

Significance
Gene duplication is thought to play an important role in the evolution of complex phenotypes. Although studies have revealed that duplicate genes are abundant, there is considerable controversy about how they are maintained throughout evolution. In this study, we develop an approach for comparing genome-wide expression profiles of closely related species to disentangle the evolutionary forces operating on duplicate genes. Application of our approach to pairs of young duplicate genes in Drosophila reveals that nearly all duplicates are retained by the evolution of a novel function in one copy. Further analysis reveals that, although young genes are primarily expressed in testes, their expression broadens as they age, illustrating how new genes become integrated into diverse functional networks over time.
Abstract
Gene duplication is a key source of genetic innovation that plays a role in the evolution of phenotypic complexity. Although several evolutionary processes can result in the long-term retention of duplicate genes, their relative contributions in nature are unknown. Here we develop a phylogenetic approach for comparing genome-wide expression profiles of closely related species to quantify the roles of conservation, neofunctionalization, subfunctionalization, and specialization in the preservation of duplicate genes. Application of our method to pairs of young duplicates in Drosophila shows that neofunctionalization, the gain of a novel function in one copy, accounts for the retention of almost two-thirds of duplicate genes. Surprisingly, novel functions nearly always originate in younger (child) copies, whereas older (parent) copies possess functions similar to those of ancestral genes. Further examination of such pairs reveals a strong bias toward RNA-mediated duplication events, implicating asymmetric duplication and positive selection in the evolution of new functions. Moreover, we show that young duplicate genes are expressed primarily in testes and that their expression breadth increases over evolutionary time. This finding supports the “out-of-testes” hypothesis, which posits that testes are a catalyst for the emergence of new genes that ultimately evolve functions in other tissues. Thus, our study highlights the importance of neofunctionalization and positive selection in the retention of young duplicates in Drosophila and illustrates how duplicates become incorporated into novel functional networks over evolutionary time.
Gene duplication produces two copies of an existing gene. Evolutionary theory predicts that functional redundancy of duplicate genes causes one copy to undergo a brief period of relaxed selection after duplication (1). In nearly all cases, this should result in an accumulation of deleterious mutations and pseudogenization of the copy within a few million years (2). However, most sequenced eukaryotic genomes contain many functional duplicates, some of which are hundreds of millions of years old (3–8), suggesting that duplicate genes play important roles in evolution.
Four processes can result in the evolutionary preservation of duplicate genes: conservation, neofunctionalization, subfunctionalization, and specialization. Under conservation, ancestral functions are maintained in both copies, likely because increased gene dosage is beneficial (1). Under neofunctionalization, one copy retains its ancestral functions, and the other acquires a novel function (1). Under subfunctionalization, mutations damage different functions of each copy, such that both copies are required to preserve all ancestral gene functions (9, 10). Finally, under specialization, subfunctionalization and neofunctionalization act in concert, producing two copies that are functionally distinct from each other and from the ancestral gene (11). Theoretical work has shown that different conditions can result in the retention of duplicate genes by any one of these processes (9, 12–17), and empirical studies have uncovered numerous examples of each (11, 18–23).
However, no genome-wide studies have attempted to distinguish among these processes and, thus, their relative roles in nature remain unknown. One difficulty of such a study is defining biological function on a genomic scale. To address this problem, we used relative gene expression levels in different tissues (i.e., gene expression profiles) as proxies for function. Gene expression profiles are ideal for assessing biological function because of the availability of high-throughput expression data for multiple tissues in a number of species, correlations to different measures of gene function (24–27), and simple quantitative interpretation relative to alternative functional metrics such as protein structure or interaction networks. A second obstacle to studying evolutionary processes underlying the retention of duplicate genes is the lack of methods for distinguishing among processes. To disentangle these evolutionary processes, we developed a phylogenetic approach for comparing expression divergence between duplicate genes (parent and child copies) in one species and their ancestral single-copy ortholog in a closely related sister species. Our approach combines gene expression profiles with phylogenetic relationships among gene copies to classify the evolutionary processes driving the preservation of young duplicate genes.
Results
Development of an Approach for Classifying Evolutionary Processes Underlying the Retention of Duplicate Genes.
Distinguishing among different evolutionary processes that drive preservation of duplicates requires quantification of divergence between gene expression profiles. There are two commonly used metrics for assessing differences between gene expression profiles: Euclidian distance and Pearson’s correlation coefficient (28). However, in contrast to Pearson’s correlation coefficient, Euclidian distance is robust to measurement error and does not detect divergence between genes with conserved uniform patterns of expression (28). In addition, Euclidian distance can incorporate information about gene expression levels, and its squared value increases linearly with time (28). Thus, we estimated functional divergence between genes by computing Euclidian distances between their expression profiles. In particular, we calculated Euclidian distances between expression profiles of parent and ancestral copies (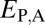 ), between expression profiles of child and ancestral copies (
), between expression profiles of child and ancestral copies (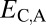 ), and between the combined parent–child expression profile and that of the ancestral copy (
), and between the combined parent–child expression profile and that of the ancestral copy (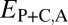 ; Fig. 1A). To establish a baseline divergence level for genes, we also calculated Euclidian distances between expression profiles of single-copy genes present in both sister species (
; Fig. 1A). To establish a baseline divergence level for genes, we also calculated Euclidian distances between expression profiles of single-copy genes present in both sister species (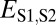 ).
).
Fig. 1.

Classification of the evolutionary processes maintaining pairs of duplicate genes. (A) A phylogenetic representation of the relationships among ancestral (A), parent (P), child (C), and combined parent–child (P+C) expression profiles. Euclidian distances corresponding to those listed in Table 1 are depicted by colored branches. 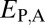 is blue,
is blue, 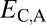 is red, and
is red, and 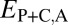 is purple. (B) Distributions of
is purple. (B) Distributions of 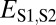 (black),
(black), 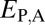 (blue),
(blue), 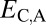 (red), and
(red), and 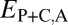 (purple). The vertical dashed line represents the semi-interquartile range from the median of
(purple). The vertical dashed line represents the semi-interquartile range from the median of 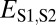 , which was used as a cutoff for identifying evolutionary processes maintaining individual pairs of duplicate genes.
, which was used as a cutoff for identifying evolutionary processes maintaining individual pairs of duplicate genes.
Assuming that 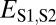 represents expected distances between expression profiles of genes in sister species, we can define a set of rules for classifying cases of conservation, neofunctionalization, subfunctionalization, and specialization via comparisons of
represents expected distances between expression profiles of genes in sister species, we can define a set of rules for classifying cases of conservation, neofunctionalization, subfunctionalization, and specialization via comparisons of 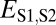 with
with 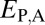 ,
, 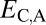 , and
, and 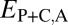 (Table 1). In particular, under conservation, the expression profiles of parent, child, and ancestral copies should be similar and, thus, we expect
(Table 1). In particular, under conservation, the expression profiles of parent, child, and ancestral copies should be similar and, thus, we expect 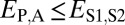 and
and 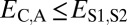 . Under neofunctionalization, the expression profile of the ancestral copy should be similar to that of either the parent or child copy and different from that of the alternate copy. Hence, we expect
. Under neofunctionalization, the expression profile of the ancestral copy should be similar to that of either the parent or child copy and different from that of the alternate copy. Hence, we expect 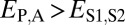 and
and 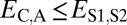 when the parent copy is neofunctionalized, and
when the parent copy is neofunctionalized, and 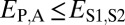 and
and 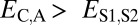 when the child copy is neofunctionalized. Under subfunctionalization, the expression profiles of parent and child copies should both be different from that of the ancestral copy, whereas the combined parent–child expression profile should be similar to that of the ancestral copy. Thus, we expect
when the child copy is neofunctionalized. Under subfunctionalization, the expression profiles of parent and child copies should both be different from that of the ancestral copy, whereas the combined parent–child expression profile should be similar to that of the ancestral copy. Thus, we expect 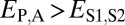 ,
, 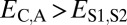 , and
, and 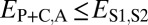 . Finally, under specialization, the expression profile of the parent copy, expression profile of the child copy, and combined parent–child expression profile should all be different from that of the ancestral copy, so we expect
. Finally, under specialization, the expression profile of the parent copy, expression profile of the child copy, and combined parent–child expression profile should all be different from that of the ancestral copy, so we expect 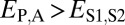 ,
, 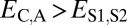 , and
, and 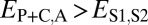 .
.
Table 1.
Rules for classifying evolutionary processes by Euclidian distances between gene expression profiles
| Classification | 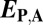 |
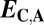 |
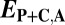 |
| Conservation | ≤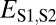
|
≤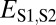
|
— |
| Neofunctionalization of parent copy | >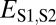
|
≤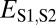
|
— |
| Neofunctionalization of child copy | ≤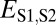
|
>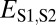
|
— |
| Subfunctionalization | >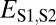
|
>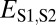
|
≤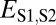
|
| Specialization | >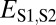
|
>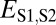
|
>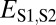
|
Classification of Evolutionary Processes Retaining Young Duplicates in Drosophila.
We applied our phylogenetic approach to 281 pairs of young duplicate genes in Drosophila, for which child copies arose in either the Drosophila melanogaster (108 pairs) or the Drosophila pseudoobscura (173 pairs) lineage after their divergence 25–46 Mya (29) (see Materials and Methods for details). In addition, we identified 8,576 single-copy genes present in D. melanogaster and in D. pseudoobscura. We computed 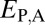 ,
, 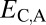 ,
, 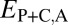 , and
, and 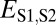 between expression profiles derived from RNA-seq data for six tissues.
between expression profiles derived from RNA-seq data for six tissues.
Comparison of the distributions of these distances (Fig. 1B) revealed that, in general, distances between parent and ancestral copies are small (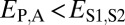 ; P = 5.22 × 10−3), whereas distances involving child copies are elevated (
; P = 5.22 × 10−3), whereas distances involving child copies are elevated (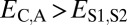 and
and 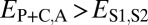 ; P = 3.70 × 10−56 and P = 1.40 × 10−68) relative to those of single-copy genes. Thus, pairs of duplicate genes in Drosophila appear to be maintained primarily by neofunctionalization of child copies. However, the distributions of
; P = 3.70 × 10−56 and P = 1.40 × 10−68) relative to those of single-copy genes. Thus, pairs of duplicate genes in Drosophila appear to be maintained primarily by neofunctionalization of child copies. However, the distributions of 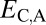 and
and 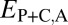 are bimodal, indicating that retention of duplicates occurs via other evolutionary processes as well.
are bimodal, indicating that retention of duplicates occurs via other evolutionary processes as well.
To identify evolutionary processes responsible for the preservation of individual pairs of duplicate genes, it was necessary to explicitly define expected expression divergence. Application of several cutoffs to Euclidian distances produced similar classifications across genes (Materials and Methods, Table S1, and Fig. S1); however, we chose to use the semi-interquartile range from the median of 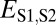 (Fig. 1B) to define expected expression divergence because of its insensitivity to extreme values. This cutoff yielded 53 cases of conservation, 183 cases of neofunctionalization (16 of parent copies, 167 of child copies), 3 cases of subfunctionalization, and 42 cases of specialization. Because distributions of classifications do not differ significantly among pairs in which child copies arose after different evolutionary divergence times (Tables S2 and S3), our analysis was not confounded by ongoing pseudogenization of very young genes or by multiple processes affecting duplicate genes over time. Hence, as expected from our global analysis (Fig. 1B), the majority of young pairs of duplicates in Drosophila are maintained by neofunctionalization, with a strong bias toward neofunctionalization of child copies. Although there are also large contributions by both conservation and specialization, subfunctionalization appears to be rare in Drosophila. Because of the small contribution of subfunctionalization, as well as the possibility that cases we identified were false positives (Fig. S2), we did not perform any further analyses on genes classified as subfunctionalized.
(Fig. 1B) to define expected expression divergence because of its insensitivity to extreme values. This cutoff yielded 53 cases of conservation, 183 cases of neofunctionalization (16 of parent copies, 167 of child copies), 3 cases of subfunctionalization, and 42 cases of specialization. Because distributions of classifications do not differ significantly among pairs in which child copies arose after different evolutionary divergence times (Tables S2 and S3), our analysis was not confounded by ongoing pseudogenization of very young genes or by multiple processes affecting duplicate genes over time. Hence, as expected from our global analysis (Fig. 1B), the majority of young pairs of duplicates in Drosophila are maintained by neofunctionalization, with a strong bias toward neofunctionalization of child copies. Although there are also large contributions by both conservation and specialization, subfunctionalization appears to be rare in Drosophila. Because of the small contribution of subfunctionalization, as well as the possibility that cases we identified were false positives (Fig. S2), we did not perform any further analyses on genes classified as subfunctionalized.
Classification by an Alternative Approach Using Expression Localization Patterns.
To validate our classifications of evolutionary processes, we developed and tested an alternative binary approach based on expression localization patterns. In particular, we assumed that the acquisition of a novel function is specifically linked to changes in where a gene is expressed. Hence, rather than computing differences between expression levels, we assessed divergence (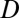 ) by comparing the tissues in which genes are expressed. Genes expressed in all of the same tissues were classified as having conserved functions (
) by comparing the tissues in which genes are expressed. Genes expressed in all of the same tissues were classified as having conserved functions (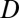 = 0), whereas those differing in their expression localization patterns were classified as having divergent functions (
= 0), whereas those differing in their expression localization patterns were classified as having divergent functions (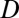 = 1). For each triplet of genes (parent, child, and ancestral), we assessed divergence between expression localization patterns of parent and ancestral copies (
= 1). For each triplet of genes (parent, child, and ancestral), we assessed divergence between expression localization patterns of parent and ancestral copies (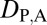 ), between expression localization patterns of child and ancestral copies (
), between expression localization patterns of child and ancestral copies (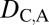 ), and between the combined parent–child expression localization patterns and those of the ancestral copy (
), and between the combined parent–child expression localization patterns and those of the ancestral copy (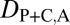 ). Then, using Table 1 as a template, we constructed an analogous set of rules for classifying evolutionary processes by divergence in expression localization patterns (Table S4; see Materials and Methods for details). Application of this alternative approach to our dataset yielded 56 cases of conservation, 161 cases of neofunctionalization (11 of parent copies, 150 of child copies), 0 cases of subfunctionalization, and 64 cases of specialization. These numbers are consistent with those obtained from our original approach, providing strong support for the prevalence of neofunctionalization, as well as for the relative contributions of different evolutionary processes in the retention of duplicate genes in Drosophila.
). Then, using Table 1 as a template, we constructed an analogous set of rules for classifying evolutionary processes by divergence in expression localization patterns (Table S4; see Materials and Methods for details). Application of this alternative approach to our dataset yielded 56 cases of conservation, 161 cases of neofunctionalization (11 of parent copies, 150 of child copies), 0 cases of subfunctionalization, and 64 cases of specialization. These numbers are consistent with those obtained from our original approach, providing strong support for the prevalence of neofunctionalization, as well as for the relative contributions of different evolutionary processes in the retention of duplicate genes in Drosophila.
Comparison of Evolutionary Rates and Functions of Genes in Different Classes.
We used two separate methods to determine whether our classifications reflect true evolutionary phenomena. First, we examined protein evolutionary rates of parent and child copies maintained by different evolutionary processes. Under neofunctionalization, the copy with a novel function, which is under relaxed constraint (or possibly positive selection), is expected to evolve faster at the sequence level than the functionally conserved copy, which is under stronger selective constraint (1). To test for asymmetric evolutionary rates between duplicate genes, we compared the branch-specific Ka/Ks (see Materials and Methods) of parent and child copies in conserved, neofunctionalized, and specialized pairs. Indeed, gene copies of pairs that underwent neofunctionalization display unequal rates of protein sequence evolution (Fig. 2A). In particular, protein sequences of copies with new functions evolve faster than those with ancestral functions, and this difference is statistically significant for pairs with neofunctionalized child copies. In contrast, protein sequences of parent and child copies in conserved and specialized pairs evolve at comparable rates.
Fig. 2.

Sequence and functional support for classifications of the evolutionary processes maintaining young duplicate genes in Drosophila. (A) Distributions of branch-specific Ka/Ks for parent (P) and child (C) copies. Significance was tested by comparing distributions of parent and child copies. (B) Distributions of numbers of protein–protein interactions for ancestral (A), parent, and child copies. The horizontal dashed line depicts the median number of protein–protein interactions for single-copy genes. Significance was tested by comparing each distribution to that of single-copy genes. (C) Distributions of tissue specificities (τ) for ancestral, parent, and child copies. High τ indicates tissue-specific expression, and low τ indicates broad expression. The horizontal dashed line depicts the median τ for single-copy genes. Significance was tested by comparing each distribution to that of single-copy genes. (D) Heat maps illustrating mean relative expression levels in six tissues of single-copy (S), ancestral, parent, and child genes. Relative expression ranges from 0% to 46%, with darker colors indicating higher values. *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we examined three metrics of biological function to determine whether their patterns are consistent with our classifications. In particular, we compared numbers of protein–protein interactions, tissue specificities, and relative expression levels across tissues (see Materials and Methods for details) among ancestral, parent, and child genes in different categories, using observations for single-copy genes as a baseline for comparison (Fig. 2 B–D). As expected, ancestral, parent, and child copies of conserved pairs have similar numbers of protein–protein interactions, tissue specificities, and expression patterns. Although there are no significant trends for pairs with neofunctionalized parent copies, possibly because of the small sample size, there is strong statistical support for our classification of pairs with neofunctionalized child copies. Ancestral and parent copies participate in similar numbers of protein–protein interactions, have comparable tissue specificities, and display the same broad expression patterns, whereas child copies have fewer protein–protein interaction partners, higher tissue specificities, and are primarily expressed in testes. Moreover, as expected, ancestral, parent, and child copies in the specialization class are quite distinct from one another in terms of numbers of protein–protein interactions (ancestral and parent are different from child), tissue specificities (ancestral and child are different from parent), and relative expression levels across tissues (all copies are different from one another). Therefore, all three functional metrics support our classifications, illustrating that our approach detects biologically relevant differences in gene function among duplicate pairs.
Analysis of Factors Contributing to Evolutionary Fates of Duplicates.
The surprisingly strong bias toward neofunctionalization of child copies prompts a key question: Is functional divergence triggered by the duplication process itself? A simple way to test this hypothesis is to compare proportions of DNA- and RNA-mediated duplicates in conserved, neofunctionalized, and specialized pairs. In contrast to DNA-mediated duplication, RNA-mediated duplication generates child copies that lack parental cis-regulatory sequences, thus making it unlikely for such genes to retain ancestral functions. Therefore, we expected overrepresentations of DNA-mediated duplicates among pairs in which child copies maintained some or all ancestral functions (those that are conserved, specialized, or have neofunctionalized parent copies) and overrepresentations of RNA-mediated duplicates among pairs in which child copies are neofunctionalized. Comparisons of observed and expected numbers of DNA- and RNA-mediated duplicates in each class support this hypothesis; conserved and specialized child copies typically arose via DNA-mediated duplication, whereas neofunctionalized child copies generally arose via RNA-mediated duplication (Table 2). Although there is also an overrepresentation of DNA-mediated duplicates in pairs with neofunctionalized parent copies, it is not statistically significant, possibly because of the small sample size. However, it is important to note that, although neofunctionalized child copies tend to be produced by RNA-mediated duplication, neofunctionalization of child copies is also the most common evolutionary process maintaining DNA-mediated duplicates.
Table 2.
Numbers of DNA- and RNA-mediated duplicate genes by evolutionary process
| Classification | DNA-mediated | RNA-mediated | P |
| Conservation | 37 (30.36) | 10 (16.64) | 0.0428 |
| Neofunctionalization of parent copy | 11 (9.04) | 3 (4.96) | 0.2741 |
| Neofunctionalization of child copy | 85 (100.12) | 70 (54.88) | 0.0111 |
| Specialization | 32 (24.69) | 6 (13.31) | 0.0130 |
Expectations are shown in parentheses.
Aside from duplication mechanism, we hypothesized that another factor that may influence the evolutionary fates of young duplicates is the functional diversity of their ancestral genes. In particular, duplicate genes with highly specialized ancestral functions may have a lower capacity to evolve novel functionality than those with multiple ancestral functions. Consistent with the idea that highly specialized genes have less evolutionary potential, ancestral copies of conserved pairs participate in few protein–protein interactions and are highly tissue-specific, with expression primarily localized to testes (Fig. 2 B–D). In contrast, ancestral genes of pairs with neofunctionalized child copies have diverse functions. They participate in many protein–protein interactions, have low tissue specificities, and are expressed more broadly across tissues than ancestral genes in any other class. In specialized pairs, ancestral functional diversity is intermediate to those of conserved and neofunctionalized child pairs; ancestral genes participate in typical numbers of protein–protein interactions, are relatively tissue-specific, and are highly expressed in both carcass and ovary tissues. Thus, our analysis suggests that the evolutionary fates of young duplicates are limited by their ancestral functional diversities. Ancestral functions tend to be narrow for conserved pairs, moderate for specialized pairs, and broad for neofunctionalized pairs.
Examination of the “Out-of-Testes” Hypothesis for the Origin of New Genes.
Despite ancestral differences among classes, a common observation is that one gene copy, typically the child, is highly expressed in testes. Indeed, high testis expression in young genes has been uncovered in a variety of species, leading to the proposition of the out-of-testes hypothesis for the emergence of new genes (30). Testes provide an ideal environment for young genes to become established because of two inherent properties. First, transcription in testes may be more promiscuous because of open chromatin states during meiosis, as well as because relatively simple promoters are required for transcription. Hence, testes may facilitate initial transcription of young genes lacking regulatory elements, such as those produced by RNA-mediated duplication. Second, because of strong selective pressures, testes are the fastest-evolving tissues, possibly making them most receptive to accommodating evolutionary innovations such as new genes. Thus, although new genes originate in testes, they may ultimately acquire expression in other tissues as well (30). To test this hypothesis, we compared numbers of protein–protein interactions, tissue specificities, and relative expression levels across tissues between young pairs of duplicates and 301 old pairs that are shared between D. melanogaster and D. pseudoobscura. Although both young and old genes participate in fewer protein–protein interactions, are more tissue-specific, and are expressed more highly in testes than single-copy genes, old duplicate genes tend to have significantly more protein–protein interaction partners, lower tissue specificities, and broader expression patterns than young genes (Fig. 3). Therefore, our results support the idea that testes are a general conduit for gene origination and provide an entry point for the evolution of novel gene functions in other tissues.
Fig. 3.
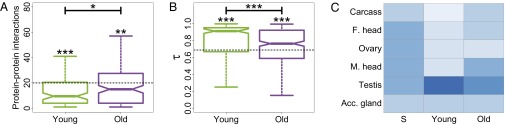
Comparison of functional diversity between young and old duplicate genes. (A) Distributions of numbers of protein–protein interactions for young and old duplicates. The horizontal dashed line depicts the median number of protein–protein interactions for single-copy genes. Significance was tested by comparing each distribution to that of single-copy genes (asterisks above boxplots), as well as by comparing distributions of young and old duplicates (asterisks above horizontal bars). (B) Distributions of τ values for young and old duplicates. The horizontal dashed line depicts the median τ value for single-copy genes. Significance was tested by comparing each distribution to that of single-copy genes (asterisks above boxplots), as well as by comparing distributions of young and old duplicates (asterisks above horizontal bars). (C) Heat maps illustrating mean relative expression levels in six tissues of single-copy (S), young, and old genes. Relative expression ranges from 0% to 46%, with darker colors indicating higher values. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Previous studies comparing gene expression levels of duplicate genes revealed that expression divergence between copies occurs rapidly (31–38) and can be asymmetric (32, 35, 38). However, our analysis is unique in that it uses expression data and phylogenetic relationships among genes to explicitly classify the evolutionary processes underlying the retention of duplicates on a genome-wide scale. Classifications obtained by our approach are robust to different cutoffs for expression divergence and are supported by an alternative approach based on expression localization patterns, patterns of protein evolutionary rates, and three metrics of biological function (numbers of protein–protein interactions, tissue specificities, and relative expression levels across tissues).
Application of our approach to young duplicates in Drosophila revealed neofunctionalization to be the primary evolutionary process maintaining duplicate genes and showed that it is primarily child copies that evolve new, often testis-specific, functions. Although a significant fraction of pairs are also maintained by either conservation or specialization, we did not uncover any evidence of subfunctionalization, which is thought to be an important process driving retention of young duplicate genes (2). This may be attributed to one of two reasons. First, previous studies have shown that duplicate genes produced by large-scale events, such as whole-genome duplications, typically have similar functions, whereas those produced by small-scale events often have divergent functions (37, 39). Because subfunctionalization requires that both gene copies start out with the same function, these findings suggest that subfunctionalization may be an important process maintaining duplicate genes produced by large-scale, but not small-scale, duplication events. Second, the probability of subfunctionalization decreases with increasing effective population size (Ne) and is approximately zero when Ne ≥ 106 (40), as is the case for both D. melanogaster and D. pseudoobscura (29, 41). Therefore, our lack of support for subfunctionalization of duplicate genes in Drosophila matches theoretical predictions, and subfunctionalization is expected to play a more dominant role in species with smaller Ne, such as mammals. Furthermore, selection for new functions is more efficient in species with larger Ne and, thus, patterns of functional divergence between duplicate genes might mimic those of protein evolution, for which there is widespread evidence of adaptive protein evolution in Drosophila, but little in mammals (42–44).
Although our analysis links asymmetry in evolution at sequence and expression levels of duplicate genes, correlations between the two metrics are small (r = 0.14; P < 2.2 × 10−16), indicating that functional divergence between duplicate genes cannot be attributed entirely to changes in protein-coding sequences. This imperfect correlation is not limited to duplicate genes and is a key argument for the crucial role of regulatory changes in functional evolution of all genes (45). The importance of regulatory evolution in functional divergence of duplicate genes is also highlighted by our finding that neofunctionalized child copies are often RNA-mediated duplicates, which must acquire regulatory motifs to become functional. Because it is unlikely that these newly formed regulatory regions are identical to those of parent genes, RNA-mediated duplicates typically do not have the same functions after duplication, undermining theoretical predictions that assume redundancy between copies. Therefore, in many cases, the duplication process itself creates a gene with a novel, potentially beneficial function, greatly increasing the probability that this gene will be fixed by positive selection and retained in the population (46, 47). Thus, fixation of RNA-mediated duplicates can proceed rapidly, particularly in species for which Ne is large and natural selection is efficient, such as Drosophila.
Whereas young duplicates are limited in their abilities to acquire expression in new tissues, comparison of young and old duplicates illustrated that the evolutionary potential of duplicate genes increases over time. In particular, we showed that although young duplicates tend to have very narrow functions that are often restricted to testes, old duplicates generally have diverse functions that are distributed across multiple tissues. Hence, our analysis supports the out-of-testes hypothesis of new gene emergence, which states that new genes arise in testes and evolve broader functions over time (30). Although it may seem counterintuitive for genes to first narrow and then broaden their functions again, testes are an ideal environment for young genes because of their rapid evolution and may enable new genes to become established and maintained rather than undergoing pseudogenization. Once established, such a transition may provide duplicate genes with opportunities to diversify from their ancestral states and create unique functional niches. In this scenario, duplicate genes can evolve essential functions by first being incorporated into a rapidly evolving tissue that is highly susceptible to evolutionary innovation (testis) and later becoming integrated into more slowly evolving functional networks.
Materials and Methods
Identification of Pairs of Duplicate Genes.
We downloaded D. melanogaster and D. pseudoobscura protein sequences from www.flybase.org and performed BLAST searches (48) to identify duplicate genes in each species. Matches with identity ≥50%, length ≥35%, and significance <10−3 were kept, and gene families containing more than two copies were removed. D. melanogaster pairs were supplemented with those from Chen et al. (49). Quantile-normalized RNA-seq data for carcass, female head, ovary, male head, testis, and accessory gland tissues of each species were obtained as described by Assis et al. (50). We restricted our analysis to gene pairs in which both copies are expressed in at least one tissue [i.e., fragments per kilobase of exon per million fragments mapped (FPKM) ≥ 1 for D. melanogaster and FPKM ≥ 4 for D. pseudoobscura (50)]. These expression cutoffs were also used in our alternate classification approach, based on expression localization patterns (see Classification of Evolutionary Processes by Expression Localization Patterns).
Assignment of Orthologs.
We obtained orthologs for each gene from the Drosophila ortholog table downloaded from www.flybase.org. This table contains orthologs from the Drosophila 12 Genomes Consortium, which were assigned by requiring both sequence similarity and conserved synteny (51). We defined old pairs as those for which both copies are present in D. melanogaster and D. pseudoobscura, and young pairs as those for which one copy is present in both sisters (parent/ancestral) and the other is not present in one of the two sisters or in any outgroups (child). We used similar parsimony rules to date the emergence of child copies along the Drosophila phylogeny.
Identification of Processes Maintaining Individual Pairs of Duplicate Genes.
We selected the semi-interquartile range from the median as a cutoff for 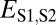 because it is affected very little by extreme values and is thus a robust measure of spread for skewed distributions (see Fig. S2). To determine its biological relevance, we tested a number of alternative cutoffs, including the mean, standard deviation from the mean, median, median plus the median absolute deviation with different constants, and various quantiles (Table S1). Application of any of these cutoffs yields neofunctionalization as the dominant evolutionary process identified, illustrating that selection of a particular cutoff does not alter our main result. Although smaller cutoffs result in fewer pairs classified under neofunctionalization and more under specialization, examination of relative expression levels in different tissue of ancestral, parent, and child genes revealed that, with smaller cutoffs, both classes have similar distributions that look like the neofunctionalized child class in Fig. 2D. In contrast, larger cutoffs yield more cases of conservation, but relative expression levels in different tissues of ancestral, parent, and child genes are not similar and thus do not support conservation of function. Hence, the semi-interquartile range from the median appears to be the most appropriate cutoff tested for
because it is affected very little by extreme values and is thus a robust measure of spread for skewed distributions (see Fig. S2). To determine its biological relevance, we tested a number of alternative cutoffs, including the mean, standard deviation from the mean, median, median plus the median absolute deviation with different constants, and various quantiles (Table S1). Application of any of these cutoffs yields neofunctionalization as the dominant evolutionary process identified, illustrating that selection of a particular cutoff does not alter our main result. Although smaller cutoffs result in fewer pairs classified under neofunctionalization and more under specialization, examination of relative expression levels in different tissue of ancestral, parent, and child genes revealed that, with smaller cutoffs, both classes have similar distributions that look like the neofunctionalized child class in Fig. 2D. In contrast, larger cutoffs yield more cases of conservation, but relative expression levels in different tissues of ancestral, parent, and child genes are not similar and thus do not support conservation of function. Hence, the semi-interquartile range from the median appears to be the most appropriate cutoff tested for 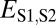 .
.
Classification of Evolutionary Processes by Expression Localization Patterns.
To classify evolutionary processes by expression localization patterns, we devised a set of rules (Table S4) similar to those used for Euclidian distances (Table 1). In particular, under conservation, the expression localization patterns of parent, child, and ancestral copies should be the same and, hence, we expect 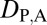 = 0 and
= 0 and 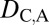 = 0. Under neofunctionalization, the expression localization patterns of the ancestral copy should be similar to those of either the parent or child copy and different from those of the other copy. Therefore, we expect
= 0. Under neofunctionalization, the expression localization patterns of the ancestral copy should be similar to those of either the parent or child copy and different from those of the other copy. Therefore, we expect 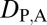 = 1 and
= 1 and 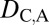 = 0 when the parent copy is neofunctionalized, and
= 0 when the parent copy is neofunctionalized, and 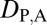 = 0 and
= 0 and 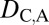 = 1 when the child copy is neofunctionalized. Under subfunctionalization, the expression localization patterns of parent and child copies should both be different from those of the ancestral copy, whereas the combined parent–child expression localization patterns should be similar to those of the ancestral copy. Thus, we expect
= 1 when the child copy is neofunctionalized. Under subfunctionalization, the expression localization patterns of parent and child copies should both be different from those of the ancestral copy, whereas the combined parent–child expression localization patterns should be similar to those of the ancestral copy. Thus, we expect 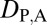 = 1,
= 1, 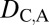 = 1, and
= 1, and 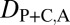 = 0. Finally, under specialization, the expression localization patterns of the parent copy, expression localization patterns of the child copy, and combined parent–child expression localization patterns should all be different from those of the ancestral copy, so we expect
= 0. Finally, under specialization, the expression localization patterns of the parent copy, expression localization patterns of the child copy, and combined parent–child expression localization patterns should all be different from those of the ancestral copy, so we expect 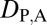 = 1,
= 1, 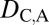 = 1, and
= 1, and 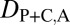 = 1.
= 1.
Estimating Evolutionary Rates of Duplicate Gene Copies.
D. melanogaster and D. pseudoobscura CDS sequences were downloaded from www.flybase.org. Sequences for single-copy genes, as well as for ancestral, parent, and child copies of duplicate gene pairs, were aligned by multiple alignment of coding sequences accounting for frameshifts and stop codons (MACSE) (52). Phylogenetic analysis by maximum likelihood (PAML) (53) was used to estimate branch-specific substitution rates at synonymous (Ks) and nonsynonymous (Ka) sites of parent and child genes. To be conservative, we removed genes with Ks ≥ 3 from our analysis. There are 20 such genes, and their exclusion did not alter Ka/Ks distributions.
Distinguishing Between DNA- and RNA-Mediated Duplications.
Exons for parent and child copies in each duplicate pair were downloaded from www.flybase.org. DNA-mediated duplication was inferred when parent and child copies each have multiple orthologous exons. RNA-mediated duplication was inferred when the parent copy has multiple exons, and the child copy has only one. For cases in which both parent and child copies have single exons, the mechanism was considered unknown.
Functional Analyses of Duplicate Genes.
Protein–protein interaction data were downloaded from the Drosophila Interactions Database (DroID) at www.droidb.org and from FlyBase at www.flybase.org. Numbers of interactions were estimated by concatenating the datasets (eight in total) and counting unique interaction partners for each gene. Because data are only available for D. melanogaster, D. pseudoobscura genes were excluded from this analysis. The tissue specificity index (τ) for each gene was obtained as described by Assis et al. (50). τ ranges from 0 to 1, where low τ values indicate broad expression and high τ values indicate tissue-specific expression. Relative expression levels across tissues were determined by calculating the mean relative expression in each tissue for single-copy, ancestral, parent, and child genes. To enable comparison between species, we normalized D. pseudoobscura expression in each tissue by the mean D. melanogaster expression in the corresponding tissue of single-copy genes.
Statistical Analyses.
Mann–Whitney U tests were used to compare distributions of Euclidian distances, Ka/Ks estimates, numbers of protein–protein interactions, and τ values. χ2 tests were used to compare observed and expected numbers of DNA- and RNA-mediated duplications for different classifications, as well as observed and expected proportions of genes retained by each evolutionary process for different phylogenetic ages. For each analysis, expected numbers were assumed to be proportional to those observed in the entire dataset. All statistical analyses were performed in the R software environment (54).
Supplementary Material
Acknowledgments
We thank Kevin Thornton for his comments on the manuscript. This work was funded by a National Institutes of Health (NIH) fellowship F32 GM100673-02 (to R.A.); NIH Grants R01GM076007 and R01GM093182 (to D.B.); and a Packard Fellowship (to D.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1313759110/-/DCSupplemental.
References
- 1.Ohno S. Evolution by Gene Duplication. Berlin: Springer; 1970. [Google Scholar]
- 2.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 3.Ferris SD, Whitt GS. Evolution of the differential regulation of duplicate genes after polyploidization. J Mol Evol. 1979;12(4):267–317. doi: 10.1007/BF01732026. [DOI] [PubMed] [Google Scholar]
- 4.Lundin LG. Evolution of the vertebrate genome as reflected in paralogous chromosomal regions in man and the house mouse. Genomics. 1993;16(1):1–19. doi: 10.1006/geno.1993.1133. [DOI] [PubMed] [Google Scholar]
- 5.Sidow A. Gen(om)e duplications in the evolution of early vertebrates. Curr Opin Genet Dev. 1996;6(6):715–722. doi: 10.1016/s0959-437x(96)80026-8. [DOI] [PubMed] [Google Scholar]
- 6.Brookfield JFY. Genetic redundancy. Adv Genet. 1997;36:137–155. doi: 10.1016/s0065-2660(08)60308-9. [DOI] [PubMed] [Google Scholar]
- 7.Nadeau JH, Sankoff D. Comparable rates of gene loss and functional divergence after genome duplications early in vertebrate evolution. Genetics. 1997;147(3):1259–1266. doi: 10.1093/genetics/147.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Postlethwait JH, et al. Vertebrate genome evolution and the zebrafish gene map. Nat Genet. 1998;18(4):345–349. doi: 10.1038/ng0498-345. [DOI] [PubMed] [Google Scholar]
- 9.Force A, et al. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151(4):1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoltzfus A. On the possibility of constructive neutral evolution. J Mol Evol. 1999;49(2):169–181. doi: 10.1007/pl00006540. [DOI] [PubMed] [Google Scholar]
- 11.He X, Zhang J. Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics. 2005;169(2):1157–1164. doi: 10.1534/genetics.104.037051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154(1):459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walsh JB. How often do duplicated genes evolve new functions? Genetics. 1995;139(1):421–428. doi: 10.1093/genetics/139.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh B. Population-genetic models of the fates of duplicate genes. Genetica. 2003;118(2-3):279–294. [PubMed] [Google Scholar]
- 15.Kondrashov FA, Koonin EV. A common framework for understanding the origin of genetic dominance and evolutionary fates of gene duplications. Trends Genet. 2004;20(7):287–290. doi: 10.1016/j.tig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Innan H. Population genetic models of duplicated genes. Genetica. 2009;137(1):19–37. doi: 10.1007/s10709-009-9355-1. [DOI] [PubMed] [Google Scholar]
- 17.Burki F, Kaessmann H. Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat Genet. 2004;36(10):1061–1063. doi: 10.1038/ng1431. [DOI] [PubMed] [Google Scholar]
- 18.Duarte JM, et al. Expression pattern shifts following duplication indicative of subfunctionalization and neofunctionalization in regulatory genes of Arabidopsis. Mol Biol Evol. 2006;23(2):469–478. doi: 10.1093/molbev/msj051. [DOI] [PubMed] [Google Scholar]
- 19.Escriva H, et al. Neofunctionalization in vertebrates: The example of retinoic acid receptors. PLoS Genet. 2006;2(7):e102. doi: 10.1371/journal.pgen.0020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perry GH, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39(10):1256–1260. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sackton TB, et al. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 2007;39(12):1461–1468. doi: 10.1038/ng.2007.60. [DOI] [PubMed] [Google Scholar]
- 22.Kleinjan DA, et al. Subfunctionalization of duplicated zebrafish pax6 genes by cis-regulatory divergence. PLoS Genet. 2008;4(2):e29. doi: 10.1371/journal.pgen.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shapiro JA, et al. Adaptive genic evolution in the Drosophila genomes. Proc Natl Acad Sci USA. 2007;104(7):2271–2276. doi: 10.1073/pnas.0610385104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29(4):482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- 25.Bhardwaj N, Lu H. Correlation between gene expression profiles and protein-protein interactions within and across genomes. Bioinformatics. 2005;21(11):2730–2738. doi: 10.1093/bioinformatics/bti398. [DOI] [PubMed] [Google Scholar]
- 26.Zhou X, Kao MC, Wong WH. Transitive functional annotation by shortest-path analysis of gene expression data. Proc Natl Acad Sci USA. 2002;99(20):12783–12788. doi: 10.1073/pnas.192159399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.French L, Pavlidis P. Relationships between gene expression and brain wiring in the adult rodent brain. PLOS Comput Biol. 2011;7(1):e1001049. doi: 10.1371/journal.pcbi.1001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereira V, Waxman D, Eyre-Walker A. A problem with the correlation coefficient as a measure of gene expression divergence. Genetics. 2009;183(4):1597–1600. doi: 10.1534/genetics.109.110247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beckenbach AT, Wei YW, Liu H. Relationships in the Drosophila obscura species group, inferred from mitochondrial cytochrome oxidase II sequences. Mol Biol Evol. 1993;10(3):619–634. doi: 10.1093/oxfordjournals.molbev.a040034. [DOI] [PubMed] [Google Scholar]
- 30.Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010;20(10):1313–1326. doi: 10.1101/gr.101386.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu Z, Nicolae D, Lu HH, Li WH. Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet. 2002;18(12):609–613. doi: 10.1016/s0168-9525(02)02837-8. [DOI] [PubMed] [Google Scholar]
- 32.Wagner A. Asymmetric functional divergence of duplicate genes in yeast. Mol Biol Evol. 2002;19(10):1760–1768. doi: 10.1093/oxfordjournals.molbev.a003998. [DOI] [PubMed] [Google Scholar]
- 33.Makova KD, Li WH. Divergence in the spatial pattern of gene expression between human duplicate genes. Genome Res. 2003;13(7):1638–1645. doi: 10.1101/gr.1133803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu Z, Rifkin SA, White KP, Li WH. Duplicate genes increase gene expression diversity within and between species. Nat Genet. 2004;36(6):577–579. doi: 10.1038/ng1355. [DOI] [PubMed] [Google Scholar]
- 35.Gu X, Zhang Z, Huang W. Rapid evolution of expression and regulatory divergences after yeast gene duplication. Proc Natl Acad Sci USA. 2005;102(3):707–712. doi: 10.1073/pnas.0409186102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li WH, Yang J, Gu X. Expression divergence between duplicate genes. Trends Genet. 2005;21(11):602–607. doi: 10.1016/j.tig.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 37.Casneuf T, De Bodt S, Raes J, Maere S, Van de Peer Y. Nonrandom divergence of gene expression following gene and genome duplications in the flowering plant Arabidopsis thaliana. Genome Biol. 2006;7(2):R13. doi: 10.1186/gb-2006-7-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ganko EW, Meyers BC, Vision TJ. Divergence in expression between duplicated genes in Arabidopsis. Mol Biol Evol. 2007;24(10):2298–2309. doi: 10.1093/molbev/msm158. [DOI] [PubMed] [Google Scholar]
- 39.Fares MA, Keane OM, Toft C, Carretero-Paulet L, Jones GW. The roles of whole-genome and small-scale duplications in the functional specialization of Saccharomyces cerevisiae genes. PLoS Genet. 2013;9(1):e1003176. doi: 10.1371/journal.pgen.1003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lynch M, O’Hely M, Walsh B, Force A. The probability of preservation of a newly arisen gene duplicate. Genetics. 2001;159(4):1789–1804. doi: 10.1093/genetics/159.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen JD, Bachtrog D. Characterizing the influence of effective population size on the rate of adaptation: Gillespie’s Darwin domain. Genome Biol Evol. 2011;3:687–701. doi: 10.1093/gbe/evr063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carroll SB. Evolution at two levels: On genes and form. PLoS Biol. 2005;3(7):e245. doi: 10.1371/journal.pbio.0030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Britten RJ. Rates of DNA sequence evolution differ between taxonomic groups. Science. 1986;231(4744):1393–1398. doi: 10.1126/science.3082006. [DOI] [PubMed] [Google Scholar]
- 44.Moriyama EN. Higher rates of nucleotide substitution in Drosophila than in mammals. Jpn J Genet. 1987;62:139–147. [Google Scholar]
- 45.Sharp PM, Li WH. On the rate of DNA sequence evolution in Drosophila. J Mol Evol. 1989;28(5):398–402. doi: 10.1007/BF02603075. [DOI] [PubMed] [Google Scholar]
- 46.Lynch M, Katju V. The altered evolutionary trajectories of gene duplicates. Trends Genet. 2004;20(11):544–549. doi: 10.1016/j.tig.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Innan H, Kondrashov FA. The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet. 2010;11(2):97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- 48.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 49.Chen S, Zhang YE, Long M. New genes in Drosophila quickly become essential. Science. 2010;330(6011):1682–1685. doi: 10.1126/science.1196380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Assis R, Zhou Q, Bachtrog D. Sex-biased transcriptome evolution in Drosophila. Genome Biol Evol. 2012;4(11):1189–1200. doi: 10.1093/gbe/evs093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Drosophila 12 Genomes Consortium, et al. (2007) Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218. [DOI] [PubMed]
- 52.Ranwez V, Harispe S, Delsuc F, Douzery EJP. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE. 2011;6(9):e22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 54.R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.