

Summary
BRAF is the most prevalent oncogene and an important therapeutic target in melanoma. In some cancers BRAF is activated by rearrangements that fuse its kinase domain to 5’ partner genes. We examined 848 comparative genomic hybridization profiles of melanocytic tumors and found copy number transitions within BRAF in 10 tumors, of which six could be further characterized by sequencing. In all, the BRAF kinase domain was fused in-frame to six different N-terminal partners. No other mutations were identified in melanoma oncogenes. One of seven melanoma cell lines without known oncogenic mutations harbored a similar BRAF fusion, which constitutively activated the MAP-kinase pathway. Sorafenib, but not vemurafenib, could block MAP-kinase pathway activation and proliferation of the cell line at clinically relevant concentrations, whereas BRAFV600E mutant melanoma cell lines were significantly more sensitive to vemurafenib. The patient from whom the cell line was derived showed a durable clinical response to sorafenib.
Keywords: Melanoma, oncogenes, BRAF, kinase, translocation
Introduction
The majority of melanocytic neoplasms harbor activating point mutations in BRAF (Davies et al. 2002; Pollock et al. 2003), NRAS (Padua et al. 1984), HRAS (Bastian et al. 2000), KIT (Curtin et al. 2006), GNAQ (Van Raamsdonk et al. 2009), or GNA11 (Van Raamsdonk et al. 2010). These mutations are found in a mutually exclusive pattern in early stage disease, including benign melanocytic nevi. They activate key oncogenic pathways such as the MAP-kinase pathway, which, in the case of BRAF and KIT mutations, can be directly targeted therapeutically. The equivalent mutations remain to be discovered for a considerable proportion of melanomas.
Rearrangements resulting in chimeric proteins in which the BRAF kinase is fused to a variety of 5’ partners have been reported in multiple cancers including pilocytic astrocytoma (Ciampi et al. 2005; Cin et al. 2011; Jones et al. 2008; Palanisamy et al. 2010). The only fully characterized BRAF rearrangement in melanocytic tumors described to date was found in a congenital nevus, in which the BRAF kinase domain was fused to FCHSD1. Another congenital nevus reported in the same study was positive using a fluorescent in situ hybridization (FISH) assay with a BRAF break-apart probe set suggesting a potential BRAF rearrangement (Dessars et al. 2007). Also using break-apart FISH, 1 of 131 melanoma cases tested positive in another study (Palanisamy et al. 2010). In both FISH-positive cases the nature of the BRAF rearrangement and the fusion partner could not be further characterized due to lack of biological material. The identification of BRAF fusions in melanoma would have clinical implications, as several RAF inhibitors are clinically available.
Using comparative genomic hybridization (CGH), we identified recurrent focal amplifications involving the kinase domain of the BRAF gene, which in some cases resulted in copy number transitions within the BRAF locus. We show here that these copy number transitions reflect a rearranged BRAF gene, in which the kinase domain is fused in-frame to a wide range of 5’ prime partners. We previously reported a patient with metastatic melanoma without known genetic alterations who was empirically treated with sorafenib and showed a sustained response over 9 months before dying of pulmonary edema (Passeron et al. 2011). We discovered here that the patient-derived melanoma cell line expressed a constitutively active AGK-BRAF fusion which can be inhibited by the type II BRAF inhibitor sorafenib, while it is relatively resistant to the type I BRAF inhibitor vemurafenib, compared to melanoma cell lines with BRAFV600E mutations.
Results
Recurrent BRAF rearrangements in spitzoid tumors
We analyzed the copy number profiles of 848 melanocytic neoplasms for copy number transitions within the BRAF locus for which array CGH was performed as part of the diagnostic workup. These 848 cases spanned the spectrum of ambiguous melanocytic tumors and included cases with a possible diagnosis of atypical blue nevus, deep penetrating nevus, ancient nevus, Spitz nevus or dysplastic nevus. A majority of the cases demonstrated spitzoid cytomorphology, and most of these cases occurred in children or young adults. We identified ten cases with relative gain of the 3’ portion of the gene compared to the 5’ portion (Cases 1 to 10 in Supplementary Figure 1). Sufficient material was left over to perform targeted next generation sequencing for six cases. The targeted regions included introns 7, 8, 9,and 10 of BRAF and the exons of BRAF, NRAS, HRAS, KIT and GNAQ. In all six, we identified fusions of the 3’ end of BRAF supported by greater than 10 reads over the genomic breakpoint to six different N-terminal fusion partners (Figure 1 and Supplementary Table 1). The N-terminal fusion partners included the transcription factor SOX6, the mitotic spindle assembly checkpoint protein MAD1L1, the stabilizer of dynein intermediate chain NUDCD3, the zinc finger protein ZKSCAN5, the potassium channel tetramerisation domain-containing protein KCTD7, and the mannose-6-phosphate receptor interacting protein PLIN3 (Figure 2). In all 6 cases, the array CGH data revealed copy number increase of the chromosomal region harboring the N-terminal partner (Supplementary Figure 1). The melanoma oncogenes BRAF, NRAS, HRAS, KIT or GNAQ did not show any mutations in any of the six cases. The array CGH profile of one of the 4 cases with copy number transitions in BRAF locus that had insufficient material for sequencing (Case 10 in Table 1) also demonstrated a copy number transition within the SOX6 locus with co-amplification of the 5’ portion of SOX6 and the 3’ end of BRAF similar to case 1, suggesting the presence of a SOX6-BRAF fusion.
Figure 1. Representative case with MAD1L1-BRAF fusion.
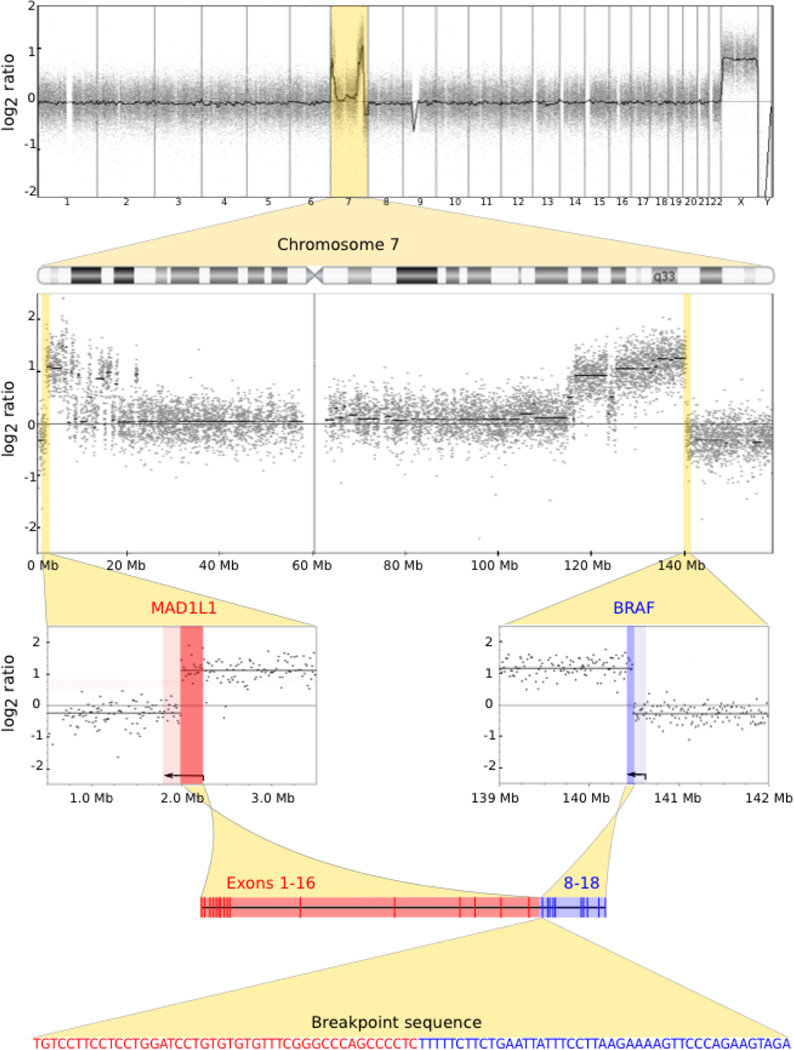
Array CGH demonstrates copy number changes limited to chromosome 7 (top row and second row). There is a copy number transition within both MAD1L1 and BRAF (third row). The y-axes represent the average log2 ratio of tumor to normal fluorescence intensity and the genomic coordinates are on the x-axis. The copy number data supports a fusion between the 5’ end of MAD1L1 and the 3’ end of BRAF as they have the same copy number level. The predicted gene model fuses intron 16 of MAD1L1 and intron 7 of BRAF (fourth row) as supported by the breakpoint sequence.
Figure 2. Predicted BRAF fusion proteins.
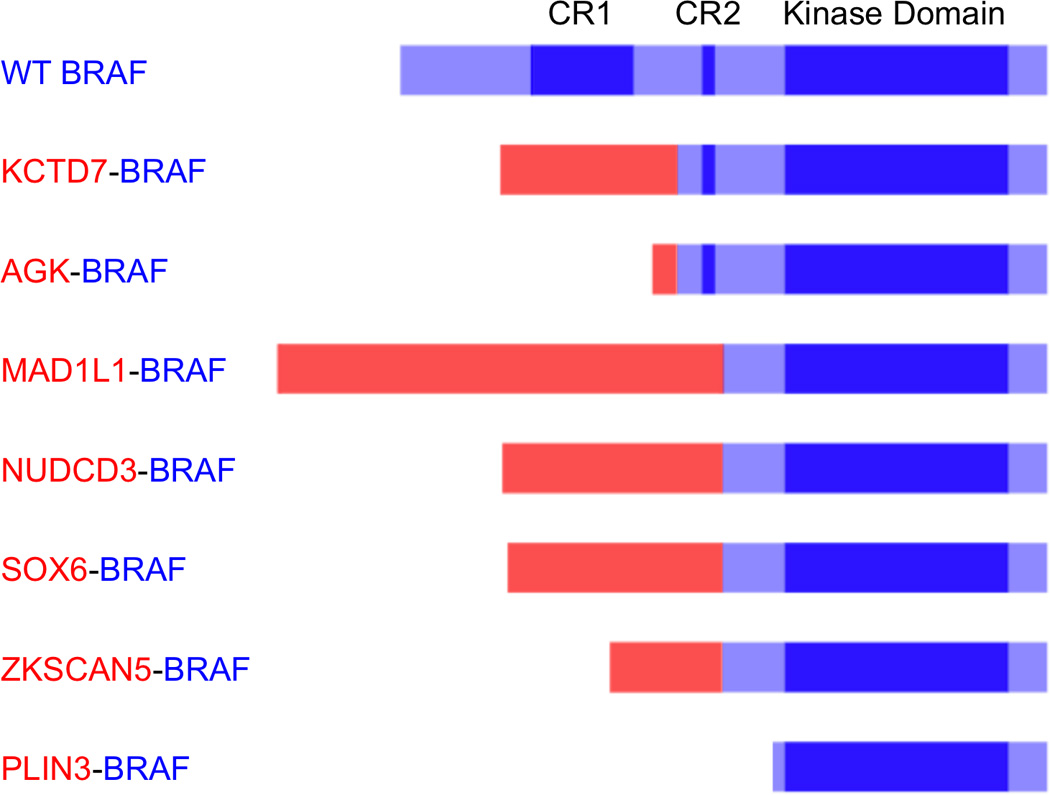
Conserved region 1 (CR1), conserved region 2 (CR2) and the kinase domain of BRAF are highlighted in dark blue. For each fusion protein, the contribution from 5’ partner gene is depicted in red and the contribution from the BRAF gene is depicted in blue. In all cases, the kinase domain of BRAF is intact and the auto-inhibitory domain CR1 is lost. The PLIN3-BRAF fusion leads to a truncated version of BRAF.
Table 1. Clinical characteristics of cases with copy number transitions in BRAF.
The final diagnosis in each case was based on evaluation of the histopathologic features in combination with the array CGH findings.
| # | Age at diagnosis (years) |
Sex | Location | Diagnosis | Fusion genes |
|---|---|---|---|---|---|
| 1 | 4 | F | Buttock | Atypical Spitz tumor | SOX6-BRAF |
| 2 | 39 | F | Back | Spitzoid melanoma | MAD1L1-BRAF |
| 3 | 22 | F | Back | Atypical Spitz tumor | NUDCD3-BRAF |
| 4 | 24 | F | Scalp | Worrisome for melanoma | PLIN3-BRAF |
| 5 | 20 | F | Thigh | Spitzoid proliferation, consistent with desmoplastic Spitz nevus | KCTD7-BRAF |
| 6 | 45 | F | Buttock | Intradermal melanocytic proliferation | ZKSCAN5-BRAF |
| 7 | 19 | M | Chest | Spitzoid melanoma | Not sequenced |
| 8 | 53 | F | Calf | Melanoma | Not sequenced |
| 9 | 23 | F | Thigh | Atypical Spitz tumor | Not sequenced |
| 10 | 13 | F | Calf | Spitzoid melanoma | not sequenced, SOX6 by array CGH |
| 11 | 17 | F | Scalp | Spitzoid melanoma | AGK-BRAF |
To identify a melanoma cell line for possible in vitro studies of the BRAF fusions we sequenced seven cutaneous melanoma cell lines without mutations in BRAF, NRAS, or KIT. Sequencing revealed a BRAF fusion in the C0902 cell line (case 11 in Table 1) that was caused by an approximately 0.8 megabase pericentric inversion of chromosome 7, which fused the first two exons of the acylglycerol kinase AGK with exons 8–18 of BRAF (Figure 2). By array CGH, the cell line demonstrated a gain of the entire chromosome 7 without any copy number transitions within the BRAF locus (case 11 in Supplementary Figure 1). Fluorescence in situ hybridization with dual colored BRAF break-apart probes did not reveal any significant split of the two signals, presumably due to the small size of the inversion (Supplementary Figure 2).
In the six melanocytic tumors and the C0902 cell line, the rearrangement of BRAF resulted in the removal of the conserved region 1 (CR1), which keeps the kinase in an inactive state when not bound to activated RAS proteins (Mercer & Pritchard 2003). The breakpoints were in intron 7 (two cases) or 8 (five cases), predicted to result in in-frame fusions harboring an intact kinase domain, similar to BRAF fusions observed in other cancers (Supplementary Figure 3) (Ciampi et al. 2005; Cin et al. 2011; Jones et al. 2008; Palanisamy et al. 2010). The 5’ partners contributed varying amounts to the fusion protein, with the notable exception of the PLIN3-BRAF fusion, in which PLIN3 only contributed the 5’ untranslated region, resulting in a truncated BRAF protein that was predicted to use a cryptic translation start site at BRAF codon 438, without any protein contribution from PLIN3.
Interestingly, all seven melanocytic tumors, including the melanoma from which the C0902 cell line was derived, demonstrated a spitzoid cellular morphology, with a predominance of large spindled or epithelioid melanocytes. While all tumors with BRAF fusions occurred in young patients, the average age was not significantly different than that of our overall cohort of ambiguous melanocytic tumors, which was highly enriched for spitzoid tumors. The predominance of females (10 of 11 tumors with demonstrated fusions or predicted fusions based on CGH) was not statistically significant (p=0.0633, Fisher exact test, two-tailed) (Supplementary Table 2).
The AGK-BRAF fusion protein constitutively activates the MAP kinase pathway
The C0902 cell line demonstrated constitutive activation of MAP-kinase pathway with elevated levels of p-ERK and p-MEK similar to two BRAFV600E mutant melanoma cell lines (A375 and SK-MEL-28) (Figure 3a). All three cell lines showed 2 to 3 bands, between 70 and 100 kDa corresponding to known BRAF isoforms (Barnier et al. 1995). By contrast, an additional protein of 53 kDa corresponding to the predicted molecular weight of the AGK-BRAF fusion protein was detected with two different antibodies exclusively in C0902, confirming that the fusion protein was expressed (Figure 3a and Supplementary Figure 4). Exogenous expression of the AGK-BRAF fusion in 293FT cells resulted in a band of similar size and activation of the MAP-kinase signaling pathway (Figure 3b).
Figure 3. Activation of the MAP kinase pathway by AGK-BRAF and increased sorafenib sensitivity of AGK-BRAF fusion containing cell line compared to BRAFV600E mutant melanoma cell lines.
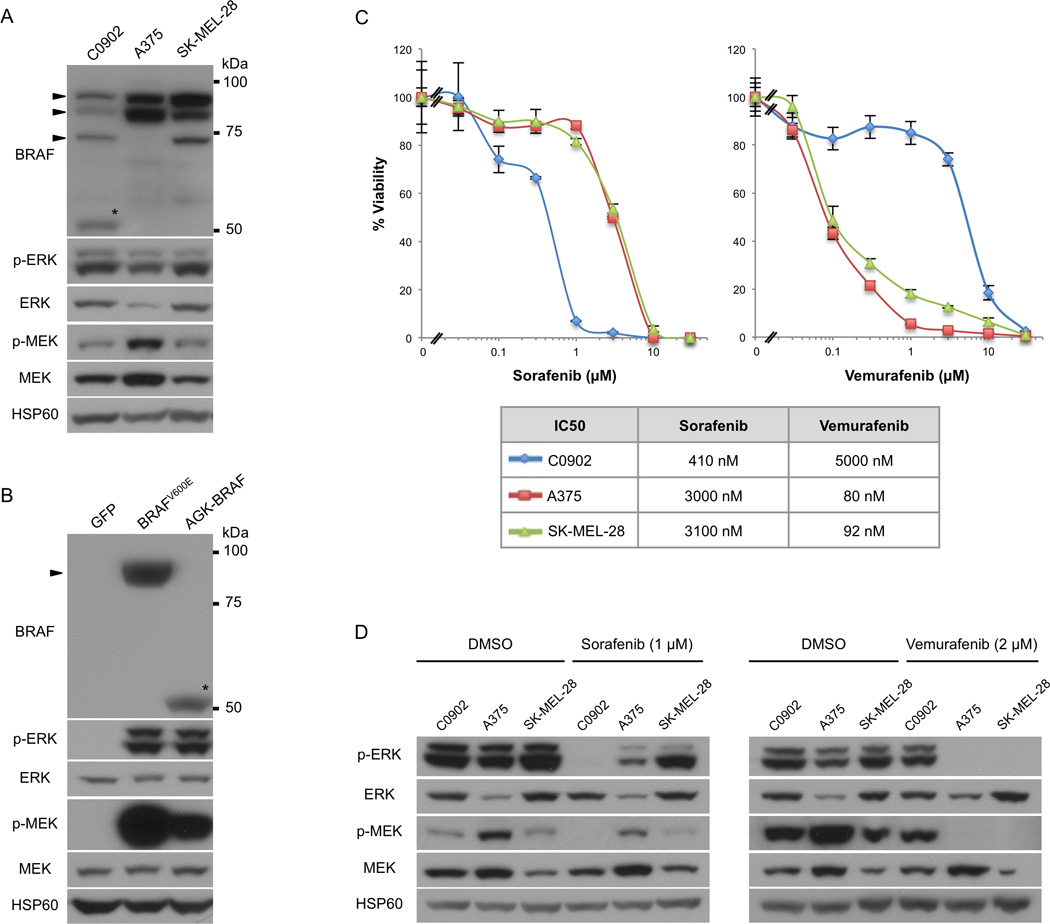
a) Comparable activation of the MAP kinase pathway in the C0902 cell line, which harbors an AGK-BRAF kinase fusion to two BRAFV600E mutant melanoma cell lines. The AGK-BRAF fusion is detected exclusively in the C0902 cell line at its predicted molecular weight of 53 kDa (asterisk). The bands corresponding to the various known isoforms of BRAF are indicated by arrowheads.
b) Transient transfection of the AGK-BRAF fusion in 293FT cells activates the MAP kinase pathway.
c) Dose response curves for sorafenib and vemurafenib for the C0902 and two BRAFV600E mutant melanoma cell lines (5 days of treatment).
d) Western Blot analysis of MAP-kinase pathway status. Cell lysates were prepared after serum starvation and HSP60 was used as the loading control.
The AGK-BRAF fusion protein can be inhibited by sorafenib but is resistant to vemurafenib
The AGK-BRAF expressing C0902 cell line showed increased sensitivity to the type II RAF inhibitor sorafenib (IC50 = 410 nM) compared to the BRAFV600E mutant melanoma cell lines A375 (IC50 = 3.0 µM) and SK-MEL-28 (IC50 = 3.1 µM). In contrast, the C0902 cell line showed relative resistance to the type I inhibitor vemurafenib (IC50 = 5.0 µM) compared to the BRAFV600E mutant cell lines (IC50 = 80–92 nM) (Figure 3c).
Treatment with 1 µM sorafenib resulted in decreased p-ERK and p-MEK in the C0902 cell line, whereas this dose did not significantly suppress p-ERK or p-MEK levels in A375 and SK-MEL-28. By contrast, vemurafenib treatment (2 µM) completely extinguished p-ERK and p-MEK levels in A375 and SK-MEL-28, while it only marginally suppressed p-ERK and p-MEK levels in the C0902 cell line (Figure 3d).
We did not observe any paradoxical activation of the MAP kinase pathway in the C0902 cell line in response to vemurafenib (Supplementary Figure 5).
Discussion
We report recurrent BRAF kinase fusions in borderline melanocytic tumors and melanomas. We identified cases with fusions based on the presence of copy number transitions within the BRAF locus by array CGH, followed by confirmatory genomic sequencing. All of the cases with copy number transitions within the BRAF locus were found to show in-frame fusions by sequencing, demonstrating that the presence of a breakpoint within BRAF with relative gain of the 3’ end of the gene is relatively specific for an in-frame BRAF fusion. The sensitivity of this approach likely underestimates the true frequency of BRAF fusions in melanocytic neoplasms. It would not detect balanced translocations that were not followed by gain of the BRAF fusion kinase or loss of the reciprocal fusion. Amplification of the BRAF fusion does not occur in pilocytic astrocytomas (Jones et al. 2008) and is thus not required for oncogenic transformation. In addition, increased copy number of the BRAF kinase fusion may not result in copy number transition in BRAF if there is a balanced gain of the reciprocal fusion. For example, in case 11 of our series, the AGK-BRAF fusion arose from a paracentric 0.8 Mb inversion. While there was increased copy number of chromosome 7 containing AGK-BRAF fusion, there was no copy number transition within the BRAF locus by array CGH.
We observed a range of different 5’ partners, none of which has been previously described to participate in oncogenic fusion kinases. As the expression of the fusion kinase is controlled by the promoter of the 5’ partner we reviewed existing gene expression data sets for the 5’ partners involved in the BRAF fusions. All seven N-terminal partners were found to be expressed in human melanocytes (Smith et al. 2005). Differences in the transcription levels of the 5’ partners may determine whether increasing the gene dosage of the fusion kinases by copy number increases is necessary to reach expression levels of the fusion kinases that are oncogenic. It is likely that BRAF fusions are also present in some of the melanocytic neoplasms of series that did not have copy number transitions through the BRAF locus. In these, the expression levels of the 5’ partner may be high enough without additional copy number increase of the fusion gene.
The diversity of 5’ partners involved in BRAF rearrangements occurring in melanocytic tumors might complicate their detection by impairing the design of targeted FISH test as suitable for pilocytic astrocytoma (Tian et al. 2011). Similarly, conventional BRAF break-apart FISH test might not be able to resolve all rearrangements as suggested by the test run on the AGK-BRAF case.
The common feature of the rearrangements found was the loss of conserved region 1 (CR1) of BRAF, which exerts an auto-inhibitory effect on kinase activity, when not bound to GTP-bound RAS (Mercer & Pritchard 2003). The loss of CR1 results in RAS-independent constitutive activation of the kinase, as has been demonstrated for KIAA1549-BRAF, a rearrangement found in about two-thirds of pilocytic astrocytomas (Jones et al. 2008). The KIAA1549-BRAF rearrangements in astrocytoma are not typically accompanied by similar copy number changes that revealed the BRAF fusions in our study.
The patient-derived AGK-BRAF expressing melanoma cell line demonstrated an increased sensitivity to sorafenib compared to melanoma cell lines with BRAFV600E mutation, but was comparatively resistant to vemurafenib. The resistance to vemurafenib is unlikely to be due to paradoxical activation as was observed when KIAA1549-BRAF transduced cells were treated with PLX4720 (Sievert et al. 2013), since there was no notable activation of the MAP kinase signaling pathway in our studies using identical dose ranges of vemurafenib. While we cannot formally rule out that the sensitivity to the different inhibitors is due to differences in their off-target effects, the effect is more likely due to their different binding properties to BRAF. The AGK-BRAF fusion contains the first 33 amino acids of AGK and the C-terminal portion of BRAF starting from position 328 of BRAF with loss of the CR1 domain. The kinase domain of AGK-BRAF does not contain any mutation, in contrast to BRAFV600E. Sorafenib binds to RAF family members in their inactive conformation, whereas vemurafenib binds to the active conformation, favored by the BRAFV600E mutation. Therefore, sorafenib may be more effective to prevent activation of the wild-type conformation by contrast with vemurafenib which blocks the pre-activated kinase conformation. Sorafenib was also active against RAF fusion proteins reported in prostate cancer (Palanisamy et al. 2010).
Importantly, the patient from whose melanoma the C0902 cell line was derived showed a significant durable response to sorafenib, demonstrating that the in vitro results were clinically relevant (Passeron et al. 2011).
It is of interest that all BRAF fusions found in our study were detected in lesions with spitzoid morphology. While we cannot rule out that this is due to bias, as our cohort of borderline lesions with ambiguous histomorphology is enriched for spitzoid melanocytic neoplasms, the finding could indicate an increased frequency of BRAF fusions in this type of tumor. The frequency of BRAF fusions across different clinical and histopathologic subtypes of melanoma remains to be determined.
In summary, our findings indicate BRAF kinase fusions as a recurrent genetic alteration in melanocytic neoplasms without mutations in known melanoma oncogenes, and that these fusions can be targeted by certain types of RAF inhibitors such as sorafenib.
Patients and methods
Study Population
We analyzed a database of 848 melanocytic neoplasms, for which array CGH was performed as part of the diagnostic assessment at the Dermatopathology Section of the Departments of Dermatology and Pathology at the University of California, San Francisco over a twenty-six month period during 2010 through 2012. The majority of these cases are borderline lesions with histopathological features that overlap those of benign nevus and melanoma, for which CGH was performed as an ancillary diagnostic test. The study was approved by the Committee on Human Research and was conducted according to the Declaration of Helsinki. We also analyzed DNA from 7 melanoma cell lines without mutations in known melanoma oncogenes (WM1963, WM3912, WM8, WM3622, WM3918, NB2107, all kindly provided by Dr. Meenhard Herlyn, Wistar Institute, Philadelphia, PA, and C0902).
Comparative Genomic Hybridization
DNA was extracted from formalin-fixed, paraffin-embedded (FFPE) tumor as described previously (Bastian et al. 1998). Array CGH was carried out with 500–1000 ng of genomic DNA on Agilent 4x180k microarrays (Agilent, Santa Clara, CA). The raw microarray images were processed with Agilent Feature Extraction software, and analyzed using Nexus Copy Number Software version 6.0 (Biodiscovery, El Segundo, CA). Cases demonstrating copy number transitions with copy number increase of the 3’ portion of BRAF and sufficient residual DNA or tissue were selected for sequencing.
Massively Parallel Sequencing of captured DNA libraries
Multiplex library preparation was performed using the Ovation Ultralow Library System according to the manufacturer’s specifications (NuGEN, San Carlos, CA). Hybridization-capture of pooled libraries was performed using a custom-designed bait library (Nimblegen SeqCap EZ Choice) spanning 1.4 Mb of the genome including the exons of BRAF, NRAS, HRAS, KIT, GNAQ and introns 7, 8, 9, and 10 of BRAF. Captured libraries were sequenced as paired-end 100 reads on a HiSeq-2000 instrument (Illumina). Sequence reads were mapped to the reference human genome (hg19) using the Burrows-Wheeler aligner (BWA) (Li & Durbin 2009). Recalibration of reads and variant calling were performed using the Genome Analysis Toolkit (GATK) (McKenna et al. 2010). Variant annotation was performed with Annovar (Wang et al. 2010). For fusion detection, read pairs with at least one read unaligned by BWA were aligned using BWA-SW (Li & Durbin 2010) and read pairs with insert sizes greater than 1000 bp or with soft clipping of at least one read were used as input to CREST (Wang et al. 2011). Structural variants were confirmed in the sequencing data by visual inspection in the Integrative Genomics Viewer (Thorvaldsdóttir et al. 2013). We predicted the resulting fusion transcripts by joining the exon directly upstream from the genomic breakpoint with the exon directly downstream. Predicted protein sequences were then determined from the predicted transcripts.
Plasmid construction and transient transfection
The cDNA of the AGK-BRAF fusion flanked by the bacteriophage λ-derived recombination sequences attL1 and attL2 sites was directly synthesized (GENEWIZ, Inc.) and cloned by LR Clonase II (Life Technologies) into the pLenti6.3/TO/V5-Dest backbone (Life Technologies). For transient transfection experiments, 7 µg of pLenti6.3 AGK-BRAF, pLenti6.2 BRAFV600E or the control vector pLenti6.2 GFP were transfected into 293FT cells using Lipofectamine 2000 (Invitrogen). Cell were lysed and assayed for their protein content 48 hours after transfection.
Western Blotting
Cell lysates were prepared in RIPA buffer supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific). Equal amounts of protein, as measured by BCA protein assay, were resolved in 4–12% Bis-Tris NuPage gradient gels (Life Technologies). Antibodies: anti-phospho-Erk (#9101), anti-phospho-BRAF (Ser445) (#2696), anti-phospho-MEK (Ser217/221) (#9121) from Cell Signaling Technologies, anti-HSP60 (sc-1722), anti-MEK (sc-436), anti-BRAF (sc-166) and anti-ERK2 (sc-1647) from Santa Cruz Biotechnology.
Cell proliferation
BRAFV600E mutant melanoma cells A375 (CRL-1619) and SK-MEL-28 (HTB-72) were purchased from ATCC and propagated as recommended (Manassas, VA). Cells were plated in 6-well plates and 24 hours later were treated as indicated with a range of concentrations of sorafenib (LC Laboratories, Woburn, MA) or vemurafenib (PLX4032, Selleckchem, Houston, TX). Five days post-treatment, cells were detached, stained with trypan blue and counted using a TC10 Automated Cell Counter (Bio-Rad).
Supplementary Material
Significance.
BRAF fusions are recurrent oncogenic drivers in melanocytic tumors and represent novel therapeutic targets.
Acknowledgements
This work was supported by National Institutes of Health Grants R01 CA131524 and P01 CA025874.
Abbreviations
- CGH
comparative genomic hybridization
- FISH
fluorescent in situ hybridization
Footnotes
Conflict of Interest
Authors’ disclosure of potential conflicts of interest
Elizabeth A. Burton and Gideon Bollag are employees of Plexxikon, and hold equity in the company.
Author contributions
TB, IY and BCB designed research. TB, IY, TAN, SSV, MCG, EAB performed research. MA, SR, PB, THM, PEL, GB, RB provided critical reagents and advice. TB, IY, BCB wrote the manuscript.
References
- Barnier JV, et al. The Mouse B-raf Gene Encodes Multiple Protein Isoforms with Tissue-specific Expression. Journal of Biological Chemistry. 1995;270(40):23381–23389. doi: 10.1074/jbc.270.40.23381. [DOI] [PubMed] [Google Scholar]
- Bastian BC, et al. Chromosomal Gains and Losses in Primary Cutaneous Melanomas Detected by Comparative Genomic Hybridization. Cancer Research. 1998;58(10):2170–2175. [PubMed] [Google Scholar]
- Bastian BC, LeBoit PE, Pinkel D. Mutations and Copy Number Increase of HRAS in Spitz Nevi with Distinctive Histopathological Features. The American Journal of Pathology. 2000;157(3):967–972. doi: 10.1016/S0002-9440(10)64609-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampi R, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. Journal of Clinical Investigation. 2005;115(1):94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cin H, et al. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathologica. 2011;121(6):763–774. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- Curtin JA, et al. Somatic Activation of KIT in Distinct Subtypes of Melanoma. Journal of Clinical Oncology. 2006;24(26):4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dessars B, et al. Chromosomal Translocations as a Mechanism of BRAF Activation in Two Cases of Large Congenital Melanocytic Nevi. Journal of Investigative Dermatology. 2007;127(6):1468–1470. doi: 10.1038/sj.jid.5700725. [DOI] [PubMed] [Google Scholar]
- Jones DTW, et al. Tandem Duplication Producing a Novel Oncogenic BRAF Fusion Gene Defines the Majority of Pilocytic Astrocytomas. Cancer Research. 2008;68(21):8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2003;1653(1):25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- Padua RA, Barrass N, Currie GA. A novel transforming gene in a human malignant melanoma cell line. Nature. 1984;311(5987):671–673. doi: 10.1038/311671a0. [DOI] [PubMed] [Google Scholar]
- Palanisamy N, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nature Medicine. 2010;16(7):793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passeron T, et al. Signalling and chemosensitivity assays in melanoma: is mutated status a prerequisite for targeted therapy? Experimental Dermatology. 2011;20(12):1030–1032. doi: 10.1111/j.1600-0625.2011.01385.x. [DOI] [PubMed] [Google Scholar]
- Pollock PM, et al. High frequency of BRAF mutations in nevi. Nature Genetics. 2003;33(1):19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk CD, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, et al. Mutations in GNA11 in Uveal Melanoma. New England Journal of Medicine. 2010;363(23):2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievert AJ, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(15):5957–5962. doi: 10.1073/pnas.1219232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AP, Hoek K, Becker D. Whole-genome expression profiling of the melanoma progression pathway reveals marked molecular differences between nevi/melanoma in situ and advanced-stage melanomas. Cancer biology & therapy. 2005;4(9):1018–1029. doi: 10.4161/cbt.4.9.2165. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, et al. Detection of KIAA1549-BRAF Fusion Transcripts in Formalin-Fixed Paraffin-Embedded Pediatric Low-Grade Gliomas. The Journal of Molecular Diagnostics : JMD. 2011;13(6):669–677. doi: 10.1016/j.jmoldx.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature Methods. 2011;8(8):652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38(16):e164–e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.