

Significance
Understanding molecular mechanisms that underlie the processes for myelin synthesis and maintenance has been an intensely investigated topic. Concurrently, recent advances in noncoding RNAs (ncRNAs) have uncovered unique insights into many biological processes, and ncRNAs have become recognized as major players for epigenetic regulation. We generated a murine model overexpressing microRNA-23a (miR-23a) to investigate its role in myelin regulation. In addition, we used this mouse model to identify two targets of miR-23a: one is a protein-coding gene phosphatase and tensin homologue and the other is a long ncRNA (lncRNA) 2700046G09Rik. Our study demonstrated a complex network comprising a protein-encoding gene, a miRNA, and a lncRNA that is central to the fine tuning and maintenance of healthy myelin.
Abstract
Demyelinating disorders including leukodystrophies are devastating conditions that are still in need of better understanding, and both oligodendrocyte differentiation and myelin synthesis pathways are potential avenues for developing treatment. Overexpression of lamin B1 leads to leukodystrophy characterized by demyelination of the central nervous system, and microRNA-23 (miR-23) was found to suppress lamin B1 and enhance oligodendrocyte differentiation in vitro. Here, we demonstrated that miR-23a–overexpressing mice have increased myelin thickness, providing in vivo evidence that miR-23a enhances both oligodendrocyte differentiation and myelin synthesis. Using this mouse model, we explored possible miR-23a targets and revealed that the phosphatase and tensin homologue/phosphatidylinositol trisphosphate kinase/Akt/mammalian target of rapamycin pathway is modulated by miR-23a. Additionally, a long noncoding RNA, 2700046G09Rik, was identified as a miR-23a target and modulates phosphatase and tensin homologue itself in a miR-23a–dependent manner. The data presented here imply a unique role for miR-23a in the coordination of proteins and noncoding RNAs in generating and maintaining healthy myelin.
MicroRNAs (miRNAs) play an important role in regulating a large number of developmental processes and diseases (1–3) through fine tuning biological networks (4, 5). Expression levels of miRNAs in oligodendroglia vary according to their differentiation stages, indicating a possible role for miRNAs in regulating developmental processes among migratory, proliferating, and myelinating oligodendrocytes (OLs) (6–9). Disruption of miRNA biogenesis by Dicer ablation in oligodendroglia at postdevelopmental stages results in a neurodegenerative phenotype including demyelination, inflammation, and axon loss (10), suggesting that miRNAs are also important for myelin maintenance at later developmental stages. miR-23 is among the most abundant miRNAs in OLs (6, 7). Previously, we reported that in the presence of excess miR-23 in vitro, a greater proportion of cells express mature markers of OLs that are paralleled by multipolar morphological appearance with increased levels of mature myelin proteins, indicating that miR-23 can enhance oligodendrogenesis (11). In contrast, excessive lamin B1, a nuclear envelope protein and target of miR-23, leads to lower numbers of cells expressing mature markers with reduced levels of mature myelin proteins both in vitro and in vivo, suggesting defective differentiation of OLs. Importantly, the adverse effects of lamin B1 on OL cells can be abrogated by overexpressing miR-23, which functions as a negative regulator of lamin B1.
Here, we use mice in which miR-23a (one of the two miR-23 isoforms: miR-23a and b) overexpression is driven by an OL-specific promoter [2′,3′-cyclic nucleotide 3′-phophodiesterase (Cnp)] to investigate the effects of miR-23a on OL differentiation and myelin synthesis in vivo. We demonstrated that in addition to the previously identified target, lamin B1, miR-23a also directly modulates the expression of two targets—phosphatase and tensin homolog on chromosome 10 (PTEN) and a long noncoding RNA (lncRNA), 2700046G09Rik (RIKEN cDNA 2700046G09 gene). Through modulating expression of these molecules in myelinating glia cells, miR-23a fine tunes activities of the serine-threonine protein kinase Akt/mTOR (mechanistic target of rapamycin) and mitogen-associated protein kinase (MAPK) pathways that promote expression of myelin genes. Our results indicate that myelination requires tightly regulated multilayer signaling pathways partly converging downstream of PIP3 (phosphatidylinositol-3,4,5-trisphosphate) with coordinated nuclear changes such as transcription to trigger myelin gene expression, which then leads to proper membrane wrapping of axons by OLs.
Results
Generation of Transgenic Mice Overexpressing miR-23a in Oligodendrocytes.
We have previously shown that miR-23 overexpression enhances OL maturation in an established culture system (11). In addition, miR-23a and miR-23b are both up-regulated in OL under differentiation conditions compared with progenitor status under proliferation conditions (Fig. S1A). Knockdown of both miR-23a and miR-23b leads to significant reductions, whereas knockdown of either miR-23a or miR-23b individually does not cause obvious changes in expression levels of myelin genes (Fig. S1B), suggesting that miR-23a and miR-23b can compensate for each other in regulating OL maturation. Immunoreactivity of myelin basic protein (MBP) also demonstrated that miR-23a and miR-23b together had stronger effects on MBP expression than either one alone (Fig. S1 C and D). Given that ectopic expression of miR-23 can promote transcription of myelin genes in cultured glia and purified OLs, we investigated the impact of overexpressing miR-23a on myelin formation in vivo. Northern blot analyses demonstrated that miR-23a displays reduced expression in Dicer1-ablated neurons, oligodendrocytes, and astrocytes (Fig. S2A), indicating ubiquitous expression of miR-23a in the CNS. Murine Cnp promoter was used to generate miR-23a transgenic mice (Fig. S2 B and C), as Cnp is highly expressed in developing OLs (12). Seven Cnp–miR-23a founder lines demonstrated miR-23a overexpression (Fig. S2D) and three of these lines with different expression levels were selected for further characterization. Quantitative RT-PCR (qRT-PCR) of oligodendrocyte progenitor cells (OPCs) purified from two lines display moderate-to-high levels of miR-23a in proliferative medium and a further significant increase in differentiation medium (Fig. S2E). Cnp–miR-23a mice carrying one transgene allele were not overtly distinguishable from control littermates but mice carrying two transgene alleles developed a notable unilateral hindlimb paralysis as early as postnatal day 5 (P5) (Fig. 1A). In addition, these Cnp–miR-23a mice exhibited abnormal axial muscle tone (kyphosis), puffy eyes, and hindlimb ataxia from P42 (Fig. 1A).
Fig. 1.

Cnp–miR-23a mice exhibited impaired motor function. (A) Morphological abnormalities displayed in Cnp–miR-23a mice: hindlimb paralysis (Upper Left ), loss of foot extensor tone (Lower Left), kyphosis (Upper Right ) and puffy eyes (Lower Right). Mice at the age of P150 were analyzed with balanced beam test (B), horizontal wire hanging test (C), and coat hanger test (D and E). Measurement of time for Cnp–miR-23a mice to traverse balance beams (B) and to hang in horizontal wire (C) and measurement of latency to reach the diagonal bar (D) and successful rate to reach diagonal bar (E) are shown. Data are presented as means ± SEM, n = 7 per genotype, *P < 0.05, unpaired Student t test. Filled bars represent Cnp–miR-23a and open bars denote WT mice.
Because there is no standard behavioral analysis specifically designed for myelin assessment (13), we used several tests to examine the Cnp–miR-23a mice for neurological dysfunction. We examined general locomotor activity of mice at 20 wk of age. Cnp–miR-23a transgenic mice showed motor impairment by requiring more time to traverse the balance beam (5 mm and 11 mm) (Fig. 1B) and a higher rate of hindlimb slips and dragging (Fig. S3 A and B). The hanging wire test was next used to examine whether the motor impairments were related to loss of muscle power, and significant differences were observed (Fig. 1C). Additionally, Cnp–miR-23a mice required more time to traverse the diagonal bar (Fig. 1D) and displayed a lower success rate (Fig. 1E) as well as reduced duration on the coat hanger (Fig. S3C), supporting that Cnp–miR-23a mice exhibited impaired motor function, which is likely a neurological consequence of miR-23a overexpression in OLs.
miR-23a Regulates Oligodendrocyte Differentiation.
To determine whether overexpressing miR-23a affects OL differentiation in vivo, oligodendroglia from transgenic mice under proliferation or differentiation conditions were used to analyze the expression levels of myelin genes. Interestingly, many of the myelin genes [particularly late phase genes such as myelin-associated glycoprotein (Mag), transferrin (Trf), and myelin oligodendrocyte glycoprotein (Mog)] are simultaneously up-regulated in miR-23a OLs compared with wild-type (WT) control cultures (Fig. 2A). Early OL gene such as proteolipid protein (Plp) and Mbp were also induced to a mild extent by miR-23a overexpression under proliferation conditions (Fig. S3D). miR-23a overexpression not only increased the number of OLs expressing MBP and MAG (Fig. 2B and Fig. S3E), but also increased the overall expression of PLP and MBP compared with control oligodendroglia (Fig. 2C), supporting miR-23a as a key regulator of OL differentiation.
Fig. 2.

Overexpressing miR-23a promotes OPC differentiation. (A) Expression of OL markers in cells purified from P7 Cnp–miR-23a mice was assessed using qRT-PCR. Data are presented as means ± SEM, *P < 0.05, **P < 0.01, unpaired Student t test. n = 4 per genotype. Filled bars represent Cnp–miR-23a OLs and open bars denote WT OLs. (B) Quantification of MBP- or MAG-positive cells in OPCs purified from P7 WT and Cnp–miR-23a mice cultured for 4 d in vitro (DIV) in differentiation medium. The total numbers of cells were determined by DAPI staining. Data are presented as ratio ± SEM from three independent experiments. *P < 0.05. (C) Representative Western analyses showed increased expression of PLP and MBP in Cnp–miR-23a OLs relative to control. Protein lysates were purified from OPCs cultured at 4 DIV in differentiation media. GAPDH was used as a loading control.
Overexpression of miR-23a Leads to Enhanced Myelination in the Central Nervous System.
Increased myelination in Cnp–miR-23a mice compared with WT control was observed in the corpus callosum stained with myelin-specific dye and CNP/MBP antibodies (Fig. 3A). The size of the corpus callosum was increased in Cnp–miR-23a mice at P90. Expression of myelin proteins, such as CNP, MBP, and MAG was elevated (Fig. 3B). Electron microscopy (EM) analysis revealed an increased number of axons exhibiting focal myelin pathology such as aberrant myelin outfoldings caused by hypermyelination at P180 (Fig. 3 C and D, yellow and green arrowheads) or “invaginating” recurrent loops (Fig. 3D, red arrowhead), which showed similar features with the myelin sheath from which they had originated. We quantified the myelin abnormalities by comparing electron microscopy of rostral corpus callosum from Cnp–miR23a and controls at 6 mo of age. G ratios for the transgenic (TG)+/WT− or TG+/TG+ Cnp–miR-23a mice are 0.6144 (±0.0043) and 0.6366 (±0.0045), respectively, which are significantly different from controls (0.7088 ± 0.0038) (Fig. 3E, Upper Right, P < 0.01). Increased myelin thickness was evident for axons of small calibers (Fig. 3E, Upper Left), but not all fibers were visibly hypermyelinated. The overall size distribution of callosal axons was similar in transgenic and control mice (Fig. 3E, Lower). EM demonstrated increased layers of myelin sheath wrapping in either small or large caliber axons with increased membrane wraps (Fig. 3F). Together, these data implicate miR-23a in the regulation of myelin thickness and proper myelin folding in the central nervous system (CNS).
Fig. 3.

Enhanced myelination in Cnp–miR-23a mice. (A) Corpus callosum in WT and Cnp–miR-23a mice at P90 showing enhanced myelination by Luxol Fast blue and CNP/MBP immunoreactivities. (B) Quantification of MBP, CNP, and MAG in corpus callosum by Western blot analysis (n = 3, *P < 0.05, unpaired Student t test). (C and D) Myelin abnormalities in corpus callosum of Cnp–miR-23a mice at P180, including hypermyelination (green arrowheads), myelin debris (red arrows), aberrant outfoldings (yellow arrowheads), and invaginating recurrent loops in axons (red arrowhead). [Scale bar for C, 1 μm (Left) or 2 μm (Right)]. (E) Quantitation of myelin thickness by modified G-ratio analysis and axon size distribution for the corpus callosum (age, P180; n = 2 per genotype; control, WT littermates; TG+/WT, one allele of transgene; TG+/TG+, two alleles of transgene). (Upper Left) Scatter plots comparing G ratios from TG+/WT (gray), TG+/TG+ (red), and age-matched controls (black) in relation to myelin sheath inner diameter. (Upper Right) Myelin thickness is significantly increased in Cnp–miR-23a mice. (Lower) Axonal calibers are comparable between Cnp–miR-23a mice and controls. (**P < 0.01, one-way ANOVA). (F) Hypermyelination in Cnp–miR-23a mice (Right) is caused by additional membrane wraps, as visualized by ultrastructure and periodicity of myelin sheaths. [Scale bar, 500 nm (Upper) and 100 nm (Lower)].
Unbiased Search for miR-23a Targeted Molecules and Mechanisms.
To further investigate the mechanisms of miR-23a in OL development and myelination, we set out to identify other relevant targets that are regulated by miR-23a. A total of 1,179 genes were identified to demonstrate differential expression between Cnp–miR-23a and WT mice [absolute fold change (FC) >1.5, multitest adjusted P value ≤0.05 correspondent to unadjusted P value ≤0.013 by cufflink] (Fig. 4A and Datasets S1 and S2). This included many known myelin-formation–associated genes, which were highly expressed in Cnp–miR-23a (Fig. 4B). Additionally, we found that genes specific to early stages of oligodendroglia development, such as Pdgfra and Lmnb1, displayed reduced levels in OLs purified from Cnp–miR-23a.
Fig. 4.

Differentially expressed transcriptome in miR-23a–overexpressing oligodendroglia. (A) Hierarchical clustering and analysis of overall expressed genes in cultured OLs overexpressing miR-23a. (B) Differential expression of known myelin-associated genes plotted on a color scale (green, low expression; red, high expression). (C) Volcano plot and kernel density estimation demonstrate the differences in the expression patterns of genes known to be enriched at different stages of oligodendrocyte differentiation (OPC, progenitors; OL, myelinating OL; MOG+, mature OL expressing MOG) in response to miR-23a overexpression.
Next, we compared the differentially expressed genes with the reference lists of genes that are enriched in astrocytes, neurons, and OLs (14). The OL-enriched genes are mostly up-regulated in Cnp–miR-23a, whereas the neuron-enriched genes tend to be down-regulated (Fig. S4 C and D). Upon further dissection of miR-23a on promoting OL differentiation, we found that genes enriched in more mature OLs, including OLs and the most mature MOG+ OLs (14), are mostly up-regulated (Fig. 4C and Fig. S4E). Collectively, the results of RNA sequencing (RNA-Seq) suggest a role for miR-23a in promoting the progression of less-differentiated OPCs into myelinating and mature OLs, likely by promoting the expression of mature OL-enriched genes. This regulation may be accompanied by repression of neuron-enriched and (to a lesser extent) astrocyte-enriched genes.
miR-23a Target Molecules: PTEN and 2700046G09Rik.
In silico prediction (miRANDA and TargetScan) followed by luciferase reporter assay was carried out to identify potential direct targets of miR-23a for regulating CNS myelination. We reasoned that true miR-23a–targeted genes would display positive correlation between RNA-Seq and luciferase reporter analysis. Among 35 candidates examined in this study, PTEN and 2700046G09Rik (Fig. S5A) displayed positive correlation. In addition, their genomic locations are rather close, which raised a possibility for the lncRNA (2700046G09Rik) to exert cis-regulatory effect on the neighboring PTEN gene (15). Therefore, we investigated them further. The repression of PTEN and enhancement of 2700046G09Rik by miR-23a were validated by mutagenizing their respective miR-23a binding elements (Fig. S5 B–D) miR-23 exhibits a gradually increased expression pattern during postnatal development (11), and the true direct target genes of miR-23a should display expression patterns correlating with miR-23a expression. Indeed, the protein levels of PTEN display a gradual decrease from postnatal day 1 to 10 months of age (Fig. 5A), whereas the levels of 2700046G09Rik display a gradual increase beginning at postnatal day 60 (Fig. 5B). We next validated the interaction of miR-23a and PTEN or 2700046G09Rik by UV–cross-link RNA immunoprecipitation (RIP) (16). FLAG-AGO2 immunoprecipitation was conducted in HEK293 cells transiently coexpressing miR-23a and its targets. By RIP-qPCR, PTEN or 2700046G09Rik coprecipitating with AGO2 was specifically enriched in miR-23a–transfected cells (Fig. 5C), indicating that miR-23a is facilitating AGO2 association with PTEN or 2700046G09Rik. Furthermore, using a biotin-coupled miR-23a mimic, we observed a significant enrichment of PTEN or 2700046G09Rik in miR-23a–captured fraction compared with control (Fig. 5D). Consistently, the levels of PTEN are decreased (Fig. 5E), whereas 2700046G09Rik are increased (Fig. 5F) in spinal cord and cerebellum of Cnp–miR-23 mice compared with WT mice. Taken together, these results strongly imply that direct interactions exist between miR-23a and PTEN or 2700046G09Rik and that PTEN and 2700046G09Rik are true miR-23a targets.
Fig. 5.

miR-23a regulates PTEN and 2700046G09Rik. (A) Western analysis of PTEN, MAG, MBP, and GAPDH from C57BL/6 brain at indicated ages. (B) qRT-PCR of 2700046G09Rik from C57BL/6 brain. **P < 0.01 compared with P0, one-way ANOVA. (C) Immunoprecipitation of FLAG-tagged AGO2 from HEK293 transfected with PTEN 3′-UTR or 2700046G09Rik and FLAG-AGO2 plus vector or miR-23a. PTEN and 2700046G09Rik levels were quantified by qRT-PCR, and the relative immunoprecipitate (IP)/input ratios were plotted. n = 4, ***P < 0.001, unpaired Student t test. (D) The 3′-end biotinylated miR-23a duplexes were transfected into HEK293. After streptavidin capture, input and bound fractions were evaluated by qRT-PCR. n = 4, *P < 0.05. (E) Western analysis of PTEN in spinal cord or cerebellum from WT and Cnp–miR-23a mouse brain at P90. (F) qRT-PCR of 2700046G09Rik transcript using spinal cord or cerebellum from control and Cnp–miR-23a mice. n = 3, *P < 0.05. Filled bars represent Cnp–miR-23a and open bars denote WT mice. (G) Luciferase activity of firefly reporter gene fused with the 3-kb 2700046G09Rik promoter in the presence of indicated transcription factors. n = 4, **P < 0.01 compared with control, one-way ANOVA. All data are presented as ratio of means ± SEM.
2700046G09Rik Participates in Myelin Regulation.
To understand the role of 2700046G09Rik in oligodendroglial differentiation and myelin production, we examined its expression in various cells. RNA levels of 2700046G09Rik were significantly higher in cultured OLs from WT mice under differentiation conditions, whereas astrocytes displayed a comparable level to cultured OPCs under proliferative conditions (Fig. S6A). Congruently, its level is increased in OLs from Cnp–miR-23a compared with WT OLs. Overexpression of 2700046G09Rik in cultured OLs led to moderately increased expression of MAG protein (Fig. S6B). In addition, promoter region reporter assays revealed that two important oligodendrocyte-associated transcription factors for OPCs differentiation to OLs, YY1 and Nkx2.2 (17, 18), display positive effects on the expression of 2700046G09Rik (Fig. 5G). Together, these data support a potential unique role for 2700046G09Rik in the regulation of myelination.
Interplays Among miR-23a, 2700046G09Rik, and PTEN.
The competitive endogenous RNA hypothesis (19) suggested that coding and noncoding transcripts share common miRNA binding elements and lead to altered transcriptome homeostasis. To test whether PTEN, 2700046G09Rik, and miR-23a have interplay in regulating OL and myelin, we first investigated the possible regulatory effects of 2700046G09Rik on PTEN in cell culture. The coding region plus 2 kb 3′-UTR of PTEN was cotransfected into HEK293 with either miR-23a or 2700046G09Rik and Western blot analysis revealed that miR-23a and 2700046G09Rik both exert repressive effects on PTEN (Fig. 6A). Next, luciferase reporters carrying full-length (6 kb) 3′-UTR (20) of PTEN was coexpressed with miR-23a and/or 2700046G09Rik (Fig. 6B). 2700046G09Rik did not significantly alter the level of PTEN, whereas miR-23a displayed moderate repression. Interestingly, the level of PTEN was reduced by 2700046G09Rik only in the presence of the PTEN coding region together with 2 kb 3′-UTR (Fig. 6 A and C). This effect was abrogated by mutating the miR-23a binding motif of 2700046G09Rik (Fig. 6C, lane 4), suggesting that an intact miR-23a binding motif is necessary for the full repressive effect of 2700046G09Rik on PTEN and that there is a possible interplay between miR-23a and 2700046G09Rik for regulation of PTEN. As expected, the level of PTEN was further reduced by the presence of both miR-23a and 2700046G09Rik (Fig. 6C, lane 5).
Fig. 6.

miR-23a and 2700046G09Rik repress PTEN. (A) Representative Western analysis of PTEN from HEK293 coexpressing PTEN coding sequences fused to 2-kb PTEN 3′-UTR with either miR-23a, 2700046G09Rik, or both. (B) Luciferase activity of firefly reporter gene fused with the 6-kb full-length PTEN 3′-UTR (without coding region) in the presence of indicated RNA. WT, 2700046G09Rik with normal miR-23a binding elements. Mut, 2700046G09Rik with mutated miR-23a binding elements (M123, Fig. S5). Data presented as ratio of means ± SEM *P < 0.05 compared with control; †P < 0.05 compared with WT 2700046G09Rik; one-way ANOVA. (C) Representative Western analysis of PTEN from HEK293 coexpressing PTEN coding sequences fused to 2-kb 3′-UTR (containing WT miR-23a binding elements) together with miR-23a and/or 2700046G09Rik. (D) qRT-PCR analysis of miR-23a in HEK293 transfected with miR-23a plus vector (gray), 2700046G09Rik carrying WT (black), or Mut (blue) miR-23a binding elements followed by actinomycin D treatment. n = 6, *P < 0.05 compared with vector control. One-way ANOVA analysis with Newman–Keuls test at 24 h. (E) qRT-PCR analysis of 2700046G09Rik transcript in HEK293 transfected with miR-23a plus 2700046G09Rik carrying WT (black) or Mut (blue) miR-23a binding elements followed by actinomycin D treatment. n = 6, NS, nonsignificant. (F) Immunoprecipitation of DCP1 from HEK293 cells transfected with vector, miR-23a, 2700046G09Rik, or miR-23a plus 2700046G09Rik. miR-23a levels were quantified by qRT-PCR, and the relative immunoprecipitate (IP)/input ratios were plotted. n = 4, NS, nonsignificant, *P < 0.05, †P < 0.05.
We next investigated the possible effect of 2700046G09Rik and miR-23a on each other. Inhibition of de novo transcription by actinomycin D treatment in HEK293 cells showed miR-23a has a longer half-life compared with U6 small nuclear RNA (snRNA) (Fig. 6D). Cotransfection of 2700046G09Rik containing perfect miR-23a binding elements with miR-23a led to higher expression of miR-23a in HEK293 cells following 24-h treatment of actinomycin D, whereas miR-23a did not alter stability of 2700046G09Rik regardless of the presence of miR-23a binding elements (Fig. 6E). These results suggested that 2700046G09Rik enhances miR-23a stability. HEK293 cells expressing miR-23a, 2700046G09Rik, or both were then accessed by RIP-qPCR following immunoprecipitation of DCP1 (decapping enzyme 1) for P body (processing bodies). Coexpression of miR-23a and 2700046G09Rik significantly increased miR-23a in DCP1 immunoprecipitant (Fig. 6F), indicating enrichment of miR-23a in P bodies by 2700046G09Rik. Altered stability and cellular localization of miR-23a by 2700046G09Rik suggest a unique role for noncoding transcripts in the regulation of PTEN transcript. Together, these results demonstrate a network of regulatory pathway, including miR-23a, 2700046G09Rik, and PTEN in the regulation of OL development and myelin gene expression.
Signaling Pathways Modulated by miR-23a Overexpression.
Because PTEN is a lipid phosphatase that negatively regulates the phosphatidylinositol 3-kinase (PI3K) signaling pathway (21), and accumulating evidence indicates that the PI3K/Akt/mTOR pathway regulates CNS myelination (22, 23), we investigated the possibility that miR-23a has a role in modulating the PI3K/Akt/mTOR signal transduction cascade. Western blot analysis revealed that the level of phosphorylated Akt was higher in miR-23a brain homogenate (Fig. 7A), indicating activation of Akt signaling. In addition, levels of PI3K signaling and MAPK activity, but not protein kinase A (PKA), were also elevated (Fig. S7 A–C). Ectopic expression of PTEN in cultured OLs purified from control and Cnp–miR-23a mice at P7 reduced expression levels of MAG compared with vector control (Fig. 7B). Overexpressing dominant-negative Akt (AKT-DN) dramatically reduced expression of myelin proteins in Cnp–miR-23a (Fig. 7C), consistent with the hypothesis that Akt acts downstream of miR-23a to mediate myelin formation. Rapamycin, the mTOR inhibitor, treatment reduced expression levels of several myelin proteins (PLP, MOG, and MAG) in cultured OLs isolated from Cnp–miR-23a mice (Fig. 7D). Collectively, these results confirm that PTEN/PI3K/Akt/mTOR is part of the cascade in miR-23a–mediated regulation and that mTOR acts downstream of miR-23a to mediate myelin production.
Fig. 7.
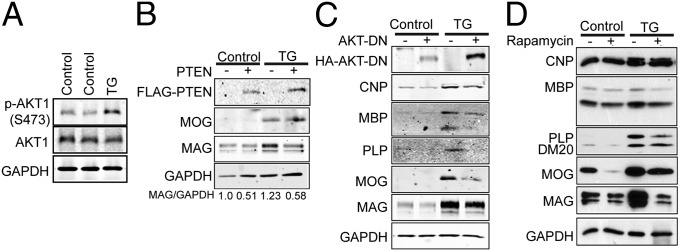
miR-23a overexpression leads to activation of the Akt/mTOR pathway. (A) Western analysis of phosphor-AKT (S473) expression in corpus callosum of P90 WT or Cnp–miR-23a mice. (B–D) Representative Western analyses of myelin proteins in mouse OPCs from WT or Cnp–miR-23a mice cultured for 4 DIV in differentiation media. Purified OPCs were overexpressing PTEN (B) or dominant-negative AKT1 (AKT-DN) (C). (D) Purified OPCs were treated with 15 nM rapamycin.
Discussion
The deposition of a precise amount of myelin around axons is necessary for proper impulse transmission, whereas too much or too little myelin surrounding axons causes nerve dysfunction in various neurological diseases. We have generated a mouse model overexpressing miR-23a that produces excessive myelin protein and myelin formation in the CNS. These mice display severe motor deficits beginning in postnatal life. Because Cnp expresses in both central and peripheral nervous systems (PNS), we cannot exclude the possibility that the motor deficits observed in Cnp–miR-23a mice were caused, at least in part, by peripheral myelin abnormality. Using this mouse model, we also produced a profile for differentially expressed genes that are associated with miR-23a overexpression. This miR23a-myelin transcriptome offers a useful resource for future investigation in understanding the signaling networks and factors that are required for the regulation of OL development/myelination and other miR-23a–regulated biological functions.
Our demonstration of miR-23a overexpression in OLs resulting in hypermyelination of mouse brain establishes a regulatory role for miR-23a in myelin production. Overexpression of miR-23a leads to up-regulation of genes in OL clusters, but substantial down-regulation of genes in neuronal clusters. This suggests that miR-23a not only functions to enhance OL lineage progression and promote myelin proteins but also safeguards against expression of genes for other cell lineages that might interfere with the progression of OL maturation and myelin sheath formation. This is consistent with previous reports that miRNAs function as guardians to enhance lineage-related protein identity and to repress other nonlineage protein expression (6, 9).
Elevated PIP3 signaling or loss of PTEN in myelinating glia has been shown to cause hypermyelination in the CNS (21, 24) and PNS (25). PTEN antagonizes PI3K signaling and negatively regulates the ERK1/2-MAPK pathway (26). Akt and mTOR, downstream effectors of PI3K signaling, promotes OL differentiation and myelin generation (22, 27, 28). Transgenic mice overexpressing constitutively active Akt kinase in OLs enhance myelin formation in the CNS but not PNS (22), and this is mediated through downstream mTOR signaling (23). The present study revealed that elevated level of miR-23a in OLs is sufficient to promote formation of myelin that can last until older age (at least 1-y old) partly through the Akt/mTOR and MAPK signaling pathways by targeting PTEN. A recent study reported that the cAMP-response element-binding protein (CREB) promotes glioma formation by up-regulating miR-23a, leading to down-regulation of its direct target, PTEN (29). Thus far, we have not observed obviously increased incidence of glioma formation in murine brains overexpressing miR-23a in myelinating glia.
Duplications of LMNB1 (which encodes lamin B1) have been identified to cause adult-onset autosomal dominant leukodystrophy (30). Excessive lamin B1 expression reduces occupancy of Yin Yang 1 (YY1) transcription factor on the promoter region of PLP, thus leading to down-regulation of PLP abundance, conferring myelin loss in the mouse brain (31). Reduced levels of both PTEN and LMNB1 by miR-23a are likely to participate in hypermyelination observed in the present study.
To date, long noncoding RNAs have been shown to function as regulators of gene expression transcriptionally and posttranscriptionally through working at the DNA level (32), modulation of chromatin modifications, or transcriptional interference by antisense transcription (1). miRNAs bind to the coding sequences or 3′-UTRs of target transcripts, thus leading to impaired translation or increased degradation of transcripts (33). Interestingly, 2700046G09Rik, one of the miR-23a targets identified in this study, is a lncRNA in cis with the neighboring PTEN gene. miR-23a up-regulates the 2700046G09Rik transcription, and 2700046G09Rik in turn lengthens the half-life of miR-23a, thus potentiating its repressive effects. 2700046G09Rik also can lead to a reduced level of PTEN expression. This down-regulation is independent of miR-23a–responsive elements (MREs) on PTEN, but requires the miR-23a MREs on 2700046G09Rik. Therefore, repressive effects on PTEN can either occur with miR-23a alone or in coordination with 2700046G09Rik. It is possible that 2700046G09Rik targets the PTEN with the assistance of miR-23a. In addition, 2700046G09Rik may aid in the cellular recompartmentation of miR-23a into P bodies, which could also contribute to the regulation of PTEN level. Interplay of miR-23a and 2700046G09Rik in this study infers additional molecular processes in regulating mRNA decay (Fig. S8). lncRNAs are tightly controlled by environmental cues and inducible functions (34). Intriguingly, we discovered that promoter of 2700046G09Rik can be activated by two important transcription factors (YY1 and Nkx2.2) (17, 18, 35) in OL development. Our results are consistent with a previous report that dynamic changes of lncRNA transcriptome are important for glia differentiation (36). We propose that the presence of 2700046G09Rik in oligodendroglia potentiates and signals the activation of the miR-23a/PTEN/Akt/mTOR and MAPK cascades in the correct developmental stage, thus regulating the expression of myelin genes in OLs.
Noncoding RNAs (ncRNAs) have emerged as a major component in epigenetic regulation, specifically in orchestrating neural gene expression and function and gene–environment interactions (2, 37). Elevated levels of miR-23a are identified in severe demyelinated regions of brains derived from the murine model of autosomal dominant adult-onset leukodystrophy (Fig. S9) and active multiple sclerosis lesions in human brains (38, 39). Interestingly, increased frequency of miR-23a rs(refSNP) 3745453 C allele has been identified as a potential risk factor of multiple sclerosis (39). Therefore, miR-23a may participate in not only modulating genes in normal myelin regulation but also in myelin repair. Through identification of miR-23a targets, we revealed that miR-23a and some lncRNAs (2700046G09Rik) interact in a complex regulatory network to modulate genes that are important for myelin formation/maintenance. Understanding the layers of complexity in the molecular mechanisms that underlie the elaboration of myelin synthesis and maintenance has undergone intensive investigation and recent advances have uncovered unique insights into these processes. Identifying targets of miR-23a and developing future demyelinating experiments using this murine model will provide the opportunity to better understand RNA regulatory networks and offer future therapeutic approaches against demyelinating diseases.
Materials and Methods
Details for generation of Cnp–miR-23a mice oligodendrocyte culture, RNA-Seq analysis, immunoblotting, immunostaining, and qRT-PCR are provided in SI Materials and Methods. Cnp-miR-23a mice were generated and maintained in C57BL/6 background. All animal experiments were performed under approval by the Animal Care and Use Committee at the University of California, San Francisco.
Supplementary Material
Acknowledgments
We acknowledge V. Gallo for the Cnp-promoter constructs; M. Kato for the PTEN-luciferase construct; Y. Zhao, L.-Y. Hsu, H.-E. Liang, and J. Pollack for suggestions in mouse generation and statistical analysis; D. H. Rowitch and J. R. Chan for helpful discussions; and B. Barres for implementing primary culturing methods, S.-Y. C. Chong for editing the manuscript, and members of the Y.-H.F. and L.J.P. laboratories for discussions. This work was supported by National Institutes of Health Grant NS062733 (to Y.-H.F.) and the Sandler Neurogenetics Fund (Y.-H.F. and L.J.P.). L.J.P. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317182110/-/DCSupplemental.
References
- 1.Pauli A, Rinn JL, Schier AF. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet. 2011;12(2):136–149. doi: 10.1038/nrg2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci. 2012;13(8):528–541. doi: 10.1038/nrn3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 4.Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137(4):647–658. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 5.Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci. 2009;12(4):399–408. doi: 10.1038/nn.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau P, et al. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J Neurosci. 2008;28(45):11720–11730. doi: 10.1523/JNEUROSCI.1932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budde H, et al. Control of oligodendroglial cell number by the miR-17-92 cluster. Development. 2010;137(13):2127–2132. doi: 10.1242/dev.050633. [DOI] [PubMed] [Google Scholar]
- 8.Dugas JC, et al. Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron. 2010;65(5):597–611. doi: 10.1016/j.neuron.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao X, et al. MicroRNA-mediated control of oligodendrocyte differentiation. Neuron. 2010;65(5):612–626. doi: 10.1016/j.neuron.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin S, et al. MicroRNAs are significantly influenced by p53 and radiation in HCT116 human colon carcinoma cells. Int J Oncol. 2009;34(6):1645–1652. doi: 10.3892/ijo_00000295. [DOI] [PubMed] [Google Scholar]
- 11.Lin ST, Fu YH. miR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis Model Mech. 2009;2(3–4):178–188. doi: 10.1242/dmm.001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belachew S, Yuan X, Gallo V. Unraveling oligodendrocyte origin and function by cell-specific transgenesis. Dev Neurosci. 2001;23(4–5):287–298. doi: 10.1159/000048712. [DOI] [PubMed] [Google Scholar]
- 13.Hu X, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9(12):1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- 14.Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci. 2008;28(1):264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460(7254):479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He Y, et al. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55(2):217–230. doi: 10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi Y, et al. Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Development. 2001;128(14):2723–2733. doi: 10.1242/dev.128.14.2723. [DOI] [PubMed] [Google Scholar]
- 19.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato M, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11(7):881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goebbels S, et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J Neurosci. 2010;30(26):8953–8964. doi: 10.1523/JNEUROSCI.0219-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flores AI, et al. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28(28):7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29(21):6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrington EP, et al. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann Neurol. 2010;68(5):703–716. doi: 10.1002/ana.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goebbels S, et al. Genetic disruption of Pten in a novel mouse model of tomaculous neuropathy. EMBO Mol Med. 2012;4(6):486–499. doi: 10.1002/emmm.201200227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pezzolesi MG, Zbuk KM, Waite KA, Eng C. Comparative genomic and functional analyses reveal a novel cis-acting PTEN regulatory element as a highly conserved functional E-box motif deleted in Cowden syndrome. Hum Mol Genet. 2007;16(9):1058–1071. doi: 10.1093/hmg/ddm053. [DOI] [PubMed] [Google Scholar]
- 27.Barros CS, et al. Beta1 integrins are required for normal CNS myelination and promote AKT-dependent myelin outgrowth. Development. 2009;136(16):2717–2724. doi: 10.1242/dev.038679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tyler WA, et al. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. 2009;29(19):6367–6378. doi: 10.1523/JNEUROSCI.0234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan X, et al. cAMP response element-binding protein promotes gliomagenesis by modulating the expression of oncogenic microRNA-23a. Proc Natl Acad Sci USA. 2012;109(39):15805–15810. doi: 10.1073/pnas.1207787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Padiath QS, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38(10):1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- 31.Heng MY, et al. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J Clin Invest. 2013;123(6):2719–2729. doi: 10.1172/JCI66737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ørom UA, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143(1):46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartels CL, Tsongalis GJ. MicroRNAs: Novel biomarkers for human cancer. Clin Chem. 2009;55(4):623–631. doi: 10.1373/clinchem.2008.112805. [DOI] [PubMed] [Google Scholar]
- 34.Geisler S, Lojek L, Khalil AM, Baker KE, Coller J. Decapping of long noncoding RNAs regulates inducible genes. Mol Cell. 2012;45(3):279–291. doi: 10.1016/j.molcel.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berndt JA, Kim JG, Tosic M, Kim C, Hudson LD. The transcriptional regulator Yin Yang 1 activates the myelin PLP gene. J Neurochem. 2001;77(3):935–942. doi: 10.1046/j.1471-4159.2001.00307.x. [DOI] [PubMed] [Google Scholar]
- 36.Mercer TR, et al. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. 2010;11:14. doi: 10.1186/1471-2202-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Junker A, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. 2009;132(Pt 12):3342–3352. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 39.Ridolfi E, et al. Expression and genetic analysis of microRNAs involved in multiple sclerosis. Int J Mol Sci. 2013;14(3):4375–4384. doi: 10.3390/ijms14034375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.