

Significance
Exosome-mediated intercellular transfer of proteins and nucleic acids has attracted considerable attention as exosomes may promote the development of cancer and other pathological conditions; however, the mechanism of exosome uptake by target cells and how this may be inhibited remain as important questions. We provide evidence that heparan sulfate proteoglycans (HSPGs) function as receptors of cancer cell-derived exosomes. Importantly, our data indicate that the HSPG-dependent uptake route is highly relevant for the biological activity of exosomes, and thus a potential target for inhibition of exosome-mediated tumor development. Given that several viruses have previously been shown to enter cells through HSPGs, our data implicate HSPG as a convergence point during cellular uptake of endogenous vesicles and virus particles.
Keywords: endocytosis, tumor, glioma
Abstract
Extracellular vesicle (EV)-mediated intercellular transfer of signaling proteins and nucleic acids has recently been implicated in the development of cancer and other pathological conditions; however, the mechanism of EV uptake and how this may be targeted remain as important questions. Here, we provide evidence that heparan sulfate (HS) proteoglycans (PGs; HSPGs) function as internalizing receptors of cancer cell-derived EVs with exosome-like characteristics. Internalized exosomes colocalized with cell-surface HSPGs of the syndecan and glypican type, and exosome uptake was specifically inhibited by free HS chains, whereas closely related chondroitin sulfate had no effect. By using several cell mutants, we provide genetic evidence of a receptor function of HSPG in exosome uptake, which was dependent on intact HS, specifically on the 2-O and N-sulfation groups. Further, enzymatic depletion of cell-surface HSPG or pharmacological inhibition of endogenous PG biosynthesis by xyloside significantly attenuated exosome uptake. We provide biochemical evidence that HSPGs are sorted to and associate with exosomes; however, exosome-associated HSPGs appear to have no direct role in exosome internalization. On a functional level, exosome-induced ERK1/2 signaling activation was attenuated in PG-deficient mutant cells as well as in WT cells treated with xyloside. Importantly, exosome-mediated stimulation of cancer cell migration was significantly reduced in PG-deficient mutant cells, or by treatment of WT cells with heparin or xyloside. We conclude that cancer cell-derived exosomes use HSPGs for their internalization and functional activity, which significantly extends the emerging role of HSPGs as key receptors of macromolecular cargo.
Cells are known to communicate via soluble ligands and through direct cell–cell and cell–matrix interactions. Recent data suggest an intriguing role of extracellular vesicles (EVs), including exosomes and microvesicles, in various physiological and pathophysiological processes through intercellular transfer of mRNA, miRNA, and signaling proteins (1–8). EVs have attracted considerable attention as studies implicate this new mode of intercellular communication in, e.g., immune system regulation, atherosclerosis, and tumor development. EVs have a size range of approximately 50 to 1000 nm, and are released from the cell surface as microvesicles, or, in the case of exosomes, through fusion of multivesicular bodies with the plasma membrane. Notably, EVs display the same surface topology as the plasma membrane, with extracellular domains of proteins at the surface, and enclosing cytosolic contents in their lumen (1–9).
Given the suggested functional role of EVs in cancer and other pathophysiological processes, they emerge as potential targets of therapeutic intervention. The complex molecular architecture of EVs should motivate studies aimed at targeting general mechanisms of EV-dependent functional effects, i.e., EV formation and entry into recipient cells. Recent studies have implicated the small GTPase Rab27a and a syndecan-syntenin-ALIX–mediated pathway in exosome biogenesis and secretion (10–12). Although the functional effects of EVs mostly rely on internalization and subsequent release of EV contents in recipient cells, the elucidation of EV uptake mechanisms and how these may be targeted remains an important challenge.
Heparan sulfate (HS) proteoglycans (PGs; HSPGs) are a family of proteins substituted with glycosaminoglycan (GAG) polysaccharides, which are extensively modified by sulfation, that largely determine their functional interactions (13–15). In the context of the present study, it is of interest that various types of virus particles, peptide–nucleic acid complexes, and lipoproteins may use HSPGs for cell-surface adsorption and internalization (13, 16, 17). Here, we have investigated the potential role of HSPG as a functional entry pathway of cancer cell-derived EVs.
Results
Exosomes Consume HSPG at the Cell Surface and Colocalize with HSPG in the Cytoplasm.
We chose to study EVs from the well-characterized U-87 MG cell line established from a glioblastoma multiforme (GBM) patient tumor (18). These cells produce significant amounts of EVs, as shown by EM and immunoblotting analysis for established markers of EVs (Fig. 1). The size distribution (approximately 150 nm) and positive staining for CD63 and tissue factor (Fig. 1A), the enrichment of the tetraspanins CD63 and CD81, and presence of RAB5 in isolated vesicles (Fig. 1B) indicate a vesicle population with exosome-like characteristics (1–9). Moreover, the cis-Golgi marker GM130 and the cytoskeletal protein α-tubulin were depleted in vesicles compared with cells (Fig. 1B). Exosomes are generally too small to be distinguished from cell debris and larger vesicles by direct flow cytometry analysis; however, flow cytometry demonstrated binding of isolated vesicles to anti-CD63 conjugated beads, whereas control beads conjugated with a control antibody were negative (Fig. S1A). These results lend further support of the isolation of exosome-like EVs (hereafter referred to as exosomes). To study the uptake of isolated exosomes, we used a fluorescent dye (PKH) with long aliphatic tails that are incorporated into the lipid membrane of exosome vesicles. By using flow cytometry analysis, we found dose-dependent and saturable uptake of PKH-labeled exosomes (Fig. 1C) that increased with incubation time (Fig. S1B). Confocal microscopy and flow cytometry analyses supported the specificity of PKH-exosome uptake, which was efficiently inhibited by incubation at 4 °C (Fig. S1C), and the presence of excess, unlabeled exosomes (Fig. 1D). Notably, the transfer of free PKH between membranes may limit the accuracy of exosome uptake experiments. To evaluate the redistribution of PKH between membranes, we performed analyses of premixed unlabeled and PKH-labeled exosomes. There was no apparent decrease of fluorescent signal in PKH-labeled exosomes and, accordingly, only a minor fraction (approximately 0.0025) of the fluorescence of PKH-labeled exosomes was transferred to unlabeled vesicles (Fig. S1D). Further, nonspecific PKH transfer would not exhibit concentration dependent and saturable kinetics (Fig. 1C), and cells treated with HSPG inhibitors (as described later) displayed down to approximately 10% remaining PKH fluorescence compared with untreated controls (see Fig. S3C). Thus, the PKH signal of recipient cells is largely associated with the specific uptake of PKH-labeled exosomes, with only a minor contribution by free PKH transfer.
Fig. 1.

Exosome-like EVs from GBM cells colocalize with cell-surface HSPGs. (A) Representative EM image shows EVs with a typical diameter of approximately 150 nm and positive staining for CD63 (Upper) and tissue factor (TF; Lower). (Scale bar, 100 nm.) (B) Equal amounts of total protein from EV and donor cell lysates were analyzed by immunoblotting for RAB5, CD63, CD81, α-tubulin, and GM130. (C) GBM cells were incubated with the indicated concentrations of PKH-labeled exosomes. Flow cytometry analysis shows dose-dependent and saturable uptake of exosomes. (D) Confocal microscopy analysis of GBM cells incubated with PKH26-labeled exosomes (10 µg/mL) in the absence (Left) or in the presence (Center) of 100 µg/mL unlabeled exosomes. (Right) Quantitative data from similar experiment (Left) analyzed by flow cytometry. (E) GBM cells transfected with syndecan-2–GFP (Sdc2-GFP; Upper) or glypican-1–GFP (Gpc1-GFP; Lower) encoding plasmid were incubated with PKH-labeled exosomes (10 µg/mL) for 30 min, washed with 1 M NaCl, and analyzed by confocal microscopy. Merged images of the indicated areas show colocalization of exosomes and Sdc2-GFP and Gpc1-GFP, respectively. (Scale bar, 20 µm.) (F) GBM cells were untreated (Ctrl) or incubated with exosomes or HIV-Tat peptide, followed by extensive washing to remove remaining cell surface-bound ligand, followed by staining with anti-HS antibody and flow cytometry analysis. (G) GBM cells were grown in the absence (Ctrl) or presence of 5 mM DFMO. Cells were then incubated with PKH-labeled exosomes (40 µg/mL) for 1 h and analyzed for uptake by flow cytometry. Data shown represent the mean ± SD from three independent experiments, each performed in triplicate (C) or duplicate (D and G). (F) Data are presented as fold of untreated control cells, and are the mean ± SD from three independent experiments, each performed in triplicate (*P < 0.05).
There are two major classes of cell surface HSPGs: the glypican family of glycosyl-phosphatidyl-inositol (GPI)-linked proteins and the syndecan family of transmembrane proteins (13, 19, 20). We performed exosome uptake studies in cells transfected to express syndecan or glypican-GFP fusion protein. Internalized exosomes colocalized with syndecan-GFP– and glypican-GFP–positive vesicles (Fig. 1E). HIV-Tat peptide enters cells through an HSPG-dependent pathway (21, 22), and the anti-HS antibody AO4B08 recognizes the internalizing population of cell-surface HSPG (23). Accordingly, incubation with HIV-Tat peptide consumed the AO4B08 HS epitope at the cell surface (Fig. 1F). Interestingly, following exosome internalization, the AO4B08 HS epitope was reduced to a similar extent as with HIV-Tat peptide (Fig. 1F). Moreover, treatment with the polyamine synthesis inhibitor α-difluoromethylornithine (DFMO), which has been shown to induce structural alterations of HS and to up-regulate HSPG-dependent uptake of HIV-Tat peptide (24, 25), significantly increased cellular uptake of exosomes (Fig. 1G). These results suggest that HIV-Tat peptide and exosomes may share a similar, HSPG-dependent entry pathway.
Exogenous HS Inhibits Exosome Uptake in a Dose-, Size-, and Charge-Dependent Manner.
To further study the interplay between exosomes and HSPG, we next performed exosome uptake experiments in the presence of heparin (an HS mimetic). Interestingly, heparin dose-dependently inhibited exosome uptake, and, at the highest concentration used (10 μg/mL), uptake was reduced by approximately 55% compared with untreated cells (Fig. 2A, Left). This is further supported by confocal microscopy studies (Fig. 2A, Right). The closely related polysaccharide chondroitin sulfate (CS) showed no inhibition of exosome uptake (Fig. 2B). Highly sulfated HS (HS-6) as well as less sulfated HS (HS-2) displayed significant inhibition of exosome uptake (Fig. 2C). Again, two different preparations of CS, sulfated either at the C-4 (CS-4) or the C-6 (CS-6) position of the hexosamine, had no effect on exosome uptake (Fig. 2C). Notably, HS-2 that exhibits almost 50% less charge density (total sulfate/hexosamine, approximately 0.56) compared with CS-4 and CS-6 (total sulfate/hexosamine, 1.0) (26) significantly inhibited exosome uptake at 10 μg/mL, whereas CS-4 and CS-6 were inefficient even at 100 μg/mL. These data indicate that exosome uptake through HS involves structural specificity of HS vs. CS. However, the observation that HS-6 (total sulfate/hexosamine, approximately 1.63) (26) is a more potent inhibitor compared with HS-2 (Fig. 2C) suggested that exosome uptake inhibition by HS depends on its overall sulfation and negative charge. Accordingly, heparin devoid of N-sulfation, as well as completely desulfated heparin, failed to inhibit exosome uptake (Fig. 2D). As shown in Fig. 2E, low molecular weight heparins (LMWHs) reduced exosome uptake in a dose-dependent manner, and their effects were similar to those of full-length heparin. However, smaller heparin oligosaccharides had no or very limited effects on exosome uptake (Fig. 2F), indicating a size-dependent effect.
Fig. 2.

Effects of free heparin, HS, and CS chains on exosome uptake. (A) (Left) GBM cells were incubated with PKH-labeled exosomes (40 µg/mL) for 1 h in the absence (Ctrl) or in the presence of 1, 5, or 10 µg/mL heparin, and then analyzed for exosome uptake by flow cytometry. (Right) Representative confocal microscopy images of cells incubated with exosomes without (Ctrl; Upper) or with 10 µg/mL heparin (Lower). (B) Same experiments as in A without (Ctrl) or with 1, 10, or 100 µg/mL CS. (C) Same experiment as in A without (Ctrl) or with 10 µg/mL heparin, HS-6, HS-2, 4-O-sulfated CS (CS-4), or 6-O-sulfated CS (CS-6). (D) Same experiment as in A without (Ctrl) or with 10 µg/mL heparin, N-desulfated heparin (NDS), or completely desulfated heparin. (E) Same experiment as in A without (Ctrl) or with 1, 10, or 100 µg/mL full-length heparin, or either of LMHWs enoxaparin, dalteparin, or tinzaparin. (F) Same experiment as in A without (Ctrl) or with 1, 10, or 100 µg/mL heparin, or heparin oligosaccharides, as indicated. (G and H) Heparin-agarose and PKH-labeled exosomes were incubated with (G) or without (H) 2 mM CaCl2, and the fraction of nonbinding and binding (1 M and 2 M NaCl) exosomes was determined by fluorescence analyses. (I) Uptake of PKH-labeled exosomes with or without 2 mM CaCl2 was determined by flow cytometry analysis. Data shown represent the mean ± SD from three independent experiments, each performed in triplicate (A–D) or duplicate (G–I). (E and F) Data are presented as fold of untreated control cells, and are the mean ± SD from three independent experiments, each performed in triplicate (*P < 0.05).
We found that the majority of isolated exosomes were strongly bound to heparin-agarose through electrostatic interactions; elution with 2 M NaCl was required to efficiently release exosomes from heparin (Fig. 2G). The exosome–heparin interaction was largely dependent on the presence of Ca2+ (Fig. 2 G and H), and, accordingly, exosome uptake was significantly reduced in Ca2+-depleted compared with Ca2+-containing medium (Fig. 2I). These results indicate that HS chains inhibit cellular uptake of exosomes in a dose-, size-, and charge density-dependent manner through competition with cell-surface HSPGs for exosome binding.
Exosome Uptake Depends on Intact HSPG Synthesis and HS Sulfation in Target Cells.
To more directly investigate the role of HSPGs in exosome uptake, the next series of experiments were performed with WT (CHO-K1) and mutant (pgsA-745) CHO cells, deficient in xylosyl transferase (XYLT). PgsA cells express approximately 5% PG compared with WT cells (27). The inhibitory activity of heparin on exosome uptake was not restricted to GBM cells, as a comparable, dose-dependent inhibition was found in WT CHO cells (Fig. 3A). Interestingly, exosome uptake was substantially reduced in mutant, PG-deficient cells compared with WT cells (Fig. 3 B and C). Over a wide range of exosome concentrations, uptake was approximately 2- to 2.5-fold greater in WT compared with PG-deficient cells (Fig. 3B), which is in good agreement with 50% to 60% inhibition of exosome uptake by heparin (Fig. 3A). PgsA cells are pan-PG deficient, i.e., they lack PGs of the HS as well as the CS type. To specifically investigate the role of HSPGs, we next performed experiments with CHO cell mutants lacking N-sulfation (pgsE) and 2-O-sulfation (pgsF), respectively (28, 29). Both mutants displayed significantly reduced exosome uptake compared with WT cells (Fig. 3D), showing a specific role of sulfation in HSPG. Moreover, these studies indicate that the results with pgsA cells were not caused by secondary mutations unrelated to HSPG synthesis. In further support of a specific role of HSPGs, enzymatic depletion of cell-surface HSPG reduced exosome uptake by approximately 50%, whereas enzymatic digestion of CSPG rather increased exosome uptake compared with control cells (Fig. 3E). To exclude that the HSPG dependence is restricted to exosomes from U-87 MG cells, we performed experiments with two additional, patient-derived tumor cell lines (U118 MG and LN18) that previously have been shown to produce exosomes (30). In accordance with the U-87 MG data, heparin as well as LMWH efficiently inhibited uptake of exosomes from these cell lines (Fig. S2 A and C). Moreover, pretreatment of recipient cells with HS lyase similarly inhibited uptake of exosomes from these cell lines (Fig. S2 B and D). We conclude that our results are applicable to exosomes from several cell sources.
Fig. 3.

Exosome uptake depends on cell-surface HSPGs. (A) WT CHO cells were incubated with PKH-labeled exosomes (40 µg/mL) for 1 h without (Ctrl) or with 0.1, 1, or 10 µg/mL heparin, and analyzed for exosome uptake by flow cytometry. (B) WT and PG-deficient (PG-def.) mutant CHO cells were incubated with the indicated concentrations of PKH-labeled exosomes, and analyzed for exosome uptake by flow cytometry. (C) WT (Upper) and PG-deficient (Lower) CHO cells were incubated with PKH-labeled exosomes (10 µg/mL) for 30 min and analyzed by confocal microscopy. (Scale bar, 50 µm.) (D) WT, PG-deficient, HS N-deacetylase sulfotransferase (NDST)–deficient (NS-def.), and HS 2-O-sulfotransferase–deficient (2OS-def.) mutant CHO cells were incubated with PKH-labeled exosomes (40 µg/mL) for 1 h and analyzed by flow cytometry. (E) GBM cells were untreated (Ctrl) or pretreated with chondroitinase ABC lyase (ABC’ase) or heparinase I and III lyases (HS’ase) to deplete cell-surface CSPG and HSPG, respectively. Uptake of PKH-labeled exosomes (40 µg/mL) for 1 h was determined by flow cytometry. Data shown represent the mean ± SD from three independent experiments, each performed in triplicate (A, B, and E). (D) Data are presented as fold of WT cells, and are the mean ± SD from three independent experiments, each performed in triplicate (*Significant decrease at P < 0.05 vs. control or WT cells; #Significant increase at P < 0.05 vs. control).
HSPGs Are Sorted to Exosomes but Exosome-Associated HSPGs Are Not Involved in Uptake.
We next studied the possible sorting of HSPGs to exosomes and, if so, their role in exosome uptake. To this end, we performed [35S]sulfate metabolic labeling experiments, followed by anion exchange chromatography, and gel filtration analysis for the presence of intact PGs as well as GAG chains and oligosaccharides in cells and corresponding exosomes. As expected, cells contained intact PGs as well as free GAGs (Fig. 4A, black squares). Here we provide biochemical evidence that exosomes indeed carry intact PGs (Fig. 4A, red squares); however, exosomes appeared relatively depleted of GAGs. We next used an antibody (3G10) specifically recognizing HSPG core protein with a remaining HS stub following treatment with HS lyase (31). Immunoblotting experiments demonstrated that glypican as well as syndecan HSPG core proteins are associated with exosomes (Fig. 4B). Together with previous studies on HSPG core protein identification (23), we conclude that exosomes may contain several types of glypican and syndecan HSPGs. Importantly, exosome-associated HSPG appear not to be involved in cellular uptake, as enzymatic depletion of exosomal HSPG had no effect on their internalization (Fig. 4C).
Fig. 4.
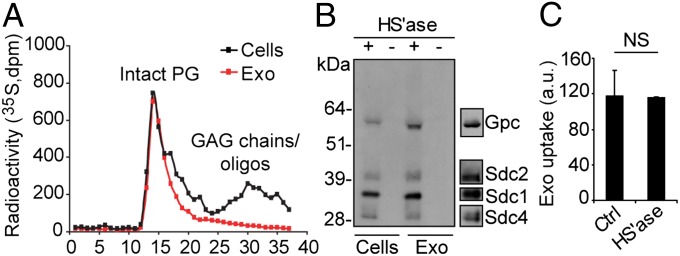
HSPGs are sorted to exosome vesicles but have no role in their uptake. (A) GBM cells were metabolically labeled with [35S]sulfate, and 35S-PGs and GAGs/oligosaccharides from cell and exosome lysates were size-fractionated on a Superose 6 column as described in SI Materials and Methods. (B) Isolated PGs from lysates of exosomes or GBM cells were untreated (−) or digested with heparinase III and ABC lyases (+). The digest products were separated by SDS page, and HSPG core proteins were visualized by immunoblotting with 3G10 anti-ΔHS antibody. Typical positions of members of the glypican and syndecan HSPG families are indicated (Right). (C) PKH-labeled exosomes (40 μg/mL) were untreated (Ctrl) or digested with heparinase I and III lyases (HS’ase), and then analyzed for uptake by GBM cells for 1 h by flow cytometry. Data shown represent the mean ± SD from three independent experiments, each performed in duplicate. NS, not significant (i.e., no statistically significant difference).
Functional Effects of Exosomes in Cancer Cells Depend on HSPGs.
The aforementioned data provide biochemical and genetic evidence that HSPGs are required for intact exosome uptake; however, a residual 40% to 50% uptake activity remained in the presence of heparin (Fig. 2) and in HSPG-deficient mutant cells as well as in cells enzymatically depleted of cell-surface HSPG (Fig. 3) compared with the respective controls. Thus, we next investigated the relevance of the HSPG-dependent entry pathway for the functional activity of exosomes. GBM cell-derived exosomes dramatically stimulated the migration of GBM as well as CHO cells (Fig. 5 A–C). Interestingly, heparin treatment and genetic HSPG deficiency efficiently attenuated exosome-dependent cell migration (Fig. 5 A and C). Consistent with the inability of CS to inhibit exosome uptake (Fig. 2), exosome-dependent stimulation of cell migration was unperturbed by CS treatment (Fig. 5B). Further, we found that ERK1/2 phosphorylation in WT CHO cells was substantially increased (to almost sixfold) by exosomes, whereas HSPG-deficient cells were virtually unresponsive (Fig. 5D). Treatment with the specific ERK1/2 inhibitor U0126 attenuated exosome-mediated cell migration, suggesting an important role of this MAPK in the functional activity of exosomes (Fig. S3A). Although these data do not distinguish between ERK1/2 activation dependent on exosome cell-surface binding and/or uptake through a specific receptor-mediated process, they clearly indicate that HSPGs are functionally relevant for exosome-mediated signaling activation.
Fig. 5.

Exosome-mediated cell migration and signaling depend on HSPG. Transwell migration of GBM cells in the absence or presence of exosomes and with or without heparin (A) or CS (B) as indicated. (C) Migration of WT and mutant PG-deficient (PG-def.) CHO cells in the absence or presence of exosomes as indicated. (Upper) Representative images of migrated cells. (Lower) Quantitative data on number of migrated cells per field (n = 9). Data shown represent the mean ± SD from three independent experiments, each performed in triplicate [*P < 0.05; NS, not significant (i.e., no statistically significant difference)]. (D) WT and PG-deficient CHO cells were treated with the indicated concentrations of exosomes for 20 min, and cell lysates were analyzed by immunoblotting for p-ERK1/2, total ERK (t-ERK), and α-tubulin. (Left) Western blot from a representative experiment. (Right) Quantitative data of p-ERK/α-tubulin ratios in WT and PG-deficient cells.
To extend these studies, we next used xylosides, i.e., small hydrophobic compounds that compete with endogenous proteins for conjugation with GAGs, resulting in pharmacological inhibition of PG biosynthesis (24, 32). Importantly, we could show that treatment with 4-nitrophenyl β-d-xylopyranoside (PNP-Xyl) resulted in approximately 50% inhibition of exosome uptake (Fig. 6A). Xyloside-mediated inhibition of exosome uptake was associated with efficient inhibition of exosome-dependent cell migration (Fig. 6B) and ERK1/2 cell signaling activation (Fig. 6 C and D). These data further support the concept of an important functional role of HSPG in exosome-dependent cell activation, and indicate that small-molecule inhibitors of HSPG biosynthesis may attenuate exosome functional activity.
Fig. 6.

Small-molecule inhibitor of PG biosynthesis reduces exosome uptake and attenuates exosome-mediated cell migration and signaling activation. (A) GBM cells were untreated (Ctrl) or pretreated with the PG biosynthesis inhibitor PNP-Xyl for 48 h, and then incubated with PKH-labeled exosomes (40 µg/mL) for 1 h, followed by flow cytometry analysis of exosome uptake. (B) Migration of untreated (Ctrl) or PNP-Xyl–pretreated GBM cells in the absence or presence of exosomes. Data shown represent the mean ± SD from three independent experiments, each performed in triplicate (*P < 0.05). (C and D) GBM cells were untreated or pretreated with PNP-Xyl and then incubated with exosomes for 20 min as indicated. Cell lysates were analyzed by immunoblotting for p-ERK1/2, total ERK (t-ERK), and α-tubulin. (C) Western blot from a representative experiment. (D) Quantitative data of p-ERK/α-tubulin ratios with the different treatments. (E) Schematic summary of major findings of the present work; 1, Exogenous HS inhibits exosome uptake in a size and charge dependent manner; 2, Enzymatic depletion of cell-surface HSPGs inhibits exosome uptake; 3, Genetic deficiency in XYLT, which catalyzes the initial step in PG formation, or NDST, which catalyses HS N-deacetylation/sulfation, or 2-O-sulfotransferase (2-OST), which catalyses HS 2-O-sulfation, all result in reduced exosome uptake; 4, Xyloside, i.e., small-molecule inhibitors of PG biosynthesis, inhibit exosome uptake; 5, Exosome-mediated ERK1/2 activation and cell migration are HSPG-dependent: and 6, Our data indicate the existence of alternative, HSPG-independent modes of internalization; however, we provide evidence that the HSPG-dependent pathway is essential for the functional activity of exosomes.
Discussion
Here, we provide evidence that HSPGs have an important role in exosome uptake. Importantly, we show that the HSPG-dependent uptake route is highly relevant for the biological response evoked by exosomes in target cells. The view of HSPG-mediated uptake of extracellular ligands was, for a long time, limited to initial HS binding followed by presentation to internalizing proteins, e.g., lipoprotein and virus receptors (13). More recent studies have established that HSPGs cointernalize with extracellular ligands, a process that may involve a specific and ubiquitous HS epitope recognized by the AO4B08 anti-HS antibody (23, 33). We show that exosome uptake depends on HS 2-O-sulfation and N-sulfation in recipient cells, which is consistent with the structural modifications of the AO4B08 epitope (34). Moreover, by using exogenous HS, pan-PG–deficient cell mutants, pharmacological inhibitors of HSPG, and HS-degrading enzyme, we convincingly show that cell-surface HSPGs are required for efficient uptake of exosomes (Fig. 6E shows a schematic summary). The findings that exosomes down-regulate the presence of HSPG at the cell surface, and that internalized exosomes reside in common vesicular structures with syndecan and glypican, indicate that HSPGs act as true internalizing receptors of exosomes rather than cell-surface attachment sites.
Several studies have reported that virus biogenesis and release converge with EV pathways, suggesting an evolutionary conserved system of virus–EV codependence (35–39). In fact, exosomes may be regarded as endogenous viruses as they have been shown to transfer various classes of nucleic acids (2–6). It is thus of interest that several viruses, e.g., HIV, herpes simplex, and adenoassociated virus, have been shown to enter cells through HSPG (16, 40). Our data suggest the intriguing possibility that HSPG represents a convergence point during cellular uptake of endogenous vesicles and viral intruders. We provide evidence of the sorting of HSPGs to exosomes, and it may be speculated that viruses become sequestered by exosome-associated HSPGs. Future studies should elucidate the potential role of exosomes as regulators of virus infectivity through the HSPG pathway.
Our finding that exosome uptake is not completely inhibited upon perturbation of HSPGs suggests the existence of additional, HSPG-independent modes of internalization. Importantly, our data indicate that the HSPG-dependent entry pathway is essential for the biological activity of exosomes. Clearly, these data do not exclude the possibility that exosomes may exert other functional effects through alternative uptake pathways. Likely candidates are other, polyanionic receptors, perhaps most importantly sialylated glycoproteins (41). However, exosome uptake was reduced by only approximately 10% in sialic acid-deficient mutant compared with parental CHO cells (Fig. S3B). Interestingly, cells pretreated with chlorate, i.e., a well-known inhibitor of the most common sulfo group donor, 3′-phosphoadenosine-5′-phosphosulfate for sulfotransferases (24), exhibited reduced exosome uptake by approximately 90% compared with controls (Fig. S3C). These results support the importance of other sulfated receptors, e.g., sulfated lipids, glycoproteins, and tyrosine sulfated proteins, in the recognition of positively charged ligands on exosomes.
The role of HSPGs in exosome uptake may be of relevance for the emerging concept of exosomes as therapeutic vehicles of RNA delivery (42). The AO4B08 antibody recognizes HSPG and is taken up by multiple cell types from various origins (23, 33), which is consistent with non–cell-specific uptake of GBM cell-derived exosomes. Thus, the identification of a cell- or tissue-specific HS for targeted delivery of therapeutic exosomes should be a great challenge. However, the specificity may be achieved at the level of exosomal cargo, e.g., an siRNA directed at a pathway essential for cancer cell survival while dispensable for normal cells. The utility of the HSPG pathway in exosome uptake could thus still prove critical for the future development of exosomes as drug delivery vehicles.
In summary, this study identifies a functionally relevant and potentially targetable entry pathway of cancer cell exosomes, and significantly extends the role of HSPG in the uptake of endogenous macromolecules.
Materials and Methods
Detailed descriptions of reagents, cell culture, enzymatic treatments, exosome isolation, EM, flow cytometry, confocal fluorescence microscopy, isolation and analysis of PGs, Western blot analysis, exosome heparin binding assay, and cell migration experiments are provided in SI Materials and Methods.
Exosome Isolation.
Exosomes were isolated by centrifugation as described in SI Materials and Methods.
EM.
Exosome samples were analyzed in a JEOL JEM 1230 electron microscope as described in SI Materials and Methods.
Exosome Uptake.
Exosomes were labeled with a PKH Fluorescent labeling kit (Sigma) as recommended by the manufacturer, and uptake was analyzed by flow cytometry and confocal fluorescence microscopy as described in SI Materials and Methods. In flow cytometry experiments, PKH-exosome uptake is expressed as arbitrary units, which represents mean cellular fluorescence subtracted by background signal caused by autofluorescence as quantified in cells not incubated with exosomes. Notably, incubation with unlabeled exosomes did not affect cellular autofluorescence (Fig. S4).
Isolation and Analysis of PGs.
The 35S-PGs and GAGs were analyzed in U-87 MG cells and corresponding exosomes by metabolic incorporation of [35S]sulfate, followed by anion exchange and size exclusion chromatography as described in SI Materials and Methods.
Western Blot Analysis.
Lysates of cells and exosomes were separated by SDS/PAGE, transferred to PVDF membranes, and incubated with antibodies as described in SI Materials and Methods.
Exosome Heparin Binding Assay.
Heparin-agarose beads were incubated with PKH-labeled exosomes, washed with PBS solution, and eluted with NaCl to obtain nonbinding and binding fractions, respectively, for fluorescence analysis as described in SI Materials and Methods.
Statistical Analysis.
Immunoblotting and imaging experiments were performed in duplicate or triplicate in at least three independent experiments. Results in flow cytometry, heparin agarose binding, and cell migration experiments are the mean ± SD from three independent experiments (n = 3), each performed in duplicate or triplicate, as indicated in the respective figure legend. In some cases, error bars were smaller than the drawn symbols. Statistical significance was evaluated with Student t test by using Microsoft Excel; a P value <0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Maria Johansson and Eva Lindqvist for excellent technical assistance. This work was supported by grants from the Swedish Cancer Fund; the Swedish Research Council; the Gunnar Nilsson, Anna Lisa and Sven Eric Lundgren, and Kamprad Foundations; the Skåne University Hospital donation funds; and governmental funding of clinical research within the National Health Services (ALF).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304266110/-/DCSupplemental.
References
- 1.Raposo G, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183(3):1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20(9):1487–1495. doi: 10.1038/sj.leu.2404296. [DOI] [PubMed] [Google Scholar]
- 3.Valadi H, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 4.Belting M, Wittrup A. Nanotubes, exosomes, and nucleic acid-binding peptides provide novel mechanisms of intercellular communication in eukaryotic cells: Implications in health and disease. J Cell Biol. 2008;183(7):1187–1191. doi: 10.1083/jcb.200810038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Nedawi K, Meehan B, Rak J. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle. 2009;8(13):2014–2018. doi: 10.4161/cc.8.13.8988. [DOI] [PubMed] [Google Scholar]
- 6.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009;19(2):43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 8.Svensson KJ, et al. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proc Natl Acad Sci USA. 2011;108(32):13147–13152. doi: 10.1073/pnas.1104261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles. 2013;2:20389. doi: 10.3402/jev.v2i0.20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostrowski M, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12(1):19–30, 1–13. doi: 10.1038/ncb2000. [DOI] [PubMed] [Google Scholar]
- 11.Bobrie A, et al. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012;72(19):4920–4930. doi: 10.1158/0008-5472.CAN-12-0925. [DOI] [PubMed] [Google Scholar]
- 12.Baietti MF, et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14(7):677–685. doi: 10.1038/ncb2502. [DOI] [PubMed] [Google Scholar]
- 13.Belting M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem Sci. 2003;28(3):145–151. doi: 10.1016/S0968-0004(03)00031-8. [DOI] [PubMed] [Google Scholar]
- 14.Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: The concept of specificity. J Cell Biol. 2006;174(3):323–327. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446(7139):1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 16.Shukla D, et al. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99(1):13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 17.Spillmann D. Heparan sulfate: Anchor for viral intruders? Biochimie. 2001;83(8):811–817. doi: 10.1016/s0300-9084(01)01290-1. [DOI] [PubMed] [Google Scholar]
- 18.Clark MJ, et al. U87MG decoded: The genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet. 2010;6(1):e1000832. doi: 10.1371/journal.pgen.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9(5):224. doi: 10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Couchman JR. Syndecans: Proteoglycan regulators of cell-surface microdomains? Nat Rev Mol Cell Biol. 2003;4(12):926–937. doi: 10.1038/nrm1257. [DOI] [PubMed] [Google Scholar]
- 21.Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. 2001;276(5):3254–3261. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- 22.Sandgren S, Cheng F, Belting M. Nuclear targeting of macromolecular polyanions by an HIV-Tat derived peptide. Role for cell-surface proteoglycans. J Biol Chem. 2002;277(41):38877–38883. doi: 10.1074/jbc.M205395200. [DOI] [PubMed] [Google Scholar]
- 23.Wittrup A, et al. ScFv antibody-induced translocation of cell-surface heparan sulfate proteoglycan to endocytic vesicles: Evidence for heparan sulfate epitope specificity and role of both syndecan and glypican. J Biol Chem. 2009;284(47):32959–32967. doi: 10.1074/jbc.M109.036129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belting M, Persson S, Fransson LA. Proteoglycan involvement in polyamine uptake. Biochem J. 1999;338(Pt 2):317–323. [PMC free article] [PubMed] [Google Scholar]
- 25.Mani K, et al. HIV-Tat protein transduction domain specifically attenuates growth of polyamine deprived tumor cells. Mol Cancer Ther. 2007;6(2):782–788. doi: 10.1158/1535-7163.MCT-06-0370. [DOI] [PubMed] [Google Scholar]
- 26.Fransson LA, Sjöberg I, Havsmark B. Structural studies on heparan sulphates. Characterization of oligosaccharides; obtained by periodate oxidation and alkaline elimination. Eur J Biochem. 1980;106(1):59–69. [PubMed] [Google Scholar]
- 27.Bai X, Crawford B, Esko JD. Selection of glycosaminoglycan-deficient mutants. Methods Mol Biol. 2001;171:309–316. doi: 10.1385/1-59259-209-0:309. [DOI] [PubMed] [Google Scholar]
- 28.Bame KJ, Zhang L, David G, Esko JD. Sulphated and undersulphated heparan sulphate proteoglycans in a Chinese hamster ovary cell mutant defective in N-sulphotransferase. Biochem J. 1994;303(pt 1):81–87. doi: 10.1042/bj3030081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai X, Esko JD. An animal cell mutant defective in heparan sulfate hexuronic acid 2-O-sulfation. J Biol Chem. 1996;271(30):17711–17717. doi: 10.1074/jbc.271.30.17711. [DOI] [PubMed] [Google Scholar]
- 30.Kucharzewska P, et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci USA. 2013;110(18):7312–7317. doi: 10.1073/pnas.1220998110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.David G, Bai XM, Van der Schueren B, Cassiman JJ, Van den Berghe H. Developmental changes in heparan sulfate expression: In situ detection with mAbs. J Cell Biol. 1992;119(4):961–975. doi: 10.1083/jcb.119.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belting M, et al. Tumor attenuation by combined heparan sulfate and polyamine depletion. Proc Natl Acad Sci USA. 2002;99(1):371–376. doi: 10.1073/pnas.012346499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wittrup A, et al. Magnetic nanoparticle-based isolation of endocytic vesicles reveals a role of the heat shock protein GRP75 in macromolecular delivery. Proc Natl Acad Sci USA. 2010;107(30):13342–13347. doi: 10.1073/pnas.1002622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurup S, et al. Characterization of anti-heparan sulfate phage display antibodies AO4B08 and HS4E4. J Biol Chem. 2007;282(29):21032–21042. doi: 10.1074/jbc.M702073200. [DOI] [PubMed] [Google Scholar]
- 35.Gould SJ, Booth AM, Hildreth JE. The Trojan exosome hypothesis. Proc Natl Acad Sci USA. 2003;100(19):10592–10597. doi: 10.1073/pnas.1831413100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meckes DG, Jr, et al. Human tumor virus utilizes exosomes for intercellular communication. Proc Natl Acad Sci USA. 2010;107(47):20370–20375. doi: 10.1073/pnas.1014194107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izquierdo-Useros N, et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood. 2009;113(12):2732–2741. doi: 10.1182/blood-2008-05-158642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadiu I, Narayanasamy P, Dash PK, Zhang W, Gendelman HE. Biochemical and biologic characterization of exosomes and microvesicles as facilitators of HIV-1 infection in macrophages. J Immunol. 2012;189(2):744–754. doi: 10.4049/jimmunol.1102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wurdinger T, et al. Extracellular vesicles and their convergence with viral pathways. Adv Virol. 2012;2012:767694. doi: 10.1155/2012/767694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shukla D, Spear PG. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J Clin Invest. 2001;108(4):503–510. doi: 10.1172/JCI13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright TC, Smith B, Ware BR, Karnovsky MJ. The role of negative charge in spontaneous aggregation of transformed and untransformed cell lines. J Cell Sci. 1980;45:99–117. doi: 10.1242/jcs.45.1.99. [DOI] [PubMed] [Google Scholar]
- 42.Koppers-Lalic D, Hogenboom MM, Middeldorp JM, Pegtel DM. Virus-modified exosomes for targeted RNA delivery; a new approach in nanomedicine. Adv Drug Deliv Rev. 2013;65(3):348–356. doi: 10.1016/j.addr.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.