

Graphical abstract

Keywords: Myeloperoxidase, Neutrophils, Oxidative stress, Suicide inhibitor, Mechanism-based inhibition, Oxidative modification
Highlights
-
•
Low H2O2 concentrations: fast but limited inactivation of MPO via Compound I.
-
•
High H2O2 concentrations: complete inactivation of MPO via Compound III.
-
•
H2O2 (⩾1000 fold molar excess) acts as a suicide substrate.
-
•
Inactivation: destruction of heme, oxidation and disruption of protein.
Abstract
Human myeloperoxidase (MPO) uses hydrogen peroxide generated by the oxidative burst of neutrophils to produce an array of antimicrobial oxidants. During this process MPO is irreversibly inactivated. This study focused on the unknown role of hydrogen peroxide in this process. When treated with low concentrations of H2O2 in the absence of reducing substrates, there was a rapid loss of up to 35% of its peroxidase activity. Inactivation is proposed to occur via oxidation reactions of Compound I with the prosthetic group or amino acid residues. At higher concentrations hydrogen peroxide acts as a suicide substrate with a rate constant of inactivation of 3.9 × 10−3 s−1. Treatment of MPO with high H2O2 concentrations resulted in complete inactivation, Compound III formation, destruction of the heme groups, release of their iron, and detachment of the small polypeptide chain of MPO. Ten of the protein’s methionine residues were oxidized and the thermal stability of the protein decreased. Inactivation by high concentrations of H2O2 is proposed to occur via the generation of reactive oxidants when H2O2 reacts with Compound III. These mechanisms of inactivation may occur inside neutrophil phagosomes when reducing substrates for MPO become limiting and could be exploited when designing pharmacological inhibitors.
Introduction
Neutrophils are the predominant white blood cells in circulation. They are highly specialized for their primary function, the phagocytosis and destruction of invading pathogens by antimicrobial proteins and reactive oxidants [1]. When stimulated, neutrophils consume oxygen in a respiratory burst that produces superoxide and hydrogen peroxide [1]. Simultaneously, these white blood cells discharge the abundant heme enzyme myeloperoxidase (MPO)1 that uses hydrogen peroxide to oxidize chloride, bromide and thiocyanate to the respective hypohalous acids and hypothiocyanite. These oxidants kill ingested bacteria but are also implicated in tissue damage associated with numerous inflammatory diseases [2]. It has been demonstrated that during phagocytosis the amount of extractable neutrophilic MPO decreases while a significant fraction of the soluble enzyme is inactivated [3,4]. The mechanism by which this occurs and its relevance to infection and inflammation have yet to be investigated in detail.
Enzyme inactivation is likely to involve reactions with either hydrogen peroxide or its products because they have the potential to oxidatively modify amino acids and the prosthetic heme group. It has been demonstrated with several heme peroxidases, including horseradish peroxidase [5,6], ascorbate peroxidase [7] and lactoperoxidase [8], that hydrogen peroxide alone can promote irreversible inactivation. There are also reports that MPO is inactivated by hydrogen peroxide in the absence of exogenous electron donors [9,10]. However, the mechanism of inactivation and the effect on its structural integrity have not been investigated.
In this study we aimed to show how hydrogen peroxide affects the activity of MPO and describe the mechanisms involved in enzyme inactivation. We demonstrate that MPO is highly sensitive to inactivation by hydrogen peroxide. Irreversible inactivation of MPO was accompanied by changes in its secondary and tertiary architecture including breakage of covalent heme linkages and disruption of its subunit structure.
Materials and methods
Reagents
Human MPO (lyophilized and highly purified, Reinheitszahl 0.84) was obtained from Planta Natural Products (http://www.planta.at) and the concentration was determined spectrophotometrically with a molar extinction coefficient of 91,000 M−1 cm−1 per heme [11]. Hydrogen peroxide (30% analytical grade) was purchased from Biolab (Aust) Ltd. and concentrations were determined spectrophotometrically using a molar extinction coefficient of 43.6 M−1 cm−1 at 240 nm [12]. TMB (3,3′,5,5′-tetramethylbenzidine), xylenol orange, D-sorbitol, diethylenetriaminepentaacetic acid (DTPA), alkyltrimethylammonium bromide (cetrimide), glucose, catalase from bovine liver, bovine superoxide dismutase, glucose oxidase from Aspergillus niger, 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p′-disulfonic acid monosodium salt hydrate (ferrozine) and l-ascorbate were purchased from Sigma. Dimethylformamide and ferrous ammonium sulfate were from J.T. Baker.
All spectrophotometric assays were performed on an Agilent 8453 diode array spectrophotometer. For enhanced chemiluminescence Amersham™ ECL Plus Western Blotting Detection System from GE Healthcare was used.
Myeloperoxidase activity assays
The residual peroxidase activity of MPO was determined by measuring its ability to oxidize TMB. MPO (32 nM) in 50 mM sodium phosphate buffer, pH 7.4, and 100 μM DTPA was incubated at various protein to hydrogen peroxide ratios and aliquots were taken over time to assess the decline in peroxidase activity. Superoxide dismutase was added to a separate number of experiments at a concentration of 20 μg/mL to investigate the requirement for superoxide in enzyme inactivation caused by hydrogen peroxide. Residual peroxidase activity was measured by adding a 25 μL aliquot to 850 μL of 200 mM sodium acetate buffer, pH 5.4, containing 0.01% cetrimide, 100 μL of 20 mM TMB in DMF (made fresh each day and kept in the dark) and 25 μL of 8 mM hydrogen peroxide. Reactions were performed at 28 ± 0.5 °C and started by addition of the MPO aliquot. TMB oxidation was followed at 670 nm and initial rates were calculated over the first 60 s of the reaction.
The halogenation activity of MPO was determined under the same conditions as described above with the exception that 10 mM bromide was present in the TMB assay. Under these conditions, bromide was the preferred substrate for MPO and was converted to HOBr, which was responsible for 80% of the oxidation of TMB with the remainder due to direct oxidation by MPO (i.e., peroxidase activity).
Determination and generation of hydrogen peroxide
Consumption of hydrogen peroxide by MPO was measured using ferrous iron-catalyzed oxidation of xylenol orange (FOX assay) [13]. The FOX reagent was composed of 1 mM ammonium ferrous sulfate, 400 μM xylenol orange and 400 mM D-sorbitol in 200 mM H2SO4. Each peroxide assay was performed by adding 70 μL of sample to 25 μL FOX reagent while vortexing. The solution was then incubated at room temperature for 45 min in the dark prior to reading the absorbance at 560 nm. The hydrogen peroxide concentration was calculated against a standard curve of the range of 0–5 nM hydrogen peroxide. Samples with concentrations higher than the standard curve range were diluted accordingly in 50 mM phosphate buffer, pH 7.4, before adding to the FOX reagent. This assay was also used to determine the rate at which glucose oxidase and glucose (1 mg/mL) produced hydrogen peroxide in 50 mM phosphate buffer, pH 7.4. The flux of hydrogen peroxide was linear over 60 min and increasing concentrations of glucose oxidase (0.1–2.5 μg/mL glucose oxidase) gave a linear increase in production of hydrogen peroxide (9.4 μM–285.5 μM per h).
Spectral analysis of myeloperoxidase
Spectra of 1.5 μM MPO in 50 mM phosphate buffer, pH 7.4, containing 100 μM DTPA were recorded after reactions were started by adding hydrogen peroxide. To determine how peroxidase or halogenation substrates influenced the degradation of the heme groups of MPO, 200 μM ascorbate or 5 mM bromide with 5 mM methionine, respectively were also added as indicated below. The involvement of superoxide was checked by adding 20 μg/mL SOD to a separate set of reactions.
SDS–PAGE analyses
To follow the impact of hydrogen peroxide on the structural integrity of myeloperoxidase, 1.5 μM MPO in 50 mM sodium phosphate buffer, pH 7.4, and 100 μM DTPA was incubated with 1.5 mM hydrogen peroxide. Aliquots were taken at 5, 10, 25, 40 and 60 min and residual hydrogen peroxide was removed by adding 20 μg/mL catalase. Samples of 15 μL were added to non-reducing and reducing sample loading buffers, respectively (final concentrations: 2% SDS, 10% glycerol, 125 mM Tris–HCl buffer, pH 6.5, and for reducing SDS–PAGE 1% β-mercaptoethanol). Samples were loaded without prior heating and resolved by 8–20% gradient SDS–polyacrylamide gel electrophoresis. Gels were stained with Coomassie Brilliant Blue R-250 for 60 min and subsequently destained. Gels were scanned using ChemiDoc® XRS (Bio-Rad).
Furthermore, 1.5 μM MPO in 50 mM sodium phosphate buffer, pH 7.4, and 100 μM DTPA was incubated with the following hydrogen peroxide: protein ratios: 1:1, 5:1, 10:1, 40:1, 100:1, 167:1, 500:1, 1000:1 and 2000:1 for 2 h at room temperature. Samples were resolved on SDS–PAGE, stained and analyzed as described above.
For detection of intact heme covalently linked to the protein, MPO was incubated with hydrogen peroxide and resolved by SDS–PAGE under the same conditions as described above and subsequently blotted onto a PVDF membrane (100 V, 60 min). Enhanced chemiluminescence (Amersham™ ECL Plus Western Blotting Detection System, GE Healthcare) was used to detect covalently bound and intact heme [14].
Analysis of free iron
Release of free iron from the heme prosthetic group of MPO was measured colorimetrically using ferrozine following a published method but with slight modifications [15]. After buffer exchange with 50 mM sodium acetate buffer, pH 7.4, using a Micro Bio-Spin chromatography column (Bio-Rad) 1.5 μM MPO was treated with 1.5 mM hydrogen peroxide and incubated at room temperature for 2 h. The volume of 1 mL was reduced to dryness and the pellet resuspended in 30 μL water. Ascorbic acid (30 μL of 1.13 mM in 0.2 M HCl) was added and left for 5 min. The protein was then precipitated by adding 30 μL of 11.3% trichloroacetic acid and samples were kept on ice for 5 min followed by a short fast spin at 4 °C. Finally, 36 μL of 10% ammonium acetate was added to the supernatant followed by 9 μL of 6.1 mM ferrozine and the absorbance at 563 nm was measured after 5 min (ε = 28,000 M−1 cm−1).
Differential scanning calorimetry
Differential calorimetric (DSC) measurements were performed using a VP-capillary DSC microcalorimeter from Microcal (cell volume: 137 μL), controlled by the VP-viewer program and equipped with an autosampler for 96 well plates. Samples were analyzed using a programmed heating scan rate of 60 °C h−1 over a temperature range of 20–110 °C and a cell pressure of approximately 60 psi (4.136 bar). The maximum temperature inside the cuvette was 95 °C. Collected DSC data were corrected for buffer baseline and normalized for protein concentration. The reaction conditions were: 5 μM MPO in 5 mM phosphate buffer, pH 7, in the absence or presence of 500 μM or 5 mM hydrogen peroxide for 60 min). Guanidinium hydrochloride (GdnHCl; 5 mM) was added before measurements were started to avoid non-specific protein aggregation at increasing temperatures.
Microcal origin software was used for data analysis. Heat capacity (Cp) was expressed in kcal mol−1 K−1 (1 cal = 4.184 J). Data points were fitted to non-two-state equilibrium-unfolding models by the Lavenberg/Marquardt (LM) non-linear least square method.
Electronic circular dichroism spectrometry
Thermal unfolding was also followed by electronic circular dichroism (ECD) spectroscopy (Chirascan, Applied Photophysics, Leatherhead, UK). The instrument was flushed with nitrogen at a flow rate of 5 L min−1 and was capable of simultaneous UV–vis and ECD monitoring. The instrument was equipped with a Peltier element for temperature control and temperature-mediated denaturation was monitored between 20 and 95 °C with stepwise increments of 1.0 °C min−1.
Single wavelength scans were performed with instrumental parameters set as follows. Visible ECD at a Soret minimum of 412 nm was performed with 5 μM MPO in 5 mM phosphate buffer, pH 7.0, containing 0.5 M GdnHCl to avoid aggregation at higher temperatures. Samples were monitored in the absence and presence of 100 and 1000-fold excesses of hydrogen peroxide over 60 min. The pathlength was 10 mm, spectral bandwidth 1 nm and the scan time per point was set at 10 s. Far-UV ECD at 222 nm was performed on the same samples with the pathlength set at 1 mm, spectral bandwidth at 3 nm and the scan time per point was set at 10 s.
The fraction α of unfolded protein was calculated according to α = (θN − θ)/(θN − θU) with θN being the ellipticity (in mdeg) at 222 nm of the protein in the native folded state, θ the ellipticity at defined temperature (T), and θU being the ellipticity at 222 nm of the completely unfolded state.
Mass spectrometric analyses
To determine which amino acid residues become oxidized during enzyme inactivation, 1.5 μM MPO was incubated with 1.5 mM hydrogen peroxide in 50 mM sodium phosphate buffer, pH 7.4. The treated sample and a control sample were reduced and alkylated prior to trypsin digestion following standard procedures. Changes in the tryptic peptides due to oxidation were then determined by LC-MS/MS at the center for Protein Research, University of Otago, Dunedin. Each sample was analyzed four times using the same instrument settings.
For LC-MS/MS analysis of tryptic peptides samples were reconstituted in 5% (v/v) acetonitrile, 0.2% (v/v) formic acid in water and injected onto an Ultimate 3000 nano-flow HPLC-System (Thermo Scientific, Dionex Co, CA) that was in-line coupled to the nanospray source of a LTQ Orbitrap XL hybrid mass spectrometer (Thermo Scientific, San Jose, CA). Peptides were loaded onto a PepMap100 trap column (Thermo Scientific, Dionex Co, CA) and separated on a PepMapRSLC analytical column (75 μm × 150 mm; Thermo Scientific, Dionex Co, CA) by a gradient developed from 5% (v/v) acetonitrile, 0.2% (v/v) formic acid to 80% (v/v) acetonitrile, 0.2% (v/v) formic acid in water at a flow rate of 400 nL/min.
Full MS in the mass range between m/z 400 and 2000 was performed in the Orbitrap mass analyzer with a resolution of 60,000 at m/z 400 and an AGC target of 5e5. Preview mode for FTMS master scan was enabled to generate precursor mass lists. The strongest five signals were selected for collision induced dissociation (CID)-MS/MS in the LTQ ion trap at a normalized collision energy of 35% using an AGC target of 2e4 and one microscan. Dynamic exclusion was enabled with one repeat counts during 60 s and an exclusion period of 180 s. Exclusion mass width was set to 0.01.
For protein identification MS/MS data were searched against a user defined amino acid sequence database containing the target sequence using the SEQUEST program operated through the Proteome Discoverer software (Thermo Scientific). The search was set up for full tryptic peptides with a maximum of two missed cleavage sites. Carboxyamidomethyl cysteine, oxidation (Cys, Met), dioxidation (Cys, Met), trioxidation (Cys) were selected as variable modifications. The precursor mass tolerance threshold was 10 ppm and the maximum fragment mass error 0.8 Da. Ion chromatograms were compared pair-wise using the SIEVE software (Thermo Scientific). The SIEVE Frame List was filtered for monoisotopic peaks and positive identifications (based on the imported SEQUEST search results). For the analysis of differential modifications the SIEVE Frame List and the Proteome Discoverer Peptide List were both integrated into a relational database (Microsoft Access) and linked by the unique combination of scan number and raw file name.
Results
Kinetics of inactivation of MPO by hydrogen peroxide
Myeloperoxidase was incubated with a given concentration of hydrogen peroxide in the absence of reducing substrate. Then, aliquots were taken and the residual enzyme activity was determined. As there were no differences between the residual halogenation and peroxidase activity, only the latter activity was measured in further experiments. The enzyme was incubated with increasing stoichiometric excesses of hydrogen peroxide and the loss of peroxidase activity was determined over time. These experiments were carried out in the absence of reducing substrates. At low ratios of hydrogen peroxide to MPO (⩽5:1), there was rapid but limited inactivation (Fig. 1A). For example, at ratio of 5:1, approximately 30% of the enzyme’s activity was lost within 10 min. No further loss in activity occurred after this time. Interestingly, the rate of inactivation decreased as the ratio of hydrogen peroxide to MPO was increased to 20:1 and then 1000:1 (Fig. 1A and B). Thereafter the rate of inactivation increased with increasing molar ratios of hydrogen peroxide to MPO (Fig. 1B).
Fig. 1.

Loss of peroxidatic activity of myeloperoxidase upon incubation with hydrogen peroxide. (A) Loss of peroxidase activity at low MPO to hydrogen peroxide ratios. 32 nM MPO reacted with hydrogen peroxide at oxidant to protein ratios of (●) 1:1, (○) 5:1 and (▾) 20:1 in 50 mM phosphate buffer, pH 7.4, containing 100 μM DTPA. The residual peroxidase activity of MPO was determined by taking aliquots over time and measuring the TMB peroxidase activity (n = 2). (B) 32 nM MPO was incubated with hydrogen peroxide at H2O2 to protein ratios of (●) 1000:1 (32 μM H2O2), (○) 5000:1 (160 μM H2O2), (▾) 10,000:1 (320 μM H2O2), in 50 mM phosphate buffer, pH 7.4, with 100 μM DTPA. Aliquots were taken and the residual TMB activitiy of MPO over time was determined (n = 3). (C) Loss of peroxidase activity at a constant hydrogen peroxide to protein ratio. MPO was incubated with a 5000-fold stoichiometric excess of hydrogen peroxide at varying enzyme concentrations: (●) 160 μM, (○) 240 μM, (▾) 320 μM, () 640 μM and (■) 1280 μM H2O2 in 50 mM phosphate buffer, pH 7.4, with 100 μM DTPA. MPO concentrations ranged accordingly from 32 to 256 nM. Residual TMB activities over time are presented (n = 3). (D) Loss of peroxidase activity when MPO was reacted with a steady flux of hydrogen peroxide. 32 nM MPO reacted with a continuous hydrogen peroxide flux of (●) 33.3 μM/60 min, (○) 153.3 μM/60 min and (▾) 286.6 μM/60 min hydrogen peroxide in 50 mM phosphate buffer, pH 7.4, and 100 μM DTPA. Residual activity over time was determined by the TMB peroxidase activity assay (n = 2).
When the ratio of hydrogen peroxide to MPO was kept constant at 5000:1 but the concentrations varied, it was evident that the rate of inactivation was dependent on the concentration of hydrogen peroxide (Fig. 1C). As an alternative to this experiment, we also generated a slow flux of hydrogen peroxide with glucose oxidase to maintain a low steady-state concentration of hydrogen peroxide but still expose the enzyme to similar doses of hydrogen peroxide as when we used bolus additions of the substrate. The rate of inactivation of MPO over the first 25 min of reaction was independent of the flux of hydrogen peroxide (Fig. 1D). The extent of inactivation, however, increased with increasing fluxes of hydrogen peroxide. Interestingly, at the lowest flux of hydrogen peroxide (i.e., 0.5 μM/min) inactivation reached a maximum of only about 40% which is similar to that obtained with a bolus addition of 160 nM hydrogen peroxide. From these data we conclude that MPO is very susceptible to low concentrations of hydrogen peroxide. Higher concentrations of hydrogen peroxide protect the enzyme from a fast inactivation event but at the highest concentrations a slower inactivation process occurs that is dependent on the concentration of hydrogen peroxide.
Next we determined whether the kinetics of inactivation at high concentrations of hydrogen peroxide conform to the equations developed for mechanism-based inhibitors [16]. Plotting the initial concentration of MPO against the concentration of hydrogen peroxide consumed over 60 min gave a straight line with the slope representing the partition ratio (r) for turnover versus inactivation (Fig. 2A). The value obtained indicates that the enzyme turned over 799 times before it was fully inactivated. However, as will be outlined below (see also Fig. 7), each mole of MPO consumes two moles of H2O2, if it follows the peroxidase cycle (r = 400), and three moles of H2O2 are consumed if Compound III is involved in turnover (r = 266).
Fig. 2.
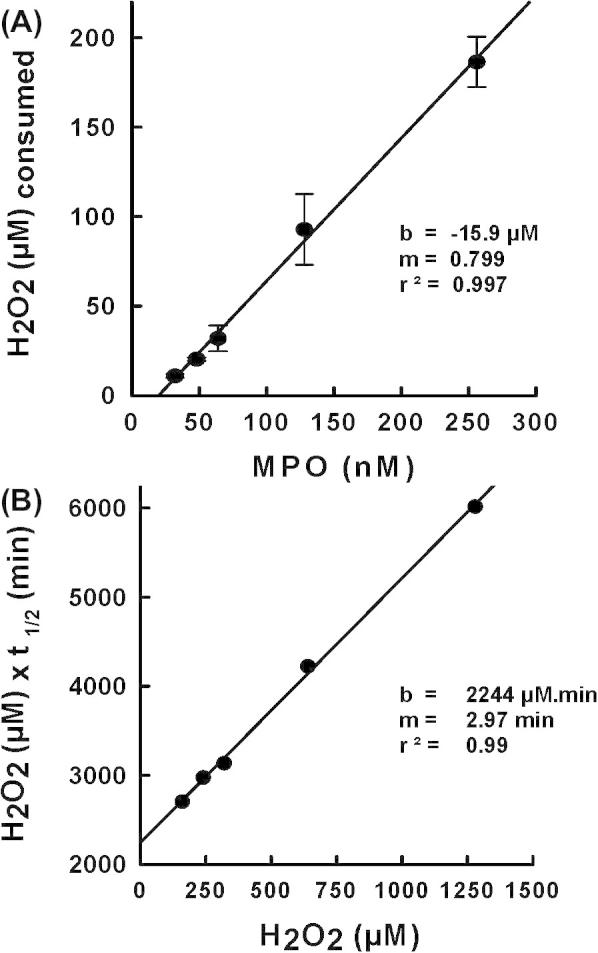
Mechanistic analysis of inactivation of myeloperoxidase by hydrogen peroxide. (A) Partition ratio for the inactivation of MPO by hydrogen peroxide. Residual hydrogen peroxide concentrations were measured at 60 min using the FOX assay when MPO reacted with increasing hydrogen peroxide concentrations at the constant hydrogen peroxide to protein ratio of 5000:1 in 50 mM phosphate buffer, pH 7.4, and 100 μM DTPA (same conditions as Fig. 1C). (B) Determination of kinetic parameters for the inactivation of MPO by H2O2. Half-times of inactivation, t1/2, were determined from Fig. 1C and initial concentrations of hydrogen peroxide were used in the secondary plot.
Fig. 7.

Proposed reaction scheme for redox interconversion of MPO in the presence of hydrogen peroxide. In the micromolar H2O2 concentration range cycling of MPO includes the ferric state, Compound I and Compound II and Reactions 1, 3b and 4b (blue cycle). Compound II accumulates. The strong electrophilicity of Compound I oxidizes the prosthetic group and the protein. Inactivation is suppressed in the presence of one- (Reactions 3a and 4a) and two-electron donors (Reaction 2). In the millimolar concentration range Compound II conversion to Compound III (dark grey intermediates) becomes relevant and cycling of MPO then includes the ferric and ferrous state, Compound I, Compound II and Compound III. The latter intermediate accumulates. Upon reaction of Compound III with excess of hydrogen peroxide reactive oxidants might be formed that destroy the heme and oxidize the protein. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Half-times for inactivation were determined from the data in Fig. 1C and plotted according to Equation (1). From the slope of the secondary plot depicted in Fig. 2B the rate constant for inactivation (kin) was determined to be 3.9 × 10−3 s−1 while the dissociation constant for hydrogen peroxide (K′) was calculated from the intercept to be 740 μM. It is evident from this analysis that inactivation of MPO conforms to the model proposed for mechanism-based inactivation [15] and hydrogen peroxide can be considered to be a suicide substrate for MPO.
| (1) |
where M = (1 + r) × [MPO]o / [H2O2]o
Spectral analysis of MPO reacting with hydrogen peroxide
MPO has numerous redox intermediates that have distinct absorption spectra [17]. To demonstrate which of these redox intermediates are involved in the turnover and inactivation of the enzyme, absorption spectra were recorded at different molar ratios of hydrogen peroxide to MPO. In these analyses the redox intermediates Compound II and Compound III were distinguished from each other by the ratio of absorbances at 625 and 456 nm (A625/A456), which are 0.17 for Compound II and 0.54 for Compound III [17]. When 1.5 μM MPO was reacted with 60 μM of hydrogen peroxide, A625/A456 changed to 0.20 within 5 s, which indicates that the enzyme was almost completely converted to Compound II (Fig. 3A). Compound II subsequently slowly decayed back to the ferric MPO and no more spectral changes could be observed after 120 min. At this time the Soret absorbance at 430 nm was decreased by 6.2 ± 2.8%. Under these conditions all the hydrogen peroxide was consumed within 2 min and the enzyme lost ∼10% activity (insets to Fig. 3A). At 250 μM hydrogen peroxide, MPO was also rapidly converted to Compound II but there was a greater loss of the absorbance at 430 nm (19.9 ± 3.2%) (Fig. 3B). Under these conditions all the hydrogen peroxide was consumed within five minutes and the enzyme lost about 30% of its activity (insets to Fig. 3B). With 1.5 mM of hydrogen peroxide, the enzyme was converted to a mixture of about 50% Compound II and 50% Compound III (A625/A456 = 0.33), with a subsequent fast decrease in absorbance at 454 nm. At 60 min the loss of absorbance at 430 nm was 78.1 ± 1.3% after which no more spectral changes were observed (Fig. 3C). Under these conditions MPO consumed all hydrogen peroxide and lost most of its activity within 20 min (insets to Fig. 3C).
Fig. 3.
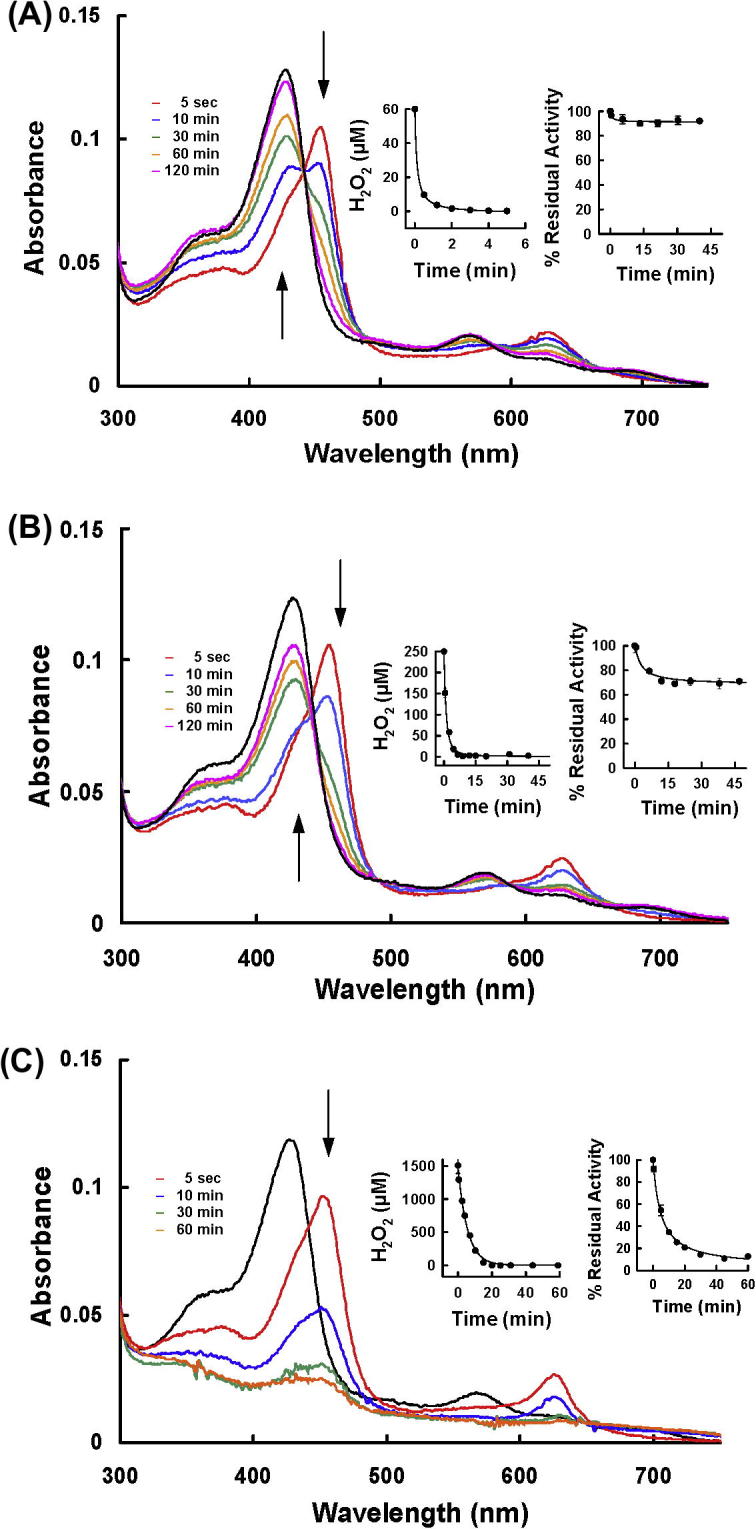
Spectral analysis of MPO reacting with hydrogen peroxide. 1.5 μM MPO was incubated with 60 μM H2O2 (A), 250 μM H2O2 (B) and 1.5 mM H2O2 (C) in 50 mM phosphate buffer, pH 7.4, 100 μM DTPA. Time resolved spectra were recorded over time. The black spectrum depicts the spectral signature of ferric MPO before addition of H2O2, arrows indicate direction of spectral changes. Insets represent consumption of hydrogen peroxide over time as measured by the FOX assay as well as the residual peroxidase activity determined with the TMB-assay.
With all concentrations of hydrogen peroxide, adding 100 μM ascorbate at the end of the reactions did not restore the absorbance at 430 nm. Consequently, because ascorbate readily reduces Compounds I and Compound II [18], it is apparent that the loss of heme absorbance was due to an irreversible oxidative modification and not incomplete conversion of the redox intermediates back to the native MPO.
From these results we conclude that MPO readily consumes hydrogen peroxide in the absence of other reducing substrates. During this process heme absorbance is lost and enzyme activity declines. As the enzyme consumed more hydrogen peroxide, there was greater heme destruction and enzyme inactivation, which was associated with conversion of the enzyme to Compound III.
Protective effect of reducing substrates on loss of heme absorbance
To determine whether reducing substrates protect the enzyme against inactivation by hydrogen peroxide, absorption spectra were recorded under the same conditions as in Fig. 3A–C but in the presence of either ascorbate or bromide plus methionine. Ascorbate is a classical peroxidase substrate that reduces Compound I and Compound II [18] with a single electron whereas bromide is a halogenation substrate that donates two electrons to Compound I and is oxidized to hypobromous acid [17]. Methionine was present in this reaction to scavenge produced HOBr. Addition of 200 μM ascorbate significantly decreased the loss of Soret absorbance at 430 nm at all concentrations of hydrogen peroxide (Table 1). Compound II was formed in the presence of ascorbate and all the hydrogen peroxide was consumed within a few minutes. Bromide also protected the enzyme but was less effective than ascorbate. In its presence, conversion of Compound II back to native MPO took much longer than with ascorbate (Table 1).
Table 1.
Protective effect of a reducing substrate on the loss of absorbance in the ferric state (430 nm). The absorbance loss of the heme Soret peak (1.5 μM in 50 mM phosphate buffer, pH 7.4) was further investigated under the same conditions as in Fig. 3A–C, with either 200 μM ascorbate or 5 mM bromide plus 5 mM methionine present. The time until no more further spectral changes were observed is shown as Δ time (min).
| H2O2 (μM) | Without substrate |
200 μM ascorbate |
5 mM bromide 5 mM methionine |
|||
|---|---|---|---|---|---|---|
| Loss A430 nm (%) | Δt (min) | Loss A430 nm (%) | Δt (min) | Loss A430 nm (%) | Δt (min) | |
| 60 | 6.2 ± 2.8 | 120 | 4.1 ± 1.5 | 0.7 | 9.6 ± 1.0 | 15 |
| 250 | 19.9 ± 3.2 | 120 | 5.5 ± 1.3 | 1 | 10.5 ± 1.6 | 40 |
| 1500 | 78.1 ± 1.3 | 60 | 18.1 ± 1.8 | 2.5 | 20.7 ± 1.5 | 60 |
Hydrogen peroxide-mediated changes to the structure of MPO
Treatment of 1.5 μM MPO with 1.5 mM of hydrogen peroxide resulted in the release of approximately 85% of the iron from the protein (Fig. 4A). This result correlated very well with the loss of heme absorbance observed under the same conditions (Fig. 3C) and suggests that the oxidative break down of the heme prosthetic group causes it to release the chelated iron.
Fig. 4.

Structural consequences of myeloperoxidase incubated with hydrogen peroxide. (A) Detection of iron release from the heme prosthetic group. 1.5 μM MPO was reacted with 1.5 mM hydrogen peroxide in 50 mM sodium acetate buffer, pH 7.4, (column 2). Column 1 shows the result for untreated MPO. Ferrozine was used to measure the release of iron from the heme group (n = 3). (B, C) SDS–PAGE: Time course of MPO reacted with hydrogen peroxide. Top panels: 1.5 μM MPO was reacted with 1.5 mM of hydrogen peroxide in 50 mM phosphate buffer, pH 7.4, and 100 μM DTPA. 15 μL aliquots were taken at 5 min (lane 2), 10 min (lane 3), 25 min (lane 4), 45 min (lane 5) and 60 min (lane 6) and added to 20 μg/mL catalase to remove remaining hydrogen peroxide. Lane 1: negative control (15 μL of untreated MPO, no catalase added). The samples were run on a 8–20% resolving SDS–PAGE under non-reducing (B) and reducing (C) conditions. Bottom panels: SDS–PAGE run under the same conditions as shown in top panels, blotted onto a PVDF membrane and detection of the heme by chemiluminescence. (D) 1.5 μM MPO was incubated with hydrogen peroxide in 50 mM phosphate buffer, pH 7.4, containing 100 μM DTPA for 2 h at various hydrogen peroxide to MPO ratios; 1:1 (lane 2), 5:1 (lane 3), 10:1 (lane 4), 40:1 (lane 5), 100:1 (lane 6), 167:1 (lane 7), 500:1 (lane 8), 1000:1 (lane 9) and 2000:1 (lane 10). 15 μL aliquots were taken and resolved on a 8–20% SDS PAGE under non-reducing conditions. Lane 1: negative control (15 μL of untreated MPO). Bottom panel: SDS–PAGE run under the same conditions as shown in top panel, blotted onto a PVDF membrane and detection of the heme by chemiluminescence.
The effect of hydrogen peroxide on the overall structure of MPO was analyzed by gel electrophoresis. Chemiluminescence [14] was used to detect changes in the heme content of the protein. Hydrogen peroxide was added to MPO at a 1000:1 ratio and aliquots were taken over time after the reaction was stopped by addition of catalase to remove residual hydrogen peroxide. Under non-reducing conditions untreated MPO ran as one single diffuse band on SDS–PAGE at the expected molecular weight of the MPO homodimer (146 kDa, Fig. 4B top panel, lane 1) consisting of two identical glycosylated heavy chain monomers (58.5 kDa) and two light chains (14.5 kDa). The heavy and light chains are connected via their covalent linkages to the heme prosthetic group. The two monomers are linked via one disulfide bridge to form the dimer. On some gels a faint band of 14.5 kDa was also visible for untreated MPO, indicating that a minor fraction of the light chain can detach from the homodimer under non-reducing conditions (Fig. 4D top panel, lane 1). After reaction with hydrogen peroxide the single band progressively changed into a doublet with a concomitant increase in the appearance of a 14.5 kDa band (Fig. 4B top panel, lanes 2–6) for the light polypeptide chain. The 60 kDa band visible in lanes 2–6 originated from the added catalase. A second identical gel was blotted on a PVDF membrane and enhanced chemiluminescence was used to detect the heme prosthetic group. The strong signal for untreated MPO at 146 kDa (Fig. 4B lower panel, lane 1) decreased over time when the enzyme was treated with hydrogen peroxide (Fig. 4B lower panel, lane 2–6). No chemiluminescence signals appeared at other molecular weights on the membrane after treatment with hydrogen peroxide. These results suggest that the heme was degraded by hydrogen peroxide while there were relatively subtle changes to the protein involving detachment of the light chain from the heme.
Under reducing SDS–PAGE conditions untreated MPO should theoretically run as one polypeptide band of the molar mass of 73 kDa for the monomer consisting of one heavy and one light subunit linked via the heme. Under reducing conditions we observed two additional bands at molar masses of approximately 60 and 14 kDa, as shown in Fig. 4C top panel, lane 1. Taking the above results under non-reducing conditions into consideration, it was concluded that the polypeptide chain at approx. 60 kDa was the heavy chain and the 14 kDa band was the light chain. When MPO was treated with hydrogen peroxide, the intensity of the 73 kDa band of the MPO monomer decreased progressively over time (Fig. 4C, lanes 2–6) with no additional bands appearing. The catalase that was added to stop the reaction ran at the same molar mass as the heavy polypeptide chain. An identical gel run under reducing conditions was blotted onto a PVDF membrane and again the heme prosthetic group was visualized using chemiluminescense. The heme group of untreated MPO showed one strong signal at the molecular weight of 73 kDa (Fig. 4C lower panel, lane 1), which upon treatment with hydrogen peroxide progressively decreased with no additional signals appearing on the blot (Fig. 4C lower panel, lanes 2–6).
MPO was also reacted with increasing hydrogen peroxide concentrations for two hours and subsequently resolved by SDS–PAGE under non-reducing condition (Fig. 4D upper panel). The same fragmentation pattern as described above in both the high and low molar mass regions was observed with increasing concentrations of hydrogen peroxide. The doublet became very apparent from lane 6 (100:1 ratio of hydrogen peroxide to protein). At a ratio of 2000:1 (lane 10) the most prominent high molecular weight band was observed at approx. 120 kDa which correlated well with the molecular weight of the homodimer that has lost both light chains (117 kDa), whereas the second prominent high molecular weight band correlated with the homodimer that has lost one light chain (131.5 kDa). At the same time a strongly increased band for the light chain at 14.5 kDa was observed. The 38 kDa fragment also visible on this gel, was observed only on some of the gels run under the same conditions and was interpreted earlier as a degradation product deriving from a specific cleavage between Met243 (that forms the sulfonium ion linkage at pyrrole ring A) and Pro244 [19]. The heme prosthetic group was detected again using chemiluminescence (Fig. 4D, lower panel). The intensity of the band decreased with increasing concentrations of hydrogen peroxide which became very noticeable from lane 7 (167:1 ratio of hydrogen peroxide to MPO) (Fig. 4D top panel, lanes 7–10). Collectively, these results indicate that as MPO is inactivated by hydrogen peroxide the heme group is destroyed with concomitant release of iron and the light chain becomes detachable from the homodimer under SDS–PAGE conditions as a consequence of the heme destruction.
Hydrogen peroxide-mediated changes to protein unfolding and the thermal stability of MPO
Next we evaluated the impact of hydrogen peroxide treatment on the thermal stability of MPO. With native MPO one single endotherm was obtained that was best fitted by a non-two-state transition with maxima at 84.0 and 88.4 °C which confirms recently published data [20]. Treatment of MPO with a 100-fold excess of hydrogen peroxide resulted in a very similar unfolding pathway (Tm1 = 83.9 and Tm2 = 86.5 °C) for the protein (Fig. 5A). This also includes the calculated calorimetric enthalpies for the two transitions (Table 2). With a 1000-fold excess of hydrogen peroxide three transitions were seen with Tm1 = 75.0 °C, Tm2 = 85.2 °C and Tm2 = 86.1 °C (Fig. 5A) reflecting protein fragmentation as observed by SDS–PAGE analyses (compare with Fig. 4D, lane 9). In all assays 0.5 M GdnHCl was present to avoid non-specific aggregation during heating, which is confirmed by the fact that the sum of calorimetric enthalpies (∑Hm) were very similar for all fitted transitions (Table 2A) [20].
Fig. 5.

Thermal stability and unfolding of human myeloperoxidase treated with hydrogen peroxide. (A) Thermal unfolding of leukocyte MPO followed by differential scanning calorimetry. Normalized DSC scans of 5 μM MPO in 5 mM phosphate buffer, pH 7.4, after pre- and post-transitional baseline subtraction in the range of 20–100 °C. Absence of H2O2 (black line), incubation with 500 μM H2O2 (blue line) and incubation with 5 mM H2O2 (red line). Experimental data and fitting to a non-two-state model (thin black lines) are shown. (B) Thermal unfolding followed by circular dichroism at 416 nm. Same conditions as in (A). (C) Thermal unfolding of leukocyte MPO followed by circular dichroism at 222 nm. The insets represent the corresponding van’t Hoff plots and linear fits. Same conditions as in (A). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 2.
(A) Calorimetric enthalpy calculated from thermal unfolding experiments (Fig. 5) using differential scanning calorimetry. Conditions: 5 μM human myeloperoxidase in 5 mM phosphate buffer, pH 7.0. (Native enzyme or incubated with 100- or 1000-fold stoichiometric amount of hydrogen peroxide for 2 h. (B) Thermodynamic data of thermal unfolding (calculated from van’t Hoff plot) for MPO without and with 100 and 1000 fold excess of H2O2 followed by electronic circular dichroism spectroscopy at 222 nm.
| MPO | MPO plus 100 eq H2O2 |
MPO plus 1000 eq H2O2 |
|
|---|---|---|---|
| (A) | |||
| ΔHm (kJ mol−1) | – | – | 38.5 |
| ΔHm2 (kJ mol−1) | 111.7 | 104.6 | 85.0 |
| ΔHm3 (kJ mol−1) | 138.7 | 135.0 | 140.3 |
| ∑ΔHm (kJ mol−1) | 250.5 | 239.6 | 263.8 |
| MPO ([θ]222) | MPO 100 eq H2O2 ([θ]222) | MPO 1000 eq H2O2 ([θ]222) | |
| (B) | |||
| Tm (°C) | 76.8 ± 0.1 | 76.1 ± 0.2 | 73.2 ± 0.1 |
| ΔHm (kJ mol-1) | 261.4 ± 16.7 | 245.7 ± 18.9 | 266.7 ± 11.5 |
| ΔSm (J mol-1) | 746.8 ± 48.7 | 703.6 ± 54.3 | 770.1 ± 33.2 |
| ΔCp (kJ mol-1 K-1) | 4.88 | 4.70 | 5.41 |
| ΔG25°C (kJ mol-1) | 18.9 | 17.4 | 17.6 |
To gain more information about the hydrogen peroxide-mediated structural changes, unfolding of MPO was also followed by electronic circular dichroism (ECD) spectroscopy in the far-UV as well as in the visible region. The far-UV ECD spectrum of MPO in the native state exhibited two minima at 208 and 222 nm, respectively (not shown) [20], which is typical for its mainly α-helical structure [17,21,22]. Upon heating the protein from room temperature to 70 °C a small fraction of ellipticity was already lost, but melting of the α-helices started above 70 °C (Tm = 76.8 °C) (Fig. 5C) concomitantly with the observed changes in the heme cavity followed at 416 nm (i.e., minimum of Soret ellipticity) (Fig. 5B). Thus unfolding of α-helices followed by ECD suggests a cooperative two-state transition, including loss of the proper heme environment. The conformational stability at room temperature (ΔG25°C) was calculated to be 18.9 kJ mol−1 (222 nm) (Fig. 5C and Table 2A). A similar unfolding pathway was seen when MPO was incubated with a 100-fold stoichiometric excess of hydrogen peroxide. Upon incubation of MPO with a 1000-fold excess of hydrogen peroxide the loss of ellipticity in the initial phase was more pronounced and the Tm-value of the main transition was lowered to 73.2 °C (Table 2B). From Fig. 5B it is also evident that incubation of MPO with hydrogen peroxide leads to loss of Soret ellipticity reflecting the loss of Soret absorbance at 430 nm (for comparison see Fig. 3).
Hydrogen peroxide-mediated modifications of amino acid residues of MPO
When MPO was reacted with various hydrogen peroxide concentrations the loss of the heme prosthetic group was detectable by following its absorbance at 430 nm but we could not identify the modified heme products by mass spectrometry. Thus, we focused our attention on modifications of the protein by analyzing tryptic peptides for oxidized sulfur containing amino acid residues. 10 out of 17 methionine residues were oxidized to methionine sulfoxide (two from the light and eight from the heavy chain) as depicted in Fig. 6A and summarized in Table 3. Oxidation of cysteine would not be detected because the sample was reduced and alkylated before digestion. Of the modified methionines only three were surface exposed (i.e., having more than 8 Å2 surface exposed). Interestingly, many of the surface exposed methionines were not modified (Fig. 6 and Table 3). This suggests that the methionine residues did not react directly with hydrogen peroxide but served as scavenger of protein radicals formed inside the protein. The methionine residue that forms the covalent sulfonium ion linkage with the vinyl group on pyrrole ring A (Met243) was not modified.
Fig. 6.

Modification of amino acids of myeloperoxidase incubated with hydrogen peroxide. (A) LC-MS/MS analysis (amino acid sequence coverage of 86%) and oxidized methionine residues of MPO after treatment with hydrogen peroxide. Mature MPO is a homodimer, each half being composed of a light chain (106 amino acids, blue) and a heavy chain (467 amino acids, black). The small excised hexa-peptide between the light and heavy subunit is depicted in grey. Important catalytic residues are highlighted in turquiose, amino acids involved in the covalent link with the prosthetic group are in green. Glycosylation sites (asparagines, N) are marked by ∗, the cysteine that forms the disulfide bridge between the subunits is marked by # and ligands of the calcium ion binding site are underlined. The amino acid numbering for MPO is based on the mature protein with the first cysteine in the small polypeptide designated as residue 1. Amino acid sequence coverage by LC-MS/MS is depicted in bold letters, oxidized methionine residues are highlighted in red. (B) 3D structure of the MPO monomer (one light and one heavy chain covalently linked via the heme group) showing the ten oxidized methionine residues which were detected by LC-MS/MS when 1.5 μM MPO was reacted with 1.5 mM of hydrogen peroxide. Surface exposed methionines that were not modified by H2O2 incubation are depicted in yellow, oxidized methionines are shown in red. For definition of surface accessibility see Table 4. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 3.
LC-MS/MS analysis of oxidized tryptic peptides of MPO when reacted with hydrogen peroxide. 1.5 μM MPO was reacted with 1.5 mM of hydrogen peroxide in 50 mM phosphate buffer pH 7.4. The reduced and alkylated protein was digested with trypsin and peptides were analyzed by LC-MS/MS. Peptides with a signal ratio of treatment versus control of ⩾3 and a p value of ⩽0.001 are presented.
| MPO amino acid # | Identified modified peptide | Oxidized residue on peptide | Charge | m/z Value | Ratio ⩾3 treatment vs control | p-value ⩽0.001 | SD |
|---|---|---|---|---|---|---|---|
| M 85 M 87 | SLmFmQWGQLLDHDLDFTPEPAAR | M3 M5 |
3 | 950.7812 | 11.36 | 1.02E-5 | 2.75 |
| M 287 | KIVGAmVQIITYR | M6 | 3 | 503.2933 | 19.68 | 1.08E-6 | 4.59 |
| M 306 | DYLPLVLGPTAmR | M12 | 2 | 731.3934 | 6.72 | 5.99E-10 | 0.85 |
| M 306 | DYLPLVLGPTAmRK | M12 | 2 | 795.4401 | 7.06 | 0.00016 | 1.41 |
| M 306 | KIVGAMVQIITYRDYLPLVLGPTAmRK | M25 | 4 | 766.4382 | 4.30 | 1.72E-5 | 1.06 |
| M 343 | YGHTLIQPFmFR | M10 | 2 | 763.3850 | 5.06 | 8.60E-6 | 0.42 |
| M 343 | YGHTLIQPFmFR | M10 | 3 | 509.2593 | 10.82 | 3.43E-5 | 1.46 |
| M 343 | IANVFTNAFRYGHTLIQPFmFR | M20 | 4 | 665.5958 | 9.76 | 1.85E-6 | 3.00 |
| M 385 | GLmATPAKLNR | M3 | 2 | 594.3316 | 9.28 | 2.38E-5 | 0.57 |
| M 385 | VVLEGGIDPILRGLmATPAKLNR | M15 | 3 | 817.1353 | 36.18 | 7.90E-6 | 6.39 |
| M 411 | LFEQVmRIGLDLPALNmQR | M6 M17 |
3 | 759.4016 | 13.59 | 0.000266 | 2.42 |
| M 422 | IGLDLPALNmQR | M10 | 2 | 678.8680 | 7.38 | 1.00E-5 | 0.53 |
| M 465 M 478 | LmEQYGTPNNIDIWmGGVSEPLKR | M2 M15 |
3 | 927.4525 | 16.01 | 2.76E-5 | 4.14 |
Discussion
In this investigation we have shown that MPO is exquisitely sensitive to a fast but limited inactivation event mediated by low concentrations of hydrogen peroxide. Higher concentrations of hydrogen peroxide protect against this fast inactivation event but lead to a slower pathway of inactivation in which the heme prosthetic groups are destroyed, and in the process their iron is released and the small polypeptide subunits detach from the heme prosthetic group.
Human myeloperoxidase is a cationic 146 kDa dimer of high conformational and thermal stability [17,20,21,22] with a single disulfide bridge between symmetry-related halves (73 kDa), each of which contains two polypeptides of 14.5 and 58.5 kDa. The heavy polypeptide is glycosylated and contains five intra-chain disulfides whereas the light polypeptide contains only one [21,22]. Each subunit of MPO contains a derivative of protoporphyrin IX in which the methyl groups on pyrrole rings A and C are linked via ester bonds to the carboxyl groups of highly conserved aspartate and glutamate residues [21,22]. In addition, the β-carbon of the vinyl group on pyrrole ring A forms a covalent bond with the sulfur atom of a fully conserved methionine [20,21]. As a consequence, the heme porphyrin ring is considerably distorted from planarity, confering unique spectral and redox properties to MPO [23,24].
The mechanisms of inactivation of MPO by hydrogen peroxide are understandable in terms of the protein’s architecture and the redox transformations it undergoes during turnover (Fig. 7). Reaction of hydrogen peroxide with ferric MPO leads to reductive heterolytic cleavage of its oxygen–oxygen bond and oxidation of the enzyme to its Compound I state (Fig. 7; Reaction 1). This form of MPO contains two oxidizing equivalents more than the resting enzyme [17]. Compound I contains an oxoiron (IV) and a π-cation radical on the porphyrin ring. It has high one- and two-electron reduction potentials [25–27], allowing the oxidation of numerous one- and two-electron donors. Compound I is either reduced directly by (pseudo-)halides (X− = Cl−, Br−, SCN−) to the resting state producing hypohalous acids (HOX = HOCl, HOBr, HOSCN) (Reaction 2) or it undergoes one-electron reduction by a typical peroxidase substrate (AH, Reaction 3a) and hydrogen peroxide (Reaction 3b) to produce Compound II and the substrate radical (A) or superoxide (O2•- or HO2•), respectively [28]. Finally, Compound II is reduced back to the resting state with either another peroxidase substrate (Reaction 4a) [29] or superoxide (Reaction 4b) [30]. Furthermore, at high concentrations of hydrogen peroxide Compound II can be converted to Compound III (Reaction 5) [31], where ferrous MPO with bound dioxygen exists in equilibrium with ferric MPO with bound superoxide. Compound III slowly decays to ferric MPO and superoxide. Alternatively, it can decay to ferrous MPO which rapidly recombines with dioxygen or reacts with hydrogen peroxide to form Compound II (Reaction 6) [31]. Based on burst phase kinetics for hydrogen peroxide consumption by MPO in the absence of chloride, it has also been proposed that the enzyme has catalatic activity, degrading hydrogen peroxide to oxygen and water. That is, it acts in an analogous manner to catalase where hydrogen peroxide also reduces Compound I directly in a two-electron reaction to produce dioxygen [32]. However, no evidence for this reaction has been demonstrated using pre-steady-state kinetic measurements [28,33]. All of these redox intermediates are relevant to the physiological activities of MPO because when the enzyme is released by neutrophils into phagosomes or the extracellular environment it reacts with both superoxide and hydrogen peroxide [34,35].
It has been demonstrated that a ten-fold molar excess of hydrogen peroxide over MPO is needed [28] for complete conversion of ferric MPO to Compound I, which is subsequently reduced to Compound II via Reaction 3b. Among heme peroxidases MPO is unique in catalyzing the one-electron oxidation of hydrogen peroxide to superoxide [28]. Compound I is likely to be the crucial redox intermediate in the fast inactivation pathway because there was irreversible loss of peroxidase activity at ratios lower than 10:1. Protection of the enzyme from this fast inactivation event by high concentrations of hydrogen peroxide and reducing substrates, including ascorbate and bromide, can be explained by their reactions with Compound I. Compound I has a very positive reduction potential [25,26] so that in the absence of exogenous electron donors it will promote radical oxidation reactions within the heme group or with the protein moiety.
At higher concentrations of hydrogen peroxide analysis of the loss of peroxidase activity at a constant oxidant to protein ratio conformed to the kinetics expected for mechanism-based inhibition [16]. The calculated partition ratio (r = 799), the inactivation rate constant (kin = 3.9 × 10−3 s−1) and the dissociation constant for hydrogen peroxide (K′ = 740 μM) suggest that at higher concentrations hydrogen peroxide acts as a very moderate suicide substrate for MPO. A turnover of 799 + 1 molecules of hydrogen peroxide per MPO is followed by irreversible inactivation of the enzyme under the conditions tested. Thus, the mechanism of inactivation under these conditions appears to be different from that at low concentrations of hydrogen peroxide.
Spectral analysis revealed that formation of Compound II was associated with minimal inactivation and heme loss. In contrast, there was extensive degradation of the heme groups and loss of activity when the concentration of hydrogen peroxide was sufficient to convert the enzyme to Compound III (Reaction 5). Previous work with lactoperoxidase suggested that its inactivation by hydrogen peroxide resulted from reaction of the oxidant with either Compound III or the ferrous enzyme to produce reactive oxygen species including singlet oxygen and hydroxyl radical [8,36]. The role of ferrous MPO is questionable since it rapidly binds O2 or reacts with H2O2 forming Compound III or Compound II, respectively. In any case, we found that ten of its methionine residues were oxidized to methionine sulfoxide at high concentrations of hydrogen peroxide. Most of the oxidized methionine residues are located in the interior of the protein whereas the majority of the surface exposed methionine residues were not modified. This result suggests that radical production within the protein was responsible for oxidation of these residues as opposed to their direct reaction of hydrogen peroxide. The protection afforded by bromide and ascorbate can be explained by their ability to promote rapid consumption of hydrogen peroxide via Reactions 3a and 4a and prevent the conversion of Compound II to Compound III.
Although MPO has high thermal and conformational stability [19], the oxidative modification induced by hydrogen peroxide were able to disrupt the proteins structural integrity. This was reflected by loss of absorbance as well as ellipticity at the Soret maximum, which suggests that the heme was degraded. This was confirmed by demonstrating that high concentrations of hydrogen peroxide promote the release of heme iron. Degradation of the heme groups explains why the light polypeptide chains became detached from the heme and subsequently from the protein under SDS–PAGE conditions. These polypeptides are linked to the heme groups via an ester bond with Asp94. Presumably this or an adjacent bond must break when the heme is degraded by hydrogen peroxide. The free iron would also be expected to contribute to further protein damage because it is likely to catalyze site specific Fenton-type reactions with hydrogen peroxide.
Conclusion
We have identified two mechanisms by which hydrogen peroxide can inactivate MPO. The first involves formation of Compound I at low concentrations of hydrogen peroxide where, in the absence of a reducing substrate, the strong electrophilicity of this redox intermediate enables it to oxidize the heme group or protein moiety. Secondly, hydrogen peroxide is likely to react with Compound III to give rise to reactive oxidants that destroy the heme and oxidize the protein. Under normal physiological conditions, such that reducing substrates are always present and the concentration of hydrogen peroxide does not accumulate, these mechanisms of inactivation are unlikely to occur in vivo. However, it is within the confines of the neutrophil phagosomes where these mechanisms of inactivation are most likely to operate. The concentration of chloride inside phagosomes has been reported to be approximately 70 mM [38]. If this declines due to its oxidation to hypochlorous acid and the conversion to chloramines [39], then Compound I may be free to oxidize itself. Hydrogen peroxide may also accumulate sufficiently to react with Compound III, which is the predominant form of the enzyme in this compartment [39]. Hypochlorous acid could also contribute to inactivation of MPO within phagosomes after other more abundant targets are quenched. These mechanisms would be reliant on continued activity of the NADPH oxidase which is also susceptible to inactivation [40]. Inactivation via Compound I could be exploited in the search for pharmacological inhibitors of MPO. Inhibitors that bind tightly to the active site of MPO and prevent binding of reducing substrates would make the enzyme susceptible to inactivation by low concentrations of hydrogen peroxide.
Table 4.
Surface exposure [37] of methionines in human myeloperoxidase. In addition, the distance of the Cα atom of the respectives methionines to the heme iron is given.
| Methionine |
Molecular surface accessibility (Å2) |
Distance (Cα) to heme iron (Å) | |||
|---|---|---|---|---|---|
| Residue | Oxidized | Total | Backbone | Sidechain | |
| 13 | no | 17.8993 | 0.5285 | 17.3707 | 26.6 |
| 85 | yes | 0.0000 | 0.0000 | 0.0000 | 14.7 |
| 87 | yes | 1.0485 | 0.1747 | 0.8737 | 10.3 |
| 175 | no | 0.1747 | 0.0000 | 0.1747 | 14.7 |
| 190 | no | 36.5191 | 9.6475 | 26.8715 | 33.1 |
| 243 | no | 0.1747 | 0.0000 | 0.1747 | 10.8 |
| 249 | no | 0.8047 | 0.1057 | 0.6990 | 15.3 |
| 287 | yes | 1.3979 | 0.0000 | 1.3979 | 22.6 |
| 306 | yes | 10.8340 | 0.1747 | 10.6593 | 23.0 |
| 343 | yes | 0.0000 | 0.0000 | 0.0000 | 15.7 |
| 353 | no | 14.6223 | 0.3495 | 14.2728 | 26.9 |
| 385 | yes | 0.0000 | 0.0000 | 0.0000 | 20.0 |
| 411 | yes | 15.8951 | 5.1581 | 10.7370 | 17.1 |
| 422 | yes | 0.0000 | 0.0000 | 0.0000 | 12.3 |
| 465 | yes | 11.4057 | 2.8131 | 8.5926 | 26.1 |
| 478 | yes | 0.0000 | 0.0000 | 0.0000 | 14.2 |
| 522 | no | 48.8422 | 2.6605 | 46.1817 | 37.4 |
| 553 | no | 31.7514 | 0.9147 | 30.8367 | 21.1 |
Acknowlegements
This project was supported by the Austrian Science Foundation, FWF [FWF-Schroedinger Fellowship J2877-B11 and doctoral program BioToP – Biomolecular Technology of Proteins (FWF W1224)] and a grant from the Health Research Council of New Zealand.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abbreviations used: MPO, myeloperoxidase; Por, porphyrin; TMB, 3,3′,5,5′-tetramethylbenzidine; DTPA, diethylenetriaminepentaacetic acid; cetrimide, alkyltrimethylammonium bromide; FOX, ferrous oxidation of xylenol orange; PVDF, polyvinylidene difluoride; CID, collision induced dissociation; ECD, electronic circular dichroism; DSC, differential scanning calorimetry; GdnHCl, guanidinium hydrochloride.
References
- 1.Klebanoff S.J., Kettle A.J., Rosen H., Winterbourn C.C., Nauseef W.M. J. Leukoc. Biol. 2013;93:185–198. doi: 10.1189/jlb.0712349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies M.J., Hawkins C.L., Pattison D.I., Rees M.D. Antioxid. Redox Signal. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 3.Bradley P.P., Christensen R.D., Rothstein G. Blood. 1982;60:618–622. [PubMed] [Google Scholar]
- 4.Edwards S.W., Nurcombe H.L., Hart C.A. Biochem. J. 1987;245:925–928. doi: 10.1042/bj2450925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnao M.B., Acosta M., del Rio J.A., Garcia-Canovas F. Biochim. Biophys. Acta. 1990;1038:85–89. doi: 10.1016/0167-4838(90)90014-7. [DOI] [PubMed] [Google Scholar]
- 6.Arnao M.B., Acosta M., del Rio J.A., Garcia-Canovas F. Biochim. Biophys. Acta. 1990;1041:43–47. doi: 10.1016/0167-4838(90)90120-5. [DOI] [PubMed] [Google Scholar]
- 7.Hiner N.P., Rodriguez-Lopez J.N., Arnao M.B., Raven E., Garcia-Canovas F., Acosta M. Biochem. J. 2000;348:321–328. [PMC free article] [PubMed] [Google Scholar]
- 8.Huwiler M., Jenzer H., Kohler H. Eur. J. Biochem. 1986;158:609–614. doi: 10.1111/j.1432-1033.1986.tb09798.x. [DOI] [PubMed] [Google Scholar]
- 9.Kettle A.J., Gedye C.A., Winterbourn C.C. Biochem. Pharmacol. 1993;45:2003–2010. doi: 10.1016/0006-2952(93)90010-t. [DOI] [PubMed] [Google Scholar]
- 10.Olorunniji F.J., Iniaghe M.O., Adebayo J.O., Malomo S.O., Adediran S.A. Open Enzym. Inhib. J. 2009;2:28–35. [Google Scholar]
- 11.Furtmüller P.G., Burner U., Obinger C. Biochemistry. 1998;37:17923–17930. doi: 10.1021/bi9818772. [DOI] [PubMed] [Google Scholar]
- 12.Beers R.F., Sizer I.W. J. Biol. Chem. 1952:133–140. [PubMed] [Google Scholar]
- 13.Gay C., Gebicki J.M. Anal. Biochem. 2000;284:217–220. doi: 10.1006/abio.2000.4696. [DOI] [PubMed] [Google Scholar]
- 14.Cheng G., Salerno J.C., Cao Z., Pagano P.J., Lambeth J.D. Free Radic. Biol. Med. 2008;45:1682–1694. doi: 10.1016/j.freeradbiomed.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter P. Anal. Biochem. 1971;40:450–458. doi: 10.1016/0003-2697(71)90405-2. [DOI] [PubMed] [Google Scholar]
- 16.Waley S.G. Biochem J. 1985;227:843–849. doi: 10.1042/bj2270843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furtmüller P.G., Zederbauer M., Jantschko W., Helm J., Bogner M., Jakopitsch C., Obinger C. Arch. Biochem. Biophys. 2006;445:199–213. doi: 10.1016/j.abb.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Marquez L.A., Dunford H.B., Van Wart H. J. Biol. Chem. 1990;265:5666–5670. [PubMed] [Google Scholar]
- 19.Taylor L.T., Pohl J., Kinkade J.M. J. Biol. Chem. 1992;267:25282–25288. [PubMed] [Google Scholar]
- 20.Banerjee S., Stampler J., Furtmüller P.G., Obinger C. Biochim. Biophys. Acta. 2011;1814:375–387. doi: 10.1016/j.bbapap.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 21.Fiedler T.J., Davey C.A. J. Biol. Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- 22.Carpena X., Vidossich P., Schroettner K., Calisto B.M., Banerjee S., Stampler J., Soudi M., Furtmüller P.G., Rovira C., Fita I., Obinger C. J. Biol. Chem. 2009;284:25929–25937. doi: 10.1074/jbc.M109.002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zederbauer M., Furtmüller P.G., Brogioni S., Jakopitsch C., Smulevich G., Obinger C. Nat. Prod. Rep. 2007;24:571–584. doi: 10.1039/b604178g. [DOI] [PubMed] [Google Scholar]
- 24.Battistuzzi G., Stampler J., Bellei M., Vlasits J., Soudi M., Sola M., Furtmüller P.G., Obinger C. Biochemistry. 2011;50:7987–7994. doi: 10.1021/bi2008432. [DOI] [PubMed] [Google Scholar]
- 25.Arnhold J., Furtmüller P.G., Obinger C. Redox Rep. 2003;8:179–186. doi: 10.1179/135100003225002664. [DOI] [PubMed] [Google Scholar]
- 26.Furtmüller P.G., Arnhold J., Jantschko W., Pichler H., Obinger C. Biochem. Biophys. Res. Commun. 2003;301:551–557. doi: 10.1016/s0006-291x(02)03075-9. [DOI] [PubMed] [Google Scholar]
- 27.Arnhold J., Furtmüller P.G., Regelsberger G., Obinger C. Eur. J. Biochem. 2001;268:5142–5148. doi: 10.1046/j.0014-2956.2001.02449.x. [DOI] [PubMed] [Google Scholar]
- 28.Furtmüller P.G., Burner U., Jantschko W., Obinger C. FEBS Lett. 2000;484:139–143. doi: 10.1016/s0014-5793(00)02143-8. [DOI] [PubMed] [Google Scholar]
- 29.Jantschko W., Furtmüller P.G., Allegra M., Livrea M., Jakopitsch C., Regelsberger G., Obinger C. Arch. Biochem. Biophys. 2002;398:12–22. doi: 10.1006/abbi.2001.2674. [DOI] [PubMed] [Google Scholar]
- 30.Kettle A.J., Anderson R.F., Hampton M., Winterbourn C.C. Biochemistry. 2007;46:4888–4897. doi: 10.1021/bi602587k. [DOI] [PubMed] [Google Scholar]
- 31.Jantschko W., Furtmüller P.G., Zederbauer M., Lanz M., Obinger C. Biochem. Biophys. Res. Commun. 2003;312:292–298. doi: 10.1016/j.bbrc.2003.10.117. [DOI] [PubMed] [Google Scholar]
- 32.Kettle A.J., Winterbourn C.C. Biochemistry. 2001;40:10204–10212. doi: 10.1021/bi010940b. [DOI] [PubMed] [Google Scholar]
- 33.Furtmüller P.G., Burner U., Jantschko W., Regelsberger G., Obinger C. Redox Rep. 2000;5:171–176. doi: 10.1179/135100000101535717. [DOI] [PubMed] [Google Scholar]
- 34.Winterbourn C.C., Garcia R., Segal A.W. Biochem. J. 1985;228:583–592. doi: 10.1042/bj2280583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kettle A.J., Sangster D.F., Gebicki J., Winterbourn C.C. Biochem. Biophys. Acta. 1988;956:58–62. doi: 10.1016/0167-4838(88)90297-x. [DOI] [PubMed] [Google Scholar]
- 36.Jenzer H., Jones W., Kohler H. J. Biol. Chem. 1986;261:15550–15556. [PubMed] [Google Scholar]
- 37.Vriend G. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 38.Painter R.G., Marrero L., Lombard G.A., Valentine V.G., Nauseef W.M., Wang G. J. Leukoc. Biol. 2010;87:933–942. doi: 10.1189/jlb.1009655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winterbourn C.C., Hampton M.B., Livesey J.H., Kettle A.J. J. Biol. Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 40.Nauseef W.M. Immunol. Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]