

Abstract
Most head and neck squamous cell carcinomas (HNSCC) overexpress epidermal growth factor receptor (EGFR) and EGFR inhibitors are routinely used in the treatment of HNSCC. However, many HNSCC tumors do not respond or become refractory to EGFR inhibitors. Autophagy, which is a stress-induced cellular self-degradation process, has been reported to reduce the efficacy of chemotherapy in various disease models. The purpose of this study is to determine if the efficacy of the EGFR inhibitor erlotinib is reduced by activation of autophagy via NOX4-mediated oxidative stress in HNSCC cells. Erlotinib induced the expression of the autophagy marker LC3B-II and autophagosome formation in FaDu and Cal-27 cells. Inhibition of autophagy by chloroquine and knockdown of autophagy pathway genes Beclin-1 and Atg5 sensitized both cell lines to erlotinib-induced cytotoxicity, suggesting that autophagy may serve as a protective mechanism. Treatment with catalase (CAT) and diphenylene iodonium (DPI) in the presence of erlotinib suppressed the increase in LC3B-II expression in FaDu and Cal-27 cells. Erlotinib increased NOX4 mRNA and protein expression by increasing its promoter activity and mRNA stability in FaDu cells. Knockdown of NOX4 using adenoviral siNOX4 partially suppressed erlotinib-induced LC3B-II expression, while overexpression of NOX4 increased expression of LC3B-II. These studies suggest that erlotinib may activate autophagy in HNSCC cells as a pro-survival mechanism, and NOX4 may play a role in mediating this effect.
Keywords: HNSCC, EGFR, Erlotinib, Autophagy, NOX4, LC3B
Introduction
The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase that activates numerous pro-survival signaling pathways (Rocha-Lima et al., 2007). EGFR is activated when growth factor ligands bind to the ligand binding domain of the receptor resulting in receptor dimerization and enhancement of intracellular tyrosine kinase activity (Olayioye et al., 2000). Receptor activation leads to recruitment and phosphorylation of several intracellular substrates resulting in pro-growth signaling and tumor promoting activities (Mendelsohn 2002, Mendelsohn and Baselga 2006). Given that EGFR signaling is upregulated in many cancers including head and neck squamous cell carcinoma (HNSCC) (Grandis and Tweardy 1993), several drugs that target EGFR have been developed and approved for cancer therapy. However EGFR-based chemotherapy has limited results in HNSCC patients due to development of resistance mechanisms including mutations in EGFR, PTEN and PI3KCA, and amplification or upregulation of other tyrosine kinase receptor signaling pathways (Harari et al., 2009, Rexer et al., 2009).
Autophagy is a self-degradation phenomenon activated under conditions of stress including nutrient deprivation, oxidative stress, chemotherapeutic insult and radiation (Mizushima 2005, Mizushima 2009). During this process a double membranous structure is formed which encloses the cytoplasm along with the components targeted for degradation, then fuses with lysosome to form an autophagolysosome where the contents are degraded and recycled for use by the cells under conditions of stress (Tanida 2011). Autophagy has been implicated as both a tumor suppressor and tumor promoting mechanism depending on the cell model used, type of stress and duration of stimuli (Bialik and Kimchi 2008, Eisenberg-Lerner and Kimchi 2009). Additionally, autophagy has been implicated in resistance and decreased response to chemotherapeutic agents due to its tumor promoting activity (Kondo et al., 2005).
Previous studies in our lab have shown that the EGFR inhibitor erlotinib increases oxidative stress via activation of NADPH oxidase 4 (NOX4). NADPH oxidases are a class of membrane bound enzymes that transfer electrons from NADPH across the membrane resulting in the formation of reactive oxygen species (ROS) in a variety of cells (Lambeth 2004). Since autophagy may be activated by oxidative stress, the purpose of this study was to determine if erlotinib activated autophagy as a protective mechanism in HNSCC cells via NOX4.
Materials and methods
Cell culture conditions
FaDu and Cal-27 human head and neck cancer squamous carcinoma cells (HNSCCs) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The cells were grown in Dulbecco's Modified Eagle's medium (DMEM) containing 10% FBS, 4 mM L-glutamine, 1 mM sodium pyruvate, 4.5 g/L glucose (DMEM) and gentamycin. Cultures were maintained in 5% CO2 and in a 37°C incubator.
Drug treatment
Chloroquine (CQ), catalase (CAT) and diphenylene iodonium (DPI) were obtained from Sigma Chemical Co. Erlotinib (ERL) was obtained from Cayman Chemicals and dissolved in DMSO. All drugs were used without further purification. Drugs were added to cells at final concentrations of 100U/mL CAT, 50 nmol/L DPI, 2, 12 and 25 μmol/L of CQ and 5 μmol/L ERL. Catalase was conjugated to polyethylene glycol (PEG) and PEG was used as the control in the appropriate studies. The required volume of each drug was added directly to complete cell culture media on cells to achieve the desired final concentrations. All cells were placed in a 37°C incubator and harvested at the time points indicated.
Western Blot analysis
Cell lysates were standardized for protein content, resolved on 4% to 12% SDS polyacrylamide gels, and blotted onto nitrocellulose membranes. Membranes were probed with rabbit anti-LC3B, anti-beclin-1, anti-Atg5, anti-β-actin (Cell Signaling Technologies), anti-NOX1, anti-NOX2, anti-NOX3, anti-NOX4, anti-DUOX2 (Abcam), anti-NOX5, anti- DUOX1 (Santa Cruz Biotechnology) antibodies. Antibody binding was detected by using an ECL Chemiluminescence Kit (Amersham).
Staining autophagosomes with GFP-LC3
Cells were transfected with GFP-LC3 (pSELECT-GFP-LC3) expressing plasmids (Invivogen) and successfully transfected cells selected in DMEM with zeocin. After treatment with erlotinib for 48 hours the cells were fixed with 4% paraformaldehyde, washed with PBS, counter stained with DAPI to visualize nuclei and the fluorescence of GFP-LC3 was viewed under a fluorescent microscope.
Immunofluorescence staining
Cells were cultured in chamber slides and treated with erlotinib for 48 hours. The media was then removed and the slides fixed in 4% paraformaldehyde for 30 minutes at room temperature. Slides were then blocked in 1% BSA in 0.05% Triton X-100/PBS for 30 minutes at room temperature and incubated with rabbit anti-human LC3B antibody (Cell Signaling Technologies, 1:400 dilution) for 1 hour. Secondary detection was done using AlexaFluor488 anti-rabbit (Invitrogen) for 1 hour. Nucleus was stained with DAPI. The chamber wells were detached from the glass slide, mounted with Vectashield Mounting media (Vector Laboratories) and observed under fluorescence microscope.
Real Time PCR analysis
Total RNA was extracted from cells after treatment at indicated time point using Qiagen RNeasy mini kit. The cDNA was amplified using iScript cDNA synthesis kit (Bio-rad). The cDNAs were subjected to RT-PCR analysis with the following 5′-3′primer sets: NOX1 (5′TGGGTTTTCTAAACTACCGTCTCT3′ and 5′TTTAATGCTGCATGACCAACA3′), NOX2 (5′GGCTTCCTCAGCTACAACATCT3′ and 5′GTGCACAGCAAAGTGATTGG3′), NOX3(5′CACACCATGTTTTCATCGTCTT3′ and 5′GTTTGGCCTCGAACAATCC3′), NOX4 (5′CTCAGCGGAATCAATCAGCTGTG3′ and 5′AGAGGAACACGACAATCAGCCTTAG3′), NOX5 (5′CGTCTGTGCCGGCTTATC3′ and 5′CCAATTCCAGATACAACATGACTG3′) (Prata et al., 2008), DUOX1 (5′CCGGAGACAAGTTGCAGTC3′and 5′TCTCCAATCTGATCCTTGTTTTC3′), DUOX2 (5′CGGAAGAAGGTGGAGATCAG3′ and 5′TGGAATTGCAGGTAGGTCACT3′) and 18S (5′CCTTGGATGTGGTAGCCGTTT3′ and 5′AACTTTCGATGGTAGTCGCCG3′). The assay was performed using 2X SYBR Green/ROX PCR master mix (Bio-Rad). Samples were run on an ABI PRISM Sequence Detection System (model 7000, Applied Biosystems). PCR conditions were 50°C for 2 min, 95°C for 2 min, 95°C for 15 s, and 60°C for 1 min for 40 cycles. Results were analyzed using the ABI PRISM 7000 SDS software. The CT values for the target genes in all of the samples were normalized on the basis of the abundance of the 18S transcript, and the fold difference (relative abundance) was calculated using the formula 2−ΔΔCT and was plotted as the mean. For analysis of miR-25 expression, RNA was isolated using mirVana microRNA isolation kit (Applied Biosystems) according to manufacturer's instructions. 250 ng of RNA was reverse transcribed using microRNA specific primers; miR-25 (Applied Biosystems; part no-001006) and RNU48 (Applied biosystems; part no-00403) using the TaqMan microRNA reverse transcription kit (Applied Biosystems). The reaction was performed using TaqMan universal PCR master mix (Applied biosystems) according to manufacturer's instructions on the ABI 7900HT. Results were analyzed using SDS 2.3 (Applied biosystems).
Clonogenic survival assay
Both floating and attached cells from the treated dishes were collected. Attached cells were collected by trypsinization. Cells were re-plated at a low density of 150-1000 cells/plate and allowed to grow for 14 days in complete media. Cells were stained with Coomassie Blue dye, and the colonies on each plate subsequently counted for analysis of clonogenic survival as previously described (Spitz et al., 1990).
Construction of NOX4 expression plasmids
NOX4 promoter: The NOX4 promoter was cloned from normal keratinocyte genomic DNA. Approximately 1500 base pairs upstream of the NOX4 transcription start site was chosen as a representative promoter region. This area possessed numerous putative transcription factor binding sites as indicated by the Transcription Element Search System (TESS, University of Pennsylvania). The promoter region was amplified using HotStarTaq Master Mix by conventional PCR and the following primers (with restriction sites added): 5′ (Sac1): GAGCTCTATTAAGAGTTGGAGCCATTTTTTAAACCTTTTG and 3′ (Xho1): CTCGAGGCTGCCCAGACGCCCAG. PCR product was gel purified and cloned into a pJet1.2 vector using the CloneJET PCR cloning kit (Thermo Scientific, USA). After sequence validation, the NOX4 promoter was subcloned into pGL4.10[luc2]; a promoterless vector with multiple cloning site (MCS) directly upstream of a luciferase construct and confirmed by sequencing.
NOX4 3′UTR Luciferase Vector Constructs: Genomic DNA was isolated using the purelink genomic DNA isolation kit (Invitrogen) from HEK293 cells (ATCC). The NADPH Oxidase 4 3′UTR was amplified using 3′TTCGTAGTCGACCTGTTGTGGACCCAATTCACTATCC5′ and 5′AGCTCCTCACTAGGGAGTTCTTGAGCGGCCGCATTTTA3′. The amplification product was cloned in to the pGEM-T Easy Vector (Promega). The vector was propagated and DNA isolated using a Plasmid Midi kit (Qiagen). A restriction digestion using SALI (NEB) and NOTI (NEB) endonucleases was carried out on the PsiCheck2 vector (Promega) and the NOX4 3′UTR plasmid at 37°C for 1hr. These products were purified using a PCR clean-up kit (Qiagen). The NOX4 3′UTR and the PsiCheck 2 vector were then ligated using T4 ligase (NEB) at 16°C overnight. This product was purified using a PCR clean-up kit (Qiagen) and transformed into DH5α Max Efficiency cells (Invitrogen). The product was then propagated and the DNA isolated with a Plasmid Midi kit (Qiagen). The miR-25 binding site was mutated from position 141 to position 149 of the NOX4 3′UTR using the Quick Change Multisite Directed Mutagenesis Kit (Agilent Technologies) and with the primer set 3′GCAGGACTCTAAAGAAGGAATTGCGAAGATTTCTAAGACT 5′ and 5′AGTCTTAGAAATCTTCGCAATTCCTTCTTTAGAGTCCTGC 3′ according to manufacturer's instructions. The amplification and final product were sequenced using the Applied Biosystems Model 3730 capillary sequencer.
Plasmid transfection and luciferase assays
The cells were plated in 12 well plates and incubated for 24 hours. Cells were transiently transfected according to the manufacturer's instructions by incubating the constructs with Lipofectamine 2000 (2 μl/well; Invitrogen), for 4 hours in case of the pGL4.10 human NOX4 promoter and 24 hours for the NOX4 3′ UTR construct. To control for transfection efficiency of NOX4 promoter plasmid, cells were cotransfected with 0.1 μg/well pRL-TK Renilla luciferase or construct. The NOX4 3′UTR Luciferase vector expressed Renilla luciferase. After transfection the cells were allowed to recover for 24 hours and then treated with either DMSO (control) or 5μM erlotinib for 48 hours. Cells were then washed twice with PBS and collected into passive lysis buffer (Promega). Luciferase activities were measured with the Dual Luciferase Reporter Assay System (Promega). Relative light units were normalized to Renilla activity.
siRNA transfection
Beclin-1, Atg-5 and control siRNA were purchased from Cell Signaling Technologies. HNSCC cells were transfected with 100 nmol/L siRNA at 80% confluence in reduced serum Eagle's Minimum Essential Medium for 24 hours. Lipofectamine 2000 (Invitrogen) was used for transfections following protocols provided by the manufacturer. Biochemical analyses were done 48 to 72 hours after transfection.
Adenoviral NOX4 manipulation
Adenoviral vectors expressing siRNA against NOX4 (AdsiNOX4) or a scrambled control were constructed, purified and provided by the University of Iowa Gene Transfer Vector Core (GTVC) as described previously (Peterson et al., 2009). Construction of Ad5.CMV.hNOX4WT (NOX4 wild-type) and Ad5.CMV.hNOX4DN(NOX4 dominant-negative) adenoviruses was done as follows. Wild-type (WT) human NOX4 cDNA (BC040105.1) was obtained from Open Biosystems (MHS1010-9204787; Huntsville, AL) in the pCMV-SPORT6 vector. cDNA for the dominant-negative (DN) form of NOX4 lacking the C-terminal NADPH binding domain was generated by introduction of a stop codon at amino acid 384 as described previously (Mahadev et al., 2004) using Stratagene Quickchange XL site-directed mutagenesis (La Jolla, CA) with forward CTGGACAGAACGATTTCGAGATTAACTACTGCCTCC and reverse GGAGGCAGTAGTTAA TCTCGAAATCGT TCTGTCCAG primers (nucleotide change underlined). The entire cDNA for the WT and DN clones were subsequently sequence verified and subcloned into the pacAd5.CMV.K-NpA shuttle vector (U Iowa GTVC) using Kpn1/Not1 restriction. Viruses were purified by the U Iowa GTVC using CsCl gradient centrifugation and dialyzed against buffer containing 3% sucrose/PBS and stored at -80°C. Each virus was tested for WT revertants and for titer by PCR and A549 plaque assay as described (Anderson et al., 2000). Cells were transfected with adenovirus (MOI-150) in serum free media for 24 hours. Media was changed into complete media and allowed to recover for 72 hours, treated with drug for indicated time period and harvested for biochemical assays.
Transmission Electron microscopy
Cells were fixed in 2.5% gluteraldehyde in 0.1 M sodium cacodylate for 45 minutes, washed and post fixed with 1% OsO4 for 30 minutes. The cells were again washed and stained for 5 minutes in 2.5% aqueous uranyl acetate. The samples were dehydrated with graded alcohol and embedded on Epon resin (Canemco Inc). Ultrathin sections were cut on a Leica EM microtome, counterstained with 5% uranyl acetate and 0.3% lead citrate and observed under electron microscope.
Measurement of intracellular ROS levels
Attached cells were trypsinized, resuspended in PBS and labeled with 5-(and-6)-carboxy-2′, 7′ -dichlorodihydrofluorescein diacetate, (DCFH, 10 μg/mL) dissolved in 0.1% DMSO for 15 minutes at 37°C. Labelled cells were analyzed by using a FACSCalibur flow cytometer [excitation 488 nm, emission 530 nm band-pass filter (DCFH). The mean fluorescence intensity (MFI) of 10,000 cells was analyzed in each sample and corrected for autofluorescence from unlabeled cells.
Statistical analysis
Statistical analysis was done using GraphPad Prism version 5 for Windows (GraphPad Software, San Diego, CA). Data were compared using two-tailed Students t-test and differences between 3 or more means were determined by one-way ANOVA with Tukey post-tests. P values less than 0.05 was considered significant.
Results
EGFR tyrosine kinase inhibitor (EGFR TKI) erlotinib decreased clonogenic survival and induced autophagy in HNSCC cells
We initially confirmed that erlotinib (5 μM, 48 hours) was capable of decreasing activated EGFR (pEGFR) expression in FaDu and Cal-27 cells by western blot (Figure 1A). The dose of 5 μM erlotinib was chosen because it is within the range of plasma levels (0.8 - 6.7 μM) found in HNSCC patients after being dosed with 150 mg Erlotinib p.o. daily for 3 weeks and therefore clinically relevant (Soulieres et al., 2004). The effect of erlotinib on clonogenic survival of FaDu and Cal-27 cells was determined by treating the cells with 5 μM erlotinib for 24, 48 and 72 hours before analysis by clonogenic survival. Erlotinib significantly decreased the clonogenic survival of these HNSCC cells at all time points compared to time 0 but there was no significant added cytotoxicity after 24 hours (Figure 1B). To prevent decreases in drug concentration due to metabolism, fresh erlotinib was added each time point but the results did not change (data not shown).
Figure 1. Erlotinib inhibits activation of EGFR, decreases clonogenic survival and activates autophagy in HNSCC cells.
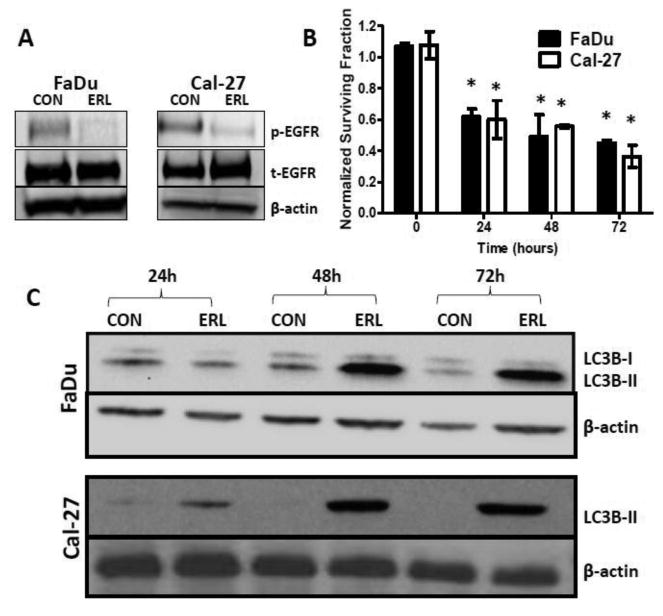
FaDu and Cal-27 cells were treated with 5μM erlotinib (ERL) for 48 hours and whole cell lysates analyzed by Western blotting for phosphorylated and total EGFR (p-EGFR and t-EGFR) levels (A). FaDu and Cal-27 cells were treated with erlotinib (ERL) for 24, 48 and 72 hours and analyzed for clonogenic survival. Data were normalized to corresponding controls (B). Cells were treated with erlotinib and analyzed for LC3B-II levels at 24, 48 and 72 hours by Western Blotting (C). β-actin used as loading control. *: p<0.05 vs Time 0.
In order to investigate autophagy induction, we investigated the effects of erlotinib on LC3B levels. Shortly after translation LC3B gets cleaved at its C-terminal exposing a glycine side chain and this form is known as LC3B-I which appears as the upper band (16kD) in a western blot. When autophagy is induced, a polyethanolamine (PE) group gets conjugated to the glycine side chain and this conjugated form is called LC3B-II which appears as the lower band (14kD). During autophagy more LC3B-II forms and gets incorporated into the autophagosomal membrane. The amount of LC3B-II is closely correlated with the number of autophagosomes, serving as a good indicator of autophagosome formation and autophagy induction. Erlotinib increased expression of LC3B-II levels using western blot in both cell lines after 48 and 72 hours (Figure 1B). Fluorescent imaging showed increased fluorescence of GFP-LC3 vacuoles in erlotinib treated FaDu and Cal-27 cells (Figure 2). Additionally, double membraned and organelle enclosing autophagosomes were identified in erlotinib treated FaDu and Cal-27 cells compared to control cells using transmission electron microscopy (Figure 3). Taken together these results suggest that the cytotoxic effect of erlotinib decreased over time and that erlotinib induces autophagy in both FaDu and Cal-27 HNSCC cells.
Figure 2. Erlotinib induced GFP-LC3 fluorescence and formation of autophagosomes in HNSCC cells.
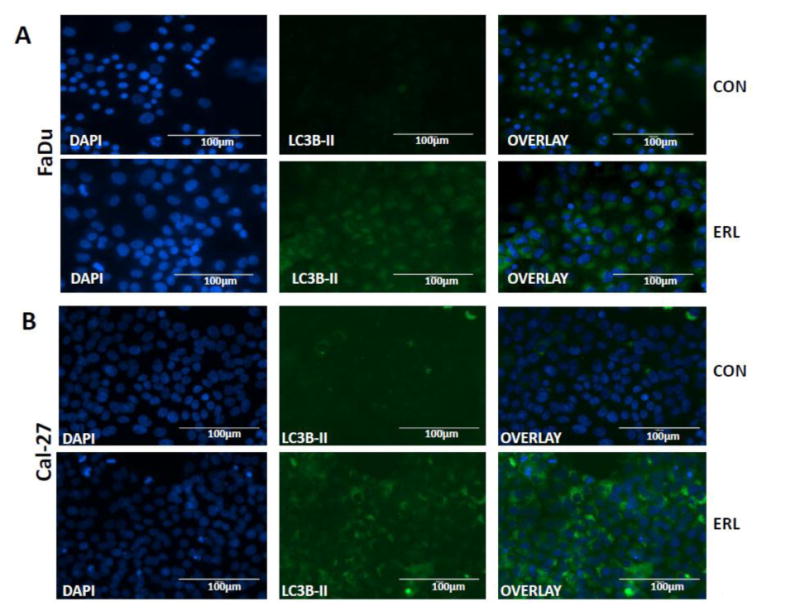
GFP-LC3 expressing FaDu cells were treated with DMSO (CON) or erlotinib (ERL) for 48 hours and LC3B-II expression determined by fluorescence microscopy (A). Cal-27 cells were treated with 5 μM ERL for 48 hours and LC3B-II expression determined by immunofluorescence assay (B). Blue- DAPI nuclear stain; Green- LC3B-II expression.
Figure 3. Erlotinib induces autophagosome formation in HNSCC cells.
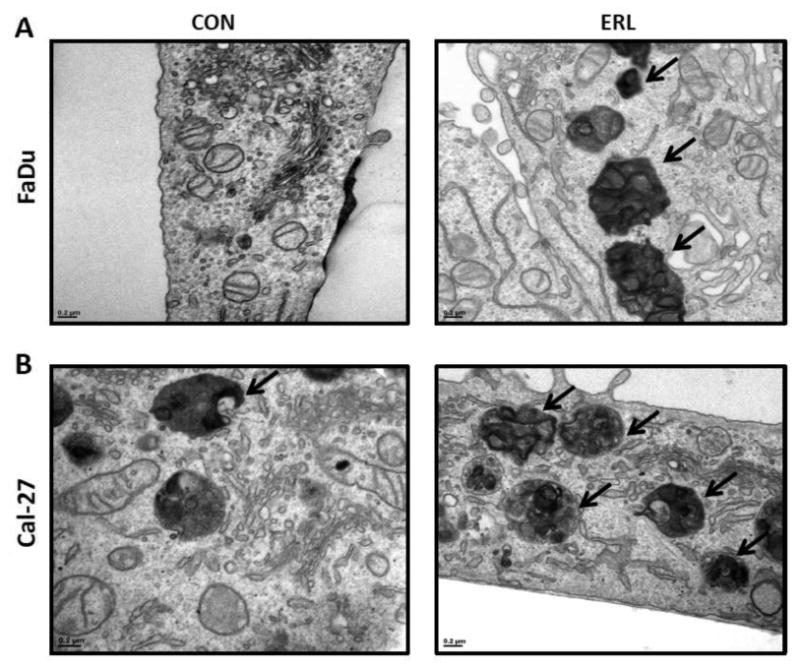
FaDu and Cal-27 cells were treated with 5 μM ERL for 48 hours and observed for autophagosome formation by transmission electron microscopy. Arrows indicate autophagosomes.
Inhibition of autophagy sensitizes HNSCC cells to Erlotinib
To examine whether inhibition of autophagy would sensitize HNSCC cells to erlotinib, chloroquine (CQ), an autophagy inhibitor was used in combination with erlotinib. FaDu and Cal-27 cells were treated with 2, 12 and 25 μM CQ for 2 hours prior to treatment with erlotinib and cytotoxicity assayed by clonogenic survival assay. CQ at doses of 12 and 25 μM caused significant cytotoxicity in both cell lines compared to controls (Figure 4). CQ at 2 and 12 μM was able to sensitize Cal-27 cells to erlotinib, while only 2 uM CQ was able to sensitize FaDu cells to erlotinib (Figure 4). To confirm that enhanced clonogenic killing by erlotinib was due to inhibition of autophagy we knocked down genes in the autophagy pathway such as beclin-1 and Atg5, with their corresponding siRNA before treatment with erlotinib. Beclin-1 and Atg5 siRNA knockdown increased the clonogenic killing by erlotinib in both FaDu and Cal-27 HNSCC cells (Figure 5A), and successfully suppressed expression of Beclin-1 and Atg5 respectively (Figure 5B). These results confirmed that erlotinib induced autophagy as a protective mechanism and inhibition of autophagy may increase efficacy to erlotinib in vitro.
Figure 4. Chloroquine sensitizes HNSCC cells to erlotinib.
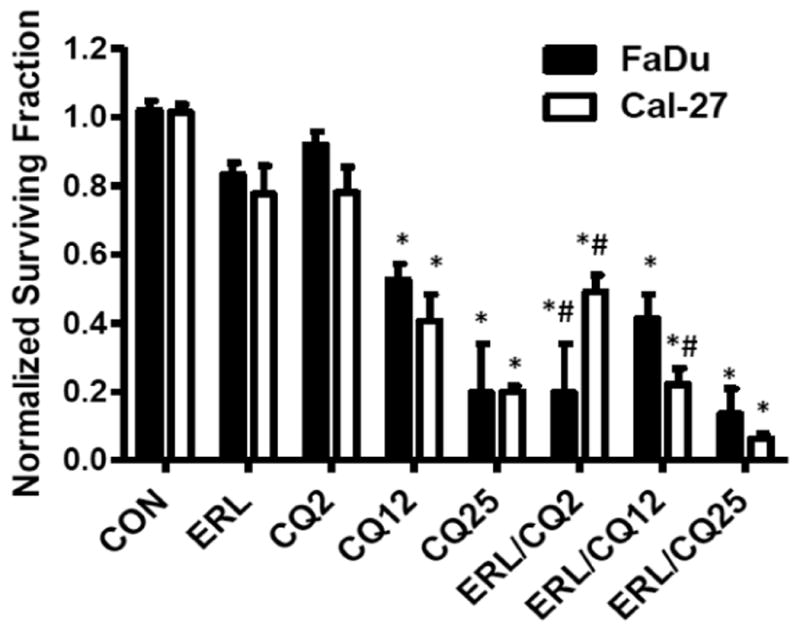
FaDu and Cal-27 cells were pre-treated with 2, 12 and 25 μM chloroquine (CQ) for 2 hours, treated with DMSO (CON) or 5 μM erlotinib (ERL) for 48 hours, then analyzed for clonogenic survival. Data were normalized to respective controls. Error bars represent the standard error of the mean for N=3 experiments. *:p<0.05 vs CON, #:p<0.05 vs ERL.
Figure 5. Knockdown of autophagy genes sensitizes HNSCC cells to erlotinib.
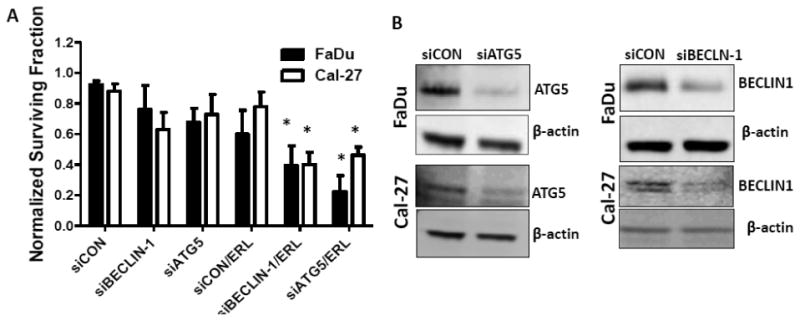
FaDu and Cal-27 cells were transfected with scrambled siRNA (siCON), or siRNA targeted to ATG5 (siATG5) or BECLIN-1 (siBEC1) for 24 hrs. The cells were then treated with DMSO or 5 μM erlotinib (ERL) for 48 hrs and analyzed by clonogenic assay (A) and analyzed for ATG5 and BECLIN-1 expression by western blot (B). β–actin used as loading control. Error bars represent standard error of the mean for n= 3 experiments, *: p<0.05 vs siCON.
NOX enzymes are involved in erlotinib induced autophagy
It has been previously reported that ROS can induce autophagy (Scherz-Shouval and Elazar 2011) and our lab has shown that erlotinib induced hydrogen peroxide (H2O2)-mediated oxidative stress via NOX4 (Orcutt et al., 2011). To confirm erlotinib induced H2O2 was responsible for autophagy, FaDu cells were pretreated with 100 U/mL pegylated catalase (CAT) for 1 hour prior to treatment with erlotinib and LC3B-II expression was measured. CAT suppressed the erlotinib-induced increase in LC3B-II levels suggesting the involvement of H2O2 in mediating autophagy in HNSCC cells (Figure 6A). To determine if NOX enzymes were involved in erlotinib-induced autophagy, FaDu and Cal-27 cells were treated with diphenylene iodonium (DPI), a flavin enzyme inhibitor and the protein expression of LC3B-II was analyzed. DPI decreased erlotinib-induced LC3B-II levels in both cell lines suggesting the role of NOX enzymes in erlotinib induced autophagy (Figure 6B-D). Out of all known NOX enzymes (NOX1-5, DUOX1, DUOX2), erlotinib increased NOX4 and DUOX2 mRNA compared to control cells (Figure 7A) but only increased NOX4 protein expression (Figure 7B). Taken together these results suggested that NOX enzymes (NOX4 in particular) may be involved in erlotinib-induced autophagy.
Figure 6. NADPH Oxidase enzymes (NOX) are involved in erlotinib-induced autophagy.
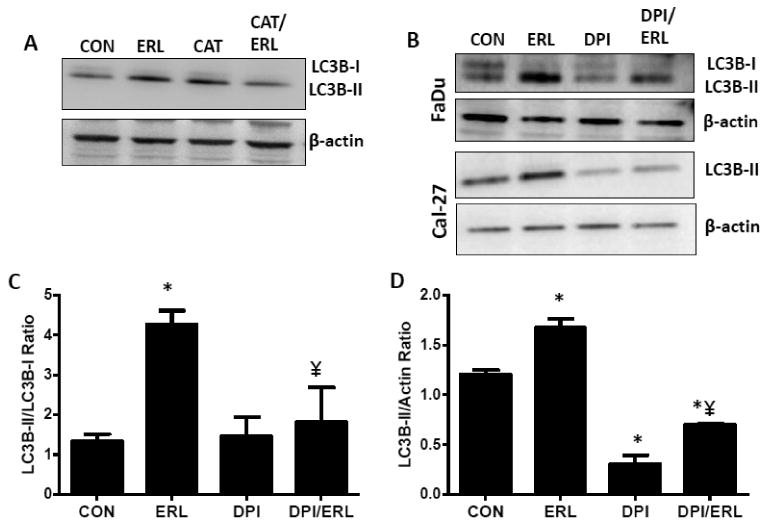
A) FaDu cells were treated with 5 μM erlotinib (ERL) and/or 100 U/mL catalase (CAT) for 48 hours and whole cell lysates analyzed for LC3B-II expression. Equal amounts of PEG was present in all treatment groups. B) FaDu and Cal-27 cells were treated with DMSO (CON), 5 μM erlotinib (ERL) and/or 0.05 μM DPI for 48 hrs. Whole cell lysates were analyzed by Western blot for LC3B-II expression. β –actin was used as a loading control. Bar graphs shown in (C) and (D) are quantifications (n=3 experiments) of FaDu and Cal-27 western blot bands in (B) respectively. *:p< 0.05 versus control; ¥:p<0.05 versus ERL.
Figure 7. NADPH oxidase 4 (NOX4) is induced by erlotinib in HNSCC cells.

FaDu and Cal-27 cells were treated with DMSO (CON) or 5 μM erlotinib (ERL) for 48 hours and mRNA expression of all NOX enzymes (NOX1-5, DUOX1,2) determined by RT-PCR (A). Data were normalized to DMSO control which was set at a value of 1. Error bars represents standard error of the mean of n=3 experiments. FaDu and Cal-27 cells were treated as mentioned above and protein levels of various NOX enzymes were analyzed by immunoblotting. β –actin was used as a loading control (B). FaDu cells were transfected with a NOX4-promoter-luciferase reporter (NOX4-luc) construct before treatment with DMSO (CON) or 5 μM erlotinib (ERL) for 48 hours and promoter activity measured by luciferase assay (C). *:p< 0.05 versus NOX4-luc+DMSO.
Erlotinib increases NOX4 expression both by promoter activation and 3′UTR stabilization mechanisms
To further examine the mechanism of induction of NOX4 by erlotinib, two luciferase expression plasmids containing the NOX4 promoter and 3′UTR of NOX4, were transfected separately into FaDu HNSCC cells and activity was measured by a luciferase assay after incubation with DMSO or erlotinib. FaDu cells were used to pursue these studies because of the greater induction of NOX4 and high transfection efficiency observed in this cell line compared to Cal-27 cells. Erlotinib caused a significant increase in NOX4 promoter (Figure 7C) and 3′UTR luciferase activity (Figure 8A) compared to control. The 3′ UTR of NOX4 contains a binding site for miR-25 which upon binding degrades the NOX4 mRNA (Fu et al., 2010). To investigate if the increase in 3′ UTR luciferase activity in the presence of erlotinib may be due to decreased endogenous miR-25 binding, a mutation in one of the miR-25 binding sites in the 3′ UTR was introduced. The mutated 3′UTR construct demonstrated increased luciferase activity of the control conditions by 20% compared to the wild-type (Figure 8B). Additionally, 3′UTR luciferase activity after erlotinib treatment was decreased in the mutated 3′UTR construct (Figure 8C). Furthermore, erlotinib decreased miR-25 expression in FaDu cells (Figure 8D). These results suggest that erlotinib mediated increases in NOX4 expression may result from both an increase in NOX4 promoter and increased mRNA stability, the latter via changes in endogenous miR-25 levels.
Figure 8. Erlotinib induces NOX4 promoter activity and mRNA stabilization.
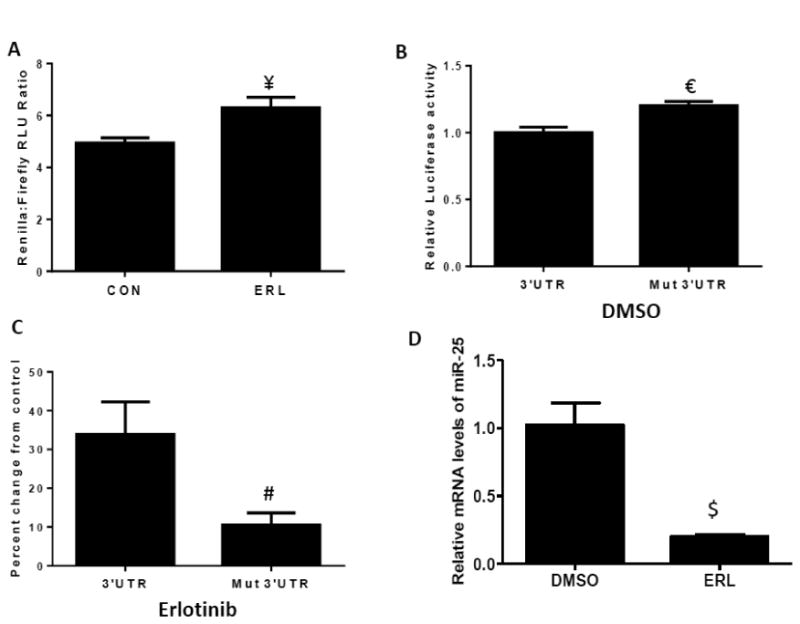
FaDu cells were transfected with NOX4 3′UTR and mutated NOX4 3′ UTR (A-C) luciferase constructs before treatment with DMSO (CON) or 5 μM erlotinib (ERL) for 48 hours. FaDu cells were treated with erlotinib (ERL) for 48 hours and mRNA levels of miR-25 analyzed by RT-PCR (D). Error bars represent the standard error of the mean (SEM) of n =3 experiments. ¥:p<0.05 vs CON; €:p<0.05 vs 3′UTR; #:p<0.05 vs 3′UTR+erlotinib; $:p<0.05 vs DMSO.
NOX4 mediates erlotinib induced autophagy
To determine if erlotinib induced autophagy via NOX4, NOX4 was knocked down with adenoviral siRNA (AdsiNOX4) in FaDu cells and levels of immunoreactive LC3B-II was analyzed by western blot. AdsiNOX4 successfully suppressed erlotinib-induced NOX4 mRNA and protein expression (Figure 9A) and additionally decreased LC3B-II expression compared to empty vector transfected cells (Figure 9B). To confirm that NOX4 activity was responsible for autophagy induction, NOX4 was overexpressed using an adenoviral wild-type NOX4 construct (AdN4WT). Overexpression of NOX4 showed that expression of NOX4 mRNA (Figure 10A), NOX4 protein (Figure 10A), LC3B-II (Figure 10B) as well as ROS production (Figure 10C) and cytotoxicity (Figure 10D) was increased compared to EMP-transfected cells. These changes were not observed with overexpression of a NOX4 dominant-negative construct (AdN4DN) although NOX4 mRNA was induced (Figure 10A-D). These results indicate that NOX4 contributed to erlotinib induced autophagy.
Figure 9. NOX4 knockdown suppresses erlotinib induced autophagy.
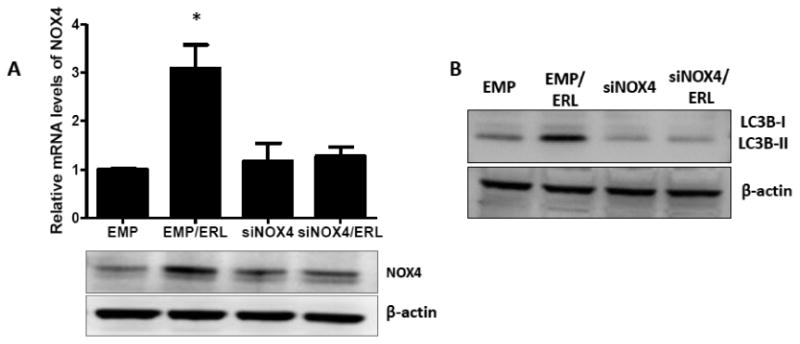
FaDu cells were transfected with an adenoviral scrambled siRNA control (EMP) or adenoviral siRNA targeted to NOX4 (siNOX4) before treatment with erlotinib (ERL) for 48 hours (A, B). NOX4 mRNA levels (A) were determined by RT-PCR, and NOX4 protein (A) and LC3B protein levels (B) were determined by western blotting. Data normalized to empty vector. β –actin was used as a loading control. Error bars represent the standard error of the mean (SEM) of n=3 experiments. *:p<0.05 vs EMP.
Figure 10. NOX4 overexpression induces autophagy.
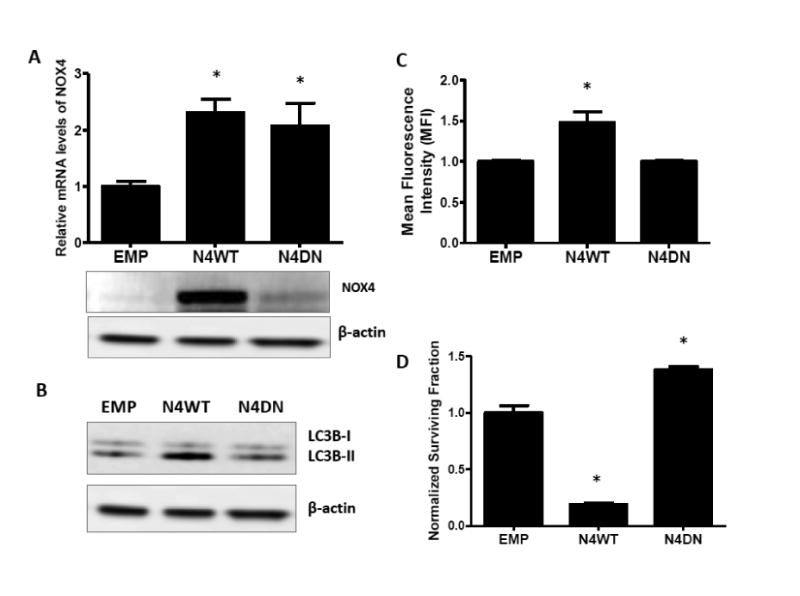
FaDu cells were transfected with an adenoviral empty vector (EMP) and adenoviral vectors expressing wildtype-NOX4 (N4WT) and dominant-negative NOX4 (N4DN) (A-D). NOX4 mRNA levels (A) were determined by RT-PCR, and NOX4 protein (A) and LC3B protein levels (B) were determined by western blotting. N4WT and N4DN-induced oxidative stress was measured by DCFH oxidation (C) and cytotoxicity measured by clonogenic assay (D). Data were normalized to EMP. β –actin was used as a loading control. Error bars represent the standard error of the mean (SEM) of n=3 experiments. *:p<0.05 vs EMP.
Discussion
These data identify NOX4-mediated autophagy as a mechanism by which HNSCC develop resistance to cytotoxic effects of erlotinib, and possibly other EGFR inhibitors. EGFR inhibitors are routinely used in chemotherapy regimens but development of drug tolerance is a common problem. In support of this we observed that the cytotoxicity of a clinically relevant dose of erlotinib decreased over time with no additional effect after 24 hours (Figure 1B). Induction of autophagy has been implicated in numerous studies to promote survival of drug resistant cancer cells following chemotherapy (Hu et al., 2012, O'Donovan et al., 2011). We have shown that erlotinib induces autophagy in our head and neck cancer cell model by immunoblot, immunofluorescence and electron microscopy (Figure 1-3) which supports earlier reports of the role of autophagy in the mechanism of action of erlotinib (Fung et al., 2012, Han et al., 2011).
It is controversial whether autophagy is primarily a pro-survival or pro-death phenomenon. Many studies have shown that autophagy promotes the survival of tumor cells under conditions of poor nutrient supply in case of rapidly growing tumors and also protects the cells from cytotoxic chemotherapeutic and radiation insults (Kondo et al., 2005). Paradoxically, many others have reported that an increase in autophagy enhances the cytotoxic effects of anticancer agents or radiation (Fung et al., 2012, Shin et al., 2012) and regress tumor volumes in small animal models (Fung et al., 2012). In our in vitro HNSCC cell model, we found that inhibiting autophagy by pharmacological or genetic manipulation sensitizes HNSCC cells to cytotoxic effects of erlotinib (Figure 4, 5) suggesting that it has a pro-survival role. However, more studies are needed in EGFR-positive tumor models to determine the consistency of these observations and to determine the safety and efficacy of the combination of erlotinib and CQ. In fact, a Phase 1 clinical trial utilizing chloroquine and erlotinib in lung cancer patients has demonstrated that this combination is safe and tolerated (Goldberg et al., 2012) suggesting that this combination should also be tested in HNSCC patients.
Previous studies from our lab have shown that erlotinib induced oxidative stress via NOX4 activation and NOX4-mediated oxidative stress was required to induce cytotoxicity of HNSCC cells (Orcutt et al., 2011). We now show that NOX4 may have both a cytotoxic and cytoprotective role in EGFR-based chemotherapy implying a dual role for NOX4 (Figure 11). In the present studies, the cytotoxicity of NOX4 was demonstrated by significantly increased clonogenic cell killing of NOX4WT-transfected cells compared to EMP and NOX4DN-transfected cells (Figure 10D). In fact overexpression of NOX4DN, which is functionally inactive (Figure 10C), significantly increased clonogenic survival compared to EMP (Figure 10D), which further supports the cytotoxic mechanism of NOX4. On the other hand, the cytoprotective role of NOX4 is demonstrated in these studies by its role in autophagy. We observed that overexpression of NOX4 increased autophagy (Figure 10B) and that knockdown of NOX4 was able to suppress erlotinib-induced autophagy (Figure 9B).
Figure 11. Hypothetical role of NOX4 in the mechanism of action of Erlotinib.
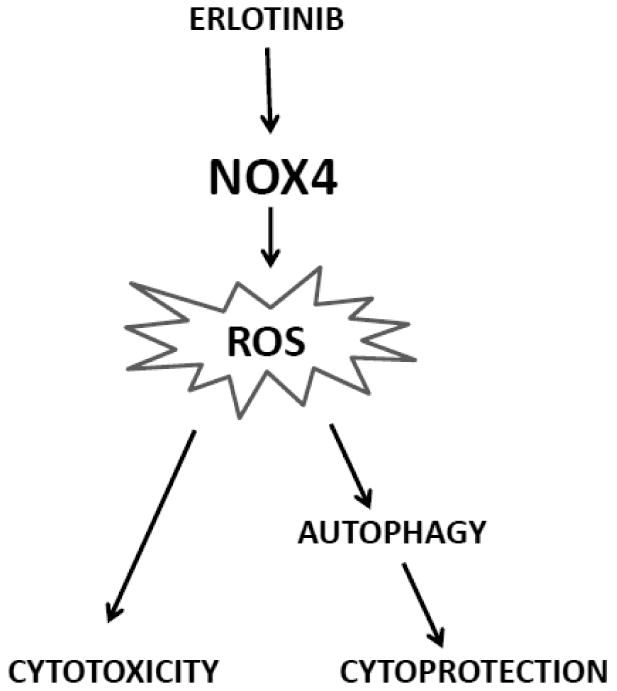
Erlotinib induces the expression and activity of NOX4 resulting in ROS production. This ROS production results in cytotoxicity in HNSCC cells but also activates autophagy as a protective mechanism.
Although NOX4 was the focus of these studies, we did observe that both NOX4 and DUOX2 mRNA was induced with erlotinib treatment in both cell lines (Figure 7). DUOX2 along with DUOX1, both produce H2O2 while the other NOX enzymes are known to produce superoxide (O2.-) (Donko et al., 2010, Ohye and Sugawara 2010). Numerous studies have shown that NOX4 may produce both H2O2 and O2.- in various cell models (Ago et al., 2010, Kuroda et al., 2010, Serrander et al., 2007) however in our previous work, we have shown that NOX4 produced H2O2 and not O2.- in FaDu and Cal-27 cells (Orcutt et al., 2011) suggesting that NOX4-mediated H2O2 may also be responsible for autophagy induction in the present studies. The role of H2O2 was also confirmed by showing that catalase was able to suppress erlotinib-induced autophagy (Figure 6A). Although DUOX2 mRNA expression was induced by erlotinib treatment, we could not detect any increase in DUOX2 protein expression (Figure 7B). It is possible that DUOX2 protein may be induced and rapidly degraded by proteosomal machinery which may explain why we do not see an increase in DUOX2 protein expression 48 hours after erlotinib treatment. However, we were unable to see an increase in protein expression as early as 1 hour after erlotinib treatment (data not shown). Nevertheless, DUOX2 may play a role in the mechanism of action of erlotinib and further studies exploring this are ongoing.
ROS is a known inducer of autophagy (Scherz-Shouval and Elazar 2011) and the results of this work suggest that NOX4-generated H2O2 induces autophagy upon erlotinib treatment. However the mechanism by which autophagy is induced is not clear. Inhibition of the mTOR pathway is known to activate autophagy but inhibition of this pathway does not appear to be the mechanism involved in our cell model (data not shown). It is possible that erlotinib-induced ROS may deactivate Atg4, which is a redox sensitive cysteine protease that delipidates LC3B-II and removes it from autophagosomal membranes (Scherz-Shouval et al., 2007). Additionally, erlotinib may induce endoplasmic reticulum (ER) stress, which may be responsible for the activation of downstream autophagy. We plan to further investigate these mechanisms in future studies.
The mechanism by which erlotinib induces NOX4 is not known. However, we have now shown that erlotinib increases NOX4 by a mechanism involving increased promoter activity and 3′UTR stabilization in FaDu cells (Figure 7C, 8A). The 3′ UTR of NOX4 contains binding sites for miR-25, which has been shown to bind to this region and degrade NOX4 mRNA (Fu et al., 2010). We have shown that the increase in 3′UTR stabilization is due to decrease in miR-25 levels on treatment with erlotinib (Figure 8D). Additionally, transfection of cells with a mutation in one of the miR-25 binding sites, suppressed the increase in 3′UTR activity induced by erlotinib, suggesting that erlotinib decreases miR-25 binding to the 3′UTR region of NOX4 (Figure 8B,C).
In summary these studies describes a possible dual role of NOX4 in EGFR-based chemotherapy. NOX4 induces oxidative stress that is critical for drug mediated cytotoxicity. Additionally, it also induces autophagy as a protective mechanism perhaps in cells that survive erlotinib treatment (Figure 11). Therefore, the induction of autophagy may be a possible mechanism of drug tolerance and supports the use of autophagy inhibitors in conjunction with EGFR inhibitors to increase the efficacy of HNSCC treatment.
Highlights.
Erlotinib increased LC3B-II and autophagosome formation in HNSCC cells.
Inhibition of autophagy sensitized HNSCC cells to erlotinib.
Erlotinib increased NOX4 promoter and 3′UTR luciferase activity.
Manipulating NOX4 decreases or increases autophagy.
Acknowledgments
The authors would like to thank Dr. Allan Shepard for the NOX4 wild-type and dominant-negative adenoviral vectors. The authors also thank Dr. Piedad Gomez Contreraz for help with the NOX4 promoter luciferase assays, Sean Martin for help with running DCFH assays and Janice Wang and Michael Knudson for helpful discussions about detection of autophagy markers. This work was supported by the Department of Pathology and grants NIH K01CA134941 and IRG-77-004-34 from the American Cancer Society, administered through the Holden Comprehensive Cancer Center at the University of Iowa.
Abbreviations
- EGFR
Epidermal Growth Factor Receptor
- ERL
Erlotinib
- LC3
Light chain 3
- NOX4
NADPH oxidase 4
- HNSCC
Head and neck squamous cell carcinoma
- ROS
Reactive Oxygen Species
- DUOX
Dual Oxidase
- WT
Wildtype
- DN
Dominant negative
- UTR
Untranslated Region
- TEM
Transmission Electron Microscopy
- GFP
Green Fluorescent Protein
- CQ
Chloroquine
- DPI
Diphenylene Iodonium
- TKI
Tyrosine Kinase Inhibitor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RD, Haskell RE, Xia H, Roessler BJ, Davidson BL. A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther. 2000;7:1034–1038. doi: 10.1038/sj.gt.3301197. [DOI] [PubMed] [Google Scholar]
- Bialik S, Kimchi A. Autophagy and tumor suppression: recent advances in understanding the link between autophagic cell death pathways and tumor development. Adv Exp Med Biol. 2008;615:177–200. doi: 10.1007/978-1-4020-6554-5_9. [DOI] [PubMed] [Google Scholar]
- Donko A, Ruisanchez E, Orient A, Enyedi B, Kapui R, Peterfi Z, de Deken X, Benyo Z, Geiszt M. Urothelial cells produce hydrogen peroxide through the activation of Duox1. Free Radic Biol Med. 2010;49:2040–2048. doi: 10.1016/j.freeradbiomed.2010.09.027. [DOI] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009;14:376–391. doi: 10.1007/s10495-008-0307-5. [DOI] [PubMed] [Google Scholar]
- Fu Y, Zhang Y, Wang Z, Wang L, Wei X, Zhang B, Wen Z, Fang H, Pang Q, Yi F. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am J Nephrol. 2010;32:581–589. doi: 10.1159/000322105. [DOI] [PubMed] [Google Scholar]
- Fung C, Chen X, Grandis JR, Duvvuri U. EGFR tyrosine kinase inhibition induces autophagy in cancer cells. Cancer Biol Ther. 2012;13 doi: 10.4161/cbt.22002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg SB, Supko JG, Neal JW, Muzikansky A, Digumarthy S, Fidias P, Temel JS, Heist RS, Shaw AT, McCarthy PO, Lynch TJ, Sharma S, Settleman JE, Sequist LV. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J Thorac Oncol. 2012;7:1602–1608. doi: 10.1097/JTO.0b013e318262de4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–3584. [PubMed] [Google Scholar]
- Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, Ge W, Feng L, Lin X, Wang X, Wang X, Jin H. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One. 2011;6:e18691. doi: 10.1371/journal.pone.0018691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari PM, Wheeler DL, Grandis JR. Molecular target approaches in head and neck cancer: epidermal growth factor receptor and beyond. Semin Radiat Oncol. 2009;19:63–68. doi: 10.1016/j.semradonc.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YL, Jahangiri A, Delay M, Aghi MK. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012;72:4294–4299. doi: 10.1158/0008-5472.CAN-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn J. Targeting the epidermal growth factor receptor for cancer therapy. J Clin Oncol. 2002;20:1S–13S. [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–385. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. doi: 10.1007/978-3-642-00302-8_3. [DOI] [PubMed] [Google Scholar]
- Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl 2):1535–1541. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- O'Donovan TR, O'Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7:509–524. doi: 10.4161/auto.7.6.15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohye H, Sugawara M. Dual oxidase, hydrogen peroxide and thyroid diseases. Exp Biol Med (Maywood) 2010;235:424–433. doi: 10.1258/ebm.2009.009241. [DOI] [PubMed] [Google Scholar]
- Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orcutt KP, Parsons AD, Sibenaller ZA, Scarbrough PM, Zhu Y, Sobhakumari A, Wilke WW, Kalen AL, Goswami P, Miller FJ, Jr, Spitz DR, Simons AL. Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res. 2011;71:3932–3940. doi: 10.1158/0008-5472.CAN-10-3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JR, Burmeister MA, Tian X, Zhou Y, Guruju MR, Stupinski JA, Sharma RV, Davisson RL. Genetic silencing of Nox2 and Nox4 reveals differential roles of these NADPH oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II. Hypertension. 2009;54:1106–1114. doi: 10.1161/HYPERTENSIONAHA.109.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prata C, Maraldi T, Fiorentini D, Zambonin L, Hakim G, Landi L. Nox-generated ROS modulate glucose uptake in a leukaemic cell line. Free Radic Res. 2008;42:405–414. doi: 10.1080/10715760802047344. [DOI] [PubMed] [Google Scholar]
- Rexer BN, Engelman JA, Arteaga CL. Overcoming resistance to tyrosine kinase inhibitors: lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle. 2009;8:18–22. doi: 10.4161/cc.8.1.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha-Lima CM, Soares HP, Raez LE, Singal R. EGFR targeting of solid tumors. Cancer Control. 2007;14:295–304. doi: 10.1177/107327480701400313. [DOI] [PubMed] [Google Scholar]
- Sanmartin-Suarez C, Soto-Otero R, Sanchez-Sellero I, Mendez-Alvarez E. Antioxidant properties of dimethyl sulfoxide and its viability as a solvent in the evaluation of neuroprotective antioxidants. J Pharmacol Toxicol Methods. 2011;63:209–215. doi: 10.1016/j.vascn.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Lim HT, Minai-Tehrani A, Noh MS, Kim JE, Kim JH, Jiang HL, Arote R, Kim DY, Chae C, Lee KH, Kim MS, Cho MH. Aerosol delivery of beclin1 enhanced the anti-tumor effect of radiation in the lungs of K-rasLA1 mice. J Radiat Res. 2012;53:506–515. doi: 10.1093/jrr/rrs005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- Spitz DR, Malcolm RR, Roberts RJ. Cytotoxicity and metabolism of 4-hydroxy-2-nonenal and 2-nonenal in H2O2-resistant cell lines. Do aldehydic by-products of lipid peroxidation contribute to oxidative stress? Biochem J. 1990;267:453–459. doi: 10.1042/bj2670453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201–2214. doi: 10.1089/ars.2010.3482. [DOI] [PubMed] [Google Scholar]