

Abstract
In Gram-positive bacteria, T-box riboswitches regulate expression of aminoacyl-tRNA synthetases (ARSs) and other proteins in response to fluctuating tRNA aminoacylation levels under various nutritional states1. T-boxes reside in the 5’-untranslated regions (UTRs) of the mRNAs they regulate, and comprise two conserved domains. Stem I harbors the specifier trinucleotide that base-pairs with the anticodon of cognate tRNA. 3’ to Stem I is the antiterminator domain, which base-pairs with the tRNA acceptor end and evaluates its aminoacylation state2. Despite high phylogenetic conservation and widespread occurrence in pathogens, the structural basis of tRNA recognition3,4 by this riboswitch remains ill-defined. Here, we demonstrate that the ~100-nucleotide T-box Stem I is necessary and sufficient for specific, high-affinity (Kd ~150 nM) tRNA binding, and report its structure in complex with cognate tRNA at 3.2 Å resolution. Stem I recognizes the overall architecture of tRNA in addition to its anticodon, something accomplished by large ribonucleoproteins (RNPs) like the ribosome or proteins such as ARSs5, but unprecedented for a compact mRNA domain. The C-shaped Stem I cradles the L-shaped tRNA forming an extended (1604 Å2) intermolecular interface. In addition to the specifier-anticodon interaction, two interdigitated T-loops near the apex of Stem I stack on the tRNA elbow in a manner analogous to those of the J11/12-J12/11 motif6 of RNase P and the L1 stalk7 of the ribosomal E-site. Since these RNPs and T-boxes are unrelated, this strategy to recognize an universal tRNA feature likely evolved convergently. Mutually induced fit of Stem I and the tRNA exploiting the intrinsic flexibility of tRNA and its conserved post-transcriptional modifications results in high shape complementarity, which in addition to providing specificity and affinity, globally organizes the T-box to orchestrate tRNA-dependent transcription regulation.
T-box riboswitches comprise conserved Stem I and antiterminator domains separated by a variable linker. In some T-boxes, this linker contains three stem-loops. In the tRNAGly-specificglyQ and glyQS T-boxes, there is only one short stem-loop and a ~13 nucleotide (nt) single-stranded segment (Extended Data Fig. 1). We analyzed the contribution of different T-box segments to tRNA binding by isothermal titration calorimetry (ITC). Full-length Bacillus subtilis glyQS T-box, a 3’ truncation leaving only the antiterminator residues that base pair with the tRNA acceptor end, and an isolated Stem I domain bind tRNAGly with comparable affinity (Fig. 1a-c). Therefore, Stem I provides essentially all the binding energy to tRNA. Proximal or distal truncations of Stem I abrogate binding (Extended Data Fig. 2). Mutagenesis shows that high-affinity binding is contingent on cognate specifier-anticodon base pairing (Extended Data Table 1). However, Stem I recognizes tRNA elements outside of the anticodon stem-loop (ASL), since an isolated ASL derived from tRNAGly does not bind appreciably (Extended Data Fig. 2). We conclude that Stem I is necessary and sufficient for specific, high affinity tRNA binding by the T-box riboswitch.
Figure 1. Overall structure of the T-Box Stem I in complex with tRNA.

a, ITC analysis of tRNA binding by full-length glyQS T-box (residues 14-182). b, Binding by a 3’-truncated T-box (residues 14-158). c, Binding by an isolated T-box Stem I (residues 14-113). d, Sequence and secondary structure of the cocrystallized glyQ Stem I and tRNAGly RNAs. Leontis-Westhof30 symbols denote non-canonical base pairs. Lines with embedded arrowheads denote chain connectivity. The tRNA (shaded) is numbered conventionally (‘t’ precedes tRNA residues). e. Cartoon of the complex structure. Color-coding as in (a); segments altered to facilitate crystallization and the YbxF protein are in white.
To understand how an RNA domain of only ~100 residues can recognize a 76 nt tRNA, we cocrystallized Stem I from the glyQ T-box riboswitch of Oceanobacillus iheyensis (Fig. 1d) with tRNAGly and the bacterial K-turn binding protein YbxF, and solved its structure at 3.2 Å resolution (Methods). Stem I folds into an irregular helix of ~110 Å contour length that cradles tRNA (Fig. 1e). Several RNA structural motifs are arranged linearly in Stem I, consistent with phylogenetic conservation1,8,9,10,11. The proximal end of Stem I contains a K-turn12, which introduces a 120° bend into the helical path. YbxF recognizes the K-turn as seen previously in its SAM-I riboswitch complex13. Immediately distal is the specifier trinucleotide, which base-pairs with the tRNA anticodon, as predicted genetically2. This is followed by a loop E motif14 with the extruded G20 forming an S-turn. The helical trajectory bends ~65° at a dinucleotide bulge (residues 29-30), allowing Stem I to track the tRNA. A bulge (but not its sequence) is conserved at this location in T-box Stem I domains11 (Extended Data Figs. 1, 3). The phylogenetically conserved secondary structure of the distal portion of Stem I contains another bulge followed by an apical loop (Extended Data Fig. 1). In our cocrystal structure, this bulge and the loop each folds into a pentanucleotide T-loop15. The two T-loops interdigitate and recognize the structurally conserved elbow of tRNA. Association with tRNAGly buries 1604 Å2 of the solvent accessible surface area of Stem I (Extended Data Fig. 3). Of these, 939 Å2 are at the specifier-anticodon interaction and three adjacent interfaces where the backbones of loop E and Stem I segments one and two helical turns distal contact backbone atoms of the tRNA anticodon stem. The remaining 665 Å2 are at the apical region of Stem I, which contacts two D-loop residues and one T-loop residue of the tRNA. The solvent accessible area of tRNA buried by the T-box is comparable to those buried by several ARSs5, but much larger than the ligand surface areas buried by most riboswitches16.
T-boxes have a conserved purine11 immediately 3’ to the three specifier nucleotides. Our structure reveals that this nucleotide (A90) stacks underneath the wobble base pair (C89βtG34; tRNA residue numbers are preceded by ‘t’) and also hydrogen bonds to A16 (Fig. 2a). A similar stabilizing interaction occurs in the P-site of the ribosome where the universally conserved C1400 of 16S rRNA stacks underneath the tRNA-mRNA wobble pair7,17. Near-symmetrically to A90, tA37 stacks on top of the specifier:anticodon duplex. A purine typically occupies tRNA position t37 and is often post-transcriptionally modified into a larger nucleobase. The nucleotide preceding the first specifier nucleotide of Stem I (A86) is unstacked from the specifier:anticodon duplex because of its participation in the sheared AβA pair that initiates the loop E motif of Stem I. This unstacking produces a pocket that could accommodate large, modified t37 nucleobases (Fig. 2a and Extended Data Fig. 4).
Figure 2. Interactions between Stem I and tRNA.

a, Specifier-anticodon interaction. Dashed lines denote hydrogen bonds. Displacement of A86 forms a pocket (dashed oval). b, The two interdigitated T-loops at the distal end of Stem I stack on the tRNA elbow. c, Stacking of the apical Stem I base triple on the tRNA elbow. Yellow dashed lines denote tRNA elbow base-pairing. d, tRNA residue tU20 flips out to stack with the Stem I C64 ribose (van der Waals surfaces of interacting residues shown). e. Mutagenesis and ITC analysis of selected Stem I-tRNA interactions. Error bars denote s.e.m. (n≥2).
The two interdigitated T-loops and adjacent nucleotides from the distal end of Stem I together form a compact structural module comprised of six stacked layers (Extended Data Fig. 5). The most distal of these is formed by three coplanar bases, C44, A56 and G63. In the Stem I:tRNA complex, this base triple stacks on the conserved tRNA base pair tG19βtC56 (Fig 2b). This conserved tertiary pair joins the apices of tRNA D- and T-loops and forms the ‘elbow’ of all elongator tRNAs. Thus, the two T-loops of Stem I stack on the T- and D-loops of tRNA employing the flat molecular surfaces formed by two interlocked loops in each RNA (Fig. 2b, c). The nucleobase of tU20, another conserved residue from the D-loop of tRNAs, extends the interface by stacking on the ribose of Stem I residue C64 (Fig. 2d), itself part of a conserved base pair. Similar extrahelical stacking of t20 has been observed in tRNA-ARS complexes18,19. Position t20 is post-transcriptionally modified in most tRNAs to dihydroudirine, which stabilizes the C2’-endo conformation of the nucleotide20, potentially enhancing stacking with the ribose of C64 in Stem I. The functional importance of several Stem I:tRNA interactions revealed by our cocrystal analysis is supported by mutagenesis and ITC analysis (Fig. 2e). In particular, alterations to the specifier, the adjacent purine (A90), the core and the distal surface of the interdigitated T-loops are all strongly detrimental.
Sequence analysis and modeling9,10,21 have previously suggested that the two interdigitated T-loops of Stem I fold and function similarly to the J11-12/J12-11 internal loops of RNase P22, and the L1 stalk in the E-site of the 50S ribosomal subunit23. Our cocrystal structure confirms this. However, the Stem I T-loops bind to the tRNA elbow in the opposite orientation from those of RNase P and the ribosome6,7 (Extended Data Fig. 6). Since there is no evidence that 23S rRNA, RNase P, and T-boxes share an ancestor, the use of interdigitated T-loops to recognize the tRNA elbow has likely evolved independently at least three times.
Association with Stem I distorts tRNA locally and globally. The tRNAGly ASL rearranges upon T-box binding likely to avoid steric clash. Its open structure free in solution24 superimposes poorly on that in the complex (Fig. 3a). Comparison with ribosome structures reveals that, similar to tRNAs engaged in translation7,25,26,27, the Stem I bound-tRNAGly bends near the t26βt44 pair at the junction of the anticodon and D stems. This bend resembles that of a P/P state tRNA (~10° relative to the free tRNAPhe), but is more pronounced (~20°; Fig. 3c, d). Thus, in addition to deforming the anticodon loop, the T-box appears to exploit the intrinsically flexible t26βt44 hinge to shift the tRNA elbow by ~11 Å, bringing it into contact with the distal base triple of Stem I (Extended Data Fig. 7).
Figure 3. Induced fit of tRNA by Stem I binding.
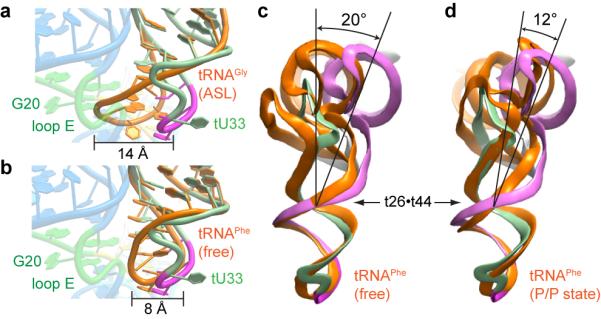
a, Superposition of ASL of free tRNAGly (PDB 2LBJ, orange) with the cocrystal structure. Steric clash between the free structure and the loop E motif (green) is evident. b, Superposition of the ASL of free tRNAPhe (PDB 1EHZ, orange) with the cocrystal structure. Note extrusion of tU33. c, Comparison of Stem I-bound tRNAGly with free tRNAPhe (orange) seen from the direction of the elbow. The approximate location of the hinge at the t26βt44 pair is indicated. d, Comparison of Stem I-bound tRNAGly with the ribosome-bound P/P tRNA (PDB 4GD2, orange).
Stem I also undergoes pronounced conformational changes upon tRNA binding. A structure21 of an apical fragment of Stem I shows good agreement of the interdigitated T-loops; however, a ~20° hinge motion proximal to the T-loops must occur upon binding to avoid steric clash with the tRNA (Fig. 4a). Comparison with the solution structure28 of a proximal fragment of a tyrS T-box shows that specifier residues rotate on average 34° out of the minor groove to base pair with the anticodon. This rotation propagates, moving the functionally critical A90 by 4 Å so it stacks under the specifier-anticodon duplex (Fig. 2e, 4b). The propagation also results in continuous stacking of the phylogenetically conserved purines29 (A8-A19) on the 5’ strand of Stem I. Markedly different from the extended structure28 exhibited in solution in the absence of tRNA, the proximal residues of Stem I fold into a canonical K-turn in the tRNA complex. This results in a dramatic contraction of the distance separating the 3’-terminus of Stem I from the acceptor end of tRNA (Fig. 4c). This distance needs to be bridged by the short linker separating the glyQS Stem I from the antiterminator (Extended Data Fig. 1) for the T-box riboswitch to function. A role for the K-turn in organizing the large-scale architecture of the T-box-tRNA complex reconciles its reported requirement in vivo8 and its lack of contribution to tRNA binding affinity in vitro (Extended Data Fig. 2 and Extended Data Table 1). Therefore, the mutually induced fit of Stem I and tRNA not only gives rise to specific, high-affinity binding, but also organizes the overall architecture of the 5’-UTR to orchestrate the aminoacylation-dependent transcriptional termination decision.
Figure 4. Stem I reorganization by tRNA binding.
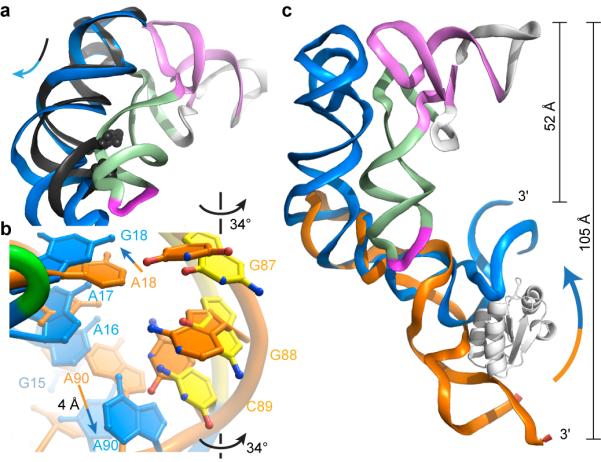
a, Superposition of an isolated Stem I distal fragment (PDB 4JRC, black) and the complex. Clash between the helix (partly shown as spheres) of the fragment and the tRNA is relieved by flexing below the T-loops in the complex. b, Superposition of the specifier free (PDB 2KZL, orange) and bound to tRNA. Specifier nucleotides rotate outward, and A90 is displaced. c, Superposition of an isolated Stem I proximal fragment (PDB 2KZL, orange) and the complex. tRNA binding induces bending of Stem I (arrow) bringing its 3’-terminus ~50 Å closer to the acceptor end of tRNA.
Despite having different sequences and being extensively modified post-transcriptionally, all elongator tRNAs share a common overall architecture as well as a flexible hinge that allows efficient transit through the ribosome. Our T-box-tRNA cocrystal structure reveals how a gene-regulatory mRNA domain exploits all these molecular features of tRNA in addition to the anticodon sequence for specific recognition. Phylogenetic conservation indicates that T-boxes use a conserved structural scaffold to recognize divergent tRNAs. Our analysis therefore provides a framework with which to understand the coevolution of interacting RNA molecules.
Methods Summary
O. iheyensis glyQ T-box Stem I was modified for crystallization by deleting one base pair from the distal side of the K-turn (above the A11βG95 pair) and introducing three base pairs at its proximal end. O. iheyensis tRNAGlyGCC was circularly permuted and its acceptor stem was capped with a GAAA tetraloop (Fig. 1d). The two RNAs and one equivalent of selenomethionyl B. subtilis YbxF [200 μM complex in a buffer comprised of 50 mM HEPES-KOH pH 7.0, 100 mM KCl, 20 mM MgCl2, and 5 mM tris(2-carboxyethyl)phosphine (TCEP)] were cocrystallized at 21°C by vapor diffusion by mixing with an equal volume of a reservoir comprised of 50 mM Bis-Tris (HCl) pH 6.5, 300 mM Li2SO4, and 20% (w/v) PEG3350, supplemented with 0.2% (w/v) low melting point agarose. Cocrystals with the symmetry of space group C2221 were incubated in a harvesting solution comprised of 50 mM Bis-Tris (HCl) pH 6.5, 100 mM KCl, 20-50 mM SrCl2, 40-45% (w/v) PEG3350, and 5 mM TCEP for 16 hours prior to flash-freezing in liquid nitrogen. Diffraction data were collected at 100 K at beamlines 5.0.1 and 5.0.2 of the Advanced Light Source (ALS), and beamlines 24-ID-C and 24-ID-E of the Advanced Photon Source (APS). Single wavelength anomalous dispersion (SAD) phases from selenomethionine and Sr2+ ions were combined with molecular replacement (MR) phases obtained using tRNA and a K-turn-YbxF complex as search models to produce initial electron density maps. The structure, which comprises two Stem I-tRNAGly-YbxF ternary complexes per crystallographic asymmetric unit (a.u.), has been refined to Rfree = 25.4% and has a mean coordinate precision of 0.48 Å (Extended Data Table 2). The two ternary complexes in the a.u. superimpose closely. Biochemical and ITC analysis of T-box:tRNA interaction was performed employing RNAs derived from the B. subtilis glyQS sequence (Extended Data Fig. 1).
Methods
RNA and protein preparation
Biochemical and ITC analysis of T-box:tRNA interaction was performed employing RNAs derived from the B. subtilis glyQS sequence (Extended Data Fig. 1). For crystallization, the O. iheyensis glyQ T-box Stem I RNA was modified by deleting one base pair from the distal side of the K-turn (above the A11:G95 pair) and introducing three base pairs at its proximal end. The O. iheyensis tRNAGlyGCC was circularly permuted and its acceptor stem was capped with a GAAA tetraloop (Fig. 1d). These alterations did not adversely affect Stem I-tRNA association (Extended Data Fig. 2h, Extended Data Table 1). B. subtilis glyQS T-box and tRNAGlyconstructs, O. iheyensis glyQ T-box Stem I RNA (A89C), and O. iheyensis tRNAGly (Fig. 1d) were transcribed in vitro using T7 RNA polymerase from double-stranded DNA templates generated by PCR, as described31, except that 2 mL of PCR reaction product were employed for each 5 mL transcription. RNAs were purified by electrophoresis on 8% polyacrylamide, 8M urea gels (29:1 acrylamide:bisacrylamide), electroeluted, washed once with 1 M KCl, desalted by ultrafiltration, and stored at 4°C. Selenomethionyl B. subtilis YbxF was expressed and purified as described13.
Isothermal titration calorimetry (ITC)
To suppress oligomerization, tRNAGly was heated to 90°C in water for 3 min and cooled to 4°C over 2 min immediately prior to use. RNA samples for ITC experiments were equilibrated in a buffer comprised of 100 mM KCl, 50 mM HEPES-KOH pH 7.0, 10 mM MgCl2 by ultrafiltration. ITC experiments were performed at least in duplicate at 20°C with 20 μM T-box RNA in the cell and 200 μM tRNA or ASL in the syringe, using a MicroCal iTC200 microcalorimeter (GE). Initial data analysis was performed as described32,33,34; then, data from replicate experiments were fitted globally35,36 using SEDPHAT and NITPIC to arrive at the dissociation constants and uncertainties in Extended Data Table 1.
Cocrystallization and diffraction data collection
Engineered O. iheyensis tRNAGly (75 nt) was heated to 90°C in water for 3 min and cooled to 4°C over 2 min, mixed with one equivalent of Stem I RNA and incubated in the presence of 50 mM HEPES-KOH pH 7.0, 100 mM KCl, 20 mM MgCl2, and 5 mM TCEP at 50°C for 10 min and then at 37°C for 30 min. One equivalent of selenomethionyl B. subtilis YbxF was then added. The solution was adjusted to 200 μM complex, 2 mM spermine, 0.2% (w/v) low melting point agarose and held at 37°C. For crystallization, the complex was mixed 1:1 with a reservoir solution comprised of 50 mM Bis-Tris (HCl) pH 6.5, 300 mM Li2SO4, and 20% PEG3350. Plate-shaped crystals grew in 1-4 weeks to maximum dimensions of 300 × 300 × 50 μm3. After incubation in 50 mM Bis-Tris (HCl) pH 6.5, 100 mM KCl, 20-50 mM SrCl2, 40-45% PEG3350, 5 mM TCEP for 16 hours, crystals were dissected out of the agarose using MicroSaws (Mitegen), mounted on 90° bent MicroLoops (Mitegen), and vitrified by plunging into liquid nitrogen. Cocrystals have the symmetry of space group C2221. Unit cell dimensions are in Extended Data Table 2. Single-wavelength anomalous dispersion (SAD) data were collected at the selenium K edge (0.9792 Å) at 100 K at beamlines 24-ID-C and 24-ID-E of the APS, and indexed, integrated, and scaled using the NE-CAT RAPD pipeline which employs XDS37 and Scala38. Additional data were collected at beamlines 5.0.1 and 5.0.2 of the ALS, and processed with HKL200039. Data collection statistics are summarized in Extended Data Table 2.
Structure determination and refinement
Four selenium sites were identified from Crystal I data using SHELX-D40 through HKL2MAP41. The resulting SAD phases were combined with model phases resulting from placing two truncated search models each of tRNA42 (PDB 3L0U) and K-turn bound by YbxF13 (PDB 3V7E) using MOLREP43, in a strategy similar to that described44. Subsequently, the composite ensemble (two copies each of tRNA and the K-turn-YbxF complex) was located in a merged, high-redundancy dataset (Crystals I-II-III) using PHASER45, allowing completion of the anomalous atom substructure, which is comprised of eight selenium atom sites and three Sr2+ ions. SAD (mean overall figure of merit = 0.36) and model phases were combined using the MR-SAD pipeline implemented in PHENIX46 and density-modified using RESOLVE47 producing a substantially improved electron density map (Extended Data Fig. 8) with which manual model building could commence. Iterative rounds of manual model building48, molecular replacement, and phase-combination produced a near-complete model. This model was then subjected to additional manual building interspersed with iterative rounds of rigid-body, simulated-annealing, and individual isotropic B-factor refinement against Crystal I data using PHENIX. Refinement statistics are summarized in Extended Data Table 2. The maximum likelihood coordinate precision of the current model is 0.47 Å. Ramachandran analysis shows that 92.4% of the amino acid residues are in the most favored regions, 7.0% in allowed regions, and 0.6% (one residue) in disallowed regions. The two Stem I and tRNA molecules in the a.u. superimpose closely (r.m.s.d. of 1.1 and 0.7 Å, respectively). In the crystals, the flat surface formed by the interdigitated T-loops on face opposite the tRNA elbow stacks on both, the apical nucleobase of the GAAA tetraloop of the engineered tRNA, and the terminal (most proximal) base pair of Stem I from symmetry-related molecules.
Comparisons were performed against the following structures: tRNAGly ASL (PDB ID: 2LBJ, ref. 24); tRNAPhe (PDB ID: 1EHZ, ref. 49); the 70S ribosome with a P/P tRNA (PDB ID: 4GD2, ref. 26); a distal fragment of Stem I (PDB ID: 4JRC, ref. 21); a proximal fragment of Stem I (PDB ID: 2KZL, ref. 28); the loop E motif of 5S rRNA (PDB ID: 354D, ref. 50); RNase P holoenzyme bound to tRNA (PDB ID: 3Q1Q, ref. 6); The L1 Stalk of the 50S ribosomal subunit (PDB ID: 1MZP, ref. 23); and the 70S ribosome with an E/E tRNA (PDB ID: 1VSA, ref. 7). The superposition shown in Fig. 4a employed the interdigitated T-loops of the two structures. Those shown in Fig. 4b and 4c were generated by overlaying the loop E residues of the two structures. In both cases, r.m.s.d. was less than 1 Å. Solvent accessible surface areas were calculated with a probe radius of 1.4 Å. Structural figures were prepared with PyMol51, using chains A (YbxF), B (tRNAGly), and C (glyQ Stem I).
Supplementary Material
Extended Data Figure 1. Sequence of the B. subtilis glyQS T-box. Unlike some other T-boxes, tRNAGly-responsive T-boxes (such as the B. subtilis glyQS T-box and the closely related O. iheyensis glyQ T-box) do not contain Stems II and IIA/B in the linker region. Conservation across known T-boxes is indicated in orange and yellow circles (from ref. 11). Base pairing between the specifier sequence in Stem I and the antiterminator domain of the T-box and the anticodon and acceptor end, respectively, of tRNAGly is indicated. The functionally important K-turn8 is boxed. The sequences of tRNAGly of B. subtilis and O. iheyensis are identical.
Extended Data Figure 2. Representative isothermal calorimetric titrations. Full-length tRNAGly (a–e) and an isolated tRNAGly anticodon stem-loop (ASL, f) were titrated into various truncated B. subtilis glyQS T-box Stem I constructs (Methods). In these panels, the specifier trinucleotide GGC is in bold. g, Superposition of the experimental data and fits for the experiments shown in Fig. 1 a-c, color-coded to correspond to schematic RNA construct depictions. h, Isothermal titration calorimetric analysis of binding by the engineered tRNAGly construct employed for cocrystallization. Results of all fits (Methods) are in Extended Data Table 1.
Extended Data Figure 3. Intermolecular interface of the Stem I-tRNA complex and phylogenetic conservation. a, The residue 29-30 bulge allows the O. iheyensis Stem I to bend and track the tRNA backbone. b, Structure21 of the equivalent region of the Geobacillus kaustophilus T-box, which folds as a C-loop52. c, Solvent accessible surface colored according to area buried from blue (no burial) to red (> 75 Å2 per residue). d, Open-book view. c, Surfaces colored by phylogenetic conservation of Stem I residues (see Extended Data Fig. 1). Conservation of tRNA is not indicated. e, Open-book view. For comparison, RNase P binding to pre-tRNA buries6 2959 Å2.
Extended Data Figure 4. T-box can accommodate bulky post-transcriptional modifications at tRNA position 37. A86 of the T-box Stem I is unstacked from the duplex formed by pairing of the tRNA andticodon and the T-box specifier by its participation in the sheared AβA pair at the base of the loop E motif. This results in a pocket (dotted line) that could accommodate the large, modified nucleobases (e.g., the three-ring heterocycle wybutine in yeast tRNAPhe) often present at position 37 of tRNAs (Fig. 2a). Note the single hydrogen bond between tU32 and tA38.
Extended Data Figure 5. Structural organization of the interdigitated T-loop motifs of the T-box Stem I. a, Six base-paired layers form the Stem I distal domain core (view as in Fig. 2b.) Dashed black lines denote base-base hydrogen boding. Dashed yellow lines denote Sr2+ ion-water (green and red spheres, respectively) interactions. Leontis-Westhof symbols30 depict non-canonical pairing. b, Orthogonal view. c, View from the tRNA elbow. d–i, Detail of layers 1-6. The position of each nucleotide within the pentaloop (1-5) or outside of it (negative and positive denote 5’ and 3’ directions, respectively) is in parentheses (Supplementary Discussion).
Extended Data Figure 6. Comparison of the interdigitated T-loops of RNase P, the L1 stalk the T-box. RNase P (a) and the L1 stalk (b) employ equivalent surfaces formed by two purines, namely, nucleotide −2 of T-loop 1 and nucleotide 3 of T-loop 2 (*), to stack on the elbows of pre-tRNA and the E-site tRNA, respectively. In contrast, the T-box (c) employs the opposite surface, using a triple (Fig. 2c) formed by nucleotide 3 of T-loop 1 (†), nucleotide −2 from T-loop 2, and nucleotide +1 from T-loop 2 to stack on the tRNA elbow.
Extended Data Figure 7. Displacement of the tRNA elbow induced by T-box binding. While the anticodon stem of the free tRNAPhe (orange) superimposes well on that of the T-box-bound tRNAGly (T-loop and acceptor stem in violet; D-loop and ASL in pale green, Fig. 3b, c), the elbow (tG19-tC56) clashes with the Stem I distal domain. T-box binding induces bending about the t26βt44 hinge (Fig. 3c) that displaces the tRNA elbow and acceptor stem away from the distal region of Stem I by 11 Å (arrows), allowing the tRNA elbow to associate with the T-box. a, side view. b, top view.
Extended Data Figure 8. Representative electron density maps. Portions of electron density maps corresponding to the interdigitated Stem I T-loops and the tRNA elbow (oriented like Fig. 2b). a, Unbiased, 3.2 Å-resolution density-modified SAD electron density (1.0 standard deviations above mean peak height, s.d.) shown as gray mesh superimposed on the refined model. b, Electron density resulting from combination of SAD and molecular replacement (using a search model comprised of two copies each of tRNAPhe and a K-turn in complex with YbxF, Methods) phases followed by density modification (1.5 s.d.) c, Composite simulated anneal-omit 2|Fo|-|Fc| electron density calculated using the final model (2.0 s.d.).
Extended Data Table 1. Thermodynamic parameters derived from isothermal titration calorimetric analysis.
* Residue numbers are for O. iheyensis glyQ T-box. Structurally equivalent B. subtilis glyQS T-box numbers are in parentheses.
† All experiments were performed at 293 K.
‡ This experiment corresponds to the titration shown in Fig. 1a.
§ This experiment corresponds to the titration shown in Fig. 1b.
∥ This experiment corresponds to the titration shown in Fig. 1c and Extended Data Fig. 2a.
¶ This experiment corresponds to the titration shown in Extended Data Fig. 2b.
# Engineered crystallization construct.
Extended Data Table 2. Data collection and refinement statistics.
One crystal was used for each of the three datasets.
*Highest resolution shell is shown in parenthesis.
Acknowledgements
We thank the staff at beamlines ID-24-C and ID-24-E of APS and 5.0.1 and 5.0.2 of the ALS for crystallographic data collection support, G. Piszczek (National Heart, Lung and Blood Institute, NHLBI) for ITC support, R. Levine and D.-Y. Lee (NHLBI) for help with mass spectrometry, K. Perry and K.R. Rajashankar of the Northeastern Collaborative Access Team (NE-CAT) of the APS for assistance with initial crystallographic phasing, and N. Baird, T. Hamma, C. Jones, M. Lau, J. Posakony, A. Roll-Mecak, O. Uhlenbeck, and K. Warner for discussions. This work is partly based on research conducted at the APS on the NE-CAT beamlines, and at the ALS on the Berkeley Center for Structural Biology beamlines, which are both supported by the National Institute for General Medical Sciences, National Institutes of Health (NIH). Use of ALS and APS was supported by the U.S. Department of Energy. This work was supported in part by the intramural program of the NHLBI, NIH.
Footnotes
Supplementary Information consists of Supplementary Discussion and Supplementary References, and is linked to the online version of the paper at www.nature.com/nature.
Methods and any associated references are available in the online version of the paper.
References
- 1.Green NJ, Grundy FJ, Henkin TM. The T box mechanism: tRNA as a regulatory molecule. FEBS Lett. 2010;584:318–324. doi: 10.1016/j.febslet.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grundy FJ, Henkin TM. tRNA as a positive regulator of transcription antitermination in B. subtilis. Cell. 1993;74:475–482. doi: 10.1016/0092-8674(93)80049-k. [DOI] [PubMed] [Google Scholar]
- 3.Yousef MR, Grundy FJ, Henkin TM. Structural transitions induced by the interaction between tRNAGly and the Bacillus subtilis glyQS T box leader RNA. J. Mol. Biol. 2005;349:273–287. doi: 10.1016/j.jmb.2005.03.061. [DOI] [PubMed] [Google Scholar]
- 4.Yousef M, Grundy F, Henkin T. tRNA requirements for glyQS antitermination: a new twist on tRNA. RNA. 2003;9:1148–1156. doi: 10.1261/rna.5540203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perona JJ, Hadd A. Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry. 2012;51:8705–8729. doi: 10.1021/bi301180x. [DOI] [PubMed] [Google Scholar]
- 6.Reiter NJ, et al. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature. 2010;468:784–789. doi: 10.1038/nature09516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korostelev A, Trakhanov S, Laurberg M, Noller H. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126:1065–1077. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 8.Winkler WC, Grundy FJ, Murphy BA, Henkin TM. The GA motif: an RNA element common to bacterial antitermination systems, rRNA, and eukaryotic RNAs. RNA. 2001;7:1165–1172. doi: 10.1017/s1355838201002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winkler W. PhD Dissertation. Ohio State University; 2002. RNA Elements Required for T box Antitermination; pp. 1–270. [Google Scholar]
- 10.Lehmann J, Jossinet F, Gautheret D. A universal RNA structural motif docking the elbow of tRNA in the ribosome, RNase P and T-box leaders. Nucleic Acids Res. 2013;41:5494–5502. doi: 10.1093/nar/gkt219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez-Preciado A, Henkin TM, Grundy FJ, Yanofsky C, Merino E. Biochemical features and functional implications of the RNA-based T-Box regulatory mechanism. Microbiol Mol Biol Rev. 2009;73:36–61. doi: 10.1128/MMBR.00026-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein DJ, Schmeing TM, Moore PB, Steitz TA. The kink-turn: a new RNA secondary structure motif. EMBO J. 2001;20:4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baird NJ, Zhang J, Hamma T, Ferré-D’Amaré AR. YbxF and YlxQ are bacterial homologs of L7Ae and bind K-turns but not K-loops. RNA. 2012;18:759–770. doi: 10.1261/rna.031518.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leontis NB, Westhof E. The 5S rRNA loop E: chemical probing and phylogenetic data versus crystal structure. RNA. 1998;4:1134–1153. doi: 10.1017/s1355838298980566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan CW, Chetnani B, Mondragón A. Structure and function of the T-loop structural motif in noncoding RNAs. Wiley Interdiscip Rev RNA. 2013 doi: 10.1002/wrna.1175. doi:10.1002/wrna.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards TE, Klein DJ, Ferré-D’Amaré AR. Riboswitches: small-molecule recognition by gene regulatory RNAs. Curr Op Struct Biol. 2007;17:273–279. doi: 10.1016/j.sbi.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Selmer M, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 18.Rock FL, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing eite. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 19.Fukai S, et al. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103:793–803. doi: 10.1016/s0092-8674(00)00182-3. [DOI] [PubMed] [Google Scholar]
- 20.Dalluge JJ, Hashizume T, Sopchik AE, McCloskey JA, Davis DR. Conformational flexibility in RNA: the role of dihydrouridine. Nucleic Acids Res. 1996;24:1073–1079. doi: 10.1093/nar/24.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grigg JC, et al. T box RNA decodes both the information content and geometry of tRNA to affect gene expression. Proc Natl Acad Sci U S A. 2013;110:7240–7245. doi: 10.1073/pnas.1222214110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krasilnikov AS, Yang X, Pan T, Mondragón A. Crystal structure of the specificity domain of ribonuclease P. Nature. 2003;421:760–764. doi: 10.1038/nature01386. [DOI] [PubMed] [Google Scholar]
- 23.Nikulin A, et al. Structure of the L1 protuberance in the ribosome. Nature Struct Biol. 2003;10:104–108. doi: 10.1038/nsb886. [DOI] [PubMed] [Google Scholar]
- 24.Chang AT, Nikonowicz EP. Solution nuclear magnetic resonance analyses of the anticodon arms of proteinogenic and nonproteinogenic tRNAGly. Biochemistry. 2012;51:3662–3674. doi: 10.1021/bi201900j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valle M, et al. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nature Struct Biol. 2003;10:899–906. doi: 10.1038/nsb1003. [DOI] [PubMed] [Google Scholar]
- 26.Dunkle JA, et al. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmeing TM, et al. The Crystal Structure of the Ribosome Bound to EF-Tu and Aminoacyl-tRNA. Science. 2009;326:688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Nikonowicz EP. Solution structure of the K-turn and specifier loop domains from the Bacillus subtilis tyrS T-Box leader RNA. J Mol Biol. 2011;408:99–117. doi: 10.1016/j.jmb.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vitreschak AG, Mironov AA, Lyubetsky VA, Gelfand MS. Comparative genomic analysis of T-box regulatory systems in bacteria. RNA. 2008;14:717–735. doi: 10.1261/rna.819308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods and Extended Data References
- 31.Xiao H, Edwards TE, Ferré-D’Amaré AR. Structural basis for specific, high-affinity tetracycline binding by an in vitro evolved aptamer and artificial riboswitch. Chem Biol. 2008;15:1125–1137. doi: 10.1016/j.chembiol.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baird NJ, Ferré-D’Amaré AR. Idiosyncratically tuned switching behavior of riboswitch aptamer domains revealed by comparative small-angle X-ray scattering analysis. RNA. 2010;16:598–609. doi: 10.1261/rna.1852310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kulshina N, Edwards TE, Ferré-D’Amaré AR. Thermodynamic analysis of ligand binding and ligand binding-induced tertiary structure formation by the thiamine pyrophosphate riboswitch. RNA. 2010;16:186–196. doi: 10.1261/rna.1847310. doi:10.1261/rna.1847310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baird NJ, Ferré-D’Amaré AR. Modulation of quaternary structure and enhancement of ligand binding by the K-turn of tandem glycine riboswitches. RNA. 2013;19:167–176. doi: 10.1261/rna.036269.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuck P. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophysical J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keller S, et al. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal Chem. 2012;84:5066–5073. doi: 10.1021/ac3007522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabsch W. XDS. Acta Crystallogr D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans P. Scaling and assessment of data quality. Acta Crystallogr D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Processing of diffraction data collected in oscillation mode. Meth. Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 41.Pape T, Schneider TR. HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Cryst. 2004;37:843–844. [Google Scholar]
- 42.Byrne RT, Konevega AL, Rodnina MV, Antson AA. The crystal structure of unmodified tRNAPhe from Escherichia coli. Nucleic Acids Res. 2010;38:4154–4162. doi: 10.1093/nar/gkq133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- 44.Xiao H, Murakami H, Suga H, Ferré-D’Amaré AR. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature. 2008;454:358–361. doi: 10.1038/nature07033. [DOI] [PubMed] [Google Scholar]
- 45.McCoy A, et al. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi H, Moore PB. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: a classic structure revisited. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Correll CC, Freeborn B, Moore PB, Steitz TA. Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell. 1997;91:705–712. doi: 10.1016/s0092-8674(00)80457-2. [DOI] [PubMed] [Google Scholar]
- 51.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; 2002. [Google Scholar]
- 52.Lescoute A, Leontis NB, Massire C, Westhof E. Recurrent structural RNA motifs, isostericity matrices and sequence alignments. Nucleic Acids Res. 2005;33:2395–2409. doi: 10.1093/nar/gki535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended Data Figure 1. Sequence of the B. subtilis glyQS T-box. Unlike some other T-boxes, tRNAGly-responsive T-boxes (such as the B. subtilis glyQS T-box and the closely related O. iheyensis glyQ T-box) do not contain Stems II and IIA/B in the linker region. Conservation across known T-boxes is indicated in orange and yellow circles (from ref. 11). Base pairing between the specifier sequence in Stem I and the antiterminator domain of the T-box and the anticodon and acceptor end, respectively, of tRNAGly is indicated. The functionally important K-turn8 is boxed. The sequences of tRNAGly of B. subtilis and O. iheyensis are identical.
Extended Data Figure 2. Representative isothermal calorimetric titrations. Full-length tRNAGly (a–e) and an isolated tRNAGly anticodon stem-loop (ASL, f) were titrated into various truncated B. subtilis glyQS T-box Stem I constructs (Methods). In these panels, the specifier trinucleotide GGC is in bold. g, Superposition of the experimental data and fits for the experiments shown in Fig. 1 a-c, color-coded to correspond to schematic RNA construct depictions. h, Isothermal titration calorimetric analysis of binding by the engineered tRNAGly construct employed for cocrystallization. Results of all fits (Methods) are in Extended Data Table 1.
Extended Data Figure 3. Intermolecular interface of the Stem I-tRNA complex and phylogenetic conservation. a, The residue 29-30 bulge allows the O. iheyensis Stem I to bend and track the tRNA backbone. b, Structure21 of the equivalent region of the Geobacillus kaustophilus T-box, which folds as a C-loop52. c, Solvent accessible surface colored according to area buried from blue (no burial) to red (> 75 Å2 per residue). d, Open-book view. c, Surfaces colored by phylogenetic conservation of Stem I residues (see Extended Data Fig. 1). Conservation of tRNA is not indicated. e, Open-book view. For comparison, RNase P binding to pre-tRNA buries6 2959 Å2.
Extended Data Figure 4. T-box can accommodate bulky post-transcriptional modifications at tRNA position 37. A86 of the T-box Stem I is unstacked from the duplex formed by pairing of the tRNA andticodon and the T-box specifier by its participation in the sheared AβA pair at the base of the loop E motif. This results in a pocket (dotted line) that could accommodate the large, modified nucleobases (e.g., the three-ring heterocycle wybutine in yeast tRNAPhe) often present at position 37 of tRNAs (Fig. 2a). Note the single hydrogen bond between tU32 and tA38.
Extended Data Figure 5. Structural organization of the interdigitated T-loop motifs of the T-box Stem I. a, Six base-paired layers form the Stem I distal domain core (view as in Fig. 2b.) Dashed black lines denote base-base hydrogen boding. Dashed yellow lines denote Sr2+ ion-water (green and red spheres, respectively) interactions. Leontis-Westhof symbols30 depict non-canonical pairing. b, Orthogonal view. c, View from the tRNA elbow. d–i, Detail of layers 1-6. The position of each nucleotide within the pentaloop (1-5) or outside of it (negative and positive denote 5’ and 3’ directions, respectively) is in parentheses (Supplementary Discussion).
Extended Data Figure 6. Comparison of the interdigitated T-loops of RNase P, the L1 stalk the T-box. RNase P (a) and the L1 stalk (b) employ equivalent surfaces formed by two purines, namely, nucleotide −2 of T-loop 1 and nucleotide 3 of T-loop 2 (*), to stack on the elbows of pre-tRNA and the E-site tRNA, respectively. In contrast, the T-box (c) employs the opposite surface, using a triple (Fig. 2c) formed by nucleotide 3 of T-loop 1 (†), nucleotide −2 from T-loop 2, and nucleotide +1 from T-loop 2 to stack on the tRNA elbow.
Extended Data Figure 7. Displacement of the tRNA elbow induced by T-box binding. While the anticodon stem of the free tRNAPhe (orange) superimposes well on that of the T-box-bound tRNAGly (T-loop and acceptor stem in violet; D-loop and ASL in pale green, Fig. 3b, c), the elbow (tG19-tC56) clashes with the Stem I distal domain. T-box binding induces bending about the t26βt44 hinge (Fig. 3c) that displaces the tRNA elbow and acceptor stem away from the distal region of Stem I by 11 Å (arrows), allowing the tRNA elbow to associate with the T-box. a, side view. b, top view.
Extended Data Figure 8. Representative electron density maps. Portions of electron density maps corresponding to the interdigitated Stem I T-loops and the tRNA elbow (oriented like Fig. 2b). a, Unbiased, 3.2 Å-resolution density-modified SAD electron density (1.0 standard deviations above mean peak height, s.d.) shown as gray mesh superimposed on the refined model. b, Electron density resulting from combination of SAD and molecular replacement (using a search model comprised of two copies each of tRNAPhe and a K-turn in complex with YbxF, Methods) phases followed by density modification (1.5 s.d.) c, Composite simulated anneal-omit 2|Fo|-|Fc| electron density calculated using the final model (2.0 s.d.).
Extended Data Table 1. Thermodynamic parameters derived from isothermal titration calorimetric analysis.
* Residue numbers are for O. iheyensis glyQ T-box. Structurally equivalent B. subtilis glyQS T-box numbers are in parentheses.
† All experiments were performed at 293 K.
‡ This experiment corresponds to the titration shown in Fig. 1a.
§ This experiment corresponds to the titration shown in Fig. 1b.
∥ This experiment corresponds to the titration shown in Fig. 1c and Extended Data Fig. 2a.
¶ This experiment corresponds to the titration shown in Extended Data Fig. 2b.
# Engineered crystallization construct.
Extended Data Table 2. Data collection and refinement statistics.
One crystal was used for each of the three datasets.
*Highest resolution shell is shown in parenthesis.