

Abstract
Purpose of review
This review provides a concise summary of significant research progress of SIRT1 deacetylase in leukemia in the past year. SIRT1 is a multi-functional protein and recent studies demonstrate that SIRT1 plays a crucial role in myeloid leukemogenesis and drug resistance.
Recent findings
SIRT1 expression is typically low in normal adult hematopoietic stem/progenitor cells, but is increased in the leukemic stem/progenitor cells of chronic myeloid leukemia (CML). SIRT1 activation is mediated by both BCR-ABL tyrosine kinase-dependent and independent manners. SIRT1 activation promotes resistance of CML stem cells to tyrosine kinase inhibitors and acquisition of BCR-ABL mutations for acquired resistance.
Summary
Based on current findings, SIRT1 inhibition in combination with BCR-ABL tyrosine kinase inhibitors can be explored as a novel approach to eradicate leukemic stem cells and residual disease in chronic phase CML. SIRT1 inhibition may also help prevent acquired resistance through genetic mutations of advanced phases of CML and extend remission.
Keywords: SIRT1, chronic myeloid leukemia, BCR-ABL, leukemic stem cells, acquired resistance
Introduction
SIRT1 is a member of mammalian sirtuins that are homologs of yeast silent information regulator 2 (Sir2) encoding a NAD-dependent histone deacetylase.[1,2] SIRT1 shares the highest homology with yeast Sir2 and is the most extensively studied sirtuins. SIRT1 preferentially deacetylates histone H4K16 and H1K26,[2,3] and is involved in gene silencing and heterochromatin formation.[4] SIRT1 regulates a wide variety of biological processes and cellular functions through deacetylating a growing list of non-histone proteins, including transcriptional factors p53, FOXO1, FOXO3a, NF-κB, c-MYC, N-MYC and E2F1 for regulating cell cycle progression and survival; DNA repair factors Ku70, RAD51, NBS1, APE1, XPA/C and WRN; and nuclear receptor, circadian clock and related factors LXR, FXR, ERα, AR, PPARγ, PGC1α, CLOCK and PER2 for regulating metabolism, as detailed in previous reviews.[5,6] This review will present an overview of SIRT1 functions in normal hematopoiesis followed by SIRT1 roles in leukemogenesis, in particular, chronic myeloid leukemia (CML), a hematopoietic stem cell malignancy caused by oncogenic fusion gene BCR-ABL.[7]
Functions of SIRT1 in normal hematopoiesis
In the past year, Yuan et al[8**] and Li et al[9**] demonstrated that SIRT1 gene expression is low in CD34+ normal human hematopoietic stem/progenitor cells as compared to CML counterparts. Similarly, SIRT1 expression is generally low in mouse bone marrow hematopoietic progenitor cells, or whole mononuclear cells.[8**] This poor expression of SIRT1 in normal hematopoietic stem/progenitor may explain why SIRT1 homozygous knockout with deletion of exons 5–7 in BALB/c mice [8**] or deletion of exon 4 in C57BL/6 mice [10*] does not significantly change adult mouse hematopoiesis except for anemia in older knockout mice that is likely due to reduced production of erythropoietin. These studies are in line with a previous study showing no significant in vivo impact of SIRT1 homozygous knockout on hematopoiesis in young C57BL/6 mice.[11]
However, SIRT1 knockout notably affects hematopoiesis studied in vitro. The impact of SIRT1 loss on mouse embryonic hematopoiesis has been examined using mouse embryonic stem cells (mESCs).[12] Mouse SIRT1 gene expression is higher in mESCs than in several somatic tissues[13]. High levels of SIRT1 help maintain self-renewal of mESCs by deacetylating p53 and preventing its translocation to the nucleus. p53 is a transcriptional repressor of Nanog, and sequestration of p53 in the cytoplasm leads to activation of Nanog expression for stem cell self-renewal.[14] mESCs are typically maintained under normoxia in a medium containing 2-mecaptoethanol (2-ME), an anti-oxidant that prevents buildup of endogenous reactive oxygen species (ROS). The maintenance of self-renewal by SIRT1 comes at the price of increasing exposure of mESCs to ROS as cytoplasm-localized p53 fails to activate several anti-oxidant genes, which is manifested by the withdrawal of 2-ME.[14] Culturing mESCs under hypoxic conditions prevents ROS buildup even in the absence of 2-ME, and thus increases survival of mESCs. Using in vitro differentiation of mESCs (derived from R1 ESC line of 129 strain) to hematpoietic cells, Ou et al [12] showed that SIRT1 homozygous knockout delays in vitro hematopoiesis phenotypically and molecularly. Ou et al [12] further showed that SIRT1−/−, but not SIRT1+/−, yolk sac forms less primitive erythroid colonies under normoxia culture than SIRT1+/+ yolk sac. Recently, Matsui et al [15] showed that when cultured in vitro, SIRT1−/− fetal liver cells differentiate more apparently and produce less c-Kit+Sca1+Lin− progenitors than SIRT1+/+ fetal liver cells; but there is no difference before in vitro culture.
In addition, Ou et al [12] also showed that SIRT1 knockout mouse bone marrow cells harvested from a mixed 129/FVB/Black Swiss strain background form fewer hematopoietic progenitor CFU colonies than wild type cells, the effect more pronounced for cells from older mice and cultured under lower oxygen as SIRT1 knockout cells do not respond to enhanced growth condition under lowered oxygen. Surprisingly, Ou et al found that SIRT1 heterozygous knockout is as nearly effective as homozygous knockout on affecting CFU potential; however, no in vivo mouse hematological data has been shown for comparison. The precise reason for this finding is not clear; but, it is known that the SIRT1 knockout in the 129/FVB/Black strain background [16] has the most severe embryonic lethality phenotype than in other strains reported so far.
Activation of SIRT1 gene expression in leukemia
SIRT1 expression is subjected to multiple layers of regulation from transcription, translation to protein functions.[17] In hematopoietic cells, SIRT1 gene expression is robustly activated by BCR-ABL and KRAS oncogenes.[8**] Yuan et al showed that SIRT1 transcription is upregulated by BCR-ABL in CML cells in part through signal transducer and activator of transcription 5 (STAT5) that binds on two sites of the SIRT1 promoter.[8**] STAT5 is a key BCR-ABL tyrosine kinase-dependent signaling molecule in CML, and is also activated in certain forms of acute myeloid leukemia (AML).[18] It is possible that SIRT1 can be similarly activated by other oncogenic events in certain AML through STAT5. In fact, it is reported that SIRT1 expression is increased in certain AML patients.[19] Interestingly, however, even with nearly complete inhibition of tyrosine kinase activity, Yuan et al found[8] that SIRT1 is only down by about half in CML cells,[8**] suggesting likely existence of BCR-ABL kinase-independent activation of SIRT1. The same was seen with SIRT1 inhibition in primary CML progenitor cells.[9**] However, mechanisms of BCR-ABL kinase-independent activation of SIRT1 remain to be identified. As shown in Fig. 1, SIRT1 activation by BCR-ABL affects multiple aspects of CML molecular pathobiology as detailed below.
Fig. 1. A model for roles of SIRT1 in CML molecular pathobiology.
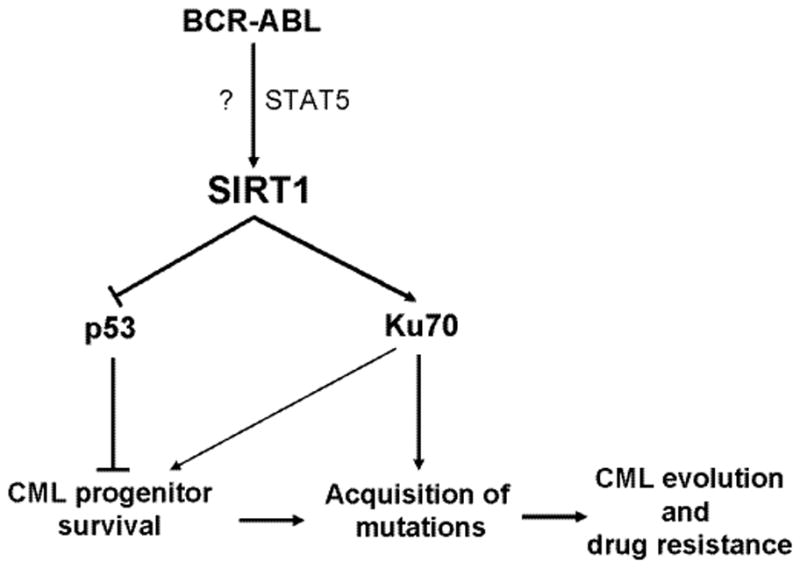
SIRT1 is activated by BCR-ABL transformation in hematopoietic stem cells, in both kinase-dependent (through STAT5) and independent (indicated by ?) manners. SIRT1 activation suppresses functions of tumor suppressor p53 and promotes survival of CML stem cells. Meanwhile, SIRT1 activates Ku70 functions to promote error-prone DNA damage repair, leading to acquisition of BCR-ABL mutations for CML evolution and drug resistance.
Activation of SIRT1 by STAT5 may also bridge SIRT1 functions to non-oncogenic physiological regulation. STAT5 is a cytokine signaling molecule for stem/progenitor cells, epithelial cells, and the immune system.[20] It is noted that incubation of mouse bone marrow cells with cytokine and growth factor cocktails elevates SIRT1 expression in culture.[8**] We speculate that such regulation may have important roles in certain physiological conditions, but constitutive SIRT1 activation by oncogenic transformation disrupts signaling balance and renders substantial cell proliferation and survival advantage leading to leukemogenesis and cancer resistance described next.
SIRT1 promotes BCR-ABL mediated leukemogenesis
Roles of SIRT1 in cancer have been controversial. Previously, it was shown that SIRT1 promotes genome stability and loss of SIRT1 predisposes mice to tumorigenesis in the presence of heterozygous deletion of p53.[16] Conditional SIRT1 over-expression reduces intestinal tumor formation in APC+/min mice,[21] and thymic lymphoma in p53+/− mice.[22] Herranz et al showed that transgenic mice with SIRT1 over-expression under its own promoter have reduced spontaneous carcinomas and sarcomas, and as well as reduced incidence of carcinogen-induced liver cancer.[23] These studies support a role of SIRT1 in tumor suppression.
In contrast to the above mouse studies, numerous reports have shown upregulation of SIRT1 gene expression in both human and mouse cancers, and SIRT1 inhibition suppresses tumor growth as summarized in a recent review.[17] Using a bone marrow transduction/transplantation model of CML, Yuan et al[8**] demonstrated that SIRT1 gene knockout significantly inhibits development of CML, providing the first genetic evidence that SIRT1 dysregulation by the oncogenic event is not merely a consequence of tumorigenesis, but is crucial for leukemia progression. Because BCR-ABL transduction of SIRT1−/− bone marrow cells was conducted ex vivo, the effect of SIRT1 loss on inhibiting CML development is considered cell-autonomous.
By examining SIRT1 over-expression in transgenic mice (SIRT1tg) with PTEN+/− background, Herranz et al[24] recently found that SIRT1 over-expression increases incidence of thyroid carcinomas and their lung metastasis, suggesting that increased SIRT1 gene expression plays a role in promoting tumor progression. In addition, Herranz et al[24] showed that PTEN+/− SIRT1tg mice also develop prostate carcinomas in situ, whereas PTEN+/− or SIRT1tg cohort mice did not,[24] indicating that SIRT1 over-expression may also have a role in promoting tumor initiation. Together, these studies provide strong genetic evidence that SIRT1 activation can not only facilitate BCR-ABL mediated leukemogenesis but also development of solid tumors.
SIRT1 promotes leukemia stem cells survival and resistance to BCR-ABL inhibitors
Several leukemias including CML are initiated and propagated by small populations of leukemia stem cells (LSC). LSC can resist elimination by standard therapies that target more mature cells, and persist after treatment as potential sources of relapse.[25] Although BCR-ABL tyrosine kinase inhibitors imatinib mesylate is effective in inducing clinical remissions and prolonging survival in CML patients, primitive LSC are retained in patients achieving remission with TKI treatment.[26,27] This is related to the resistance of primitive, quiescent CML LSC to apoptosis following TKI treatment despite effective inhibition of BCR-ABL kinase activity. Disease recurrence is usually seen following cessation of drug treatment, even in patients with undetectable BCR-ABL expression by q-PCR. CML patients currently need to take TKI treatment indefinitely, with risks of toxicity, lack of compliance, drug resistance, relapse and associated expense.
Li et al[9**] have shown that SIRT1, a stress-related NAD+-dependent deacetylase, is overexpressed in CML compared to normal CD34+CD38− stem cells. SIRT1 inhibition, using RNAi or the pharmacological inhibitor Tenovin-6,[28] selectively decreased survival and proliferation of CP and BC CML LSC and sensitizes them to TKI treatment. In vivo treatment with Tenovin-6 reduced CML LSC numbers, reduced LSC engraftment in secondary recipients, and prolonged survival after completion of treatment. These observations support an important role for SIRT1 in maintenance of CML LSC and their resistance to TKI treatment and suggest that SIRT1 inhibition may represent a potential strategy to enhance elimination of LSC in CML patients treated with TKI.
The p53 gene is activated in response to genotoxic and oncogenic stress to induce cell cycle arrest or apoptosis. Loss of p53 function is a common feature of human malignancies and contributes to early stages of cancer development.[29] Recent studies indicate that p53 inactivation is also required to maintain proliferation and survival of established malignancies, in the setting of continued oncogenic stress.[9**] SIRT1 knockdown using RNAi or the small molecule inhibitor Tenovin-6 increased p53 acetylation and nuclear localization, and enhanced p53 target gene expression in CML progenitors. Knockdown of p53 or expression of an acetylation-defective p53 prevented SIRT1-mediated inhibition of CML progenitors.[9**] These results suggest that deacetylation of p53 by SIRT1 inhibits p53 activity and contributes to increased proliferation and preservation of CML LSC. These studies raise the possibility that other strategies to activate p53 could potentially be of benefit in targeting CML LSC. Since SIRT1 affects multiple targets, other potential mechanisms such as activation of Foxo transcription factors or alteration in DNA repair mechanisms must also be considered.
SIRT1 promotes CML acquired resistance through mutations
Another important progress last year was the discovery that SIRT1 promotes acquisition of BCR-ABL mutations in CML cells for resistance to tyrosine kinase inhibitors.[30**] Although imatinib remarkably increases 5-year survival of chronic phase CML patients,[31] the drug has only limited effect on patients in accelerated and blast crisis phases of the disease who frequently relapse with acquisition of BCR-ABL kinase domain mutations.[32,33] These mutations alter BCR-ABL conformation and prevent imatinib from access to its ATP binding pocket.[34] Acquisition of genetic mutations is a major mechanism underlying cancer acquired resistance with different molecular targets in a variety of cancers including CML, lung cancer, colon cancer, ovarian and breast cancer, and gastrointestinal cancer.[35] However, precise molecular mechanisms how resistant mutations are actually acquired during cancer therapy are largely unknown.
Recently, Yuan et al[36] have developed a novel culture model of CML acquired resistance based on a blast crisis CML cell line KCL-22. In this model, following initial apoptotic response, the cells rapidly acquire T315I BCR-ABL kinase domain mutation upon treatment with therapeutic concentrations of imatinib, recapitulating features of clinical BCR-ABL mutation acquisition. Yuan et al showed that most, if not all, BCR-ABL mutations are acquired de novo in this model. Using this model, Wang et al[30**] showed that SIRT1 inhibition by gene knockdown or small molecules blocks acquisition of BCR-ABL mutations in CML cells and prevents CML cell relapse from tyrosine kinase inhibitors. SIRT1 inhibition also effectively suppresses de novo mutation acquisition of hypoxanthine phosphoribosyl transferase (HPRT) gene in CML and prostate cancer cells upon treatment of chemotherapeutic agent camptothecin. Wang et al[30**] showed that suppression of mutation acquisition by SIRT1 inhibition does not require increased cell killing, and that SIRT1 promotes mutation acquisition in association with its ability to enhance error-prone DNA damage repair, particularly non-homologous end joining (NHEJ), through deacetylating Ku70.
The findings by Wang et al are surprising given that SIRT1 maintains genome integrity [16,22] and SIRT1 inhibition is expected to increase genetic instability and mutagenesis. However, it is important to note that although genetic instability is shown previously in SIRT1−/− embryonic stem cells,[16,22] impaired DNA damage response is similarly observed in SIRT1−/− mouse embryonic fibroblasts[16] and in SIRT1 knockdown CML cells.[30**] SIRT1 knockdown reduces the efficiency of NHEJ and homologous recombination repair in both CML [30**] and osteosarcoma cells.[22] Therefore, a consensus role of SIRT1 has emerged for genome maintenance in both normal and cancer cells. But, there may be an important distinction of the consequence of SIRT1-regulated DNA repair in normal versus cancer cells. In normal cells, DNA repair is typically a high fidelity process, and SIRT1 promotes repair and reduce genetic lesions, leading to genome stability. Contradictorily, in cancer cells, DNA repair fidelity is compromised,[37,38] and increased repair by SIRT1 may help cancer cell survival by avoiding catastrophic genomic events such as deletion of key oncogenes, but simultaneously allow accumulation of non-fatal genetic lesions and point mutations. By doing so, increased DNA repair mediated by SIRT1 may provide an incubation bed for accelerating cancer evolution and allow rapid acquisition of genetic mutations for drug resistance when cancer cells are under therapeutic stress.[17,39] In this regard, it is important to note a recent study by Chakraborty et al[40*] showing that CML stem and progenitor cells harbor chromosomal instability mediated by increased NHEJ repair. As SIRT1 expression increases further from chronic CML to advanced phases of the disease,[8**] SIRT1-regulated error prone DNA repair may underlie CML evolution to higher malignant state. More studies are needed to further elucidate precise molecular mechanisms of SIRT1 in acquisition of BCR-ABL mutations for CML drug resistance and evolution. Such studies may shed novel insight on cancer acquired resistance in general.
Conclusions
Several studies in the past year have revealed crucial roles of SIRT1 dysregulation in tumorigenesis, in particular, chronic myeloid leukemia in which SIRT1 is activated by BCR-ABL. SIRT1 activation promotes BCR-ABL-mediated leukemogenesis and CML stem cells resistance to imatinib, and facilitates acquisition of BCR-ABL mutations for CML acquired resistance. SIRT1 is a promising novel target for eradicating CML leukemic stem cells and residual disease with tyrosine kinase inhibitor treatment. Inhibition of SIRT1 may also prevent acquisition of BCR-ABL mutations in advanced phases of CML to inhibit acquired resistance and extend remission.
Bullet Summary.
BCR-ABL activates SIRT1 through kinase-dependent STAT5 signaling and kinase-independent manner.
SIRT1 knockout suppresses BCR-ABL mediated leukemogenesis.
SIRT1 promotes resistance of CML leukemic stem cells to tyrosine kinase inhibitors.
SIRT1 promotes acquisition of BCR-ABL mutations for CML acquired resistance.
Acknowledgments
The authors would like to acknowledge the research support from the National Cancer Institute of the National Institutes of Health under award number R01 CA143421, and the State of California Tobacco Related Disease Research Program (TRDRP) award 20XT-0121 to W.Y.C.; and by NIH grants R01 CA95684, a Samuel Waxman Cancer Research Foundation research grant, and a Translational Research grant from the Leukemia and Lymphoma Society to R.B. The contents are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest Statement:
The authors declare no conflict of interest with respect to the authorship and/or publication of this article.
References
- 1.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 2.Imai S, Armstrong CM, Kaeberlein M, et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 3.Vaquero A, Scher M, Lee D, et al. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 4.Vaquero A. The conserved role of sirtuins in chromatin regulation. Int J Dev Biol. 2009;53:303–322. doi: 10.1387/ijdb.082675av. [DOI] [PubMed] [Google Scholar]
- 5.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nature Reviews Molecular Cell Biology. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489–5504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- 7.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–453. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- **8.Yuan H, Wang Z, Li L, et al. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 2012;119:1904–1914. doi: 10.1182/blood-2011-06-361691. This is the first paper providing genetic evidence that loss of SIRT1 inhibits BCR-ABL mediated leukemogenesis, and also showing BCR-ABL activation of SIRT1 in part through STAT5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **9.Li L, Wang L, Wang Z, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266–281. doi: 10.1016/j.ccr.2011.12.020. This is the first paper showing that SIRT1 is crucial for resistance of CML leukemic stem cells to imatinib by deacetylating and inactivating p53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *10.Leko V, Varnum-Finney B, Li H, et al. SIRT1 is dispensable for function of hematopoietic stem cells in adult mice. Blood. 2012;119:1856–1860. doi: 10.1182/blood-2011-09-377077. This paper provides detailed analysis of hematological phenotypes of adult SIRT1 knockout mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narala SR, Allsopp RC, Wells TB, et al. SIRT1 acts as a nutrient-sensitive growth suppressor and its loss is associated with increased AMPK and telomerase activity. Mol Biol Cell. 2008;19:1210–1219. doi: 10.1091/mbc.E07-09-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ou X, Chae HD, Wang RH, et al. SIRT1 deficiency compromises mouse embryonic stem cell hematopoietic differentiation, and embryonic and adult hematopoiesis in the mouse. Blood. 2011;117:440–450. doi: 10.1182/blood-2010-03-273011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McBurney MW, Yang X, Jardine K, et al. The absence of SIR2alpha protein has no effect on global gene silencing in mouse embryonic stem cells. Mol Cancer Res. 2003;1:402–409. [PubMed] [Google Scholar]
- 14.Han MK, Song EK, Guo Y, et al. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsui K, Ezoe S, Oritani K, et al. NAD-dependent histone deacetylase, SIRT1, plays essential roles in the maintenance of hematopoietic stem cells. Biochem Biophys Res Commun. 2012;418:811–817. doi: 10.1016/j.bbrc.2012.01.109. [DOI] [PubMed] [Google Scholar]
- 16.Wang RH, Sengupta K, Li C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Chen WY. Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes & Cancer. 2013 doi: 10.1177/1947601912473826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Etten RA. Aberrant cytokine signaling in leukemia. Oncogene. 2007;26:6738–6749. doi: 10.1038/sj.onc.1210758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradbury CA, Khanim FL, Hayden R, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 20.Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Firestein R, Blander G, Michan S, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oberdoerffer P, Michan S, McVay M, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herranz D, Munoz-Martin M, Canamero M, et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:3. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herranz D, Maraver A, Canamero M, et al. SIRT1 promotes thyroid carcinogenesis driven by PTEN deficiency. Oncogene. 2012 doi: 10.1038/onc.2012.407. [DOI] [PubMed] [Google Scholar]
- 25.O’Hare T, Zabriskie MS, Eiring AM, et al. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12:513–526. doi: 10.1038/nrc3317. [DOI] [PubMed] [Google Scholar]
- 26.Chu S, McDonald T, Lin A, et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood. 2011;118:5565–5572. doi: 10.1182/blood-2010-12-327437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konig H, Holtz M, Modi H, et al. Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia. 2008;22:748–755. doi: 10.1038/sj.leu.2405086. [DOI] [PubMed] [Google Scholar]
- 28.Lain S, Hollick JJ, Campbell J, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheok CF, Verma CS, Baselga J, et al. Translating p53 into the clinic. Nat Rev Clin Oncol. 2011;8:25–37. doi: 10.1038/nrclinonc.2010.174. [DOI] [PubMed] [Google Scholar]
- **30.Wang Z, Yuan H, Roth M, et al. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene. 2013;32:589–598. doi: 10.1038/onc.2012.83. This is the first paper showing that SIRT1 promotes acquisition of BCR-ABL mutations for CML acquired resistance to tyrosine kinase inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 32.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 33.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 34.Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 35.Baselga J. Targeting tyrosine kinases in cancer: the second wave. Science. 2006;312:1175–1178. doi: 10.1126/science.1125951. [DOI] [PubMed] [Google Scholar]
- 36.Yuan H, Wang Z, Gao C, et al. BCR-ABL gene expression is required for its mutations in a novel KCL-22 cell culture model for acquired resistance of chronic myelogenous leukemia. J Biol Chem. 2010;285:5085–5096. doi: 10.1074/jbc.M109.039206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nowicki MO, Falinski R, Koptyra M, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 38.Slupianek A, Nowicki MO, Koptyra M, et al. BCR/ABL modifies the kinetics and fidelity of DNA double-strand breaks repair in hematopoietic cells. DNA Repair (Amst) 2006;5:243–250. doi: 10.1016/j.dnarep.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen W. Accelerating cancer evolution: a dark side of SIRT1 in genome maintenance. Oncotarget. 2012;3:363–364. doi: 10.18632/oncotarget.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *40.Chakraborty S, Stark JM, Sun CL, et al. Chronic myelogenous leukemia stem and progenitor cells demonstrate chromosomal instability related to repeated breakage-fusion-bridge cycles mediated by increased nonhomologous end joining. Blood. 2012;119:6187–6197. doi: 10.1182/blood-2011-05-352252. This paper provides extensive studies of chromosomal instability of chronic CML and shows a central role of NHEJ in mediating such instability. [DOI] [PMC free article] [PubMed] [Google Scholar]