1 release and the ensuing ETA receptor activation mediate the CIH-induced carotid body hypersensitivity to hypoxia, but the ETA signaling pathway is not associated with sensory LTF elicited by CIH.
Key worlds: Recurrent apnea, endothelin-converting enzyme, endothelin-1 release, ETA receptors
1. INTRODUCTION
Chronic intermittent hypoxia (CIH) is a hallmark manifestation of sleep disordered breathing with apnea. Recurrent apnea patients exhibit persistent elevation of sympathetic nerve activity and are prone to develop hypertension (Nieto et al., 2000; Kara et al., 2003). Carotid bodies are the primary sensory organs for detecting arterial blood O2 levels, and the ensuing chemo reflex regulates breathing and sympathetic nerve activity during hypoxia. The augmented sympathetic nerve activity in recurrent apnea patients has been attributed to heightened carotid body chemo reflex (Narkiewicz et al., 1999; Kara et al., 2003). Studies on experimental animals have shown that CIH sensitizes the carotid body response to hypoxia (Peng & Prabhakar, 2004; Rey et al., 2004), and induces long-lasting increase in baseline sensory activity, which was termed as “sensory long-term facilitation” or sensory LTF (Peng et al., 2003; Peng et al., 2006). It was proposed that CIH-induced sensitization of the hypoxic response leads to hyperactive chemo reflex resulting in instability of breathing and greater number of apneas, whereas the sensory LTF contributes to reflex activation of the sympathetic nervous system (Prabhakar, 2013). Given the significance of chemo reflex, it is of importance to understand the mechanisms by which CIH affects the carotid body function.
Carotid bodies express a variety of neurotransmitters or modulators including biogenic amines, peptidergic modulators, and gasotransmitters, which play important roles in the chemo sensory response to hypoxia (Kumar & Prabhakar, 2012). Iturriaga and co-workers reported that CIH increases endothelin-1 (ET-1) expression in the carotid body and an ET-1 receptor antagonist prevents CIH-induced sensitization of the chemo sensory response to hypoxia (Rey et al., 2004; Rey et al., 2006; Rey et al., 2007; Rey et al., 2008). However, the mechanism(s) by which CIH up regulates ET-1 expression in the carotid body is not known. It was proposed that the transcriptional activator, hypoxia-inducible factor-1 (HIF-1) might contribute to CIH-induced ET-1 up regulation in the carotid body (Lam et al., 2006). We previously reported that CIH activates HIF-1 and CIH-induced sensitization of the chemo sensory response to hypoxia was absent in mice partially deficient in HIF-1α (Peng et al., 2006). Therefore, one of the objectives of the present study was to test whether transcriptional activation of the preproendothelin-1 gene contributes to ET-1 up regulation by CIH.
Release of ET-1 by hypoxia is pre-requisite for the activation of ET-1 receptors. Whether CIH facilitates ET-1 release by hypoxia from the carotid body, however, has not been established. Biological actions of ET-1 are mediated by ETA and ETB receptors (Pollock, 2005, Schneider et al., 2007). Although previous studies documented that bosentan, a pan ET-1 receptor antagonist reduces the heightened chemosensory response to hypoxia in CIH exposed cat carotid body (Rey et al., 2004; Rey et al., 2006), the relative contribution of ETA and ETB receptors is not known. CIH–induced carotid body sensory LTF requires NADPH oxidase (Nox), especially Nox 2 (Peng et al., 2009). ETA and ETB receptor subtypes are known to activate Nox isoforms (Dong et al., 2005; Capone et al., 2012). Whether ET-1 receptors also contribute to sensory LTF of the carotid body has not been investigated. Therefore, the other objectives of the present study were to determine the effects of CIH on hypoxia-evoked ET-1 release from the carotid body and assess the relative contributions of ETA and ETB receptors to CIH-induced sensitization of the hypoxic response and sensory LTF of the carotid body.
2. MATERIALS AND METHODS
Experiments were performed on male Sprague-Dawley rats weighing 250–320 g. The Institutional Animal Care and Use Committee of the University of Chicago approved the experimental protocols.
2.1. Exposure to chronic intermittent hypoxia (CIH)
Awake rats were exposed to a CIH paradigm consisting of 15 s of hypoxia followed by 5 min of room air, 9 episodes/hr, 8 hr/day as described previously (Peng & Prabhakar, 2004; Peng et al., 2009). Briefly, animals housed in feeding cages were placed in a chamber for exposure to CIH. The animals were unrestrained, freely mobile, and fed ad libitum. The chamber was flushed with alternating cycles of N2 combined with compressed room air so that inspired O2 levels reached 5% O2 during hypoxia within 68–75 s and 21% O2 during room air within 70–85 s. Ambient O2 levels in the chamber were continuously monitored with an O2 analyzer (Beckman model OM-11). A continuous vacuum was created within the chamber to balance the pressure between the inflow- and outflow of gases. Inspired CO2 levels were maintained at 0.2–0.5% and were monitored continuously by an infrared analyzer (Beckman model LB-2). The duration of the gas flow during each hypoxic and normoxic episode was regulated by timer controlled solenoid valves. Animals exposed to alternating cycles of room air instead of hypoxia for 10 days served as controls. Animals were subjected to either intermittent hypoxia or normoxia between 9:00 AM and 5:00 PM for 10 consecutive days. In the experiments involving antioxidant treatment, rats received manganese (III) tetrakis(1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP; Alexis), a membrane permeable antioxidant via an intraperitoneal route each morning (5 mg·kg−1·day−1) before they were subjected to daily regimen of intermittent hypoxia. MnTMPyP was given for 10 days.
2.2. Acute experiments involving anesthetized animals
Rats were anesthetized with Urethane (1.2g/kg, IP) after terminating the 10th day of CIH exposure. Carotid bodies were harvested for immunocytochemical, neurophysiological, mRNA and biochemical analyses.
2.3. Measurements of ET-1 immunofluorescence in the carotid body
Carotid bifurcations harvested from anesthetized rats were placed in 4% paraformaldehyde for 4h at room temperature, washed in PBS, and cryoprotected in 30% sucrose/PBS at 4°C for 24h. Tissue specimens were frozen in OCT compound (Tissue Tek, Fisher Scientific), serially sectioned at a thickness of 8 μm and stored at −80°C. For assessing ET-1 like immunoreactivity, carotid body sections were incubated at 37°C for 2h with polyclonal rabbit anti-ET-1 (1:200 dilution, Peninsula Laboratories, LLC, San Carlos, CA) and a monoclonal tyrosine hydroxylase (TH) antibody (1:4000 dilution, Sigma, St. Louis, MO), an established marker of glomus cells. After washing with PBS, sections were incubated for 1h with Alexa 555 (red)-conjugated goat anti-rabbit and Alexa 488 (green) conjugated goat anti-mouse (1:250, Molecular Probes, Oregon, USA) antibodies. Sections were visualized using a Nikon fluorescent microscope (Eclipse E600).
2.4. Measurements of ET-1 content and release
For monitoring ET-1 content, carotid bodies were harvested from anesthetized rats and homogenized in 10 volumes of a mixture of 1 M acetic acid/20 mM HCl. The homogenate was boiled at 100°C and centrifuged at 13, 000 g for 10 min at 4°C. The supernatant was removed and stored at −80°C until further analysis. The protocols for assessing ET-1 release from the carotid bodies were essentially the same as described previously (Pawar et al., 2009). Briefly, the carotid bodies harvested from anaesthetized rats were incubated in 100 μl of Ca2+/Mg2+ free Krebs Ringer bicarbonate (KRB) medium pre-equilibrated with 95% O2 – 5% CO2 (hyperoxia) for 30 min at 4°C. Subsequently, tissues were transferred to a reaction vial and incubated at 37°C with 60 μl of normal KRB medium equilibrated with appropriate gas mixtures (containing 5% CO2) resulting in medium PO2 of either 142 ± 4 mmHg (normoxia) or 38 ± 3 mmHg (hypoxia) for 30 min. We chose 30 min hypoxic exposure because the sensitivity of enzyme immunoassay was inadequate for detecting ET-1 release during 3 min of acute hypoxic exposure. The medium and the carotid bodies were removed and stored at −80°C until further analysis.
ET-1 levels were determined with a commercially available ET-1 enzyme immunoassay (EIA) kit (Assay Designs, Michigan, USA) following the manufacturer’s instructions. All measurements were performed in duplicate. The detection limit of EIA was 0.41 pg/ml. ET-1 content was expressed as picogram per milligram of protein and ET-1 release was expressed as picogram per milliliter per carotid body. Protein concentration in the samples was determined by Bio-Rad DC protein assay with bovine serum albumin as the standard.
2.5. Measurement of mRNA expression
The carotid bodies were harvested from anesthetized rats, homogenized, and RNA was exacted using TRIZOL (Invitrogen) according to the manufacturer’s instructions. Primer sequences for real-time RT-PCR amplification were as follows: preproendothelin-1 (133bp), forward CCGAGCCCAAAGTACCATGC and reverse GCTGATGGCC-TCCAACCTTC; ETA receptor (119bp), forward CTTCTGCATGCCCTTGGTGT and reverse CTCGACG-CTGCTTGAGGTGT; ETB receptor (117bp), forward AAGTCGTGTTTGTGCTGCTGGTG and reverse GCTGGAGCGGAAGTTGTCGT; and 18S rRNA (151bp), forward GTAAC-CCGTTGAACCCCATT and reverse CCATCCAATCGGTAGTAGCG. Real-time RT-PCR was carried out using a MiniOpticon system (Bio-Rad Laboratories, Hercules, California, USA) with SYBR Green as a fluorogenic binding dye (Invitrogen). The reactions were initiated by incubating at 50°C for 2 min (action of uracil DNA glycosylase) then at 95°C for 8 min and 30s (uracil DNA glycosylase inactivation and DNA polymerase activation), followed by 40 two-step cycles of 15s at 95°C and 1 min at 60°C. Relative mRNA quantification was calculated using the comparative threshold (CT) method using the formula ‘2−ΔCT’ where ΔCT is the difference between the threshold cycle of the given target cDNA between control and CIH. The CT value was taken as a fractional cycle number at which the emitted fluorescence of the sample passes a fixed threshold above the baseline. Values were compared with an internal standard gene 18S. Purity and specificity of all products were confirmed by omitting the template and by performing a standard melting curve analysis.
2.6. Measurement of endothelin-converting enzyme (ECE) activity
Carotid bodies pooled from 3 rats were homogenized in 100 μl of 50 mM HEPES buffer (pH 7.4) containing 100 mM NaCl and 20 mM CHAPS (3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate) and incubated at 4°C overnight. The extract was centrifuged at 18,000 g for 30 min at 4°C and the supernatant was immediately used for ECE activity assay. ECE activity was determined using the procedure as described previously (Warner et al., 1992) with few modifications. Briefly, tissue extract equivalent to 1–2 microgram protein was incubated with 100 pmol of prepro ET-1 (Big ET 1), 50 mM HEPES buffer (pH 7.4) containing 100 mM NaCl and 20 mM CHAPS at 37°C for 4h. At the end of incubation, the reaction was stopped by the addition of phosphoramidon (10 μM). In parallel experiments, same amount of tissue extract was pre-incubated with phosphoramidon (10 μM) for 15 min prior to the addition of ET-1 precursor. The amount of ET-1 formed in both reactions was determined using commercially available ET-1 assay kit (Assay designs, Ann Arbor, MI). ECE activity was expressed as picogram of phosphoramidon-inhibitable ET-1 formed per hour per milligram of protein.
2.7. Carotid body chemoreceptor sensory activity
Sensory activity from carotid bodies ex vivo was recorded as described previously (Peng & Prabhakar, 2004; Peng et al., 2009; Peng et al., 2011). Briefly, carotid bodies along with the sinus nerves were harvested from anesthetized rats. After cleaning the connective tissue, the carotid body along with the sinus nerve was placed in a recording chamber (volume 250 μl) and superfused at a rate of 2.5 ml/min with warm physiological saline (36°C) containing the following composition (mM): NaCl (125), KCl (5), CaCl2 (1.8), MgSO4 (2), NaH2PO4 (1.2), NaHCO3 (25), D-Glucose (10), Sucrose (5), bubbled with 95% O2/5% CO2. Hypoxia was accomplished by bubbling the perfusate with 5% O2 balanced with 5% CO2. Carotid bodies were challenged with hypoxia for 3min. PO2, PCO2 and pH values in the perfusates were determined by a blood gas analyzer (ABL-5, Radiometer, Copenhagen, Denmark). To facilitate recording of clearly identifiable action potentials, the sinus nerve was treated with 0.1% (Wt/Vol) collagenase for 5 min. Action potentials (2–4 active units) were recorded from one of the nerve bundles with a suction electrode, amplified (AC-preamplifier, Grass Instrument, P511K; bandwidth of 30–3000 Hz), displayed on an oscilloscope (Tektronix 5B12N), and stored in a computer via a data acquisition system (PowerLab/4P, AD Instruments Pty Ltd., Australia). “Single” units were selected based on the height and duration of the individual action potentials using a spike discrimination program (Spike Histogram Program, Power Lab, AD Instruments). Two to three chemoreceptor units were analyzed in each experiment.
2.8. Measurements of carotid body sensory LTF
Carotid body sensory LTF was determined as described previously (Peng et al. 2003). Briefly, baseline sensory discharge was recorded from ex vivo carotid bodies for 15 min. Subsequently, carotid bodies were challenged with 10 episodes of acute intermittent hypoxia (AIH), wherein each episode of AIH consisted 30s of 5% O2 (PO2 =38 mmHg) followed by 5 min of 95% O2+ 5% CO2. The sensory activity was continuously monitored during the 10 episodes of AIH and 60 min of the post-AIH period.
2.9. Experimental protocols
Series 1: ET-1-like immunoreactivity in the carotid bodies from control and CIH treated rats was determined (n=3 rats in each group). Series 2: mRNA levels of preproendothelin-1, ETA and ETB receptors, and 18S (housekeeping gene) were determined by real-time RT-PCR in carotid bodies from control and CIH treated rats (n= 3 individual experiments each; 6 carotid bodies/experiment). Series 3: Endothelin-converting enzyme (ECE) activity, and ET-1 contents of the carotid body were determined in control, CIH, CIH+MnTMPyP treated rats (n=3 individual experiments each; 6 carotid bodies/experiment). Series 4: Basal and hypoxia-evoked ET-1 release from carotid bodies were determined in control and CIH treated rats (n=4 individual experiments each; 6 carotid bodies/experiment). Series 5: The effects of BQ-610 (Phoenix Pharmaceuticals, Burlingame, CA) and BQ-788 (Alexis Biochemicals, San Diego, CA), ETA and ETB receptor antagonists, respectively, on the carotid body sensory response to hypoxia in control and CIH treated rats were determined (n=6 rats and 10–11 carotid bodies each). We chose 1μM BQ-610 or BQ-788 because our preliminary experiments showed that both these compounds at concentrations greater than 1μM suppressed the carotid body response to 30 mM KCl, suggesting non-specific effects. Chemoreceptor activity was quantified during baseline and hypoxia, and expressed as the average activity during 3 min of hypoxia minus the baseline (Δimp/s). Series 6: The effect of 1μM BQ-610 or vehicle on sensory LTF of carotid body was examined in CIH exposed rats (n=6 rats and 11–12 carotid bodies each of BQ-610 and vehicle). For the analysis of sensory LTF, chemoreceptor activity was averaged every 5 min under baseline conditions for 15 min, and during the 60 min of the post-AIH period. The data were expressed as percentage of baseline values.
2.10. Data analysis
All data were expressed as mean ± S.E.M. Statistical significance was assessed by one-way analysis of variance (ANOVA). P values less than 0.05 were considered significant.
3. RESULTS
3.1. Effects of CIH on ET-1 expression in the carotid body
The effect of CIH on ET-1 expression in the carotid body was examined by immunocytochemistry. Carotid body sections were stained with an anti-ET-1 antibody and anti-tyrosine hydroxylase (TH) antibody, a marker of glomus cells. In control rats, few cells in the carotid body expressed ET-1 like immunoreactivity (ET-1-IR) whereas TH-IR was seen in many cells (Fig. 1, left panels). After CIH exposure, a greater number of cells in the carotid body exhibited ET-1-IR, and many ET-1-IR expressing cells were also positive for TH-IR. However, ET-1-IR was absent in some of the TH-positive cells (Fig. 1; at arrows in the merged immunofluorescence of the enlarged region shown in the bottom right panel). ET-1 IR was also seen in vasculature (Fig. 1, middle panels, CIH, at arrow).
Figure 1.

Effect of chronic intermittent hypoxia (CIH) on endothelin-1 (ET-1) expression in the rat carotid body. Carotid body sections were stained with polyclonal ET-1 and monoclonal tyrosine hydroxylase (TH) antibodies. Representative examples of ET-1-like immunoreactivity (ET-1-IR) and TH-IR in the carotid bodies from control (Control; left panels) and CIH-exposed (CIH; middle and right panels) rats are shown (n=3 rats in each group). The scale bar in the control panel represents 50 μm. White arrow in the CIH panel represent ET-1-IR in the blood vessel. The right panels represent enlarged images corresponding to the regions enclosed by the boxes in the CIH panels. The scale bar in the magnified images represents 20 μm. The arrows in the merged panel indicate the absence of ET-1-IR in TH-IR positive cells from CIH-exposed carotid bodies.
3.2. Effect of CIH on preproendothelin-1 mRNA levels
To assess whether CIH activates preproendothelin-1 gene, preproET-1 mRNA levels were determined in carotid bodies from control and CIH exposed rats by real-time RT-PCR assay. There was no significant difference in preproET-1 mRNA expression between control and CIH exposed carotid bodies (P>0.05, Fig. 2A).
Figure 2.
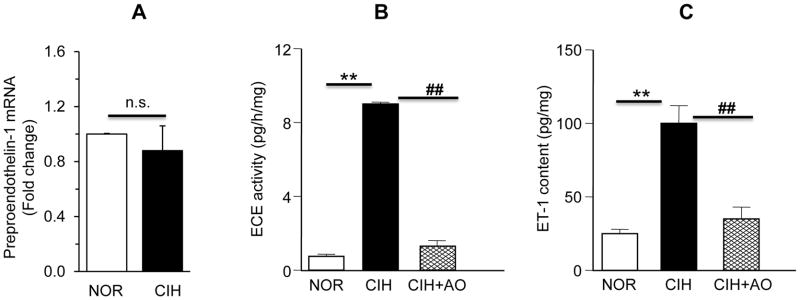
Effect of CIH on preproendothelin mRNA expression, endothelin-converting enzyme (ECE) activity and ET-1 levels in rat carotid bodies. A. Average data of preproendothelin-1 mRNA expression normalized to 18S in CIH exposed carotid bodies is expressed as the fold change from control (NOR) carotid bodies. Data presented are mean ± SEM from 3 individual experiments performed in triplicate with 6 carotid bodies in each experiment. n.s. denotes P >0.05 (NOR vs. CIH). B&C. Average data of ECE activity (B) and ET-1 content measured by enzyme immunoassay (C) in control (NOR), CIH, and CIH+antioxidant (AO; MnTMPyP; 5 mg·kg−1·day−1) treated rat carotid bodies are shown. Data are mean ± SEM from 3 individual experiments performed in triplicate with 6 carotid bodies in each experiment. ** and ## denote P<0.01 and n.s. = not significant, P>0.05.
3.3. Reactive oxygen species (ROS)-dependent activation of endothelin-converting enzyme (ECE) by CIH
ECE catalyzes the processing of biologically active ET-1 from its precursor protein, preproendothelin-1 (Xu et al., 1994). Whether increased processing of ET-1 by ECE contributes to increased ET-1 expression by CIH was determined. In control carotid bodies ECE activity averaged 0.75±0.05 picogram/milligram protein/hr. CIH increased ECE activity by ~12-fold as compared to controls (P<0.01, Fig. 2B). The increased ECE activity was associated with a ~4.5-fold increase in ET-1 levels in CIH exposed carotid bodies as compared to controls (ET-1 levels: control= 22±1.2 picogram/milligram of protein vs. CIH= 99.8±3.2 picogram/milligram of protein; P<0.01; Fig. 2C).
Previous studies reported that CIH increases ROS levels in the carotid body and anti-oxidant treatment prevents CIH -induced sensitization of the carotid body response to hypoxia (Peng & Prabhakar, 2004; Del Rio et al., 2010), and sensory LTF (Peng et al., 2003). We therefore examined whether antioxidant treatment prevents ECE activation and ET-1 up regulation in CIH exposed rats. Rats were given MnTMPyP, a membrane permeable antioxidant every day (5 mg·kg−1·day−1; I.P) prior to exposing them to daily regimen of CIH. MnTMPyP treatment prevented ECE activation and ET-1 up regulation evoked by CIH in carotid bodies (Fig. 2 B &C).
3.4. Effect of CIH on ET-1 release from the carotid body
ET-1 has to be released from the carotid body in order to affect the sensory nerve activity. We therefore examined the effect of CIH on ET-1 release from carotid bodies as described previously (Pawar et al., 2009). Basal ET-1 release under normoxia (medium PO2 = 142±4 mmHg) was comparable between control and CIH exposed carotid bodies (P>0.05, Fig. 3). Hypoxia (medium PO2 = 38±3 mmHg) facilitated ET-1 release by ~87% in CIH exposed carotid bodies, as compared to baseline value (P<0.01, Fig. 3). However, hypoxia had no effect on ET-1 release from control carotid bodies (P> 0.05, Fig. 3).
Figure 3.
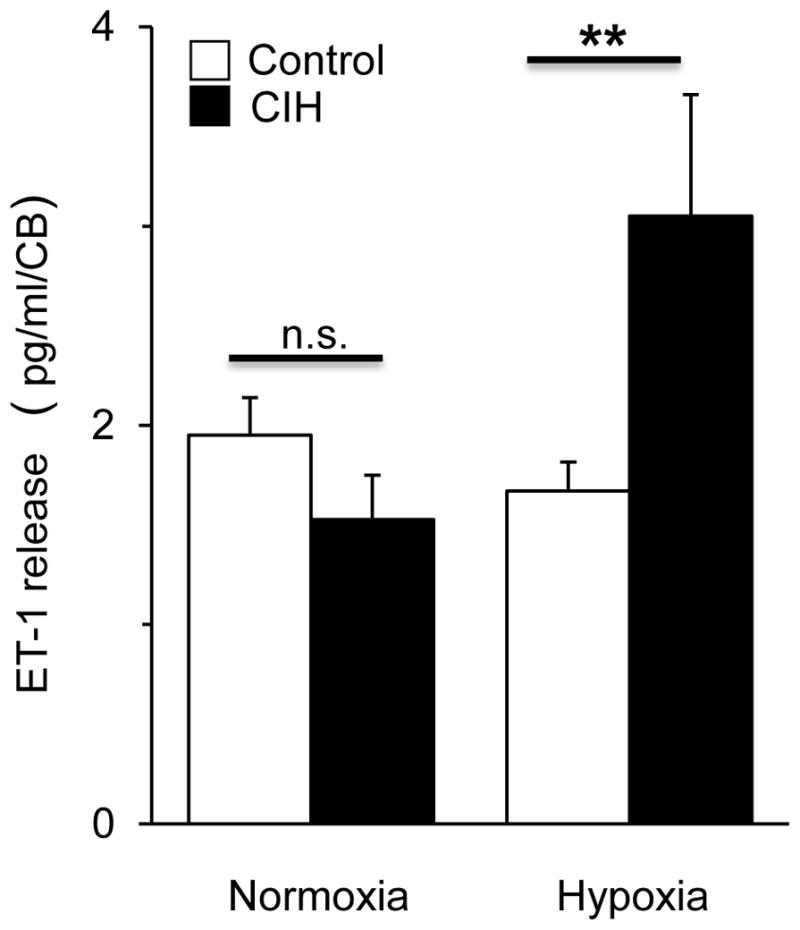
Effect of CIH on ET-1 release from rat carotid bodies. Average data of ET-1 release during normoxia (medium PO2 = 142±4mmHg) and hypoxia (PO2 = 38±3 mmHg) from carotid bodies harvested from controls (NOR) and CIH-treated (CIH) rats. Data are mean ± SEM from 4 individual experiments with 6 carotid bodies used in each experiment. ** denote P < 0.01 and n.s. = not significant, P>0.05.
3.6. Roles of ETA and ETB receptors in CIH-induced sensitization of the carotid body response to hypoxia
Carotid body sensory response to hypoxia (PO2 = 39 ± 3 mmHg) was determined in the presence of 1 μM BQ-610, an ETA receptor specific antagonist. In control carotid bodies, BQ-610 had no significant effect on the sensory response to hypoxia (P > 0.05; Fig. 4 A & C). In CIH exposed carotid bodies hypoxic sensory response was augmented as compared to controls and BQ-610 prevented this effect (P< 0.01; Fig. 4 B& C). CIH resulted in a modest but significant increase in ETA mRNA levels in carotid bodies (P< 0.05; Fig. 4D).
Figure 4.

Role of ETA receptors in CIH-induced carotid body response to hypoxia. A&B. Representative examples of sensory responses to hypoxia in ex vivo carotid bodies from control (NOR, A) and CIH-exposed (B) rats in the presence of vehicle (left panels) or 1μM BQ-610, an antagonist of ETA receptors (right panels). Black bar represents the duration of the hypoxic challenge (Hx, medium PO2=~39 mmHg). Superimposed action potentials of a single fiber from which the data were derived are shown in insets. C. Average data of the sensory responses to hypoxia (medium PO2 =39 ± 3 mmHg) in carotid bodies from control (NOR) and CIH-exposed (CIH) rats. Data presented are mean ± SEM from 10~11 carotid bodies from 6 rats in each group. ** denote P<0.01, n.s = not significant, P>0.05. D. Average data of mRNA expression of ETA receptors normalized to 18S in carotid bodies from control (NOR) and CIH exposed rats. Data presented are absolute values (mean ± SEM) from 3 individual experiments performed on 6 carotid bodies per experiment run in triplicate. * denote P < 0.05.
1 μM BQ-788, which is a selective ETB receptor antagonist, had no effect on the hypoxic response in either control or CIH exposed carotid bodies (P>0.05; Fig. 5A–C). Basal ETB mRNA levels were significantly higher as compared to ETA mRNA (ETA mRNA = 9.3±0.3 (10−6) vs ETB mRNA = 17.6±1.7 (10−6) 2– Δ CT, P< 0.01). However, ETB mRNA levels were significantly down regulated in CIH exposed carotid bodies (P<0.01; Fig. 5 D).
Figure 5.

Role of ETB receptors in CIH-induced carotid body response to hypoxia. A&B. Representative examples of sensory responses to hypoxia in ex vivo carotid bodies from control (NOR, A) and CIH-exposed (B) rats in the presence of vehicle (left panels) or 1μM BQ-788, an antagonist of ETB receptors (right panels). Black bar represents the duration of the hypoxic challenge (Hx, medium PO2=~39 mmHg). Superimposed action potentials of a single fiber from which the data were derived are shown in insets. C. Average data of the sensory responses to hypoxia (medium PO2 =39 ± 3 mmHg) in carotid bodies from control (NOR) and CIH-exposed (CIH) rats. Data presented are mean ± SEM from 10–11 carotid bodies from 6 rats in each group. n.s. = not significant., P>0.05. D. Average data of mRNA expression of ETB receptors normalized to 18S in carotid bodies from control (NOR) and CIH exposed rats. Data presented are absolute values (mean ± SEM) from 3 individual experiments performed in triplicate. ** denote P < 0.01.
3.7. Role of ETA receptors in CIH-induced sensory LTF of the carotid body
NADPH oxidases (Nox), especially the Nox2 is required for evoking sensory LTF (Peng et al., 2009) and ETA receptors activates Nox2 (Capone et al., 2012). Therefore, we hypothesized that ETA receptor antagonist will prevent sensory LTF of the carotid bodies. Representative examples of sensory LTF in the presence of either vehicle or BQ-610 are shown in Figure 6A. In the presence of vehicle, AIH lead to progressive increase in sensory activity, which remained elevated above the baseline level during the post-AIH period, indicating the induction of sensory LTF (Fig. 6A, upper panel). In the presence of BQ-610, the sensory excitation during each episode of hypoxia was reduced, whereas the sensory LTF was unaffected (Fig. 6A, bottom panel). Average data showed that in the presence of vehicle, sensory LTF (i.e., sensory activity during the entire 60 min. of the post-AIH period) was 252±39% above the baseline activity (Baseline = 2.1±0.3 vs. post-AIH 5.3±0.4 impulses/s; P< 0.01). In the presence of BQ-610, sensory LTF was 240±23% (Baseline activity= 2.0±0.2 vs. post-AIH 5±0.5 impulses/s; P<0.01), which was the same as in vehicle treated controls (P>0.05, Fig. 6B).
Figure 6.

Effect of ETA receptor antagonist on CIH-induced carotid body sensory LTF. A. Examples of acute intermittent hypoxia (AIH)-induced sensory LTF in ex vivo carotid bodies from CIH-exposed rats in the presence of vehicle (upper panel) or 1μM BQ-610, an ETA receptor antagonist (bottom panel). Arrows denote AIH challenge. Superimposed action potentials of a single fiber from which the data were derived are shown in insets. White horizontal line represents baseline activity. B. Average data of the carotid body sensory LTF (cumulative sensory activity during 60 min of the post-AIH period) in the presence of vehicle and BQ-610. Data are presented as percentage of baseline activity. Values are mean ± SEM from n = 11~12 carotid bodies from 6 rats in each group; n.s. = not significant, P>0.05 compared with vehicle treated controls.
DISCUSSION
The overall objectives of the present study were to delineate the mechanism(s) by which CIH up regulates ET-1 in the carotid body and assess the roles of ET-1 receptor subtypes in the remodeling of carotid body function by CIH. Our results demonstrate that: a) ROS-dependent activation of ECE contributes to up regulation of ET-1 by CIH, b) CIH facilitates hypoxia-evoked ET-1 release, c) ETA receptor activation mediate the CIH-induced carotid body hypersensitivity to hypoxia, and d) ETA signaling pathway is not associated with sensory LTF elicited by CIH.
Consistent with previous studies on cats (Rey et al., 2006; Rey et al., 2007; Rey et al., 2008) and rats (Lam et al., 2006; Del Rio et al., 2011), we found that CIH increases ET-1 expression in glomus cells of the adult rat carotid body, as evidenced by co-localization with TH-IR, a marker of this cell type and in blood vessels. A study by Rey et al (2008) based on quantification of immunofluorescence concluded that CIH increases ET-1 expression by 10 fold (Rey et al., 2008), whereas our results with quantification of the peptide levels by enzyme immunoassay revealed a ~4.5 fold elevation of ET-1 levels in the carotid body. Based on previous studies (Lam et al., 2006; Peng et al., 2006), we hypothesized that transcriptional activation of the preproendothelin 1 gene contributes to ET-1 up regulation in the carotid body CIH. However, the increased ET-1 expression was not associated with any significant alteration in preproendothelin-1 mRNA levels, suggesting that transcriptional activation per se does not account for ET-1 up regulation. The activity of ECE, which catalyzes the conversion of preproendothelin-1 protein to biologically active ET-1 peptide (Xu et al., 1994), however, was markedly elevated with concomitant increase in ET-1 levels in CIH exposed carotid bodies. Remarkably, MnTMPyP, a membrane permeable antioxidant prevented both the ECE activation and ET-1 up regulation by CIH. Given that CIH increases ROS levels in the carotid body (Peng et al., 2003), these findings demonstrate that ROS-dependent activation of ECE contributes to CIH-induced ET-1 up regulation in the rat carotid body. These observations are reminiscent of an earlier report showing that CIH-induced up regulation of neuropeptide Y (NPY) in the rat adrenal medulla is also mediated by ROS-dependent activation of processing of NPY from its precursor protein (Raghuraman et al., 2011). The mechanism(s) by which ROS activates ECE, however, requires further investigation, which is beyond the scope of the present study.
Previous studies have shown that a pan ET-1 receptor antagonist reduces carotid body hypersensitivity in CIH exposed adult cats (Rey et al., 2004; Rey et al., 2006; Rey et al., 2008). The results of the present study confirm these observations and further establish the relative contributions of ETA and ETB receptor subtypes to CIH-induced sensitization of the carotid body response to hypoxia. The following findings demonstrate that ETA but not ETB receptors mediate the hypersensitivity of the carotid body to hypoxia evoked by CIH. First, BQ-610, an ETA receptor antagonist, but not the BQ-788, an ETB receptor antagonist abolished the hypoxic hypersensitivity of the CIH exposed carotid body. Second, CIH up regulated ETA receptor mRNA but down regulated the ETB receptor mRNA. Although previous studies (Rey et al., 2008) showed up regulation of ET-1 by CIH, whether hypoxia releases ET-1 has not been investigated. We found that hypoxia releases ET-1 from CIH-treated carotid body, a requisite for activation of ET-1 receptors; whereas hypoxia had no effect on ET-1 release in the control carotid bodies. Thus, these findings suggest that ECE-dependent increased production of ET-1 coupled with hypoxia-evoked ET-1 release and the ensuing activation of ETA receptors contribute to the CIH-induced sensitization of the carotid body response to hypoxia. However, CIH lasting more than a week has been shown to reduce the intensity of ET-1-like immunoreactivity in the carotid body (Del Rio et al., 2011). Further studies are needed to assess whether decreased ECE activity contribute to reduced ET-1 expression with prolonged CIH.
Interestingly, the effects of CIH on the expression of ET-1 receptor subtypes in the carotid body seem to vary with species. It was reported that CIH up regulates ETB receptor protein in the cat carotid body (Rey et al., 2007). However, we found a decrease in ETB receptor mRNA expression in CIH exposed rat carotid body. ETB receptor protein was not determined in the rat carotid body because of the relatively small size of the tissue compared to cats, which is a limitation. It is possible that CIH differentially regulates ETB receptors in cat versus rat carotid bodies or alternatively, changes in mRNA are not translated to corresponding alterations in the protein. It has been proposed that ET-1 receptors contribute to CIH-induced heightened hypoxic sensitivity by affecting the carotid body vasculature (Rey et al., 2006). However, our studies were performed on an ex vivo superfused carotid body preparation, wherein vascular changes are absent. Yet, ETA receptor antagonist abolished CIH-induced hypoxic hypersensitivity, suggesting a direct action of ETA receptors on hypoxic sensing elements in the carotid body other than the vasculature.
ETA receptors activate Nox 2 in cerebral vasculature of CIH exposed mice (Capone et al., 2012). Given that activation of Nox2 in glomus cells is necessary for CIH-induced sensory LTF of the carotid body (Peng et al., 2009), we reasoned that ETA receptor antagonist will also prevent sensory LTF. Contrary to our expectation, ETA receptor antagonist virtually had no effect on CIH-induced sensory LTF. It is possible that ETA receptor signaling pathway is insufficient to activate Nox2 in glomus cells or alternatively, the mechanisms of Nox2 activation in cerebral vasculature differ from glomus cells. None-the-less, these results suggest that distinctly different mechanisms mediate CIH-evoked sensitization of the hypoxic response and sensory LTF of the carotid body.
The heightened hypoxic sensitivity of the carotid body and the ensuing hyperactive chemo reflex has been proposed to mediate breathing instability and greater number of apneas in recurrent apnea patients experiencing CIH (Prabhakar, 2013). Whether ETA receptor specific antagonists can be used to alleviate the respiratory co-morbidity associated with sleep disordered breathing remains an attractive possibility.
New Findings.
What is the central question of this study?
What mechanisms mediate chronic intermittent hypoxia-induced increases in endothelin-1 in the carotid body?
What is the main finding and its importance?
Endothelin-1 up regulation by chronic intermittent hypoxia results from reactive oxygen species-dependent activation of endothelin converting enzyme and is not the result of augmented endothelin-1 gene transcriptional activity. The resultant increased endothelin-1 acts via endothelin-1A receptors to induce hypoxic hypersensitivity of the carotid body but does not contribute to the sensory long term facilitation observed in this condition. These findings provide mechanistic insight into how chronic intermittent hypoxia alters carotid body function.
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute Grants HL-76537 and HL-90554.
References
- Capone C, Faraco G, Coleman C, Young CN, Pickel VM, Anrather J, Davisson RL, Iadecola C. Endothelin 1-dependent neurovascular dysfunction in chronic intermittent hypoxia. Hypertension. 2012;60:106–113. doi: 10.1161/HYPERTENSIONAHA.112.193672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Iturriaga R. Carotid body and cardiorespiratory alterations in intermittent hypoxia: the oxidative link. Eur Respir J. 2010;36:143–150. doi: 10.1183/09031936.00158109. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Iturriaga R. Differential expression of pro-inflammatory cytokines, endothelin-1 and nitric oxide synthases in the rat carotid body exposed to intermittent hypoxia. Brain Res. 2011;1395:74–85. doi: 10.1016/j.brainres.2011.04.028. [DOI] [PubMed] [Google Scholar]
- Dong F, Zhang X, Wold LE, Ren Q, Zhang Z, Ren J. Endothelin-1 enhancesoxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: role of ETB receptor, NADPH oxidase and caveolin-1. Br J Pharmacol. 2005;145:323–333. doi: 10.1038/sj.bjp.0706193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara T, Narkiewicz K, Somers VK. Chemoreflexes--physiology and clinical implications. Acta Physiol Scand. 2003;177:377–384. doi: 10.1046/j.1365-201X.2003.01083.x. [DOI] [PubMed] [Google Scholar]
- Kumar P, Prabhakar NR. Peripheral chemoreceptors: function and plasticity of the carotid body. Comprehensive Physiology. 2012;2:141–219. doi: 10.1002/cphy.c100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SY, Tipoe GL, Liong EC, Fung ML. Hypoxia-inducible factor (HIF)-1alpha and endothelin-1 expression in the rat carotid body during intermittent hypoxia. Adv Exp Med Biol. 2006;580:21–27. doi: 10.1007/0-387-31311-7_4. discussion 351–359. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D’Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA. 2000;283:1829–1836. doi: 10.1001/jama.283.14.1829. [DOI] [PubMed] [Google Scholar]
- Pawar A, Nanduri J, Yuan G, Khan SA, Wang N, Kumar GK, Prabhakar NR. Reactive oxygen species-dependent endothelin signaling is required for augmented hypoxic sensory response of the neonatal carotid body by intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R735–742. doi: 10.1152/ajpregu.90490.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Prabhakar N. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. Journal of Applied Physiology. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Peng Y-J, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1 alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. Journal of Physiology-London. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci. 2009;29:4903–4910. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Raghuraman G, Khan SA, Kumar GK, Prabhakar NR. Angiotensin-II evokes sensory long-term facilitation of the carotid body via NADPH oxidase. J Appl Physiol. 2011;111:964–970. doi: 10.1152/japplphysiol.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock DM. Endothelin, angiotensin, and oxidative stress in hypertension. Hypertension. 2005;45:477–480. doi: 10.1161/01.HYP.0000158262.11935.d0. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Sensing hypoxia: physiology, genetics and epigenetics. J Physiol. 2013;591:2245–2257. doi: 10.1113/jphysiol.2012.247759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman G, Kalari A, Dhingra R, Prabhakar NR, Kumar GK. Enhanced neuropeptide Y synthesis during intermittent hypoxia in the rat adrenal medulla: role of reactive oxygen species-dependent alterations in precursor peptide processing. Antioxid Redox Signal. 2011;14:1179–1190. doi: 10.1089/ars.2010.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Corthorn J, Chacon C, Iturriaga R. Expression and immunolocalization of endothelin peptides and its receptors, ETA and ETB, in the carotid body exposed to chronic intermittent hypoxia. J Histochem Cytochem. 2007;55:167–174. doi: 10.1369/jhc.6A7079.2006. [DOI] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Iturriaga R. Contribution of endothelin-1 to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Brain Res. 2006;1086:152–159. doi: 10.1016/j.brainres.2006.02.082. [DOI] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Iturriaga R. Contribution of endothelin-1 and endothelin A and B receptors to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Adv Exp Med Biol. 2008;605:228–232. doi: 10.1007/978-0-387-73693-8_40. [DOI] [PubMed] [Google Scholar]
- Schneider MP, Boesen EI, Pollock DM. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu Rev Pharmacol Toxicol. 2007;47:731–759. doi: 10.1146/annurev.pharmtox.47.120505.105134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TD, Mitchell JA, D’Orleans-Juste P, Ishii K, Forstermann U, Murad F. Characterization of endothelin-converting enzyme from endothelial cells and rat brain: detection of the formation of biologically active endothelin-1 by rapid bioassay. Mol Pharmacol. 1992;41:399–403. [PubMed] [Google Scholar]
- Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, Yanagisawa M. ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78:473–485. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]