

Abstract
Cold atmospheric-pressure plasmas are currently in use in medicine as surgical tools and are being evaluated for new applications, including wound treatment and cosmetic care. The disinfecting properties of plasmas are of particular interest, given the threat of antibiotic resistance to modern medicine. Plasma effluents comprise (V)UV photons and various reactive particles, such as accelerated ions and radicals, that modify biomolecules; however, a full understanding of the molecular mechanisms that underlie plasma-based disinfection has been lacking. Here, we investigate the antibacterial mechanisms of plasma, including the separate, additive and synergistic effects of plasma-generated (V)UV photons and particles at the cellular and molecular levels. Using scanning electron microscopy, we show that plasma-emitted particles cause physical damage to the cell envelope, whereas UV radiation does not. The lethal effects of the plasma effluent exceed the zone of physical damage. We demonstrate that both plasma-generated particles and (V)UV photons modify DNA nucleobases. The particles also induce breaks in the DNA backbone. The plasma effluent, and particularly the plasma-generated particles, also rapidly inactivate proteins in the cellular milieu. Thus, in addition to physical damage to the cellular envelope, modifications to DNA and proteins contribute to the bactericidal properties of cold atmospheric-pressure plasma.
Keywords: plasma medicine, atmospheric-pressure plasma, antibacterial mechanisms, biomacromolecules, UV, reactive oxygen species
1. Introduction
Plasmas, referred to as the fourth state of matter, are generated through the ionization of gas molecules by electric fields. Naturally occurring plasmas are typically hot (the sun being a good example), but ‘cold’ plasmas can be found in nature as well, as for instance the aurora borealis. Over the past decade, there has been growing interest in the artificial ‘cold’ plasmas, particularly in atmospheric-pressure plasmas with gas temperatures below 40°C. These have found applications in the emerging field of plasma medicine (reviewed in Kong et al. [1]).
Cold atmospheric-pressure plasmas inactivate microorganisms on time-scales ranging from seconds to minutes. Several studies have assessed inactivation efficiencies for bacteria, fungi, bacterial and fungal spores, and viruses [2–5]; however, the underlying molecular mechanisms of inactivation and the contributions of the different plasma components are poorly understood. Plasmas can be modulated to preferentially produce radicals, metastables, ions or photons simply by adjusting the gas composition and treatment conditions. Thus, if the molecular mechanisms of interaction with biological samples were fully understood, then plasmas could be tuned for specific applications.
Published evidence supports a number of mechanisms: etching (the removal of matter) by accelerated ions [6]; disruption of the cell membrane by electroporation and formation of membrane ruptures [7]; and breakage of DNA or protein backbones [8–10]. Wang et al. [11] further demonstrated mutagenic properties of plasma using bacterial spores. Previous in vitro experiments showed that particles emitted from plasma cause DNA strand breaks, whereas photons emitted from plasma cross-link DNA strands [12].
1.1. Our general approach
It is our long-term goal to gain an in-depth understanding of the antibacterial mechanisms of different plasmas on a molecular level. The main challenge is that individually plasmas and bacteria are highly complex with regard to their composition and properties. In addition, both come in large varieties. To tackle the problem, on the plasma side, we chose to take a multi-step approach working our way up from a single well-characterized plasma source with constant operating conditions (step one), to the same well-characterized source operated with different gas mixtures (step two), finally to sources that are more complex in terms of their range of interactions with biological systems (step three). This study describes step one of this journey, where we use a microscale atmospheric-pressure plasma jet (µAPPJ) specifically modified to allow separate investigation of the effects of the photons and the particles emitted from the jet. This plasma source and its properties are introduced in more detail below.
On the biological side, we are interested in understanding the mechanisms that cause plasmas to inactivate microorganisms so efficiently. Bacteria are arguably the living systems of lowest complexity. Yet, these organisms are still complex enough that any number of molecules could be modified by different components of plasma. Here, we chose to gradually reduce the complexity of the model to facilitate cataloguing the molecular modifications that plasmas are capable of introducing into biological matter. For a start, we began to analyse the impact of plasma on the biomacromolecules DNA and protein, which are made of the same building blocks (nucleotides and amino acids, respectively) in all domains of life. Thus, any plasma-triggered modification of these macromolecules first catalogued in vitro could potentially also occur in the context of a living bacterial cell or in a eukaryotic cell. Of course, we expect that the occurrence of a plasma-triggered modification as such, as well as the frequency of occurrence is context-dependent: some modifications might be quenched in the cellular environment, whereas other modifications might require the context of a living cell; maybe even that of a specific cell type.
The cataloguing of plasma-triggered modifications can be performed in realms of different complexity. The characterization of modifications occurring in simplified model substrates in vitro falls into the realm of lowest complexity (first realm). Such substrates can be analysed in their entirety on a molecular level, so that all modifications can be detected, even unanticipated ones. The second realm extends to functional biomolecules that serve as models in vitro. These biomolecules can be obtained in reasonable purity, their structure and/or function (or a loss thereof) can be measured, and ideally they can still be analysed in their entirety on a molecular level. The third realm comprises functional biomolecules in bacterial model organisms. Biomolecules best suited are those whose structure and/or activity can either be measured in the highly complex context of the cell or that can be purified from the cell in a native state for characterization.
Clearly, the challenge is how to pick suitable simplified model substrates and functional biomolecules of relevance to the interaction of plasmas with living cells, not knowing what the main targets of plasma are. We decided for a top-down approach, of first identifying the challenges plasma is posing on bacterial cells, letting this guide our search for biomolecules affected, to finally choosing simplified model substrates that allow tracing the impact of plasma to molecular modifications. Thus, starting from the bacterial cell, we venture into realms three to one in this study, partially building on modifications known to occur as a result of (V)UV treatment or oxidative stress. An outline of the experiments performed is given below.
A fourth realm to be addressed in future studies covers modifications occurring in a specialized organismic context, for example in a particular pathogen, in a particular eukaryotic organism, tissue or cell line, or in dependence of a certain morphology, physiology or lifestyle.
1.2. The plasma source used
A radiofrequency-driven µAPPJ was used throughout this study. In this set-up, biological samples are not directly immersed in the plasma. Instead, they are exposed to the plasma effluent, the plasma-emitted components. In addition, in contrast to other commonly used sources such as dielectric barrier discharges, no electric current is flowing through the samples.
The modified µAPPJ was operated with He gas with an admixture of 0.6% O2. Wherever possible, samples were treated within He atmosphere to avoid the reactions of effluent components with components of ambient air. For the same reason, wherever possible, samples were treated in an air-dried state. One has to keep in mind that this does not eliminate hydration of model substrates and that approximately 80% of the weight of an air-dried bacterial cell is attributed to water. The original µAPPJ has been very well characterized with regard to the species and photons generated as well as the dependence of their densities on the distance from the jet (note: the latter was determined in ambient air). The main species generated are atomic oxygen, ozone and singlet delta oxygen metastables O2 (a1Δg), amounting to a total of 1015 cm−3 reactive oxygen species (ROS) in the effluent [13–15]. Directly at the jet nozzle (which in the original µAPPJ is identical with the point of exit of the effluent from the plasma), an atomic oxygen density of 7 × 1014 cm3 and an ozone density of 3.8 × 1014 cm3 have been measured by molecular beam mass spectroscopy (MBMS) [13]. Photons with wavelengths of 115 and 130 nm dominate, but weak emission of photons with 120 nm wavelength are detected as well [16].
In the set-up used in this study the total distance of samples from the plasma is 12 mm (8 mm inside the jet nozzle plus 4 mm in atmosphere). With 0.6% O2 admixture in the feed gas, in He atmosphere, we expect the emitted vacuum ultraviolet (VUV) and UV photons to mostly reach the samples [17]. The densities of atomic oxygen, ozone and singlet delta oxygen metastables O2 (a1Δg) that reach the samples after having passed through the jet nozzle and He atmosphere have not yet been determined. Compared with the point of exit of the effluent from the plasma, we expect the densities of atomic oxygen to decrease and the ozone densities to increase with increasing distance. In ambient air, for the original µAPPJ, maximal ozone densities were found at more than 40 mm distance from the jet [13].
We would like to stress that the operating conditions were chosen for minimal complexity of species generated. Owing to the feed gas mixture used and the jet being operated in He atmosphere, we would not expect exposure of samples to reactive nitrogen species. We have, however, found additional species stemming from gas impurities, namely protonated water clusters [17]. Atomic oxygen and ozone resulting from photochemistry in the effluent were more than three orders of magnitude lower than plasma-generated atomic oxygen and ozone and below the detection limit of MBMS [17]. The influence of water in the system is currently subject to further study. The generation of hydrogen peroxide by (V)UV photolysis of water has been described previously [18], and we would expect further ROS such as hydroxyl radicals to occur.
To study the effects of plasma-emitted photons and particles separately, and to identify potential synergies between the two, we used the X-jet geometry described previously [12,19]. A lateral He flow crossing the effluent channel steers away the particles and thus allows the separation of heavy particles (such as ions, radicals, metastables) from the (V)UV photons. To allow parallel treatment of samples, three plasma jets were mounted next to each other, each featuring a different nozzle. The nozzles were designed to treat samples with the undivided effluent (complete-jet), with plasma-emitted (V)UV photons alone (UV-jet) or with heavy particles (particle-jet; figure 1a). All three jets use the plasma conditions of the µAPPJ described above, and are therefore ideally suited to study the effects of plasma components on microorganisms and biomolecules. A clearer understanding of the roles played by these individual plasma-generated components is desirable; they probably have quite distinct effects on cells and macromolecules. Figure 1b gives a flavour of the different properties of the plasma-emitted particles and photons. It shows the etching profiles of the three jets generated with a hydrogenated amorphous carbon layer used as model substrate. No etching is observed under the UV-jet, whereas the particle-jet efficiently etches a surface area with a 3 mm radius. The complete-jet is the most efficient, suggesting synergistic effects of emitted particles and photons owing to photochemical reactions in the gas phase.
Figure 1.
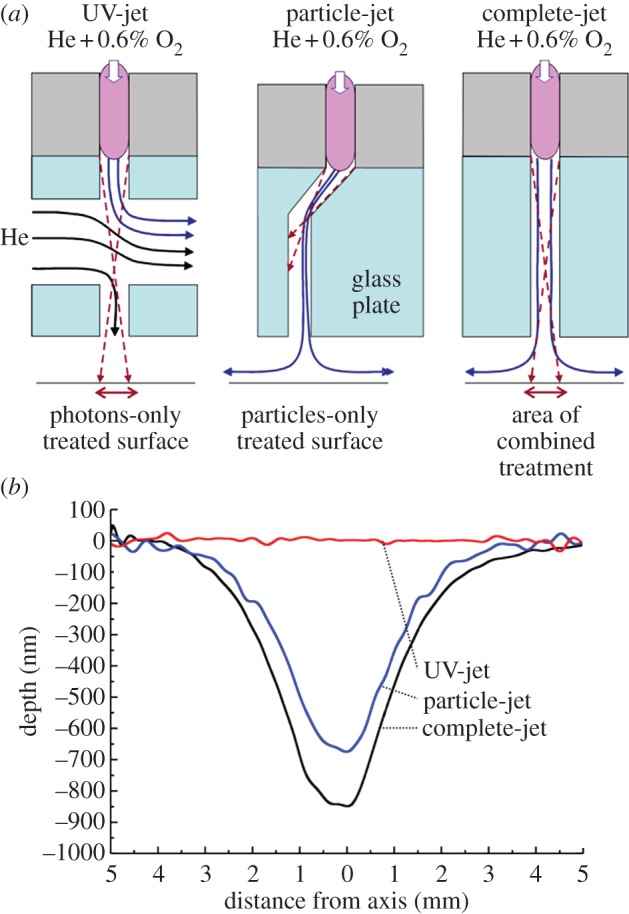
Schematic of the three µAPPJs (a) used in this study and their hydrogenated amorphous carbon (a-CH) etching profiles (b). (a) The extension of the µAPPJ plasma channel was modified to allow separate or combined treatment of the substrate with (V)UV photons and/or particles. The distance from the plasma to the substrate in all three jets was 12 mm. (b) Polymer-like a-CH films, serving as model for etching, were treated with the modified jets for 10 min. (Online version in colour.)
1.3. Outline of the experiments performed
Based on the characterization of the plasma source, we knew that bacteria are subject to etching and exposed to reactive species and (V)UV photons. The first question we posed was whether bacteria are simply killed by mechanical stress related to etching. To investigate the mechanical stress we chose scanning electron microscopy (SEM), a technique that allows imaging of the surface of live bacteria. Two model organisms frequently used to study bacterial stress responses were used in this study, namely Escherichia coli and Bacillus subtilis, representing two groups of bacteria, gram-negative and gram-positive, respectively. Both groups differ in the structure and composition of their cell envelops. All cells are enclosed in a cytoplasmic membrane serving as diffusion barrier for polar and ionic solutes. In gram-negative species, externally a thin murein layer gives mechanical stability, and a second membrane provides a further diffusion barrier for solutes. Gram-positive bacteria lack this outer membrane but are characterized by a thicker murein layer providing extraordinary mechanical stability. Since in SEM cells are imaged at 80 Pa, for this experiment, we chose the sturdier B. subtilis as model organism, assuming that if all B. subtilis died as a result of etching, E. coli would as well. As B. subtilis is one of the bacterial species capable of forming endospores to survive hostile conditions (mechanically even sturdier than vegetative B. subtilis by orders of magnitude), we used a sporulation-deficient mutant throughout the study allowing us to focus on vegetative bacterial cells. The sizes of the etching zones were compared with the zones in which bacteria die of plasma exposure, the latter being determined in inhibition zone tests using B. subtilis monolayers on agar plates.
The next question was how does plasma exposure kill the cells outside the etching zone. Building on published knowledge of the model organisms’ stress physiology, we chose reporter genes that are indicative of particular biomacromolecules and cellular structures being damaged, focusing on targets described for ROS and (V)UV photons. Reporter gene assays rely on sublethal dosing over a period of time in order to allow expression of the reporter genes. As cells directly exposed to the plasma effluent died quickly (see inhibition zone test), for this experiment, we had to treat cells in liquid culture despite several drawbacks. (V)UV does not penetrate water well leaving photon-based effects underrepresented, the liquid chemistry triggered by plasma is not well understood and the gas flow divests water that constantly needs to be replenished preventing treatment in He atmosphere, further reducing the (V)UV dose.
Reporter gene assays suggested that non-functional proteins accumulate after plasma treatment and that oxidative stress levels are high. We identified glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a suitable model to study protein inactivation by plasma by drilling down into realms three and two: its activity can be determined in vivo and in vitro, and this molecule can be analysed in its entirety by peptide-based mass spectrometry. What made GAPDH particularly attractive was that it exists in almost the same form in bacteria and in humans (being essential for life in both organisms), and it has been shown previously to be inactivated by oxidation of the active-site cysteine upon oxidative stress. To determine GAPDH activity in the bacterial cells, the cells are lysed to set free the GAPDH, which resides in the cytosol. For this assay, we chose E. coli as a model organism as it can be disrupted quantitatively, in contrast to the more sturdy B. subtilis, for which cell lysis is often incomplete. Performing Raman spectroscopy and mass spectrometry, we then investigated whether plasma-triggered inactivation of purified GAPDH is correlated to oxidation of the active-site cysteine.
In the reporter gene assay, the induction of the promoter indicative of DNA damage was weak. However, plasma has previously been shown to be capable of modifying DNA, and we assume that especially the (V)UV component of plasma is underestimated in the reporter gene assay owing to treatment of liquid culture in ambient air. In view of the vital importance of the fidelity of DNA, we thought it necessary to investigate DNA modifications following the path down through the realms of different complexity. Again referring to the inhibition zone tests, vegetative cells exposed directly to the plasma effluent died too quickly to yield surviving mutants. Thus, skipping realm three, we jumped to realm two, studying a model substrate in vitro. We chose a plasmid as substrate. Plasmids can be thought of as mini-chromosomes as they are replicated in bacteria and encode several genes. However, in contrast to the bacterial chromosomal DNA, they are small enough to be transferred back into living bacteria. Transforming E. coli with plasma-treated pUC18 plasmid, we investigated whether the DNA was still intact enough to bestow certain traits. Despite being smaller than chromosomes by three orders of magnitude, plasmids are still composed of several thousand nucleotides arranged in two large polymers that together form a double helix. The nucleotides come in four chemically distinct flavours. While the sequence of these nucleotides in a plasmid or a chromosome can be determined quite easily, these large molecules are not suitable to characterize on a molecular level the chemical modifications that plasma introduces (especially not unanticipated ones). For cataloguing plasma-induced modifications, we chose homo-oligonucleotides (DNA polymers consisting of a single strand of only few identical subunits) as simplified model substrates and analysed them by Raman spectroscopy.
2. Material and methods
2.1. Standard plasma conditions
The jets were operated with 1.4 slm He with 8.4 sccm O2. Plasma was generated at 230 Vr.m.s. with a frequency of 13.56 MHz. The UV-jet features a second gas flow of 5.6 slm He that steers particles into an exhaust. The particle-jet nozzle bends twice by 22.5° preventing exposure of samples to UV radiation. The jets were mounted in a He chamber where ambient air can be exchanged for He gas at 1.1 bar pressure. All gases used were of 5.0 grade.
We refer to the vertical axis through the jet as well as its virtual extension down to the sample as central jet axis.
2.2. Scanning electron microscopy
Non-sporulating B. subtilis 11T (ΔspoIIIC11) [20] were grown on nitrocellulose filters (Millipore) placed on Luria broth (LB) agar plates for 18 h at 37°C. Filters were removed from agar plates and placed under one of the three jets for treatment in He atmosphere. Controls were treated with He/O2 gas flow for 30 min. Filters were imaged using a scanning electron microscope (FEI Quanta 650 FEG SEM, FEI Company). Low vacuum pressure was at 80 Pa, accelerating voltage 10 kV with a working distance around 10 mm. Pictures were taken with a 12 000× magnification starting at the central jet axis and moving outwards in 1 mm steps.
2.3. Inhibition zone tests
Monolayers of B. subtilis 11T cells sprayed onto LB agar plates [12] were exposed to the plasma effluents. Cells were allowed to grow for 18 h at 37°C before measuring inhibition zones.
2.4. Reporter gene assays
Promoter regions of interest were amplified from B. subtilis 168 genomic DNA by PCR and cloned into the pDG1661 vector (from Bacillus Genetic Stock Center) in front of the promoter-less lacZ reporter gene (table 1). Plasmids were introduced into competent B. subtilis 11T cells. Stable integration of the reporter gene fusion into the amyE locus was verified by chloramphenicol resistance, streptomycin sensitivity and amylase production screening [21]. For β-galactosidase activity assays, cultures in buffered LB medium (pH 7.4) were inoculated from overnight cultures to an OD500 of 0.05 and incubated at 37°C to an OD500 of 0.4. Then, 250 µl was transferred into 96-well plates and treated with the complete-jet operated in ambient air for 30 min. Liquid levels were kept constant by continuously adding sterile aq. dest. No pH change was observed after 30 min of plasma treatment. Before performing enzyme activity assays, LB medium was exchanged for phosphate-buffered saline solution (pH 7.4). Controls were treated with He/O2 gas for 30 min.
Table 1.
Primer list.
| gene | name | sequencea |
|---|---|---|
| liaI | liaI-for-HinDIII | ATGGAAGCTTCATATGATCTCAAATATTTCGATCC |
| liaI-rev-BamHI | ATATGGATCCCAATTAATAAGAATCCGCCTATTG | |
| sigW | sigW-for-HinDIII | ATATAAGCTTCCATTATTGGGCTATAGCCAAGCGG |
| sigW-rev-BamHI | ATATGGATCCCCTCATGCACATTGCCAAGCATACG | |
| recA | recA-forb | GGCGCTAAATACGGCTGCCATTTAATC |
| recA-rev-BamHI | ATATGGATCCGCTTTCAGGACCGTATACTTC | |
| clpP | clpP-for-HinDIII | ATATAAGCTTCCTGTTGTTCTATTGGACGCGCT |
| clpP-rev-BamHI | ATATGGATCCGGATCGTATCATCATGCTTGGATC | |
| katA | katA-for-HinDIII | ACGTAAGCTTCCATCTCTGTTCTTGTAGAGAT |
| katA-rev-BamHI | ATATGGATCCCGTGAACAACACGTTCAGGAAC | |
| sigB | sigB-for-HinDIII | ATATAAGCTTCGTTTAGAGGTTATTGTGGCGGAT |
| sigB-rev-BamHI | ACGTGGATCCGCATGTCAACCAGATTTGTATACA |
aAdded restriction sites are underlined. Additional nucleotides added for cleavage efficiency are italicized.
brecA sequence carries a HinDIII restriction site directly after the primer.
2.5. In vitro GAPDH activity assay
GAPDH (Sigma Aldrich) was dissolved to a concentration of 1 mg ml−1 in aq. dest. Aliquots of 5 µl were spotted on glass coverslips (maximum spot diameter 2 mm), dried and treated in He atmosphere with the three jets. Slips were crushed into reaction tubes filled with buffer, and protein removed from the glass by agitation (10 min at 3000 r.p.m.). Protein concentrations were determined photometrically at 280 nm and using the Lambert–Beer equation. Activity assays were performed as described elsewhere [22]. In short, GAPDH samples were mixed with 50 µl of 10 mM glyceraldehyde 3-phosphate solution. The enzymatic reaction was started by adding 50 µl of 10 mM NAD+, and the increase in NADH was monitored by UV/Vis spectroscopy at 340 nm over a time course of 120 s. Enzyme activity was calculated based on the linear part of the plot. Enzyme activity per protein is expressed relative to the activity of gas-treated controls.
2.6. In vivo GAPDH activity assay
Escherichia coli K12 cells were grown in LB medium supplemented with 1% glucose to an OD580 of 0.35. Cells were harvested, resuspended in 50 mM potassium phosphate buffer pH = 7.4, and diluted to an OD540 of 0.1. Twenty microlitres was spotted on glass coverslips (maximum spot diameter 3 mm) and dried. Even distribution of cells was confirmed using bright field microscopy (Olympus BX51). Samples were treated with the different jets in He atmosphere. Afterwards, cells were washed off the coverslips with 400 µl of 50 mM potassium phosphate buffer. OD580 of all samples was measured before sonication on ice. Samples were centrifuged for 10 min to remove cell debris, and supernatant was used for activity assays (see above). Activities were corrected for their OD580 and normalized to gas-treated controls.
2.7. Mass spectrometry of GAPDH
Samples were treated for time courses between 5 and 600 s dried on glass. After treatment, samples were washed off with aq. dest. (MS grade) and protein concentration determined using Roti-Nanoquant (Roth). Sample concentrations were set to 1 µg µl−1 in 50 mM triethylammonium bicarbonate (TEAB, Sigma Aldrich) buffer containing 0.1% Rapigest (Waters) with a total volume of 25 µl. Cysteine reduction was performed with 2.5 mM tris-(2-carboxyethyl)phosphine hydrochloride (TCEP, Life Technologies) for 60 min at 60°C, followed by alkylation with 5 mM 2-iodoacetamide (IAM, Sigma Aldrich) for 15 min at 25°C in the dark. Trypsin (Sequencing grade, Promega) was added to reach a protease to substrate ratio of 1 : 100 and incubated at 37°C for 5 h. Afterwards, 2 µl of concentrated trifluoroacetic acid (Sigma Aldrich) was added to the sample, and precipitated Rapigest was removed by centrifugation. Samples were 1 : 500 diluted, and 4 µl was injected into a nanoUPLC system (Waters). Samples were loaded onto and eluted from a reversed-phase C18 column (column length 150 mm; inner diameter 75 µm; particle size 1.7 µm; pore size 130 Å; heated to 40°C) using the following gradient of solvent A (0.1% formic acid (FA, Biosolve) in aq. dest.) and B (0.1% FA in acetonitrile (Roth)) with a flow of 350 nl min−1: initial to 2 min, constant 2% B, 2–3 min, linear to 5% B; 3–45 min, linear to 50% B; 45–46, 5 min, linear to 99% B. The nanoUPLC column was coupled online to a Synapt G2-S HDMS ESI/TOF mass spectrometer (Waters) working in MSE mode with positive ionization. Spectra were recorded in resolution mode over a mass range of 50–1800 m/z with 0.5 s scan−1 using a trap collision energy ramp of 15–30 eV. The following parameters were used for the Nano Lock Spray source: capillary voltage 2.2 kV; sampling cone voltage 20 V; source temperature 100°C; desolvation temperature 200°C; cone gas flow 50 l h−1; desolvation gas flow 600 l h−1. [Glu1]-Fibrinopeptide B serving as lock mass analyte was fed through the lock spray channel with a lock mass capillary voltage of 3.0 kV.
Acquired spectra were processed in BiopharmaLynx (v. 1.3.3, Waters) to detect protein modifications. Amino acid sequence of wild-type GAPDH from rabbit muscle (UniProtKB accession number P46406) was used for spectra processing. Modifications had to be present at the same amino acids in all three replicates to be considered significant. Only peaks with an intensity of 5000 counts or more were used for modification analysis. For peptide annotation, at least two b or y ions had to be present in the tandem mass spectrum. These are the N- and C-terminal fragment ions of tryptic peptides, respectively, which occur as a result of collision-induced fragmentation at the amide bonds. Spectra were processed and searched for the following modifications (in the order of search priority): single (sulfenic acid, delta mass 17.0027 Da), triple (sulfonic acid, delta mass 47.9847 Da) and double (sulfinic acid, delta mass 31.9898 Da) oxidation of cysteines; single oxidation of methionines (delta mass 15.9949 Da); deamidation of glutamine (delta mass 0.984 Da); single oxidation of tryptophan (15.9949 Da); deamidation of asparagine and lysine (delta mass 0.984 Da); double oxidation of methionine and tryptophan (31.9898 Da); carbamido methylation at cysteines (delta mass 57.0215 Da); as well as phosphorylation at serine, threonine and tyrosine (all delta mass 79.9663 Da). All modifications were searched for as variable modifications, as carbamido methylation of cysteines caused by TCEP and IAM treatment only occurs at reduced cysteines and sulfenic acid, whereas sulfinic and sulfonic acid are not carbamido methylated.
2.8. Mutation assay
Aliquots of 500 pg of plasmid DNA (pUC18, Thermo Fisher Scientific) were spotted on glass slides (maximum spot diameter 2 mm), dried and either treated in He atmosphere with the three jets. One set of aliquots was prepared identically and treated with He/O2 gas for 10 min. Control samples were left untreated (not dried or exposed to gas or plasma). Dried samples were washed off the glass with aq. dest. and DNA concentrations determined using a Nanodrop spectrometer (Peqlab). Samples of 400 pg of DNA were transformed into MgCl2-competent E. coli DH5α cells. Dilution series of the transformed cells were plated on LB agar plates containing ampicillin and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal, Sigma Aldrich). Plates were incubated for 18 h at 37°C and stored at 4°C for additional 24 h before colony forming units (CFUs) and their colours were determined.
2.9. Raman spectroscopy
Synthetic DNA oligomers (Thermo Fischer Scientific) consisting of 18 cytosines (dC18) or guanines (dG18) were set to a concentration of 1 mM in aq. dest. Single-stranded oligomers were hybridized to form double-stranded oligomers by mixing equal amounts of dC18 and dG18, heating them to 98°C for 2 min, and hybridizing them by allowing them to cool to room temperature over 4 h. Aliquots of 20 µl were spotted on glass (maximum spot diameter 3 mm), dried and treated with the different jets for 30 min in He atmosphere. Controls were treated with He/O2 gas. Samples were analysed by confocal Raman spectroscopy with a WITec alpha300 RAS microscope (WITec). A single-frequency diode laser with a wavelength of 785 nm was used for excitation (P ∼ 100 mW). The laser beam was coupled into the microscope through a single-mode optical fibre and then focused on the sample surface with a 100× objective (NA = 0.9). The Raman scattered light was collected with the same objective and registered by a back-illuminated charged-coupled device detector (128 × 1024 pixels, kept at −60°C) after passing a multimode fibre (100 µm diameter) and a diffraction grating (300 grooves per mm). A spectral resolution of ±3 cm−1 was achieved in the spectral range from −100 to 3250 cm−1. Raman spectra were taken with a total integration time of 60 s for each spectrum (2.5 s, 24 acquisitions).
Raman datasets were managed using WITec Project 2.08 (WITec). Prior to any analysis, spectra were treated with user-controlled removal of cosmic rays. For background subtraction, areas with no Raman bands were defined in which Raman intensity values were averaged over one pixel. Other areas were interpolated linearly resulting in a background that was subtracted.
3. Results
3.1. Damage to the cellular envelope
The impact of plasma on the bacterial envelope, which is composed largely of peptidoglycan, was investigated by SEM (figure 2), which allows imaging of live cells without any fixation or staining. We set out (i) to assess the damage to the envelope directly under the jet as a function of treatment time, (ii) to correlate this envelope damage with growth inhibition around the treatment site (the ‘growth inhibition zone’) using bacterial monolayers on agar plates, and (iii) to differentiate the contribution to envelope damage by UV and the particle components of the plasma effluent. To this end, sporulation-deficient B. subtilis was grown on a nitrocellulose membrane and exposed to complete-jet, UV-jet or particle-jet for up to 30 min. The lateral extension of treatment effects was determined up to 4 mm outward from the central jet axis.
Figure 2.

Dependence of cell envelope damage on the distance from the central jet axis. Bacillus subtilis was exposed to gas flow (control) or treated with UV-jet, particle-jet or complete-jet. Photographs show inhibition zones after overnight incubation. Jet axes marked by arrows (15 × 15 mm details are shown). SEM images of B. subtilis grown on nitrocellulose membranes were taken at the jet axis moving outwards by increments of 1 mm (5 × 5 µm details are shown). (Online version in colour.)
When cells were exposed to the effluent of the complete-jet for 1 min, the first sign of damage was removal of the extracellular matrix, which became visible at the central jet axis. After 3 min of exposure, the top cell layer was removed and after 30 min the sample, approximately three cell layers thick, was etched down to the nitrocellulose membrane. Etching effects were observed in a circle with a 3 mm radius around the central jet axis, but from 4 mm outwards, the cells looked no different from gas-only treated controls. However, bacterial growth was inhibited over a much wider area, affecting cells up to 7.5 mm from the focus after a 3 min treatment and preventing growth on the entire Petri dish (43.5 mm radius) after 30 min exposure. The effects of treatment with the particle-jet were similar to those of the complete-jet although 30 min of treatment resulted in visible etching only up to 2 mm. By contrast, no etching or changes to the bacterial envelope were detected after 30 min of UV-jet treatment, although inhibition zones of 1.5 mm radius indicated that cells were inactivated around the central jet axis.
3.2. Measuring cellular stress with reporter genes
To identify the bacterial macromolecules most susceptible to inactivation in vivo, we expressed stress indicator genes using single-copy transcriptional lacZ reporter gene fusions in sporulation-deficient B. subtilis. Promoters of sigW and the liaIHGFSR operon were used to monitor general damage to the cellular envelope [23] and cell wall-specific stress [24], respectively; DNA damage was monitored using recA [25] and protein damage using clpP [26]; katA and sigB served as indicators for oxidative stress and activation of the general stress response, respectively [27,28].
Bacillus subtilis growing in liquid cultures were treated for 30 min with the complete-jet only. The jet was operated in ambient air to facilitate replenishing of the culture medium, which dries up quickly due to the gas flow. β-Galactosidase activity assays indicated that complete-jet induced strong activation of katA and clpP expression (with increases by a factor of 8.2 and 8.6, respectively), whereas the lia operon and recA were activated less remarkably (table 2). These data suggest that oxidative stress levels are high in the liquid cultures exposed to plasma, and that a significant amount of protein is damaged beyond repair. Mechanical damage to the cell envelope and DNA damage appear to be relatively less important under these treatment conditions.
Table 2.
Cellular stress response monitored with reporter gene assays.
| lacZ fusion | induced by | MU pre treatmenta | MU post treatmenta | induction factor |
|---|---|---|---|---|
| sigW | membrane stress | 9 ± 1 | 49 ± 3 | 4.7 ± 0.5 |
| liaH | cell wall stress | 11 ± 5 | 58 ± 23 | 5.4 ± 0.5 |
| recA | DNA damage | 29 ± 3 | 90 ± 6 | 3.1 ± 0.4 |
| clpP | protein stress | 81 ± 7 | 692 ± 32 | 8.6 ± 0.4 |
| katA | oxidative stress | 85 ± 11 | 704 ± 114 | 8.3 ± 0.8 |
| sigB | general stress | 11 ± 2 | 51 ± 8 | 6.0 ± 0.4 |
aAverage Miller units (MUs) and SDs representing six independent experiments.
3.3. Inactivation of protein
GAPDH was used as a model protein to investigate inactivation by plasma in vitro and in vivo. An enzyme of the central metabolism, GAPDH catalyses a reaction in glycolysis and is highly conserved from bacteria to man. Its activity can be readily measured in vitro and in vivo by monitoring the reduction of NAD+ to NADH at 340 nm.
GAPDH dried on glass was treated with the different jets to determine whether plasma components cause enzyme inactivation in vitro (figure 3a). Following exposure to the complete effluent for 10 min, only 20% of the enzymatic activity remained. By comparison, 40% and 75% of the activity was retained after treatment with the particle-jet and UV-jet, respectively. As clpP reporter gene induction indicated that protein damage occurs in vivo, GAPDH activity was determined in live E. coli K12 cells dried on glass (figure 3b). All three jets reduced GAPDH activity in the living cells much more rapidly than in vitro, with the complete-jet fully inactivating the enzyme within 10 min. The particle-jet, also, caused almost complete inactivation after 10 min, and the UV-jet reduced activity by around 60%.
Figure 3.
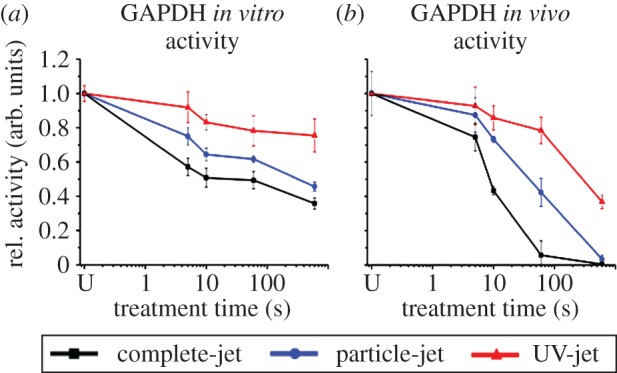
Impact of plasma on glyceraldehyde 3-phosphate dehydrogenase activity. (a) Purified GAPDH was exposed to (V)UV photons, particles or the complete plasma effluent. Relative enzyme activities were calculated and are expressed in arbitrary units. (b) In vivo GAPDH activity was determined after treatment of E. coli K12 cells. SDs for triplicate experiments are given. U denotes untreated controls. (Online version in colour.)
There was no significant loss or fragmentation of GAPDH in vitro, as assessed by SDS–PAGE (figure 4a). Analysis by Raman spectroscopy indicated cysteine overoxidation to sulfonic acid (figure 4b, peak annotation taken from [29]). Mass spectrometry revealed overoxidation of the catalytic cysteine, Cys150 (figure 5). Oxidation of this active-site cysteine is known to render GAPDH inactive under oxidative stress conditions [30] and overoxidation to sulfonic acid is irreversible [31].
Figure 4.
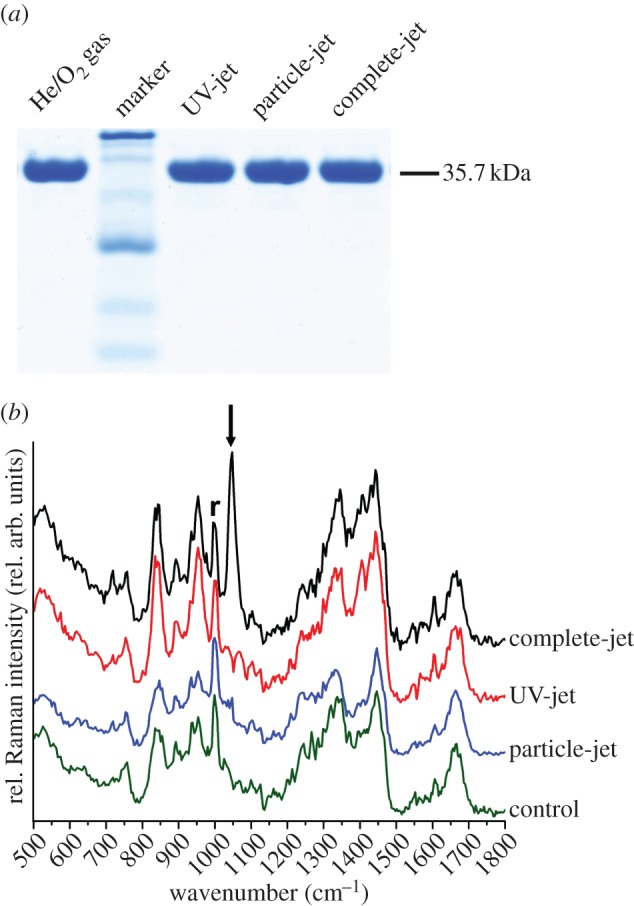
Etching and modification of dried glyceraldehyde 3-phosphate dehydrogenase. (a) SDS–PAGE of GAPDH samples after treatment with the complete-jet (C), particle-jet (P) and UV-jet (UV) for 10 min showed no loss of band intensity or fragmentation. U denotes untreated control. (b) Raman spectra are cascaded for better visibility. All relative intensity changes were normalized to the phenylalanine peak at 1003 cm−1 (r). Samples were treated for 10 min with the three different jets or with He/O2 gas-only (control). The SO2 stretch peak at 1045 cm−1 (arrow) indicates formation of sulfonic acid [29]. (Online version in colour.)
Figure 5.

Mass spectrometry reveals oxidation of the catalytic cysteine of glyceraldehyde 3-phosphate dehydrogenase. The mirror plot shows sections of the deisotoped mass spectrum of the control (top) and the spectrum of GAPDH treated for 600 s with the complete-jet (bottom). Peaks identified as tryptic peptides containing the catalytic Cys150 (bold) are annotated with their sequence and modifications. T35: tryptic peptide number 35 of GAPDH, also found in this m/z range.
3.4. Damage to DNA
To investigate the effects of plasma treatment on DNA, we used different in vitro approaches. In vivo studies were hampered by vegetative bacteria dying quickly when exposed to the plasma effluent. We were unable to find treatment conditions with the complete-jet that yielded surviving but mutated bacteria. The impact on the structure and function of large DNA molecules was tested using the pUC18 plasmid. This plasmid carries the genetic information for two enzymes, β-lactamase, which confers ampicillin resistance, and β-galactosidase (lacZ), which can convert colourless X-Gal to a blue dye. When competent E. coli DH5α cells are transformed with pUC18, growth of colonies on ampicillin-containing LB agar indicates successful transformation. Blue colonies indicate the presence of functional β-galactosidase, whereas in white colonies a functional β-galactosidase is absent.
Aliquots of pUC18 plasmid were dried on glass and treated with the three jets. Equal quantities of DNA were then transformed into E. coli, and the number of blue and white colonies determined after incubation (figure 6). About 0.5% of the colonies were white after transformation of control plasmid that was neither dried nor gas-treated. This increased to 2% after transformation of plasmid treated with the He/O2 gas mix alone for 10 min, probably owing to damage induced by drying the DNA on glass. Treatment with the complete-jet was most effective in reducing the number of CFUs. After a 10 min exposure, total CFU counts were reduced to 3.5% of the level of the gas-only treated control, and the majority of colonies were white. The highest absolute number of white colonies was observed after treatment with the UV-jet; in this case, the reduction in CFUs was roughly 50% and white CFUs increased from 25% after a 1 min exposure to 60% after 10 min. The particle-jet caused a more rapid reduction in CFUs than the UV-jet. For all three jets, the increase in white colonies indicates an increase in mutation rate in the lacZ gene. Agarose gel electrophoresis showed that the DNA structure is also altered (figure 6 inset). pUC18 plasmid DNA is relaxed by treatment with the UV-jet, indicating the induction of single-stranded breaks. Further, the loss in band intensity, determined by measuring absorption at 260 nm, indicates that the DNA is partially fragmented by treatment with the particle-jet and the complete-jet.
Figure 6.
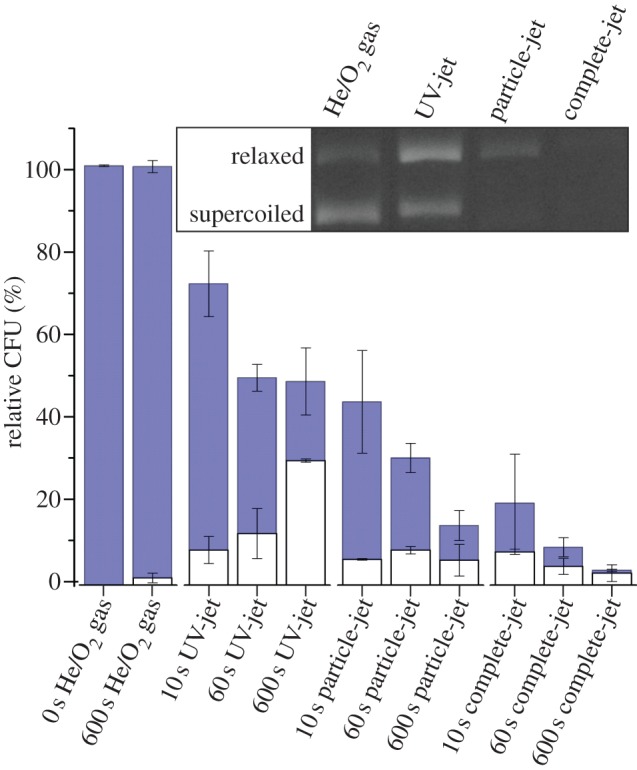
Destruction and mutation of plasmid DNA by plasma treatment. Blue colonies (carrying plasmids with intact β-galactosidase) and white colonies (in which the plasmid encoding β-galactosidase is inactive) were counted after transformation of E. coli with untreated, He/O2 gas mix-exposed or plasma-treated plasmids. CFUs were normalized to untreated controls. Experiments were repeated five times independently. Plasmid treated for 600 s was separated by agarose gel electrophoresis (figure inset). (Online version in colour.)
Next, DNA modification at the molecular level was investigated. Confocal Raman spectroscopy was used to characterize the intramolecular bonds within single-stranded DNA oligomers consisting of 18 guanines. Raman spectra of dried dG18 were taken after a 30 min treatment with the different jets. By comparing the spectra taken before plasma treatment, we identified a number of chemical modifications (figure 7a) that were assigned using annotations of structural peaks taken from references [32–34]. The peak at 920 cm−1 is generated by vibrations of coupled nucleobases: a massive loss of relative intensity after treatment with the complete-jet was observed. The peak at wavenumber 1416 cm−1 is generated by vibrations in the ribose backbone: both the particle-jet and complete-jet caused a decrease in peak height, indicating strand breaks or modifications of the DNA backbone. Treatment with the complete-jet also resulted in an intensity decrease at 674 cm−1, indicating modifications at the nucleobase, as the three peaks between 578 and 680 cm−1 are specific for purines (figure 7a). The particle-jet had a negligible effect on the nucleobase, but notable intensity changes were observed after treatment with the UV-jet.
Figure 7.
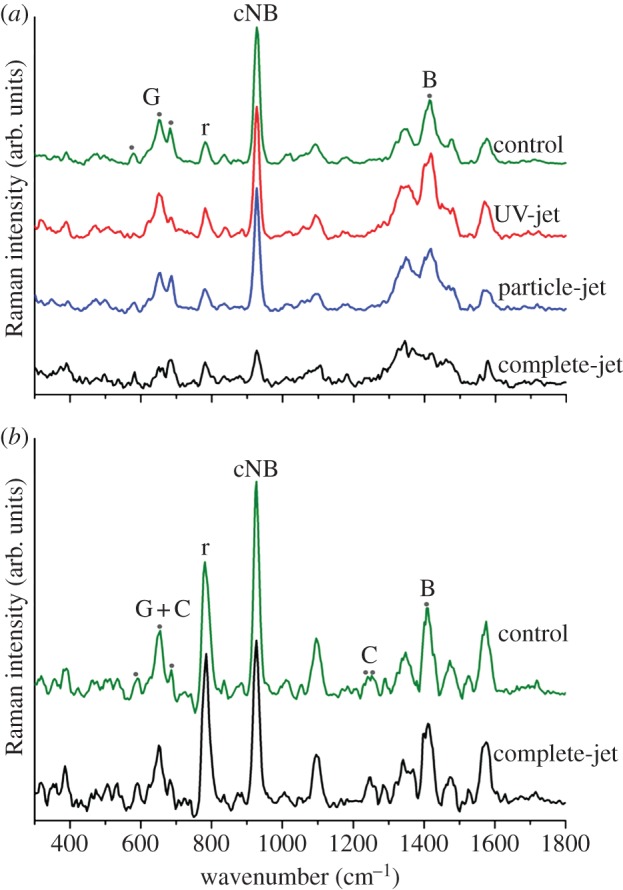
DNA modifications analysed by Raman spectroscopy. (a) The spectra of single-stranded guanine oligomers and (b) double-stranded G : C DNA oligomers are cascaded for better visibility. All relative intensity changes are compared with the ν(PO2) peak at 780 cm−1. Changes discussed in the text are highlighted by dark grey dots. G, guanine specific; C, cytosine specific; B, backbone specific; cNB, coupled nucleobases; r, reference peak. (Online version in colour.)
As intracellular DNA is usually double-stranded, we investigated the effects of plasma on double-stranded dC18 : dG18 oligomers using Raman spectroscopy (figure 7b). Only minor changes were detected: the purine nucleobase peaks were virtually unchanged, whereas certain pyrimidine-specific nucleobase peaks, such as the one at 1245 cm−1, were altered slightly, and at the 1416 cm−1 backbone peak, the shoulder at 1430 cm−1 was lost after treatment with the complete-jet.
4. Discussion
Cold atmospheric-pressure plasmas emit a cocktail of reactive particles and (V)UV photons. In this study, we modified plasma jets using a lateral He flow to split the particles from the (V)UV photons. This allowed us to study the effects on bacteria and macromolecules of emitted (V)UV photons alone, particles alone or photons and particles in combination.
When bacterial monolayers were treated with the complete-jet, the inhibition zones extended further than after treatment with either the particle-jet or the UV-jet and cells were killed further away from the jet axis than the added radii of the (V)UV and particle inhibition zones. This synergistic effect may result from a photochemical interaction between the photons and particles emitted by the combined-jet, resulting in increased toxicity of the particle mix reaching the sample (physical synergy). For example, we previously showed that UV photons emitted from a plasma jet are capable of activating oxygen [17], generating activated water clusters. An alternative or additional possibility is that the capacity of the bacteria to neutralize stress is overwhelmed when cells simultaneously face stress from (V)UV photons and reactive particles (biological synergy).
The effluent of the µAPPJ is capable of chemical etching [13]. SEM images indicate that chemical etching is an inactivation mechanism under or near the complete-jet axis. No etching or other signs of damage were seen beyond 4 mm. Although the UV-jet produced small inhibition zones in bacterial monolayers, no etching was observed by SEM, indicating that the emitted (V)UV photons are capable of inactivating bacteria without physically damaging the cell envelop. The particle-jet caused some etching but was less efficient than the complete-jet. This suggests that there are synergistic effects between particles and V(UV) photons (physical synergy) and we postulate that particles emitted from the complete-jet are activated on their way from the plasma to the sample by the co-emitted photons.
The inhibition zones extend well beyond areas of etching, implying that cells distant from the jet axis are killed by a different mechanism. The μAPPJ produces ozone [14], which is a very strong oxidant and cytotoxin. At treatment times exceeding 6 min, ozone plays a significant role in bacterial inactivation when the jets are operated in an enclosing He atmosphere [17]. In contrast to treatment in ambient air, where ozone is transported away by the buoyancy force acting on the He feed gas, in an He atmosphere ozone is transported across the sample surface. It is stable enough to reach cells even at the very edge of the Petri dishes, causing oxidative stress.
In addition to destroying the cellular envelope by etching [10], other mechanisms of cell inactivation have previously been proposed, such as damage to DNA or proteins [7,9]. We monitored the expression of different reporter gene fusions after treating cells with plasma in liquid culture for 30 min. UV-induced DNA stress was not prevalent. Note, however, that (V)UV photons penetrate liquids weakly and may inactivate bacteria more effectively on dry surfaces. katA–lacZ, an indicator of oxidative stress, and clpP–lacZ, an indicator for protein stress, were most strongly induced, suggesting that under these experimental conditions plasma primarily causes oxidative stress and protein damage, with the two likely to be connected. The jet that we used produces a variety of ROS, predominantly atomic oxygen, ozone [13], and singlet delta O2 (a1Δg) [14], but also protonated water clusters and unspecified reactive species stemming from photochemical reactions [17]. These could trigger the observed bacterial reaction by oxidizing macromolecules either directly or indirectly through liquid chemistry reactions causing formation of hydroxyl radicals or H2O2. Indeed, several proteins are highly sensitive to oxidative stress, among them the glycolytic enzyme GAPDH, which is known to be sensitive to oxidation by H2O2. The plasma-based protein inactivation indicated by the clpP reporter gene assay may, at least in part, be attributed to protein oxidation.
Previous studies showed that treatment of lysozyme with a low-frequency plasma jet causes a mass shift and protein unfolding [35], and that the µAPPJ is capable of etching protein [12]. Here, we specifically investigated protein oxidation as inactivation mechanism. The enzyme GAPDH was used as a model protein. Its enzymatic activity can be easily measured and oxidation of the cysteine in the active site (Cys150) is a well-described inactivation mechanism [36]. Dried GAPDH was inactivated to different extents by the three jets and in each case inactivation correlated with oxidation of Cys150. Using 10 s exposures, the complete-jet reduced GAPDH activity by 50%, the particle-jet resulted in 45% inactivation and the UV-jet caused 10–15% inactivation. Mass spectrometric analysis of the peptide containing Cys150 confirmed very low oxidation rates after short in vitro treatments with the UV-jet, which probably result from reactions with activated water clusters or products of photochemical reactions with gas impurities [17].
The inactivation of GAPDH is much more effective in the living cell than it is under in vitro conditions. Using dried bacteria containing GAPDH, treatment with complete-jet for 10 s caused greater inactivation rates than the combined rate for the particle-jet and UV-jet. This synergistic effect was observed only in the cellular environment and not using dried GAPDH. We therefore suggest that it might be linked to plasma-induced chemistry in the liquid cellular milieu, possibly enhanced by plasma-triggered biological processes.
Treatment with the complete-jet for 10 min fully inactivated GAPDH. At this treatment time, even the UV-jet achieved a 60% GAPDH inactivation in vivo. The (V)UV photons emitted from the UV-jet are unlikely to be responsible for GAPDH inactivation. In previous studies, even 10 min of more intense UV radiation at wavelengths better suited to protein inactivation (260–280 and 310–315 nm) failed to inactivate GAPDH in solution [37,38]. It seems likely that, in addition to secondary reactions of photons and particles with cellular components in the cellular milieu (liquid chemistry), the ozone produced by the jets plays a significant role in GAPDH inactivation at this exposure time. Ozone forms ROS [39], which can oxidize GAPDH, an effect that should not be neglected, given its biological effectiveness. The additive and synergistic effects of plasma-emitted (V)UV photons and particles, however, are best studied in an He atmosphere using treatment times of 10–60 s.
Fidelity of cellular DNA is of the utmost importance not only for bacterial survival but also for proliferation. Previous work has shown that in vitro plasma treatment damages plasmid DNA by inducing single- and double-stranded breaks [8] and by modifying nucleobases [12], and that it is mutagenic for bacterial spores [11]. We failed to establish treatment conditions that allowed vegetative bacterial cells to survive direct exposure to the plasma effluent. For that reason, DNA was exposed in vitro to the three different jets, then analysed for DNA modifications using Raman spectroscopy. In addition, dried pUC18 plasmid was treated with the three different jets and then transformed into E. coli cells. Decreases in CFUs may be due to non-functional plasmids, for example owing to backbone breaks or faulty origin of replication, or to β-lactamase mutation, resulting in antibiotic sensitivity. The observed reductions in transformation efficiencies, in combination with increases in white CFUs (which contain the plasmid but are no longer capable of converting X-Gal), indicate a mutagenic effect, at least for plasma treatment of DNA in vitro. The complete-jet and the particle-jet both reduced CFU counts effectively. The UV-jet was less efficient in reducing CFU counts and yielded the highest mutant per survivor rate. It appears that the increased efficiency of the combined-jet results from the additive effects of photons and particles. While the UV-jet predominantly induces modifications of the nucleobases and has little effect on the backbone, the particle-jet causes significant damage to the backbone and has only a minor effect on the guanine bases; the complete-jet efficiently nicks the backbone and modifies nucleobases. Raman spectroscopy showed that, in vitro, single-stranded DNA is much more sensitive to plasma treatment than double-stranded DNA.
Both the SEM studies and the GAPDH activity assay indicate that the complete-jet features synergistic inactivation properties, most notably in vivo. It is known that effluents of cold atmospheric-pressure plasmas can interact with water to create a huge variety of different radicals and toxic products [40], and of course water is always present inside and between living cells. Secondary reactions in liquid environments, which we refer to as ‘liquid chemistry’, will need to be studied in detail in the future. They seem to contribute significantly to the bactericidal properties of plasma, especially when complete-jet is used. Another possible explanation for the high efficiency of the complete-jet is that, as it contains both (V)UV and particles, it causes the greatest variety of stress to the cell and its components. The complexity of the effluent composition is further increased by reactions between the (V)UV photons and particles within the effluent, a phenomenon that we term ‘physical synergy’. The simultaneous action of many different stressors—we call this ‘biological synergy’—likely overwhelms the cellular repair and detoxification machineries so that dysfunctional macromolecules accumulate and the cells die.
Cold atmospheric-pressure plasmas are highly effective inactivators of microorganisms. Plasmas have therefore found their way into sterilization applications, disinfection and wound treatment. In particular, the treatment of eczema and chronic wounds has potential, and early clinical trials have indicated that plasma expedites the wound healing process [41–44], probably owing to a combination of stimulating the healing process and inactivating pathogens. Our studies on the mechanism of bacterial inactivation indicate that the cell envelop, proteins and DNA are all affected by plasma treatment. This multi-targeted attack appears to overwhelm the cellular repair and stress response systems more effectively than treatments that focus on any single bacterial target. An in-depth understanding of the contributions of individual plasma components to the biological effects will further inform the design and tuning of plasmas for particular clinical applications. With anti-cancer applications on the horizon [45,46], this characterization should not stop at the bacterial cell but extend to human cells and tissues to provide a molecular basis for assessing the benefits and the risks of plasma exposure. Only with a detailed knowledge of the effects of plasmas and their components on bacteria and humans will the full potential of plasma sources in medicine be discovered.
Acknowledgements
We acknowledge E. Bründermann for his help with the Raman experimental set-up. We thank him as well as P. Awakowicz, L. Leichert and F. Narberhaus for valuable comments and discussions. This manuscript was in part written at the 2013 Scientific Writing workshop of the RUB Research School, and we thank R. Gallagher for his many helpful suggestions.
Funding statement
This work was supported by the German Research Foundation (grant no. PAK728 to J.E.B. and J.B., grant no. PAK816 to M.H.N.), the Research School of the Ruhr University Bochum (grant to J.W.L.), the BMBF (grants FKZ 05KS7PC2 & FKZ 05K10PCA to M.H.N.) and the Research Department Plasma with Complex Interactions of the Ruhr University Bochum. J.E.B. acknowledges the financial support for the mass spectrometer by the state of North Rhine Westphalia (Forschungsgroßgeräte der Länder). M.H.N. acknowledges the financial support for the Raman microscope by the German Research Foundation (DFG, Großgeräte der Länder).
References
- 1.Kong MG, Kroesen G, Morfill G, Nosenko T, Shimizu T, van Dijk J, Zimmermann JL. 2009. Plasma medicine: an introductory review. New J. Phys. 11, 115012 ( 10.1088/1367-2630/11/11/115012) [DOI] [Google Scholar]
- 2.Gaunt LF. 2006. Bactericidal action of the reactive species produced by gas-discharge nonthermal plasma at atmospheric pressure: a review. IEEE Trans. Plasma Sci. 34, 1257–1269. ( 10.1109/TPS.2006.878381) [DOI] [Google Scholar]
- 3.Maisch T, Shimizu T, Isbary G, Heinlin J, Karrer S, Klämpfl TG, Li YF, Morfill G, Zimmermann JL. 2012. Contact-free inactivation of Candida albicans biofilms by cold atmospheric air plasma. Appl. Environ. Microbiol. 78, 4242–4247. ( 10.1128/AEM.07235-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raballand V, Benedikt J, Wunderlich J, von Keudell A. 2008. Inactivation of Bacillus atrophaeus and of Aspergillus niger using beams of argon ions, of oxygen molecules, and of oxygen atoms. J. Phys. D 41, 115207 ( 10.1088/0022-3727/41/11/115207) [DOI] [Google Scholar]
- 5.Yasuda H, Miura T, Kurita H, Takashima K, Mizuno A. 2010. Biological evaluation of DNA damage in bacteriophages inactivated by atmospheric pressure cold plasma. Plasma Process. Polym. 7, 301–308. ( 10.1002/ppap.200900088) [DOI] [Google Scholar]
- 6.Park BJ, Lee DH, Park J-C, Lee I-S, Lee K-Y, Hyun SO, Chun M-S, Chung K-H. 2003. Sterilization using a microwave-induced argon plasma system at atmospheric pressure. Phys. Plasmas 10, 4539–4544. ( 10.1063/1.1613655) [DOI] [Google Scholar]
- 7.Kvam E, Davis B, Mondello F, Garner AL. 2012. Non-thermal atmospheric plasma rapidly disinfects multidrug-resistant microbes by inducing cell surface damage. Antimicrob. Agents Chemother. 56, 2028–2036. ( 10.1128/AAC.05642-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Connell D, Cox LJ, Hyland WB, McMahon SJ, Reuter S, Graham WG, Gans T, Currell FJ. 2011. Cold atmospheric pressure plasma jet interactions with plasmid DNA. Appl. Phys. Lett. 98, 043701 ( 10.1063/1.3521502) [DOI] [Google Scholar]
- 9.Kalghatgi S, Fridman A, Azizkhan-Clifford J, Friedman G. 2012. DNA damage in mammalian cells by non-thermal atmospheric pressure microsecond pulsed dielectric barrier discharge plasma is not mediated by ozone. Plasma Process. Polym. 9, 726–732. ( 10.1002/ppap.201100156) [DOI] [Google Scholar]
- 10.Kim S-M, Kim J-I. 2006. Decomposition of biological macromolecules by plasma generated with helium and oxygen. J. Microbiol. 44, 466–471. [PubMed] [Google Scholar]
- 11.Wang LY, Huang ZL, Li G, Zhao HX, Xing XH, Sun WT, Li HP, Gou ZX, Bao CY. 2010. Novel mutation breeding method for Streptomyces avermitilis using an atmospheric pressure glow discharge plasma. J. Appl. Microbiol. 108, 851–858. ( 10.1111/j.1365-2672.2009.04483.x) [DOI] [PubMed] [Google Scholar]
- 12.Lackmann J-W, Schneider S, Narberhaus F, Benedikt J, Bandow JE. 2012. Characterization of damage to bacteria and bio-macromolecules caused by (V)UV radiation and particles generated by a microscale atmospheric pressure plasma jet. In Plasma for bio-decontamination, medicine and food security (eds Machala Z, Hensel K, Akishev Y.), pp. 17–29. Heidelberg, Germany: Springer. [Google Scholar]
- 13.Ellerweg D, Benedikt J, von Keudell A, Knake N, Schulz-von der Gathen V. 2010. Characterization of the effluent of a He/O2 microscale atmospheric pressure plasma jet by quantitative molecular beam mass spectrometry. New J. Phys. 12, 013021 ( 10.1088/1367-2630/12/1/013021) [DOI] [Google Scholar]
- 14.Liu DX, Rong MZ, Wang XH, Iza F, Kong MG, Bruggeman P. 2010. Main species and physicochemical processes in cold atmospheric-pressure He + O2 plasmas. Plasma Process. Polym. 7, 846–865. ( 10.1002/ppap.201000049) [DOI] [Google Scholar]
- 15.Waskoenig J, Niemi K, Knake N, Graham LM, Reuter S, Schulz-von der Gathen V, Gans T. 2010. Atomic oxygen formation in a radio-frequency driven micro-atmospheric pressure plasma jet. Plasma Sources Sci. Technol. 19, 045018 ( 10.1088/0963-0252/19/4/045018) [DOI] [Google Scholar]
- 16.Bahre H, Lange H, Schulz-von der Gathen V, Foest R. 2011. Vacuum ultraviolet (VUV) emission of an atmospheric pressure plasma jet (µ-APPJ) operated in helium-oxygen mixtures in ambient air. Acta Tech. 56, T199. [Google Scholar]
- 17.Schneider S, Lackmann J-W, Ellerweg D, Denis B, Narberhaus F, Bandow JE, Benedikt J. 2012. The role of VUV radiation in the inactivation of bacteria with an atmospheric pressure plasma jet. Plasma Process. Polym. 9, 561–568. ( 10.1002/ppap.201100102) [DOI] [Google Scholar]
- 18.Azrague K, Bonnefille E, Pradines V, Pimienta V, Oliveros E, Maurette MT, Benoit-Marquié F. 2005. Hydrogen peroxide evolution during V-UV photolysis of water. Photochem. Photobiol. Sci. 4, 406–408. ( 10.1039/b500162e) [DOI] [PubMed] [Google Scholar]
- 19.Schneider S, Lackmann J-W, Narberhaus F, Bandow JE, Denis B, Benedikt J. 2011. Separation of VUV/UV photons and reactive particles in the effluent of a He/O2 atmospheric pressure plasma jet. J. Phys. D 44, 295201 ( 10.1088/0022-3727/44/29/295201) [DOI] [Google Scholar]
- 20.Ionesco H, Michel J, Cami B, Schaeffer P. 1970. Symposium on bacterial spores: II. Genetics of sporulation in Bacillus subtilis Marburg. J. Appl. Bacteriol. 33, 13–24. ( 10.1111/j.1365-2672.1970.tb05230.x) [DOI] [PubMed] [Google Scholar]
- 21.Cutting SM, Vander Horn PB. 1990. Genetic analysis. In Molecular biological methods for Bacillus (eds Harwood CR, Cutting SM.), pp. 27–74. Chichester, UK: John Wiley & Sons. [Google Scholar]
- 22.Tanii H, Hashimoto K. 1985. Effect of acrylamide and related compounds on glycolytic enzymes of rat brain. Toxicol. Lett. 26, 79–84. ( 10.1016/0378-4274(85)90188-2) [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Frederick KL, Helmann JD. 1998. Promoter recognition by Bacillus subtilis σW: autoregulation and partial overlap with the σX regulon. J. Bacteriol. 180, 3765–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, Hamoen L, Metzler-Nolte N, Sahl HG, Bandow JE. 2012. Proteomic response of Bacillus subtilis to lantibiotics reflects differences in interaction with the cytoplasmic membrane. Antimicrob. Agents Chemother. 56, 5749–5757. ( 10.1128/AAC.01380-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayora S, Carrasco B, Cárdenas PP, César CE, Cañas C, Yadav T, Marchisone C, Alonso JC. 2011. Double-strand break repair in bacteria: a view from Bacillus subtilis. FEMS Microbiol. Rev. 35, 1055–1081. ( 10.1111/j.1574-6976.2011.00272.x) [DOI] [PubMed] [Google Scholar]
- 26.Krüger E, Witt E, Ohlmeier S, Hanschke R, Hecker M. 2000. The Clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J. Bacteriol. 182, 3259–3265. ( 10.1128/JB.182.11.3259-3265.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bol DK, Yasbin RE. 1990. Characterization of an inducible oxidative stress system in Bacillus subtilis. J. Bacteriol. 172, 3503–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hecker M, Pané-Farré J, Völker U. 2007. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu. Rev. Microbiol. 61, 215–236. ( 10.1146/annurev.micro.61.080706.093445) [DOI] [PubMed] [Google Scholar]
- 29.Rokos H, Moore J, Hasse S, Gillbro JM, Wood JM, Schallreuter KU. 2004. In vivo fluorescence excitation spectroscopy and in vivo Fourier-transform Raman spectroscopy in human skin: evidence of H2O2 oxidation of epidermal albumin in patients with vitiligo. J. Raman Spectrosc. 35, 125–130. ( 10.1002/jrs.1114) [DOI] [Google Scholar]
- 30.Bedhomme M, Adamo M, Marchand CH, Couturier J, Rouhier N, Lemaire SD, Zaffagnini M, Trost P. 2012. Glutathionylation of cytosolic glyceraldehyde-3-phosphate dehydrogenase from the model plant Arabidopsis thaliana is reversed by both glutaredoxins and thioredoxins in vitro. Biochem. J. 445, 337–347. ( 10.1042/BJ20120505) [DOI] [PubMed] [Google Scholar]
- 31.Brandes N, Schmitt S, Jakob U. 2009. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 11, 997–1014. ( 10.1089/ars.2008.2285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prescott B, Steinmetz W, Thomas GJ. 1984. Characterization of DNA structures by laser Raman-spectroscopy. Biopolymers 23, 235–256. ( 10.1002/bip.360230206) [DOI] [PubMed] [Google Scholar]
- 33.Small EW, Peticolas WL. 1971. Conformational dependence of Raman scattering intensities from polynucleotides. Biopolymers 10, 1377–1416. ( 10.1002/bip.360100811) [DOI] [PubMed] [Google Scholar]
- 34.Thomas GJ, Jr, Benevides JM, Overman SA, Ueda T, Ushizawa K, Saitoh M, Tsuboi M. 1995. Polarized Raman-spectra of oriented fibers of A-DNA and B-DNA. Biophys. J. 68, 1073–1088. ( 10.1016/S0006-3495(95)80282-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takai E, Kitano K, Kuwabara J, Shiraki K. 2012. Protein inactivation by low-temperature atmospheric pressure plasma in aqueous solution. Plasma Process. Polym. 9, 77–82. ( 10.1002/ppap.201100063) [DOI] [Google Scholar]
- 36.Hwang NR, Yim SH, Kim YM, Jeong J, Song EJ, Lee Y, Lee JH, Choi S, Lee KJ. 2009. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 423, 253–264. ( 10.1042/BJ20090854) [DOI] [PubMed] [Google Scholar]
- 37.Maloletkina OI, Markossian KA, Chebotareva NA, Asryants RA, Kleymenov SY, Poliansky NB, Muranov KO, Makeeva VF, Kurganov BI. 2012. Kinetics of aggregation of UV-irradiated glyceraldehyde-3-phosphate dehydrogenase from rabbit skeletal muscle. Effect of agents possessing chaperone-like activity. Biophys. Chem. 163–164, 11–20. ( 10.1016/j.bpc.2012.02.001) [DOI] [PubMed] [Google Scholar]
- 38.Voss P, Hajimiragha H, Engels M, Ruhwiedel C, Calles C, Schroeder P, Grune T. 2007. Irradiation of GapDH: a model for environmentally induced protein damage. Biol. Chem. 388, 583–592. ( 10.1515/BC.2007.068) [DOI] [PubMed] [Google Scholar]
- 39.Glaze WH. 1986. Reaction products of ozone: a review. Environ. Health Perspect. 69, 151–157. ( 10.1289/ehp.8669151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oehmigen K, Winter J, Hähnel M, Wilke C, Brandenburg R, Weltmann K-D, von Woedtke T. 2011. Estimation of possible mechanisms of Escherichia coli inactivation by plasma treated sodium chloride solution. Plasma Process. Polym. 8, 904–913. ( 10.1002/ppap.201000099) [DOI] [Google Scholar]
- 41.Emmert S, et al. 2013. Atmospheric pressure plasma in dermatology: ulcus treatment and much more. Clin. Plasma Med. 1, 24–29. ( 10.1016/j.cpme.2012.11.002) [DOI] [Google Scholar]
- 42.Isbary G, et al. 2012. Successful and safe use of 2 min cold atmospheric argon plasma in chronic wounds: results of a randomized controlled trial. Br. J. Dermatol. 163, 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Isbary G, Morfill G, Zimmermann J, Shimizu T, Stolz W. 2011. Cold atmospheric plasma: a successful treatment of lesions in Hailey–Hailey disease. Arch. Dermatol. 147, 388–390. ( 10.1001/archdermatol.2011.57) [DOI] [PubMed] [Google Scholar]
- 44.Heinlin J, Isbary G, Stolz W, Zeman F, Landthaler M, Morfill G, Shimizu T, Zimmermann JL, Karrer S. 2011. A randomized two-sided placebo-controlled study on the efficacy and safety of atmospheric non-thermal argon plasma for pruritus. J. Eur. Acad. Dermatol. Venereol. 27, 324–331. ( 10.1111/j.1468-3083.2011.04395.x) [DOI] [PubMed] [Google Scholar]
- 45.Brullé L, et al. 2012. Effects of a non thermal plasma treatment alone or in combination with gemcitabine in a MIA PaCa2-luc orthotopic pancreatic carcinoma model. PLoS ONE 7, e52653 ( 10.1371/journal.pone.0052653) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandamme M, et al. 2012. ROS implication in a new antitumor strategy based on non-thermal plasma. Int. J. Cancer 130, 2185–2194. ( 10.1002/ijc.26252) [DOI] [PubMed] [Google Scholar]