

Abstract
Facioscapulohumeral muscular dystrophy has been genetically linked to reduced numbers (≤8) of D4Z4 repeats at 4q35 combined with 4A(159/161/168) DUX4 polyadenylation signal haplotype. However, we have recently reported that 1.3% of healthy individuals carry this molecular signature and 19% of subjects affected by facioscapulohumeral muscular dystrophy do not carry alleles with eight or fewer D4Z4 repeats. Therefore, prognosis for subjects carrying or at risk of carrying D4Z4 reduced alleles has become more complicated. To test for additional prognostic factors, we measured the degree of motor impairment in a large group of patients affected by facioscapulohumeral muscular dystrophy and their relatives who are carrying D4Z4 reduced alleles. The clinical expression of motor impairment was assessed in 530 subjects, 163 probands and 367 relatives, from 176 unrelated families according to a standardized clinical score. The associations between clinical severity and size of D4Z4 allele, degree of kinship, gender, age and 4q haplotype were evaluated. Overall, 32.2% of relatives did not display any muscle functional impairment. This phenotype was influenced by the degree of relation with proband, because 47.1% of second- through fifth-degree relatives were unaffected, whereas only 27.5% of first-degree family members did not show motor impairment. The estimated risk of developing motor impairment by age 50 for relatives carrying a D4Z4 reduced allele with 1–3 repeats or 4–8 repeats was 88.7% and 55%, respectively. Male relatives had a mean score significantly higher than females (5.4 versus 4.0, P = 0.003). No 4q haplotype was exclusively associated with the presence of disease. In 13% of families in which D4Z4 alleles with 4–8 repeats segregate, the diagnosis of facioscapulohumeral muscular dystrophy was reported only in one generation. In conclusion, this large-scale analysis provides further information that should be taken into account when counselling families in which a reduced allele with 4–8 D4Z4 repeats segregates. In addition, the reduced expression of disease observed in distant relatives suggests that a family’s genetic background plays a role in the occurrence of facioscapulohumeral muscular dystrophy. These results indicate that the identification of new susceptibility factors for this disease will require an accurate classification of families.
Keywords: facioscapulohumeral muscular dystrophy, D4Z4 reduced allele, genotype–phenotype correlations, penetrance, disease expression
Introduction
Facioscapulohumeral muscular dystrophy (FSHD, OMIM #158900) is the third most common hereditary myopathy with prevalence of 1 in 20 000 (Mostacciuolo et al., 2009). FSHD is characterized by progressive, asymmetric atrophy and weakness of a highly selective set of muscle groups (Padberg et al., 1991; Lamperti et al., 2010; Tawil et al., 2010) and wide inter- and intra-familial variability of clinical expression (Padberg, 1982; Tawil and van der Maarel, 2006). Age-dependent penetrance based on the presence of the characteristic clinical signs was estimated >95% by age 20 (Lunt et al., 1989; Tawil et al., 2010). The mode of inheritance is considered autosomal dominant (Flanigan, 2004).
A large majority of patients with FSHD carry rearrangements occurring in a 3.3 kb tandemly repeated sequence (D4Z4) located at the 4q subtelomeric region (Wijmenga et al., 1992; Lunt et al., 1995a; Upadhyaya et al., 1997). These rearrangements result in polymorphic EcoRI alleles detected by the p13E-11 probe (Wijmenga et al., 1992; Upadhyaya et al., 1997). Early studies of small numbers of individuals observed that both de novo and familial patients with FSHD carry p13E-11 EcoRI alleles of 35 kb, corresponding to eight D4Z4 units, or shorter (Griggs et al., 1993; Lunt et al., 1995a; van Deutekom et al., 1996). For the last 20 years the clinical diagnosis of FSHD has been supported by this type of D4Z4 DNA testing, which has been considered highly sensitive and specific for disease (Lunt et al., 1995a, b; Lunt, 1998; Tawil et al., 2010). However, several studies on FSHD families describe subjects carrying D4Z4 alleles of reduced size (DRA) and no signs of the disease, defined as non-penetrant carriers (Tawil et al., 1996; Zatz et al., 1998; Ricci et al., 1999; van Overveld et al., 2000; Goto et al., 2004; Tonini et al., 2004; Sakellariou et al., 2012; Scionti et al., 2012a). As a possible explanation of some non-penetrant cases, it has been proposed that reduction of D4Z4 repeats on chromosome 4q35 is pathogenic only in certain chromosomal backgrounds, defined by ‘permissive’ specific haplotypes, namely (i) reduction of D4Z4 elements; (ii) presence of the 4A(159/161/168) haplotype; and (iii) a single nucleotide polymorphism that provides a polyadenylation signal (PAS) for the DUX4 transcript (Lemmers et al., 2002, 2007, 2010).
Nonetheless, our most recent studies (Scionti et al., 2012b) showed that although the majority of FSHD index cases (70%) carry DRA with 4–8 units, this size range is also carried by 3% of healthy subjects from the general population. Additionally, our work raised the possibility that there is little predictive value of the 4A161PAS haplotype in the absence of family history because 1.3% of healthy subjects carry this haplotype, which is a frequency similar to other common polymorphisms. Finally, we found that 19% of FSHD probands do not carry D4Z4 alleles with 1–8 repeats and only 50% of the probands carry the 4A161PAS permissive haplotype associated with DRA (Scionti et al., 2012b). These observations suggest that the genetic factors leading to FSHD might be incompletely described. Consistent with this idea, Lemmers et al. (2012) recently described mutations in SMCHD1 gene in patients with FSHD and hypothesized that these mutations influence the disease penetrance (Lemmers et al., 2012).
Here, we evaluate FSHD occurrence among relatives carrying DRA in relation to D4Z4 reduced allele size, gender, age, degree of kinship and 4q haplotype.
Materials and methods
Study design and subjects selection
The study has been performed on FSHD families accrued through the Italian National Registry for FSHD (INRF), established in 2007 by the Italian Clinical Network for FSHD (ICNF) (www.fshd.it) (Lamperti et al., 2010). The ICNF includes two diagnostic laboratories at the University di Modena and Reggio Emilia and at the Fondazione Santa Lucia in Rome and 14 clinical centres, networked within the Italian Association of Myology (www.miologia.org) and distributed across all of Italy, from northern to southern regions, including the islands.
The study was conducted from 2008 to 2012. As outlined in Fig. 1, initially the selection process regarded 418 index cases carrying DRA with 1–8 repeats and fulfilling the clinical features of FSHD (Padberg et al., 1991). One hundred and eighty-six cases were considered not eligible because they had no available relatives. Fourteen compound heterozygotes for DRA alleles were excluded from this study and analysed separately (Scionti et al., 2012a). Forty-two de novo cases, defined as single subjects with neither parent carrying DRA, were excluded because they would not be informative for this study. For each proband the clinical and molecular examinations were extended to the available relatives at various degrees of kinship. Among the 645 relatives identified, 367 were found to be carriers of DRA. All clinical and molecular data were collected in the INRF database at Miogen Laboratory of University of Modena for data analysis.
Figure 1.

(A) Preliminary selection of probands/families from the Italian National Registry for FSHD (INRF). (B) Selection of the cohort of probands and their relatives for genotype–phenotype correlation analysis.
Clinical examination
Each subject recruited during the time of the study was examined by a trained neurologist of the ICNF using the standardized FSHD clinical protocol with validated inter-rater reliability (Lamperti et al., 2010). The FSHD clinical protocol was developed by the ICNF in order to numerically define the clinical severity of the motor impairment, and is not to be used to diagnose FSHD. The FSHD scale quantifies muscle weakness through the functional evaluation of six muscle groups specifically affected in FSHD, belonging to (i) face (score 0–2); (ii) shoulder girdle (score 0–3); (iii) upper limbs (score 0–2); (iv) distal legs (score 0–2); (v) pelvic girdle (score 0–5); and (vi) abdominal muscles (score 0–1). The FSHD score, which translates disability into a number, ranges from zero, when no objective evidence of muscle functional impairment is present, to 15, when all the muscle groups tested are severely impaired (www.fshd.it) (Lamperti et al., 2010). DRA carriers who did not show an objective motor impairment received an FSHD score equal to zero and were considered clinically unaffected at the time of examination. On the basis of the FSHD score, subjects were classified as affected by mild (FSHD score 1–2), moderate (FSHD score 3–6), or severe (FSHD score 7–15) motor impairment.
Probands from 13 families were not re-evaluated as they were not alive at the time of this study.
Age at onset was estimated on the basis of patient records. When subjects did not complain of motor impairment, but a mild muscle weakness was observed, the age at examination was set as the age at onset, according to previous reports (Lunt et al., 1995b). In six subjects it was not possible to obtain information about the age at onset of motor impairment due to their poor compliance.
The study was approved by the Local Ethics Committees of all participating Institutions. Informed consent, according to the Declaration of Helsinki, was obtained from each subject enrolled in the study.
Molecular characterization
Allele sizes were estimated by Southern hybridization using probe p13E-11. Genomic DNA extracted from peripheral blood lymphocytes was digested with EcoRI, EcoRI/BlnI or XapI, electrophoresed in a 0.4% agarose gel for 45–48 h at 35 V alongside an 8–48 kb marker (Bio-Rad) as previously described (Scionti et al., 2012b). To assess the chromosomal origin of D4Z4-reduced alleles, DNA from each subject was analysed by NotI digestion and hybridization with the B31 probe (Scionti et al., 2012b). Restriction fragments were detected by autoradiography or using a Typhoon Trio system (GE Healthcare). 4qA/4qB allelic variants were defined in all 530 subjects included in the study, using HindIII-digested DNA, pulsed field gel electrophoresis electrophoresis and Southern blot hybridization with radiolabeled 4qB and 4qA probes according to standard procedures (Scionti et al., 2012b).
The Simple Sequence Length Polymorphism (SSLP) and the pLAM Single Nucleotide Polymorphism (SNP) [AT(T/C)AAA] sequences flanking the D4Z4 repeat units were defined in 294 relatives according to published procedures (Scionti et al., 2012b).
Statistical analysis
The association between the risk of being asymptomatic (FSHD score equal to zero) and D4Z4 allele size and age was evaluated by using the multivariate logistic regression model. The association between age at onset or FSHD score and D4Z4 allele size and sex among symptomatic relatives was assessed by using a general linear model. Association estimates and their relative 95% confidence intervals (CI) were also reported.
The prevalence of FSHD score, classified into two categories (0 versus 1–15), among relatives was estimated and its association with D4Z4 allele sizes was also evaluated. Univariate and multivariate logistic regression models were fitted with D4Z4 allele size, sex and family degree as predictors.
An interaction test was also carried out to assess whether the difference in terms of FSHD scores between probands and relatives varied in relation to D4Z4 allele sizes.
In order to minimize any ascertainment bias, all the genotype–phenotype correlation analyses were performed on relatives and probands separately.
In all general and generalized linear models estimated, the sandwich estimator of covariance matrix of parameters was used to take into account any clustering effect within families (Williams, 2000). Wald tests were used to evaluate the effect of predictors and to evaluate the effects of predictors on outcomes (McCullagh and Nelder, 1989).
For the cohorts of probands and family members, Kaplan-Meier survival analysis (Kaplan and Meier, 1958) was used to estimate the age-specific cumulative motor impairment incidence, with the corresponding 95% CI. For each individual, time from birth to the earliest age at onset of motor impairment was estimated. The analysis was stratified by D4Z4 allele size only for relatives, and also by gender for relatives and probands.
The risk prediction algorithm was developed and validated using established methods (Hippisley-Cox et al., 2007). The original cohort of 367 relatives was randomly split in the derivation and validation samples. The coefficients for D4Z4 allele size, sex and family degree were estimated by using the Cox proportional hazard model. The coefficients were used as weights, which were combined with the baseline survivor function to derive risk equations at age 50 years. The risk equation was applied to the validation sample and measures of discrimination were calculated (R2, D statistics and area under the receiver operating characteristic curve).
Results
We examined 530 carriers of DRA (367 relatives and 163 index cases) from 176 unrelated families, in which at least one subject was affected by FSHD (Fig. 1B). Considering the cohort of relatives carrying DRA as a whole (367 subjects, 152 males and 215 females, mean age 46.4 ± 17.2, Supplementary Fig. 1), we observed that 118 (32.2%) did not show any functional motor impairment (FSHD score equal to zero) and 249 (67.8%) displayed muscle impairment to various degrees (FSHD score ≥1) (Table 1).
Table 1.
Distribution of unaffected relatives according to D4Z4 allele size and age at examination
| Age (years) |
Total |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 18–30 |
31–55 |
56–70 |
|||||||
| D4Z4 units | n subjects | % Score = 0 | n subjects | % Score = 0 | n subjects | % Score = 0 | n subjects | % Score = 0 | P-value* |
| (n) | (n) | (n) | (n) | ||||||
| 1–3 | 8 | 12.5 (1) | 23 | 8.7 (2) | 9 | 11.1 (1) | 42 | 9.5 (4) | 0.707 |
| 4–6 | 31 | 25.8 (8) | 65 | 33.8 (22) | 29 | 27.6 (8) | 133 | 28.6 (38) | 0.461 |
| 7–8 | 42 | 54.8 (23) | 85 | 40.0 (34) | 39 | 35.9 (14) | 192 | 39.6 (76) | 0.013 |
| Total | 367 | 32.2 (118) | |||||||
*Wald test of the age’s coefficient as ordinal predictor in the logistic model adjusted by sex.
The distribution of asymptomatic relatives was also analysed based on the size of DRA. We divided subjects in three groups: subjects carrying DRA with 1–3 D4Z4 repeats; subjects carrying DRA with 4–6 D4Z4 repeats; subjects carrying DRA with 7–8 D4Z4 repeats (Supplementary Table 1 and Supplementary Fig. 1). Table 1 also shows that 9.5% (4 out of 42) of all carriers of DRA with 1–3 repeats displayed no motor impairment. This percentage increases among carriers of DRA with 4–6 and 7–8 repeats (28.6% and 39.6%, respectively).
We then calculated the distribution of asymptomatic carriers based on age at examination, separately analysing four classes: those aged 18–30 years, 31–55 years, 56–70 years, and those over 70 years of age (Supplementary Table 2). Four classes were formed. As shown in Table 1, asymptomatic carriers were found in all classes up to 70 years. In particular, almost one-third of carriers of DRA with 4–6 and 7–8 repeats (27.6% and 35.9%, respectively) were asymptomatic between 56 and 70 years of age. Since the percentage of asymptomatic carriers varies among relatives carrying DRA of different sizes, the age-related risk of developing motor impairment was evaluated in correlation with D4Z4 size on the basis of data obtained from 361 relatives. Figure 2 and Table 2 show the penetrance estimates for DRA carriers calculated with the Kaplan-Meier method. Among subjects carrying DRA with 1–3 units the risk of developing motor impairment is 64.3% by age 20, 80.1% by age 40 and 96.2% by age 60. Among subjects carrying DRA with 4–6 and 7–8 D4Z4 units these risks are 21.8% and 19.6%, respectively, by age 20, 44.8% and 42.5% by age 40, and 71.5% and 62.9% by age 60. Therefore, FSHD penetrance is almost complete by age 60 only for carriers of DRA with 1–3 units.
Figure 2.
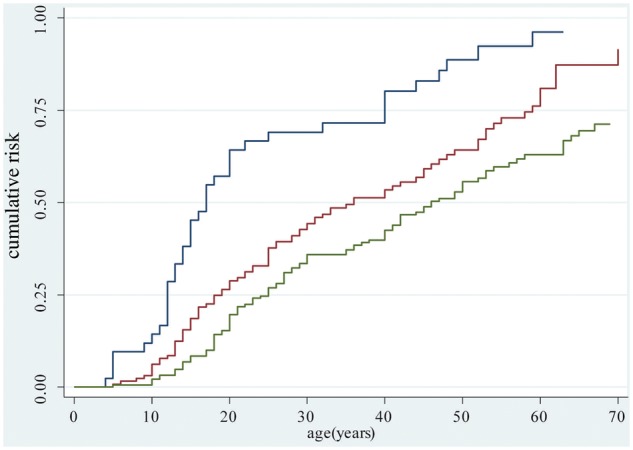
Age-specific cumulative risk of reported muscle impairment according to D4Z4 allele size. Estimates obtained on 361 relatives using the Kaplan-Meier analysis. Blue line refers to carriers of 1–3 D4Z4 units; red line refers to carriers of 4–6 D4Z4 units; green line refers to carriers of 7–8 D4Z4 units. Carriers of 7–8 versus 4–6 units Log rank test P value = 0.002.
Table 2.
Estimates of the age-specific cumulative risk obtained using the Kaplan-Meyer analysis
| Age (years) | Carriers of 1–3 D4Z4 units |
Carriers of 4–6 D4Z4 units |
Carriers of 7–8 D4Z4 units |
|||
|---|---|---|---|---|---|---|
| Risk | 95% CI | Risk | 95% CI | Risk | 95% CI | |
| 20 | 64.3 | (50.1; 78.3) | 21.8 | (21.8; 37.5) | 19.6 | (14.6; 26.0) |
| 30 | 69.1 | (55.0; 82.2) | 36.1 | (36.1; 53.4) | 35.9 | (29.4; 43.5) |
| 40 | 80.1 | (66.5; 90.8) | 44.8 | (44.8; 62.5) | 42.5 | (35.5; 50.3) |
| 50 | 88.7 | (76.3; 96.3) | 55.0 | (55.0; 73.3) | 55.7 | (47.9; 63.8) |
| 60 | 96.2 | (84.6; 99.7) | 71.5 | (71.5; 88.8) | 62.9 | (54.7; 71.2) |
| 70 | 80.3 | (80.3; 97.6) | 71.3 | (62.3; 79.7) | ||
| 80 | 82.2 | (72.1; 90.4) | ||||
We tested whether the size of DRA correlates with age at onset and disease severity. Table 3 shows that the mean age at onset is statistically lower among subjects carrying DRA with 1–3 units (20.3 years) in comparison with those carrying DRA with 4–6 and 7–8 D4Z4 repeats (29.2 and 34.6 years, respectively) (P = 0.0002). Overall, we found that 60.6% of affected relatives experienced scapular girdle onset, 19.0% facial muscle onset, 16.7% lower limbs onset, 0.9% upper limbs onset and 2.8% abdominal muscle onset (Supplementary Table 3).
Table 3.
Distribution of mean age at onset among affected relatives according to D4Z4 allele size
| Relatives |
||||
|---|---|---|---|---|
| D4Z4 units | Number of subjects | Mean age at onset (years) | 95% CI | P-value† |
| 1–3 | 38 | 20.3 | 15.5–25.2 | |
| 4–6 | 91 | 29.2 | 25.6–32.7 | |
| 7–8 | 114 | 34.6 | 30.1–39.1 | 0.0002 |
| Total | 243 | |||
†Wald test of equality to zero of D4Z4 allele size’s coefficients parametrized as categorical variable in a general linear model with age at onset as dependent variable and sex and D4Z4 allele size as predictors.
Severity is also increased among carriers of DRA with 1–3 repeats. Indeed, as shown in Table 4, affected relatives carrying DRA with 1–3 repeats had a mean FSHD score of 7.2. By contrast, individuals carrying DRA with 4–6 and 7–8 D4Z4 units had mean FSHD score of 4.4 and 4.1, respectively. This association was statistically significant (P = 0.0006).
Table 4.
Distribution of FSHD score calculated on affected relatives according to D4Z4 allele size and age at examination
| Relatives |
||||
|---|---|---|---|---|
| D4Z4 units | Number of subjects | FSHD score mean | 95% CI | P-value† |
| 1–3 | 38 | 7.2 | 5.8–8.6 | |
| 4–6 | 95 | 4.4 | 3.8–5.1 | |
| 7–8 | 116 | 4.1 | 3.5–4.7 | 0.0006 |
†Wald test of equality to zero of D4Z4 allele size and age at examination coefficients parametrized as categorical variable in a general linear model with FSHD score as dependent variable and sex, D4Z4 allele size and age at examination as predictors.
The degree of motor impairment among relatives was also evaluated in relationship to D4Z4 allele size and age at examination. Figure 3A shows that ∼40% of relatives carrying DRA with 1–3 units are severely affected (FSHD score ≥7) by age 30. In contrast, no relatives carrying DRA with 4–8 units had a FSHD score higher than 6 in this age window. Figure 3B and C shows that between age 31–55 and 56–70 a high percentage of relatives carrying DRA with 4–8 units were asymptomatic (FSHD score 0) or displayed minimal signs of functional motor impairment (FSHD score 1–2).
Figure 3.

Distribution of clinical severity among relatives carrying D4Z4 reduced allele according to D4Z4 allele size and age. Subjects were subdivided by age: (A) 18–30 years, (B) 31–55 years, (C) 56–70 years and by D4Z4 allele size: 1–3, 4–6 and 7–8 units. In each subgroup, percentages of subjects who received FSHD score equal to 0, 1–2, 3–6 and ≥ 7 are reported.
We then evaluated whether there is a correlation between the clinical status of the proband and his/her relatives. As shown in Table 5, intra-familial analysis on 163 families with 217 affected relatives revealed a positive correlation [0.72 (95%CI 0.40–1.04)] in families in which D4Z4 alleles with 1–3 D4Z4 repeats segregate. In contrast, in families with 4–6 and 7–8 D4Z4 alleles, a lower degree of correlation between the clinical status of the proband and his/her relatives was observed [0.01 (95%CI −0.23–0.26) and −0.14 (95%CI −0.35–0.07), respectively].
Table 5.
Standardized regression coefficient between FSHD score of probands and relatives
| D4Z4-allele size 1–3 |
D4Z4-allele size 4–6 |
D4Z4-allele size 7–8 |
|||||
|---|---|---|---|---|---|---|---|
| Number of subjects | Correlation coefficient | 95% CI | Correlation coefficient | 95% CI | Correlation coefficient | 95% CI | P-value |
| 217 | 0.72 | 0.40–1.04 | 0.01 | −0.23–0.26 | −0.14 | −0.35–0.07 | <0.0001 |
General linear models with FSHD score of the relative as outcome and probands’ FSHD score, age at examination, D4Z4 allele size and sex as predictors. Interaction test between proband’s FSHD score and D4Z4 allele sizes.
Remarkably, in 19 of 148 families (13%) in which 4–8 D4Z4 alleles segregate, we found affected individuals only within a single generation, and with older unaffected relatives carrying the DRA (Supplementary Fig. 2). In each of these 19 families, molecular testing excluded the presence of somatic mosaicism in the unaffected parent carrying the DRA. The finding of affected subjects in only one generation or the presence of only one affected subject in the entire family suggests that a complex mode of inheritance might be at the basis of FSHD development in these families.
To test this hypothesis we assessed whether the prevalence of disease varies among relatives according to degree of kinship with the proband (distribution is reported in Supplementary Table 4). Table 6 shows that 72.5% of first-degree relatives are affected. This percentage significantly decreases to 52.9% among relatives with lower degree of kinship (from second- to fifth-degree), irrespective of D4Z4 size allele, sex and age at examination (P = 0.018), supporting the hypothesis that beside DRA, additional genetic factors may be necessary to develop FSHD. Conversely 47.1% of second- through fifth-degree relatives was unaffected, while only 27.5% of first-degree family members did not show any motor impairment.
Table 6.
Prevalence of FSHD scores according to degree of kinship
| FSHD score |
|||||
|---|---|---|---|---|---|
| 0 |
1–15 |
||||
| Degree of kinship | Number of subjects | % | Number of subjects | % | P-value‡ |
| First | 77 | 27.5 | 203 | 72.5 | |
| Second/Fifth | 41 | 47.1 | 46 | 52.9 | 0.018 |
‡Wald test of coefficients associated to second or third degree of kinship in logistic models adjusted by D4Z4 allele size, sex and age at examination.
It has also been observed that FSHD affects males more severely and more frequently than females (Zatz et al., 1998; Tonini et al., 2004; Sakellariou et al., 2012). We thus evaluated whether gender influences expression and severity of motor impairment. We observed that the percentage of asymptomatic carriers does not significantly differ between genders (data not shown). Instead, as shown in Table 7, male relatives had a significantly higher mean FSHD score (5.4 versus 4.0, P = 0.003) and they developed motor impairment on average 7.3 years before than females (P = 0.003). Thus male relatives who develop motor impairment had a more severe disease than affected female relatives. We then calculated the risk of developing motor impairment between 20–50 years in females and males separately using the Kaplan-Meier method. As shown in Fig. 4A, the risk is higher in male relatives than females, although the difference is not statistically significant (log rank test P-value 0.113). Among probands the risk of developing motor impairment after age 20 is higher in males than in females (log rank test P-value = 0.028) (Fig. 4B). Remarkably, the risk becomes similar between genders after age 50.
Table 7.
Distribution of FSHD score and age at onset calculated on affected relatives according to sex
| FSHD Score |
Age at onset |
|||||||
|---|---|---|---|---|---|---|---|---|
| Sex | Number of subjects | FSHD score mean | 95% CI | P-value† | Number of subjects | Mean age at onset (years) | 95% CI | P-value§ |
| Male | 102 | 5.4 | 4.7–6.1 | 99 | 26.8 | (23.2; 30.5) | ||
| Female | 147 | 4.0 | 3.5–4.5 | 0.003 | 144 | 34.1 | (30.5; 37.7) | 0.003 |
| Total | 249 | 243 | ||||||
†Wald test of equality to zero of female sex coefficients in a general linear model with FSHD score as dependent variable sex, D4Z4 allele size and age at examination as predictors.
§Wald test of equality to zero of female sex coefficients in a general linear model with age at onset as dependent variable and sex and D4Z4 allele size as predictors.
Figure 4.
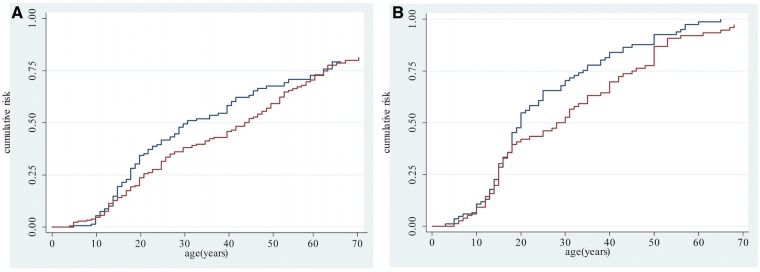
Age-specific cumulative risk of reported muscle impairment according to sex. (A) Estimates obtained on 361 relatives using the Kaplan-Meier analysis. Log-rank test P-value = 0.113. (B) Estimates obtained on 160 probands using the Kaplan-Meier analysis. Blue line refers to male; red line refers to female. Log-rank test P-value = 0.028.
All index cases and their relatives recruited in the present study carried the 4qA allele. As it has been recently proposed that FSHD occurs only when DRA at 4q35 are in combination with the 4A(159/161/168)PAS haplotype (Lemmers et al., 2010), we further characterized DNA polymorphisms flanking the D4Z4 reduced array in 294 subjects (203 affected and 91 unaffected) belonging to 133 families from the cohort selected for this study. Table 8 reports the various haplotypes detected. All were associated with the polyadenylation signal (ATTAAA) that stabilized transcripts from DUX4 gene. Notably, the 4A161PAS haplotype previously considered ‘permissive’ and the 4A166PAS haplotype previously considered ‘non-permissive’ for FSHD disease were detected in both DRA carriers with motor impairment (FSHD score ≥1) and without motor impairment (FSHD score 0). On this basis we conclude that no specific 4q haplotype can be considered as predictive of disease.
Table 8.
Distribution of haplotypes on 294 relatives
| Relatives |
|||||||
|---|---|---|---|---|---|---|---|
| 4A161 (n = 204) | 4A162 (n = 14) | 4A163 (n = 5) | 4A164 (n = 1) | 4A166 (n = 69) | 4A167 (n = 1) | ||
| FSHD score = 0 | |||||||
| Number of subjects (%) | 72 (79.1) | 5 (5.5) | 2 (2.2) | 0 (0.0) | 12 (13.2) | 0 (0.0) | |
| FSHD score ≥1 | |||||||
| Number of subjects (%) | 132 (65.0) | 9 (4.4) | 3 (1.5) | 1 (0.5) | 57 (28.1) | 1 (0.5) | |
Collectively, the statistical analysis conducted on the entire cohort of relatives carrying DRA with 1–3 or 4–8 repeats indicates that individuals carrying DRA with 1–3 repeats have a high risk of developing motor impairment by age 50 (83–93%), regardless of sex or degree of kinship. In contrast, in the group with 4–8 repeats the reduced risk of becoming symptomatic (55-63% by age 50) is also modulated by sex (males show a higher risk than females) and degree of kinship (first degree relatives show a higher risk than second-fifth degree relatives).
Discussion
Before the discovery of rearranged D4Z4 alleles, the diagnosis and counselling of FSHD families was entirely based on clinical evidence (Lunt et al., 1989). Over the years, DNA testing of the D4Z4 locus and flanking polymorphisms has been considered highly sensitive and specific and extensively used to diagnose FSHD (Tawil et al., 2010). However, two recent discoveries have challenged the current understanding of the prognostic value of D4Z4 reduced alleles (DRA) in FSHD families: (i) alleles with reduced numbers (≤8) of D4Z4 repeats at 4q35 combined with 4A(159/161/168)PAS haplotype, have a frequency of 1.3% among healthy subjects from the general population; and (ii) only 50% of FSHD probands carry the 4A161PAS permissive haplotype associated with DRA (Scionti et al., 2012b). Therefore, our understanding of the factors that cause FSHD is incomplete and we conclude that it is crucial for clinical practice to define further elements that can influence motor impairment and can support the interpretation of molecular testing in FSHD families.
The present results rely on a population-based study involving index cases recruited from all regions of Italy. In order to minimize any ascertainment bias the analyses were performed excluding index cases and the evaluation of motor impairment was based on a standardized protocol shared within the ICNF. However, beside these strengths, the study has some limitations. First, the genetic background and the socio-demographic characteristics of the Italian population might restrict the external validity of the study results. Second, even though the study has a good coverage of index cases, the involvement of relatives might be due to the presence of any symptoms with the consequence that the healthy ones might be under-represented in the study. In that case the true estimated prevalence of disease among relatives would be lower. Third, given that FSHD is a rare disease and no routinely collected diagnosis records are available (Lunt et al., 1989) the age at onset was collected retrospectively. Therefore we cannot rule out the presence of recall bias. Indeed, the perception of disease onset may be subjective and could depend on the specific motor skills required in daily activities. It is thus possible that in a number of subjects the motor impairment of limbs may be perceived as early symptom because more disabling. According to this possibility and consistent with previous works (Tawil and van der Maarel, 2006; Pastorello et al., 2012), in our cohort the most frequently complained symptom at onset was also the impairment of upper girdle (Supplementary Table 3). Nevertheless, we considered that patient’s complaints provide a reliable estimate of the time of functional disability onset related to disease. When subjects did not refer any motor impairment, but a mild muscle weakness was observed at the clinical evaluation, the age at examination was arbitrarily set as the age at onset (Lunt et al., 1995b).
Given these premises, our study shows that FSHD penetrance in DRA carriers is not complete by age 20, as previously proposed (Tawil et al., 2010), as asymptomatic carriers in all the classes of ages up to 70 years were found.
The present analysis highlights different prognostic values of DRA with 1–3 units when compared with DRA with 4–8 units. First, among carriers of DRA with 1–3 units FSHD penetrance is almost complete; in contrast, ∼30% of carriers of DRA with 4–8 units older than 55 years display no muscle weakness (Table 1). Second, the estimated risk of developing motor impairment by age 50 differs between the two classes of alleles. Carriers of DRA with 1–3 units have a risk of 88.5% of developing motor impairment by age 50; instead the risk among carriers of DRA with 4–8 units by the same age is 55% (Fig. 2). Third, 44% of carriers of DRA with 1–3 units develop severe FSHD (FSHD score ≥7) by age 55; whereas only 24% of carriers of DRA with 4–8 units develop disease with high degree of severity by the same age (Fig. 3B). Fourth, the clinical phenotype is more homogeneous in families with DRA with 1–3 units, as shown by the intra-familial analysis (Table 5). In contrast, the clinical status of probands does not seem to be predictive of disease severity in relatives carrying DRA with 4–8. Importantly, in these families with DRA with 4–8 units, the penetrance of FSHD is lower as the degree of relationship to the affected individual becomes more distant, indicating that the genetic background can affect the disease outcome.
Our study also shows that gender influences disease expression, because males are characterized by a lower mean age at onset of motor impairment (26.8 years in males versus 34.1 years in females, Table 7) and by a more severe disability in terms of FSHD score (5.4 in males versus 4.0 in females). Interestingly, the risk of developing motor impairment is higher in male relatives during adult age (range 18–55 years), whereas it is similar between males and females in childhood/teens and elderly age (Fig. 4). Overall, these data indicate that variables related to gender, including genetic, hormonal, and/or lifestyle factors, may be considered and should be further investigated. Finally, our study suggests that the predictive value of 4q haplotypes must be carefully considered because no specific 4q haplotype was exclusively associated with the presence of disease.
In summary, the genotype-phenotype correlation study presented here confirms that DRAs with 4–8 repeats have no definitive prognostic value, and that other prognostic parameters, beside DRAs, such as sex and degree of kinship with the proband should be considered. We estimated that the risk of developing the motor impairment by age 50 in FSHD family members is higher (83–93%) in subjects carrying DRA with 1–3 repeats. Instead, considering the cohort of relatives carrying DRA with 4–8 repeats, the risk of developing motor impairment is 48% for females and/or subjects with lower degree of kinship and raises to 55–63% for males and/or subjects with first degree of kinship with the proband. None of the various 4q haplotypes detected in FSHD families studied here were exclusively associated with the presence of disease, as reported in Table 8.
Interestingly, in our cohort, 19 of 148 FSHD families (13%) in which a DRA with 4–8 units segregates presented affected subjects only in one generation (Supplementary Fig. 2). In these cases the lack of autosomal dominant inheritance should prompt us to consider whether the disease develops because of the presence of additional genetic defect(s). This possibility is supported by recent observation that mutations in the SMCHD1 gene segregate independently from the FSHD permissive D4Z4 allele on chromosome 4 in FSHD subjects that do not carry a DRA, also defined as patients with FSHD2 (Lemmers et al., 2012). Therefore searches for secondary FSHD loci should be considered in all cases in which the ratio between affected and unaffected individuals expected for an autosomal dominant disease is not observed and random association between the DRA and FSHD cannot be excluded.
For all of these reasons, to define the predictive value of DRA, it is necessary to carry out clinical evaluation and collection of DNA samples of all of the proband’s family members, not only in a research setting but also in clinical practice. We believe that broadening the analysis of FSHD families may facilitate genetic counselling of patients and families with FSHD in particular when interpreting the data for prenatal diagnosis.
Supplementary Material
Acknowledgements
We are indebted to all patients with FSHD and their families for participating in this study. The Associazione Amici del Centro Dino Ferrari-University of Milan is gratefully acknowledged. We thank Dr. Hulya Gundesli, Professor Woodring E. Wright and Dr. Paul D. Kaufman for their in-depth critique of the manuscript.
Glossary
Abbreviations
- DRA
D4Z4 alleles of reduced size
- FSHD
facioscapulohumeral muscular dystrophy
Funding
This work was supported by Telethon Italy GUP08004, by Telethon Italy GUP11009, by Association Française contre les Myopathies (AFM) grant number 14339 and by National Institutes of Health (NIH)-National Institute of Neurological Disorders and Stroke (NINDS) grant number RO1 NS047584.
Supplementary material
Supplementary material is available at Brain online.
References
- Flanigan KM. Facioscapulohumeral muscular dystrophy and scapulohumeral syndrome. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2004. pp. 123–34. [Google Scholar]
- Goto K, Nishino I, Hayashi YK. Very low penetrance in 85 Japanese families with facioscapulohumeral muscular dystrophy 1A. J Med Genet. 2004;41:e12. doi: 10.1136/jmg.2003.008755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griggs RC, Tawil R, Storvick D, Mendell JR, Altherr MR. Genetics of facioscapulohumeral muscular dystrophy: new mutations in sporadic cases. Neurology. 1993;43:2369–72. doi: 10.1212/wnl.43.11.2369. [DOI] [PubMed] [Google Scholar]
- Hippisley-Cox J, Coupland C, Vinogradova Y, Robson J, May M, Brindle P. Derivation and validation of QRISK, a new cardiovascular disease risk score for the United Kingdom: prospective open cohort study. BMJ. 2007;335:136. doi: 10.1136/bmj.39261.471806.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- Lamperti C, Fabbri G, Vercelli L, D’Amico R, Frusciante R, Bonifazi E, et al. A standardized clinical evaluation of patients affected by facioscapulohumeral muscular dystrophy: the FSHD clinical score. Muscle Nerve. 2010;42:213–17. doi: 10.1002/mus.21671. [DOI] [PubMed] [Google Scholar]
- Lemmers RJ, de Kievit P, Sandkuijl L, Padberg GW, van Ommen GJ, Frants RR, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. 2002;32:235–6. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–4. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, et al. A uniying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers RJ, Wohlgemuth M, van der Gaag KJ, van der Vliet PJ, van Teijlingen CM, de Knijff P, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:884–94. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt PW, Compston DA, Harper PS. Estimation of age dependent penetrance in facioscapulohumeral muscular dystrophy by minimising ascertainment bias. J Med Genet. 1989;26:755–60. doi: 10.1136/jmg.26.12.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt PW, Jardine PE, Koch MC, Maynard J, Osborn M, Williams M, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD) Hum Mol Genet. 1995a;4:951–8. doi: 10.1093/hmg/4.5.951. [DOI] [PubMed] [Google Scholar]
- Lunt PW, Jardine PE, Koch MC, Maynard J, Osborn M, Williams M, et al. Phenotypic-genotypic correlation will assist genetic counseling in 4q35-facioscapulohumeral muscular dystrophy. Muscle Nerve. 1995b;2:S103–9. [PubMed] [Google Scholar]
- Lunt PW. 44th ENMC international workshop: facioscapulohumeral muscular dystrophy: molecular studies 19–21 July 1996, Naarden, The Netherlands. Neuromuscul Disord. 1998;8:126–30. doi: 10.1016/s0960-8966(98)00012-1. [DOI] [PubMed] [Google Scholar]
- McCullagh P, Nelder JA. Generalized linear models. 2nd edn. New York: Chapman and Hall, Inc; 1989. [Google Scholar]
- Mostacciuolo ML, Pastorello E, Vazza G, Miorin M, Angelini C, Tomelleri G, et al. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north-east Italian population sample. Clin Genet. 2009;75:550–55. doi: 10.1111/j.1399-0004.2009.01158.x. [DOI] [PubMed] [Google Scholar]
- Padberg G. Facioscapulohumeral disease. MD thesis. University of Leiden; 1982. [Google Scholar]
- Padberg GW, Lunt PW, Koch M, Fardeau M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 1991;1:231–4. doi: 10.1016/0960-8966(91)90094-9. [DOI] [PubMed] [Google Scholar]
- Pastorello E, Cao M, Trevisan CP. Atypical onset in a series of 122 cases with facioscapulohumeral muscular dystrophy. Clin Neurol Neurosurg. 2012;114:230–4. doi: 10.1016/j.clineuro.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci E, Galluzzi G, Deidda G, Cacurri S, Colantoni L, Merico B, et al. Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann Neurol. 1999;45:751–7. doi: 10.1002/1531-8249(199906)45:6<751::aid-ana9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Sakellariou P, Kekou K, Fryssira H, Sofocleous C, Manta P, Panousopoulou A, et al. Mutation spectrum and phenotypic manifestation in FSHD Greek patients. Neuromuscul Disord. 2012;22:339–49. doi: 10.1016/j.nmd.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Scionti I, Fabbri G, Fiorillo C, Ricci G, Greco F, D’Amico R, et al. Facioscapulohumeral muscular dystrophy: new insights from compound heterozygotes and implication for prenatal genetic counselling. J Med Genet. 2012a;49:171–8. doi: 10.1136/jmedgenet-2011-100454. [DOI] [PubMed] [Google Scholar]
- Scionti I, Greco F, Ricci G, Govi M, Arashiro P, Vercelli L, et al. Large scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy (FSHD) Am J Hum Genet. 2012b;90:628–65. doi: 10.1016/j.ajhg.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawil R, Forrester J, Griggs RC, Mendell J, Kissel J, McDermott M, et al. Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. The FSH-DY Group. Ann Neurol. 1996;39:744–48. doi: 10.1002/ana.410390610. [DOI] [PubMed] [Google Scholar]
- Tawil R, van der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve. 2006;34:1–15. doi: 10.1002/mus.20522. [DOI] [PubMed] [Google Scholar]
- Tawil R, van der Maarel S, Padberg GW, van Engelen BG. 171st ENMC international workshop: standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2010;20:471–5. doi: 10.1016/j.nmd.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Tonini MM, Passos-Bueno MR, Cerqueira A, Matioli SR, Pavanello R, Zatz M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD) Neuromuscul Disord. 2004;14:33–8. doi: 10.1016/j.nmd.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Upadhyaya M, Maynard J, Rogers MT, Lunt PW, Jardine P, Ravine D, et al. Improved molecular diagnosis of facioscapulohumeral muscular dystrophy (FSHD): validation of the differential double digestion for FSHD. J. Med. Genet. 1997;34:476–9. doi: 10.1136/jmg.34.6.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deutekom JC, Bakker E, Lemmers RJ, van der Wielen MJ, Bik E, Hofker MH, et al. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1. Hum Mol Genet. 1996;5:1997–2003. doi: 10.1093/hmg/5.12.1997. [DOI] [PubMed] [Google Scholar]
- van Overveld PG, Lemmers RJ, Deidda G, Sandkuijl L, Padberg GW, Frants RR, van der Maarel SM. Interchromosomal repeat array interactions between chromosomes 4 and 10: a model for subtelomeric plasticity. Hum Mol Genet. 2000;9:2879–84. doi: 10.1093/hmg/9.19.2879. [DOI] [PubMed] [Google Scholar]
- Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- Williams RL. A note on robust variance estimation for cluster-correlated data. Biometrics. 2000;56:645–6. doi: 10.1111/j.0006-341x.2000.00645.x. [DOI] [PubMed] [Google Scholar]
- Zatz M, Marie SK, Cerqueira A, Vainzof M, Pavanello RC, Passos-Bueno MR. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am J Med Genet. 1998;77:155–61. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.