

Abstract
Over the last twenty years of research on cellular mechanisms of pain hypersensitivity, long-term potentiation (LTP) of synaptic transmission in the spinal cord dorsal horn (DH) has emerged as an important contributor to pain pathology. Mechanisms that underlie LTP of spinal DH neurons include changes in the numbers, activity, and properties of ionotropic glutamate receptors (AMPA and NMDA receptors) and of voltage-gated Ca2+ channels. Here, we review the roles and mechanisms of these channels in the induction and expression of spinal DH LTP, and we present this within the framework of the anatomical organization and synaptic circuitry of the spinal DH. Moreover, we compare synaptic plasticity in the spinal DH with classical LTP described for hippocampal synapses.
1. Introduction
Long-term potentiation (LTP), an increase in the strength of synaptic transmission between neurons, has been proposed as a cellular model of learning and memory formation. Since LTP was first described for the dentate area of the hippocampal formation [1], data pertinent to mechanisms of LTP have been abundantly accumulated in diverse synapses of hippocampus and other brain areas. In contrast, investigation of LTP in the spinal dorsal horn (DH) [2] is more recent, beginning twenty years after the first description of LTP in the hippocampus, and spinal DH LTP has focused largely upon the synapses formed by primary sensory afferent fibers, because these synapses are the first checkpoint for pain signals entering the central nervous system (CNS). At these primary afferent synapses, LTP has been thought to be a cellular correlate of pain hypersensitivity and as such has been proposed as a potential target for therapeutic treatments of chronic pain.
Neurons in the spinal DH, consisting of superficial (laminae I and II) and deep (laminae III–VI) DH, receive synaptic inputs from primary afferent fibers, their cell bodies located within dorsal root ganglion (DRG) as well as those from other DH neurons, or neurons in other higher brain areas. The spinal DH neurons are considered as secondary neurons because peripheral somatosensory signals conveyed by primary sensory DRG neurons first reach these neurons. Synapses formed in these DH neurons mostly use glutamate for excitatory transmission. Generally, ionotropic glutamate receptors selectively activated by the artificial agonist α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) support the largest component of glutamatergic excitatory synaptic transmission in the CNS, while the N-methyl-D-aspartate (NMDA) receptor subtype is most important in the induction of synaptic plasticity, including LTP (see below). In addition to ligand-gated excitatory ion channels, DH neurons express various types of voltage-gated ion channels that generally contribute to neuronal excitability. Among the voltage-gated ion channels, voltage-gated Ca2+ channels (VGCCs) have been found to be involved in the control of synaptic plasticity, owing to their control of Ca2+ influx into both presynaptic nerve terminals and postsynaptic domains of neurons.
In this paper, we review the contributions of these two classes of ion channels to LTP in the spinal DH area. To provide a context for interpretation of the role of these channels in LTP, we first briefly discuss the anatomical organization and synaptic circuitry of the spinal DH and also consider synaptic transmission and plasticity in the spinal DH. For the sake of brevity, this review does not consider the roles of other types of ion channels in plasticity and pain, nor does it focus upon downstream signaling pathways known to be critical for LTP.
2. The Spinal Cord Dorsal Horn
2.1. Anatomical Organization
The DH of the spinal cord can be subdivided into six distinct layers (laminae I–VI) in the dorsal-ventral direction of the gray matter, which was first proposed in cat [3] as well as in rat [4]. The Rexed laminae I and II consist of superficial spinal DH [5], and laminae III–VI are frequently called deeper layer of the spinal cord. Due to concentrated small neurons and their processes plus a relative small number of myelinated axons, the lamina II is observed as a translucent band under the naked eye or light microscope and is called “substantia gelatinosa (SG)” [4, 6]. Lamina VI exists only in the cervical and lumbosacral enlargements [3]. Generally, the spinal DH consists of the central terminals of primary sensory neurons, projection neurons, intrinsic DH neurons, and descending nerve fibers from the brainstem and other higher brain structures. The cell bodies of the primary sensory neurons are located in the DRG. Each ganglion cell sends an axon that branches into a peripheral process and a central process. The peripheral process contributes to a peripheral nerve and terminates peripherally as a sensory receptor. The central process enters into the spinal cord through a dorsal root and further branches to numerous collaterals. Together, these two processes form primary afferent fibers that transmit encoded information from periphery to the spinal cord or trigeminal nuclei of the brain stem.
Although primary afferent fibers give off most of their collaterals to the segment of the spinal cord that they enter, they also spread in the rostrocaudal direction. The distribution of primary afferent fibers in the spinal DH is in an orderly way based on fiber size, which affects conduction velocity and sensory modality [7]. Most fine myelinated (Aδ; conduction velocity, 1–1.5 to 5–10 m/sec) or unmyelinated (C; <1–1.5 m/sec) primary afferent fibers end predominantly in laminae I and II, although a few reach laminae III–VI [8, 9]. In detail, high threshold Aδ mechanoreceptors terminate in laminae I and V, while low threshold Aδ mechanoreceptors only terminate in lamina III [9]. Most large cutaneous afferents (Aβ; >5–10 m/sec), which function as low threshold mechanoreceptors, have a characteristic pattern of termination in the deeper laminae (III–VI) of the DH [10]. Cutaneous C fibers, occupying ~80% of cutaneous primary afferent fibers [11] and the majority of which being high-threshold polymodal nociceptors in the rat [12], terminate in lamina II [13–15], although there is also a contribution to lamina I [16]. Based on neurochemical markers, the high-threshold C fibers can be divided into two major groups: peptidergic and nonpeptidergic [7]. Peptidergic C fibers are nociceptors [17] and contain neuropeptides such as calcitonin gene-related peptide (CGRP) and/or substance P and express TrkA or transient receptor potential (TRP) V1 [18]. The peptidergic substance P-containing C fibers ends mainly in lamina I and the outer layer of lamina II (IIo). It is estimated in lumbar DRG of rat that approximately half of the C fibers are peptidergic [19]. Other high-threshold C fibers do not contain peptides, but most of them can be revealed by their ability to bind the lectin Bandeiraea simplicifolia isolectin B4 (IB4) [20], and a subpopulation of the nonpeptidergic C fibers can be defined by Mas-related G-protein-coupled receptor member D (MrgprD), a sensory neuron-specific G-protein-coupled receptor [21]. Although the function of nonpeptidergic C fibers is poorly understood, this population also includes many nociceptors [22, 23] and is different from the peptidergic C fiber because ablation of the MrgprD afferents in adult mice results in a selective loss of sensitivity to noxious mechanical (but not thermal) stimuli [7, 24].
Beside the high-threshold C fibers, it should be pointed out that there are low-threshold mechanosensitive C fibers, which are nonpeptidergic and innervate specific types of hair follicles [25]. Interestingly, this type of nonpeptidergic C fiber expresses neither IB4 nor MrgprD but exclusively expresses tyrosine hydroxylase, the enzyme catalyzing L-3,4-dihydroxyphenylalanine production and participates in forming narrow unique columns in the spinal DH with other low-threshold Aδ and Aβ fibers [25].
2.2. Synaptic Circuitry
The gate control theory, proposed by Melzack and Wall [26], illustrates how pain signals are transmitted to higher brain areas via the spinal DH. In this theory, inhibitory SG neurons control presynaptically both large- and small-diameter fibers, presumably corresponding to, respectively, A and C fibers, and these in turn innervate the transmission system. Therefore, activation of SG neurons by large-diameter fibers attenuates signals conveyed via both fiber types to transmission system neurons, which corresponds to gate closing; in contrast, inhibition of SG neurons by small diameter fibers opens the gate for transmission of pain information. Although the theory proposes a prominent role for SG neurons in gating pain transmission, it assumes only a single type of SG neurons, which is certainly incorrect in regard to the synaptic organization of the DH [27]; rather, recent data demonstrate that different subtypes of SG neurons are present in spinal DH and that these subtypes make distinct contributions to the function of the complex synaptic network of the spinal DH. Thus, based on recent morphological and electrophysiological studies in the spinal DH, we attempt to assign various SG neurons to three functionally different SG neurons: inhibitory SG (iSG) neurons, excitatory SG (eSG) neurons, and transmission system-inhibiting (tiSG) SG neurons (Figure 1). Large-diameter A fibers directly, or indirectly through eSGs, activate tiSG neurons, whereas small-diameter C fibers necessarily activate iSG neurons to inhibit the tiSG neurons. The transmission system is composed of projection neurons that send pain information to higher brain centers. In future works, it will be important to more completely describe the synaptic organization of the DH, and it will be important to carefully define the three basic classes of SG neurons.
Figure 1.
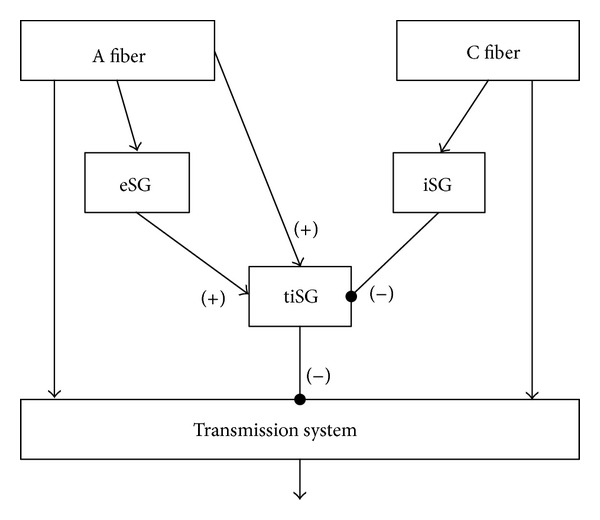
A diagram modified from the gate control theory. Both primary afferent A and C fibers directly target the transmission system that conveys the pain signals from the spinal dorsal horn to the higher brain areas. However, both fibers differentially innervate to the substantia gelatinosa (SG) neurons in the spinal DH. Although polysynaptic inputs are possible in all SG neurons from primary afferent fibers and other SG neurons, monosynaptic inputs from A fibers reach the excitatory SG neurons (eSG) and the transmission-inhibiting SG neurons (tiSG), while those from C fibers only go into the inhibitory SG neurons (iSG), not the itSG directly. The tiSG neurons receive the excitatory synaptic inputs from the eSG and the inhibitory synaptic inputs from the iSG. The main function of tiSG is inhibiting the transmission system, both presynaptically (in the gate control theory) and postsynaptically (in this diagram). In this way, the activation and inhibition of SG neurons (here, called tiSG) by large-diameter and small-diameter fibers, respectively, are possible, shown in the gate control theory.
The primary candidates for iSG neurons are γ-aminobutyric acid- (GABA-) ergic neurons in the spinal DH, an electrophysiologically heterogeneous group [28, 29] that make up approximately 30% of neurons in lamina II (SG) of the spinal cord [30]. It has been suggested, using combined single/paired whole-cell patch-clamp recordings and biocytin labeling, that ~75% of all SG neurons fall into five groups that differ in firing behavior and other electrophysiological properties and most prominently, in the structure of their dendritic arbors: islet, radial, central, medial-lateral, and vertical cells [31–36]. Among these groups, islet cells are exclusively GABAergic [37, 38] and receive monosynaptic input from C fibers and polysynaptic input from Aδ fibers [33, 35, 36]. The GABAergic nature of islet cells corresponds with the finding that, among SG neurons expressing GFP (driven by the upstream regulatory sequence of the gene encoding the GABA synthetic enzyme, glutamic acid decarboxylase (GAD) 67), 62% have islet-type morphology [29]. Because the islet cells do not directly target projection neurons [34] yet do influence the pain transmission system through other SG neuronal types, islet cells are apparently one of the major sources of SG neurons that act as iSG neurons (Figure 1).
In addition to the islet-type of SG neuron, tonically firing neurons with central-type dendritic arbors are also likely GABAergic [32]. Central neurons receiving GABAergic input from islet cells and glutamatergic input from C fibers are divided into either tonically firing neurons or transiently firing neurons [31] and are both excitatory and inhibitory [32–35]. The tonically firing central neurons synapse on vertical neurons and recurrently, on islet cells, identifying this subtype of central neuron as an additional type of iSG neuron.
As noted above, 30% of SG neurons are GABAergic so that ~2/3 of SG neurons are glutamatergic; eSG neurons are drawn from this large pool of excitatory interneurons. The eSG neurons likely include vertical and radial SG neurons because they predominantly receive monosynaptic inputs from A fibers [34, 36, 38]. In contrast, it is known that central-type SG neurons do not receive monosynaptic inputs from A fibers, thus ruling out central neurons as candidate eSG neurons.
On the other hand, considerably less is known about the types of GABAergic SG neurons that make up the tiSG circuit component, which synapse on projection neurons in lamina I and deeper laminae [7]. Neurokinin (NK) 1-positive projection neurons in laminae III-IV are known to receive inputs from GABAergic neurons that contain neuropeptide Y (NPY) [39]. Although classification of SG neuronal types by arborization pattern and electrophysiological properties remains incomplete, because NPY-expressing neurons comprise half of the GABAergic neurons of laminae I and II [40], we suggest that these NPY-positive neurons inhibit the transmission system and thus may act as tiSG neurons (Figure 1). Therefore, careful identification of the types of (1) primary afferent fibers and (2) local interneurons that send inputs to NPY-positive, GABAergic neurons, would greatly clarify the synaptic circuitry that contributes to gating of pain signals. Evidence for another type of tiSG neuron has also been obtained: “giant marginal” projection neurons, which lack NK1 receptors and express the glycine receptor-associated protein gephyrin [41], are richly contacted by GABAergic boutons that contain nitric oxide synthase (NOS) but not NPY [41]. Here again, the neuronal morphology needs to be defined for this NOS-positive neuron. Altogether, clear understanding of the synaptic circuitry underlying gating of pain signals will require a more complete description of the pattern of SG neuron connectivity with GABAergic, NPY- or NOS-positive neurons.
2.3. Synaptic Transmission
At chemical synapses in the CNS, neurotransmitters released from presynaptic nerve terminals generate graded analogue signals through the opening of ligand-gated ion channels on the plasma membrane of postsynaptic neurons. Whereas each presynaptic neuron possesses the biochemical machinery to release only one type of neurotransmitter, which can be either excitatory or inhibitory, individual postsynaptic neurons express a variety of ligand-gated ion channels that respond to different neurotransmitters so that postsynaptic neurons can, for example, exhibit both excitatory and inhibitory synaptic potentials. In the CNS, glutamate generates fast excitatory synaptic signals in postsynaptic neurons by opening any of three types of ligand-gated glutamate receptor ion channels: based on their pharmacology and structural homology, these are known as AMPA receptors (subunit: GluA1-GluA4), kainate (subunit: GluK1-GluK5) receptors, and NMDA receptors (subunit: GluN1, GluN2A-GluN2D, GluN3A, and GluRN3B) [42].
It is known from early studies of the spinal DH that glutamate-mediated fast excitatory synaptic transmission involves activation of postsynaptic ionotropic glutamate receptors [43–46]. AMPA receptors mediate the large early component of fast excitatory synaptic postsynaptic responses, whereas the more slowly opened NMDA receptors contribute only to the later component of excitatory postsynaptic responses [2, 45, 46]. Fast synaptic transmission mediated by kainate receptors is relatively small and produces slowly decaying synaptic currents [47, 48]. Although most spinal DH synapses use all three classes of ionotropic glutamate receptors, some synapses possess only NMDA receptors; these are known as “silent synapses” because, lacking AMPA-receptor-driven postsynaptic depolarization, the glutamate-activated NMDA receptors at these synapses remain blocked by Mg2+ and thus fail to generate a synaptic signal [49, 50].
Although inhibitory transmission falls outside the scope of the present review, we note that many interneurons in the spinal DH release GABA and glycine, which provide fast inhibitory transmission that is an essential feature of spinal DH circuits. Most GABAergic neurons also release glycine in the spinal DH [30, 51, 52], but GABA-mediated transmission can be distinguished from glycine-mediated transmission based on the decay kinetics of synaptic responses [53, 54].
2.4. Synaptic Plasticity
Strength of synaptic transmission in the CNS is not constant; rather, it is subject to up- or downmodulation as a consequence of patterns of presynaptic and/or postsynaptic activity. Such activity-dependent changes in synaptic strength are accomplished, in part, through long-term modulation of the properties and numbers of ion channels that mediate, affect, or respond to synaptic activity. LTP—a persistent increase in the strength of synaptic transmission [1]—can be induced by tetanic stimulation, pairing of presynaptic activity with postsynaptic depolarization, coincidence between presynaptic release of glutamate and postsynaptic depolarization, and pharmacological treatments that increase excitatory postsynaptic responsivity. Since the initial discovery of LTP, molecular and cellular mechanisms subserving this kind of plasticity have been worked out most clearly for the canonical form of NMDA receptor-dependent LTP that is found at Schaffer collateral-commissural synapses onto pyramidal neurons in the CA1 region of hippocampus. Important elements that have been identified include channel phosphorylation by protein kinases such as protein kinase A (PKA), protein kinase C (PKC), and Ca2+-calmodulin-dependent protein kinase II (CaMKII) [55–59]; consequent increases in channel opening probability and single-channel conductance [60, 61]; subunit-specific trafficking of postsynaptic AMPA receptors [62–64] to the subsynaptic membrane; and changes in glutamate release, both in probability [65] and quantal content [66] at presynaptic terminals [67]. The cellular processes most carefully worked out for hippocampal LTP [68] are generally thought to provide a fundamental basis for information processing and storage throughout CNS and particularly for learning and memory in the hippocampus [69–72].
In the spinal cord DH, early studies revealed that repetitive stimulation of dorsal root or peripheral nerve produces LTP at primary afferent synapses [2, 73, 74]. In addition to involvement of NMDA receptors and postsynaptic Ca2+ that is typical of LTP induction in hippocampus [2, 75], spinal DH studies have identified roles for NK1 [75, 76], group I metabotropic glutamate [77], and opioid [78] receptors in the induction and expression of LTP [2].
Some patterns of synaptic activity can cause a decrease in synaptic strength, referred to as “long-term depression (LTD)” [79]. This type of synaptic plasticity has also been extensively studied in various CNS regions, most particularly in the context of certain forms of information processing in the hippocampus [80, 81] and also of motor learning in cerebellum [82]. Although both high-frequency stimulation (HFS) and low-frequency stimulation (LFS) can induce LTD in the spinal DH, protein phosphatases play a role only in the induction of HFS-induced LTD [83] but not in that of LFS-LTD [84] in this area.
Because spinal LTP and LTD may play critical roles in hyperalgesia and allodynia [85, 86] and the activation of high-threshold C fibers is important to mediate many type of hyperalgesia, C fiber-mediated field potentials have been the subject of many LTP studies. LTP of C fiber-evoked field potentials is reliably produced by HFS of peripheral nerves (>3 hours), and is dependent upon the activation of NMDA receptors [75]; interestingly, LFS at C fiber intensity also induces LTP under certain conditions [87, 88]. Moreover, C fiber-mediated LTP can be induced by noxious stimulation or injury [89], revealing a contribution of this form of synaptic plasticity to induction of hyperalgesia. Although the loci of mechanisms underlying the LTP of C fiber-evoked field potential are difficult to clarify, the induction and/or maintenance of this type of LTP involve many channels and signaling molecules, including NMDA and NK1 receptors [75], N-type and P/Q-type VGCCs [90], TRPV1 channels [91], the EphB receptor tyrosine kinase [92], ryanodine receptors [93, 94], nitric oxide [95, 96], and many inflammatory agents [97].
Spinal LTP has also been studied using single (whole) cell recordings of primary afferent stimulation-evoked excitatory postsynaptic potentials or currents (EPSPs and EPSCs, resp.). In this case, Aδ fiber- and C fiber-mediated synaptic responses can be distinguished according to stimulus intensity and conduction velocity, which is advantageous in elucidating primary afferent-dependent mechanisms [87, 98, 99] or the locus of induction of LTP [93, 100]. In addition, whole cell recordings are advantageous for study of LTP in particular types of DH neurons. Combining whole cell recording from DH neurons with injection of retrograde tracers (e.g., DiI) into the parabrachial (PB) area or periaqueductal gray (PAG) has allowed researchers to determine that HFS induces LTP of C fiber-mediated EPSCs in lamina I neurons that project to PB area but not in those that project to PAG [87]; in contrast, LFS induces LTP in lamina I neurons that project to PAG area. LTP of C fiber-EPSCs by HFS in the PB-projecting lamina I neurons requires NK1 receptor-mediated signaling and activation of T-type VGCCs [99]. LFS-induced LTP of C fiber EPSCs in the PAG-projecting lamina I neurons requires nitric oxide signals [87, 93], the latter generated in response to intracellular Ca2+ rises that are slower in onset and prolonged in comparison to the kinetics of Ca2+ rises required to elicit LTP in PB-projecting lamina I neurons [87].
In CA1 of hippocampus, LTP of EPSCs can be alternatively induced by the coordinated activity of presynaptic fibers and postsynaptic neurons, which is characteristic of spike-timing [101] or pairing protocols [102] in the hippocampus. The spike-timing protocol requires low-frequency postsynaptic spikes timed within 10 ms of the onset of synaptic responses, while the pairing protocol involves persistent postsynaptic depolarization (0~+30 mV) during repetitive low-frequency presynaptic stimulation. These protocols have been applied to study LTP of EPSCs in the spinal DH as well, in an effort to overcome the low rate of success (~50%) observed for the induction of LTP by HFS [2]. Spike-timing dependent LTP is interestingly dependent on expression of the GluK2 (previously known as GluR6) kainate receptor subunit [48], along with activation of NMDA receptors and elevation of intracellular Ca2+ [103]. In spinal DH, LTP of EPSCs by the pairing protocol requires activation of extracellular signal-regulated kinase (ERK) [104].
Considering the mixture of excitatory and inhibitory interneurons and the complex synaptic circuitry in lamina II of the spinal DH (see above), the contribution of LTP to hyperalgesia will necessarily depend upon which synapses in the DH circuitry specifically undergo LTP. It is therefore critical that LTP and its pathophysiological role in pain hypersensitivity be pursued at identified types of SG neuron synapses. For example, hyperalgesia may be produced by LTP of excitatory interneurons that synapse upon neurons that, in turn, project to brain areas involved in nociception. Alternatively, hyperalgesia may be reduced by LTP of synapses onto inhibitory interneurons that target projection neurons. Hence, careful identification of the specific subtype of neuron studied will be essential to better understand the roles of spinal DH LTP in hyperalgesia and allodynia [85].
3. Contribution of Ionotropic Glutamate Receptors to LTP in the Spinal DH
3.1. AMPA Receptors
AMPA receptors consist of homo- and heterotetrameric assemblies of GluA1, 2, 3, and 4 subunits, with different assemblies of AMPA receptor subunits exhibiting distinct functional behaviors [42, 105]. Among the GluA subunits, transcripts encoding GluA2 are subject to RNA editing at position 586, which results in replacement of the neutral glutamine (Q) residue found in all other GluA subunits with a positively charged arginine (R). Position 586 is located in transmembrane segment 2 (M2), which forms the lining of the ion permeation pathway through the receptor; an arginine at this position decreases the receptor's Ca2+ permeability and also confers linear current-voltage behavior. AMPA receptors lacking GluA2 subunits are significantly more Ca2+-permeable, exhibiting a Ca2+ permeability ratio (P Ca/P Na) of 3, and they also display strong inward rectification in their current-voltage relationships [105, 106].
Because AMPA receptors are the main mediators of excitatory synaptic transmission in the CNS, they are generally considered as the final target for induction and expression of LTP, rather than as inducers or regulators. Thus, principal endpoints in LTP are phosphorylation and trafficking of specific AMPA receptor subunit subtypes, along with changes in AMPA receptor conductance [42, 105]. The GluA1 subunit, for example, can undergo phosphorylation of Ser831 by CaMKII [107] and PKC [108] and of Ser845 by PKA [108], which contributes to induction of LTP by changing the open probability and single-channel conductance of AMPA receptors containing this subunit [42]. In regard to membrane trafficking of AMPA receptors, the induction mechanism for LTP in hippocampal CA1 pyramidal neurons [64] includes increased incorporation of GluA1/GluA2-containing AMPA receptors into the synaptic surface membrane [62, 109]; however, subsequent work suggests that the newly incorporated AMPA receptors are in fact homotetrameric GluA1 complexes [110]. In accordance with these studies, the induction of LTP at hippocampal CA1 synapses is impaired in mice deficient in the GluA1 subunit [111]. Surface membrane incorporation of homomeric GluA1 receptors may result in the replacement of preexisting GluA2-containing AMPA receptors, thereby increasing the net Ca2+-permeability of the AMPA receptor population in the postsynaptic surface membrane [112]. Increased Ca2+ influx via GluA2-lacking, Ca2+-permeable AMPA receptors, is directly related to enhancement of LTP [113, 114].
Trafficking of AMPA receptors requires their interaction with transmembrane AMPA receptor regulatory proteins (TARPs) [115, 116]. Interaction of TARPs with AMPA receptors prevents AMPA receptor degradation [117], and subsequent interaction of AMPA receptors with PSD-95 results in translocation of AMPA receptors from the perisynaptic region into synaptic sites [118]. In contrast to these studies, a recent study has found that the GluA1 C-terminal tail, critical for GluA1 trafficking [109, 110], is not required for LTP [119]. This has led to the suggestion that a reserve pool of AMPA receptors, regardless of their subunit composition, is relied upon for LTP. Further studies are needed to provide a more comprehensive picture of the mechanism and role of AMPA receptor trafficking in hippocampal LTP. In addition, studies of this process in spinal DH LTP remain to be carried out.
Ca2+-permeable AMPA receptors are expressed in inhibitory interneurons [120] of lamina I and the outer layer of lamina II [121], the laminae which receive synaptic input primarily from nociceptive C fiber afferents. Ca2+-permeable AMPA receptors in layer I and II DH neurons are activated by synaptic input [122], raising the possibility that these channels play a special role in mediating sensory input by unmyelinated fibers [123]. Using GluA2 knockout mice, it has been shown that Ca2+-permeable AMPA receptors enhance HFS-evoked LTP and mediate induction of NMDA receptor-independent LTP at primary afferent-DH neuron synapses [98], which suggests that Ca2+-permeable AMPA receptors contribute significantly to LTP in the spinal DH and may substitute for NMDA receptors in LTP induction. In contrast to NMDA receptors, Ca2+-permeable AMPA receptors allow Ca2+ influx at resting membrane potential, a potential merit for induction of synaptic plasticity.
3.2. NMDA Receptors
Functional NMDA receptors are heterotetrameric assemblies composed of two GluN1 subunits and either two GluN2 subunits or a combination of GluN2 and GluN3 subunits [42]. The glutamate binding sites are located in the GluN2 subunits [124] and the glycine binding sites in the GluN1 and GluN3 [124–126]. NMDA receptors are characterized by their high permeability to Ca2+ [127], voltage-dependent block by Mg2+ [128], and slow “activation/deactivation” kinetics [129]. NMDA receptor alternative splice variants exhibit subtle differences in functional properties, thereby fine-tuning the behavior of NMDA receptors in which they are incorporated [130]. For example, NMDA receptors containing GluN2A or GluN2B subunits display high-conductance channel openings and a high sensitivity to block by extracellular Mg2+, whereas receptors composed of GluN2C or GluN2D subunits show low-conductance openings and lower sensitivity to Mg2+. Moreover, GluN1/GluN2A-containing NMDA receptor currents deactivate rapidly (time constant of tens of milliseconds), whereas GluN1/GluN2D-containing NMDA receptor currents deactivate very slowly (time constant of several seconds) [131–133]. In addition, GluN3 can also coassemble with GluN1 [134–136] to form uniquely excitatory glycine receptors [136]. These distinctive properties may provide particular NMDA receptor subtypes with specific roles in excitatory synaptic transmission/plasticity and pathology.
The role of NMDA receptors in the induction of LTP is well established for various brain synapses, particularly the Schaffer collateral input to CA1 pyramidal neurons in hippocampus [68–70]. The activation requirements for NMDA receptors—agonist (glutamate) binding and postsynaptic depolarization—are well-matched to the “Hebbian” properties of LTP induction, namely, specificity, associativity, and cooperativity [69]. Further, several recent studies show that a proper subunit composition is essential for the induction [137–139] and expression [140, 141] of LTP. These results reflect the fact that specific NMDA receptor subunits are differentially phosphorylated by various protein kinases, such as src [142, 143] and also differentially interact with other accessory and regulatory proteins [144]. In keeping with the notion of a proper NMDA subunit composition in LTP, the association of active CaMKII with GluN2B is likely required for the induction of canonical LTP at Schaffer collateral synapses on CA1 neurons [145]. Downstream of these regulatory processes, NMDA receptor-dependent protein synthesis [146–148] is needed for the expression of LTP that persists beyond ~4 hours, referred to as late-LTP [149].
In the spinal DH, in situ hybridization or immunostaining has revealed high expression of GluN1 and GluN2D [150, 151] and lower levels of GluN2A and GluN2B [152, 153]. Electrophysiological measurements of conductance ratio have shown that lamina II GABAergic interneurons express both the GluN2A/GluN2B- and GluN2C/GluN2D-containing NMDA receptors, while excitatory lamina II interneurons express primarily GluN2A/GluN2B-containing receptors [154]. In addition, outside-out patch recordings of single channel currents have shown that, at least in extrasynaptic regions, both GluN1/GluN2B (high conductance; 57 pS) and GluN1/GluN2D (low conductance; 44 pS and 19 pS) are present on spinal DH neurons [155].
In the spinal DH, induction of nearly all forms of LTP is dependent on the NMDA receptors [86]. An early report showed that HFS (100 Hz) of primary afferent fibers at C fiber-activating intensity induces LTP of EPSPs in transverse spinal cord slices in vitro; LTP was absent in the presence of the NMDA receptor antagonist D-2-amino-5-phosphonovalerate (D-AP5) [2]. In addition, LTP induction at C fiber synapses also requires activation of NMDA receptors [73, 87, 99]. Recently, LFS (2 Hz at C-fiber intensity) of sciatic nerve has been shown to induce LTP of C fiber-evoked field potentials. This LFS-induced LTP is also prevented by an NMDA receptor antagonist, MK-801 in these experiments [156]. As expected, the noble anesthetic gas xenon, which has an inhibitory effect on NMDA receptors [157], prevents induction of LTP at C fiber synapses in intact rats [158]. LTP can also be induced by chemical means, for example, by perfusion of spinal cord slices with NMDA (+ postsynaptic depolarization) [159] or by perfusion of spinal cord segments with NMDA in spinalized, deeply anesthetized adult rat [75]. Taking together, these findings indicate that the NMDA receptor is required for induction of LTP in synapses of primary afferent fibers onto spinal DH neurons.
3.3. Kainate Receptors
Kainate receptors are tetramers assembled from combinations of five different types of subunits, termed GluK1-5 (formerly, GluR5-7 and KA1-2) [42, 105, 106, 160]. Each kainate receptor monomer possesses a ligand-binding site and a distinctive amino acid sequence that forms the channel lumen. Radioligand binding assays indicate that GluK1, 2, and 3 contribute to low-affinity kainate binding sites (KD of 50–100 nM) [161], whereas GluK4 and 5 form high-affinity kainate binding sites (KD of ~4–15 nM) [162, 163]. Structural variability of kainate receptors is conferred by alternative splicing and RNA editing [160]. Alternative splice variants have been found exclusively for GluK1 (GluK1-1, 1-2a, 1-2b, and 1-2c) [164] and GluK3 (GluR3a and 3b) subunits [165] in rat; however, the mouse GluK2 exists as two splice variants that differ in their C-terminal domains [166]. RNA editing, as for GluA2 subunits, posttranscriptionally modifies a Q/R site in the M2 segment of GluK1 and GluK2 subunits. The Q-to-R substitution in GluK2 homomeric kainate receptors decreases Ca2+ permeability [167, 168] and increases Cl− permeability [169], reduces unitary conductance, and transforms channels from inwardly rectifying to linear or slightly outwardly rectifying. Mice deficient in Q/R editing in GluK1 have been found to exhibit a reduction in kainate receptor-mediated currents in DRG neurons [170], although the responses of these animals to painful stimuli are unaffected. Besides the Q/R editing site, two additional positions prone to RNA editing have been identified in the GluK1 subunit: an isoleucine (I)/valine (V) site and a tyrosine (Y)/cysteine (C) site [171], both in the M1 segment. Although the additional editing sites may modulate Q/R site control of Ca2+ permeability, the mechanism of interaction among the three editing sites remains to be elucidated.
For NMDA receptor-independent LTP at the mossy fiber-CA3 synapse in hippocampus, there is disagreement regarding the role of kainate receptors [172]. A selective antagonist for GluK1-containing kainate receptors, LY382884, blocks the induction of mossy fiber LTP [173, 174], but conflictingly, mossy fiber LTP can be elicited in the presence of the AMPA/kainate receptor antagonist, CNQX [175, 176]. Knockout mice deficient in GluK2 subunits [177] display reduced mossy fiber LTP, but mice deficient in GluK1 possess normal LTP. Although much more work is needed, the results point to a potential role for kainate receptors in mossy fiber LTP, specifically in the NMDA receptor-independent form of LTP.
For synapses of primary afferents onto spinal DH neurons, fast EPSCs have been shown to be mediated by postsynaptic kainate receptors [47]. Kainate receptor-mediated EPSCs are much smaller in peak amplitude and slower in decay kinetics than those mediated by AMPA receptors. To date, the kainate receptor subunits mediating synaptic transmission have not been well-characterized. However, low expression of GluK1 subunits, moderate expression of GluK3 and GluK4, and strong expression of GluK5 have been found for spinal DH neurons; no expression of GluK2 has been detected [150, 178]. Despite the apparent absence of GluK2 subunits from DH neurons, kainate receptor-mediated whole cell current and synaptic potentials recorded from spinal DH neurons are significantly decreased in GluK2 mutant mice [48]. In addition to expression on postsynaptic membrane in spinal DH neurons, kainate receptors are also expressed on DRG neurons, including primary afferent presynaptic terminals within the DH [179, 180]. All kainate receptor subtypes are present in DRG neurons, with GluK1 expressed at an especially high level [178, 181–183]. GluK1- or GluK2-containing presynaptic kainate receptors modulates glutamatergic transmission at Aδ and C-fiber primary afferent-activated synapses in the spinal SG [184]. Interestingly, induction of LTP is impaired in GluK2 knockout mice, while the late phase of LTP is impaired in GluK1 mutant mice [48], indicating differential involvement of kainate receptor subunits in LTP of spinal DH neurons.
4. Voltage-Gated Ca2+ Channels That Contribute to LTP in Spinal DH and to Pain
Although receptors for L-glutamate, most commonly the NMDA receptor subtype, mediate induction and expression of LTP at many synapses in the brain, some forms of LTP at hippocampal CA1 synapses, such as late-phase LTP (L-LTP), require activation of L-type VGCCs. Other VGCCs are also involved in diverse ways in LTP, as discussed below.
VGCCs consist of a pore-forming transmembrane α 1 subunit and the auxiliary β subunit and α 2 δ subunit [185]. Based on sequence homology, the ten different α 1 subunits of VGCCs are grouped into three subfamilies: two high-voltage activated subfamilies, CaV 1-2, and one low-voltage activated family, CaV3 [186]. The CaV1 subfamily carries L-type Ca2+ current, and the family members are CaV1.1 (α 1S), CaV1.2 (α 1C), CaV1.3 (α 1D), and CaV1.4 (α 1F). The CaV2 subfamily includes CaV2.1 (α 1A), CaV2.2 (α 1B), and CaV2.3 (α 1E) which correspond to P/Q-type, N-type, and R-type Ca2+ currents, respectively. The CaV3 subfamily carries T-type Ca2+ currents, and the family members are CaV3.1 (α 1G), CaV3.2 (α 1H), and CaV3.3 (α 1I) [186]. The channel's auxiliary subunits are also organized into subfamilies, and these specifically affect membrane trafficking and expression of channels, voltage-dependence of channel opening, inactivation kinetics, and sensitivities to inhibitors, thus greatly expanding the number of different subtypes of VGCCs [185, 187]. In this section, we will discuss the contribution of each major type of VGCC to LTP in the spinal DH to various forms of pain in normal and pathological states.
4.1. L-Type VGCCs
4.1.1. Contribution to LTP
L-type VGCCs are widely expressed in the CNS [188], including CA1 of the hippocampus, the preeminent region for investigations of LTP. The dendritic localization of L-type VGCCs in the CA1 area [189] implies that their activation contributes to Ca2+ signals in dendritic spines, an important step for the induction of LTP [190]. Although induction of canonical LTP in CA1 relies upon Ca2+ flux through NMDA receptors on dendritic spines and subsequent activation of Ca2+-dependent second messengers [70], several other forms of LTP in fact require activation of L-type VGCCs, and not NMDA receptors. In the hippocampal CA1 area, for example, HFS at 200 Hz [191], (higher frequency than the 100 Hz tetani typically employed for induction of NMDA receptor-dependent LTP) generates LTP that is insensitive to the NMDA receptor antagonist D-AP5 but is blocked by the L-type VGCC antagonist, nifedipine. While NMDA receptor-dependent LTP is inhibited by antagonists of serine-threonine kinases, the 200 Hz-induced, L-type VGCC-dependent LTP is blocked by antagonists of tyrosine kinases [192]. In addition, prolonged theta burst stimulation (TBS) in the CA1 area induces a form of LTP that is dependent upon L-type VGCCs [193]. L-type VGCC-dependent LTP can also be produced by application of the potassium channel blocker, tetraethylammonium (TEA) [194, 195]. Interestingly, the mechanism of induction of this type of LTP partially overlaps that of NMDA receptor-dependent LTP, particularly in regard to the timing and intensity of postsynaptic Ca2+ signals [195]. Extracellular matrix molecules, such as hyaluronic acid and tenascin-R, are important in the development of L-type VGCC-dependent LTP induced by either TBS [196] or TEA [197]. As for Schaffer collateral-CA1 pyramidal neuron synapses, L-channel-dependent LTP has been described at mossy fiber synapses onto CA3 pyramidal neurons [198] and for thalamic inputs to amygdala [199]. Induction of NMDA receptor-independent, L-channel-dependent LTP is distinctive in its reliance upon such electrical phenomena as dendritic Ca2+ spikes [200–202].
In spinal DH, although L-type VGCCs are known to be expressed in soma, dendrites, and axon terminals of neurons [203, 204], it appears that L-channels do not induce LTP during 100 Hz repetitive stimulation [98]. L-type VGCCs contribute instead to an alternative form of LTP in spinal lamina I neurons, one that is induced by postsynaptic depolarization without presynaptic stimulation (“non-Hebbian” LTP) [205]. Gabapentin, which binds to the α 2 δ subunit of VGCCs and is used to relieve neuropathic pain, does not affect C fiber-mediated basal transmission or LTP induction but does reduce C fiber-mediated transmission during the maintenance phase of LTP [206]. Thus, postsynaptic Ca2+ influx through L-type VGCCs may be critical to induce or express LTP of excitatory synaptic transmission in certain normal and/or pathological states. More extensive investigation of distinct types of LTP induced under normal or neuropathological conditions is clearly needed to better understand the contribution of L-type VGCCs to synaptic plasticity and neuropathic pain of the spinal DH.
4.1.2. Contribution to Pain
Implication of L-type VGCCs in acute and chronic pain has been controversial. Some reports show that spinal administration of L-type VGCC blockers decreases pain sensitivity to acute innocuous or noxious stimuli [207, 208], but other work has found no effect of these blockers in the hot plate test [209] or in other tests using acute mechanical and thermal stimuli [210, 211]. Furthermore, in a chronic pain model of peripheral nerve injury, intrathecal administration of a high dose of the L-type VGCC blocker, diltiazem, has no effect on paw withdrawal in response to mechanical stimulation [212].
Recently, however, it has been found that prolonged intrathecal administration of the L-channel blocker nicardipine elevates mechanical threshold in a neuropathic pain model [213], indicating the involvement of L-type VGCCs in mechanical allodynia caused by peripheral nerve injury. Along the same lines, reduced expression of L-type VGCCs in spinal DH by antisense [213] or microRNA [214] technologies suppresses the hypersensitivity of DH neurons following peripheral nerve injury. Taken together, these findings indicate that L-type VGCCs can contribute to some components of acute or chronic pain behaviors produced by tissue damage, likely reflecting the contribution of L-type VGCCs to certain forms of LTP in the spinal DH.
4.2. P/Q-Type VGCCs
4.2.1. Contribution to LTP
P/Q-type VGCCs are expressed in a subpopulation of DRG neurons [203, 215, 216] that does not respond to capsaicin and rarely expresses substance P, a marker for small high-threshold primary afferent terminals [203]; P/Q channels thus play only a small role in the control of glutamate release from small diameter, peptidergic nociceptive primary afferent fibers. In addition, it has been suggested that P/Q-type VGCCs are expressed in fewer numbers of primary afferents than are N-type VGCCs [203, 217, 218]. In the spinal DH, P/Q-type VGCCs are expressed in the laminae II–VI [203] and are preferentially involved in inhibitory neurotransmission [219, 220], indicating a limited contribution of P/Q-type VGCCs to excitatory synaptic transmission in the spinal DH.
Blockers of P/Q-type VGCCs strongly suppress induction of LTP for C fiber-evoked field potentials [90], suggesting that induction of this form of spinal DH LTP may rely in part upon P/Q-type VGCCs [221]. Similarly, in visual cortical neurons, P/Q-type VGCCs have also been proposed to contribute to the induction of LTP at the inhibitory synapses [222].
4.2.2. Contribution to Pain
In accord with the minimal contribution of P/Q-type VGCCs to glutamate release from small-diameter, high-threshold primary afferents [203], intrathecal administration of the selective P/Q channel blocker, ω-agatoxin IVA, has little or no effect on C- or Aδ fiber-mediated nociceptive transmission [223] or in tests of mechanical and thermal thresholds in neuropathic pain models [212, 224]. However, development of hyperalgesia or pathological pain is prevented by intrathecal pretreatment with blockers of P/Q-type VGCCs [209–211], as well as in animals with either a genetic deficiency [225] or spontaneous mutation [226] in P/Q-type VGCCs. These observations correlate with studies of spinal LTP that indicate a critical role for P/Q-type VGCCs in the induction of LTP of C fiber-evoked field potentials [90]. Therefore, as for L-type VGCCs, P/Q-type VGCCs may also be involved in the development or regulation of certain forms of chronic pain.
4.3. N-Type VGCCs
4.3.1. Contribution to LTP
N-type VGCCs are expressed in dorsal root ganglia, as well as in primary afferent nerve terminals in the superficial area (laminae I-II) of DH [203, 217]. In accord with these findings, glutamatergic transmission between DRG and spinal DH neurons is blocked by ω-conotoxin GVIA, a selective blocker of N-type VGCCs [227]. Many of the presynaptic nerve terminals with N-type VGCC immunoreactivity also contain substance P, suggesting that N-type channels also support the release of substance P and CGRP from peptidergic, high-threshold C fibers in the spinal DH [228, 229].
As for other VGCCs, the involvement of N-type VGCCs in synaptic plasticity, particularly LTP, appears to be specific to the synaptic pathway and induction protocol. An early study showed that when a 100 Hz induction protocol is used, N-type VGCCs are not involved in LTP of the hippocampal mossy fiber-mediated CA3 pathway [201]. However, at hippocampal CA3-to-CA1 Schaffer collateral synapses, when TBS or 200 Hz HFS is used to induce LTP, N-channel-mediated component of excitatory transmission can be identified after induction of LTP [230]. For perforant path synapses onto CA1 neurons, induction of LTP by 200 Hz HFS relies upon an increased contribution from presynaptic N-type VGCCs [231]. In the spinal DH, the N-channel blocker ω-conotoxin GVIA does not prevent induction of LTP of C fiber-field potentials, but this N channel antagonist inhibits synaptic transmission once LTP has been induced [90]. These results indicate that presynaptic N-type channels contribute to the maintenance phase of LTP in the spinal DH [231].
4.3.2. Contribution to Pain
Implication of N-type VGCCs in acute and chronic pain states correlates with the fact that N-type channels are predominantly expressed in DRG neurons, particularly small-diameter peptidergic DRG neurons and the fact that N channels may control release of neurotransmitters such as glutamate and substance P onto spinal DH neurons [228, 229]. Antagonists of N-type channels block the release of glutamate, substance P, and CGRP in the spinal DH [229, 232, 233], and in various acute, inflammatory, and neuropathic pain models, produce antinociceptive and analgesic effects [234, 235]. In addition, genetic ablation of CaV2.2 (α 1B), the pore-forming subunit of N-type channels, significantly reduces mechanical allodynia and thermal hyperalgesia in a neuropathic pain model with spinal nerve ligation [236]. Moreover, the antinociceptive effects of N-channel antagonists are enhanced in chronic pain states [237, 238], as N-type channels are upregulated after peripheral nerve injury [239]. Interestingly, these findings correlate with the observation that the block of N-channels inhibits synaptic transmission once LTP has been established [90].
Even though neuropathic pain can be reduced by blockers of N-type channels, and blockers such as ω-conotoxin MVIIA (SNX-111, ziconotide, or Prialt) have received approval by the FDA for the treatment of chronic pain; N-channel blockers are of limited use owing to side effects attributable to the fact that almost all of the presynaptic terminals in the brain express the N-type VGCCs. Thus, N-channel antagonists must be administered intrathecally, an invasive method used when other pain management options have failed. It may therefore prove useful to develop N-channel blockers that are either specific to particular CaV1.2 α 1B splice variants [240, 241] or that modulate N-type channel in selective neuronal subtypes [242].
4.4. T-Type VGCCs
4.4.1. Contribution to LTP
Although the lack of a specific antagonist prevents clean isolation of the contribution of T-type VGCCs to LTP, there is evidence indicating that T-type VGCCs are involved in the induction and/or expression of LTP in hippocampal CA1 [243] neurons and dentate gyrus [244]. The late phase of LTP in the CA1 area, which can be induced by 100 Hz HFS and is dependent on NMDA receptors, is not maintained (<120 min) in CaV3.2 T-type VGCC knockout mice [243]. LTP induction in dentate gyrus is sensitive to Ni2+, a blocker of T-type as well as R-type VGCCs, and is dependent on the induction protocol: the Ni2+-sensitive component of LTP can be induced by pairing of 1 Hz presynaptic stimulation with postsynaptic depolarization, but not by 100 Hz HFS [244]. T-type VGCCs may also be involved in LTP that is induced by TBS [245] or TEA [246] in the CA1 area, although L-channels have alternatively been reported to mediate TEA-induced LTP [195]. T-type VGCCs in CA1 are implicated in the enhancement of LTP, rather than its induction or expression, with the mechanism involving muscarinic acetylcholine receptors and phospholipase C-mediated K+ channel inhibition [247].
T-type VGCCs are expressed in the superficial DH of the spinal cord, as well as in medium- and small-diameter DRG neurons [248], raising the possibility of both pre- and postsynaptic contributions to synaptic plasticity at primary afferent-DH neuron synapses. T-type Ca2+ currents have been reported in spinal DH neurons [223, 249–253]. In addition, the contribution of T-type Ca2+ channels to LTP of C fiber-initiated EPSCs in lamina I neurons that project to the PB area has been demonstrated [99], suggesting that T channels may play a significant role in amplification of pain signals via their contribution to spinal LTP. On the other hand, LFS-induced LTP of C fiber-EPSCs in PAG-projecting lamina I neurons [87] likely involves T-type VGCCs [254]. These results underscore the idea, once again, that spinal LTP is cell type-specific.
4.4.2. Contribution to Pain
The involvement of T-type VGCCs in pain is likely specific to T-channel subtypes. Nociceptive responses induced by nerve injury are decreased after knock down of CaV3.2, but not of CaV3.1 or CaV3.3 [255], presumably reflecting the abundance of CaV3.2 in DRG neurons [255–257] and indicating that the T-channel subtype involved in spinal LTP may be CaV3.2. In support of this interpretation, genetic knockout of CaV3.2 attenuates behavioral responses to noxious stimuli such as formalin [258]. In contrast, knockout of CaV3.1 causes hypersensitivity to noxious visceral stimuli, but this involves a supraspinal mechanism [259]. Therefore, although some nonselective T-type blockers such as ethosuximide or mibefradil can reverse both tactile hypersensitivity and thermal hyperalgesia in various pain models [260, 261], development of subtype-specific antagonists of T-type channels is desirable. In this regard, downregulation of T-type channel activity and of hyperalgesia by oxidizing agents [262, 263] or by lowering levels of the endogenous gasotransmitter hydrogen sulfide [264, 265] may prove useful as leads in developing novel subtype-selective T-channel drugs for the treatment of inflammatory or neuropathic pain.
4.5. R-Type VGCCs
4.5.1. Contribution to LTP
The involvement of R-type VGCCs in LTP can be difficult to isolate, in part because a commonly-used R-type channel blocker Ni2+ is also an effective blocker of T-type channels. However, there is strong evidence that R-type channels support a presynaptic form of LTP found at parallel fiber synapses onto Purkinje cells in the cerebellum [266]. Because cerebellar granule cells (which give rise to parallel fibers) do not express T-type channels, in this system, the effects of Ni2+ block of Ca2+ current can be entirely attributed to antagonism of R-type channels. In comparison to N- and P/Q-channel-mediated Ca2+ influx at parallel fiber terminals, R-type channels contribute only modestly to bulk changes in intracellular Ca2+, suggesting that R-channel Ca2+ microdomains in presynaptic terminals are important for the induction of parallel fiber LTP [266].
On the postsynaptic side, R-type VGCCs in CA1 pyramidal neurons contribute to Ca2+ influx evoked by TBS-triggered, back-propagating dendritic action potentials [267, 268]. In turn, R-channel Ca2+ influx in distal dendrites of CA1 pyramidal neurons helps generate plateau potentials that are critical for perforant path LTP [269].
Although R-type VGCCs are expressed in a subpopulation of DRG neurons [270], it is unclear whether the primary afferent terminals or spinal DH neurons bear R-type channels. In addition, whether R-type channels are involved in synaptic plasticity in the spinal DH remains to be determined.
4.5.2. Contribution to Pain
There is evidence that R-type VGCCs are involved in the transmission and processing of inflammatory and neuropathic pain information. SNX-482, an inhibitor of R-type VGCCs (and less potently of L-type channels) [271], decreases nociceptive responses during the second phase of the formalin test [217] and inhibits neuropathic pain behavior [272]. In addition, studies using CaV2.3 knockout mice suggest a contribution of R-type VGCCs to pain transmission [217, 273].
5. Concluding Remarks
Many studies have attempted to elucidate rules and signaling mechanisms for synaptic plasticity, particularly LTP, in the spinal DH. Together, these studies show that LTP in the spinal DH shares many features with LTP in the hippocampus and with “central sensitization” in the spinal DH during hyperalgesia [274]. In this review, we have considered how inter-relationships between synaptic circuitry in the spinal DH, ionotropic glutamate receptors, voltage-gated Ca2+ channels, and induction/expression of LTP in the spinal DH are together involved in pain hypersensitivity.
Up to the present time, the complexity of synaptic circuitry in the spinal DH has hampered the understanding of LTP mechanisms in spinal DH. Generally, thorough classification of postsynaptic neurons and presynaptic fibers remains to be worked out. Although presynaptic fiber type can be identified during electrophysiological recording based upon conduction velocity and stimulus intensity, selective stimulation of a single class of primary afferent fiber remains challenging, because the range of stimulus intensities that activate C fibers overlaps the range of intensities that activates A fibers. Postsynaptically, in the spinal DH, although multiple morphological and electrophysiological criteria are available to distinguish neuronal subtypes, studying synaptic plasticity in a single type of spinal postsynaptic neuron has yet to be achieved, owing to inhomogeneities in physiological behavior, neurotransmitters, and cellular markers even within a group of neurons that carries out a similar function, such as the lamina I projection neurons [7].
In the future, progress in this field will likely rely upon studies that make use of powerful new experimental approaches, such as combining transgenic means to identify postsynaptic neurons [29, 32] and presynaptic fibers [25] with optogenetic tools to selectively activate specific fiber types [275]. This kind of approach will make it possible to study LTP at synapses between specific types of primary afferent fibers and spinal DH neurons or between specific spinal DH neurons, thereby facilitating the correlation between mechanisms of LTP and nociception in the spinal DH.
Acknowledgments
This work was supported by Kyungpook National University Research Fund, 2012, (2013, 2014) and US NIH Grant R01 HL088548.
References
- 1.Bliss TVP, Lomo T. Long lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. Journal of Physiology. 1973;232(2):331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Randić M, Jiang MC, Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. Journal of Neuroscience. 1993;13(12):5228–5241. doi: 10.1523/JNEUROSCI.13-12-05228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rexed B. The cytoarchitectonic organization of the spinal cord in the cat. Journal of Comparative Neurology. 1952;96(3):414–495. doi: 10.1002/cne.900960303. [DOI] [PubMed] [Google Scholar]
- 4.Molander C, Xu Q, Grant G. The cytoarchitectonic organization of the spinal cord in the rat. I. The lower thoracic and lumbosacral cord. Journal of Comparative Neurology. 1984;230(1):133–141. doi: 10.1002/cne.902300112. [DOI] [PubMed] [Google Scholar]
- 5.Laird JMA, Cervero F. A comparative study of the changes in receptive-field properties of multireceptive and nocireceptive rat dorsal horn neurons following noxious mechanical stimulation. Journal of Neurophysiology. 1989;62(4):854–863. doi: 10.1152/jn.1989.62.4.854. [DOI] [PubMed] [Google Scholar]
- 6.McClung JR, Castro AJ. Rexed’s laminar scheme as it applies to the rat cervical spinal cord. Experimental Neurology. 1978;58(1):145–148. doi: 10.1016/0014-4886(78)90130-9. [DOI] [PubMed] [Google Scholar]
- 7.Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nature Reviews Neuroscience. 2010;11(12):823–836. doi: 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Light AR, Perl ER. Reexamination of the dorsal root projection to the spinal dorsal horn including observations on the differential termination of coarse and fine fibers. Journal of Comparative Neurology. 1979;186(2):117–131. doi: 10.1002/cne.901860202. [DOI] [PubMed] [Google Scholar]
- 9.Light AR, Perl ER. Spinal termination of functionally identified primary afferent neurons with slowly conducting myelinated fibers. Journal of Comparative Neurology. 1979;186(2):133–150. doi: 10.1002/cne.901860203. [DOI] [PubMed] [Google Scholar]
- 10.LaMotte C. Distribution of the tract of Lissauer and the dorsal root fibers in the primate spinal cord. Journal of Comparative Neurology. 1977;172(3):529–561. doi: 10.1002/cne.901720308. [DOI] [PubMed] [Google Scholar]
- 11.Lynn B. Effect of neonatal treatment with capsaicin on the numbers and properties of cutaneous afferent units from the hair skin of the rat. Brain Research. 1984;322(2):255–260. doi: 10.1016/0006-8993(84)90115-x. [DOI] [PubMed] [Google Scholar]
- 12.Lynn B, Carpenter SE. Primary afferent units from the hairy skin of the rat hind limb. Brain Research. 1982;238(1):29–43. doi: 10.1016/0006-8993(82)90768-5. [DOI] [PubMed] [Google Scholar]
- 13.Ralston HJ, Ralston DD. Identification of dorsal root synaptic terminals on monkey ventral horn cells by electron microscopic autoradiography. Journal of Neurocytology. 1979;8(2):151–166. doi: 10.1007/BF01175558. [DOI] [PubMed] [Google Scholar]
- 14.Beal JA, Bicknell HR. Primary afferent distribution pattern in the marginal zone (lamina I) of adult monkey and cat lumbosacral spinal cord. Journal of Comparative Neurology. 1981;202(2):255–263. doi: 10.1002/cne.902020210. [DOI] [PubMed] [Google Scholar]
- 15.Nagy JI, Hunt SP. The termination of primary afferents within the rat dorsal horn: evidence for rearrangement following capsaicin treatment. Journal of Comparative Neurology. 1983;218(2):145–158. doi: 10.1002/cne.902180203. [DOI] [PubMed] [Google Scholar]
- 16.Gobel S, Falls WM, Humphrey E. Morphology and synaptic connections of ultrafine primary axons in lamina I of the spinal dorsal horn: candidates for the terminal axonal arbors of primary neurons with unmyelinated (C) axons. Journal of Neuroscience. 1981;1(10):1163–1179. doi: 10.1523/JNEUROSCI.01-10-01163.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawson SN, Crepps BA, Perl ER. Relationship of substance P to afferent characteristics of dorsal root ganglion neurones in guinea-pig. Journal of Physiology. 1997;505(1):177–191. doi: 10.1111/j.1469-7793.1997.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molliver DC, Wright DE, Leitner ML, et al. IB4-binding DRG neurons switch from NGF to GDNF dependence in early postnatal life. Neuron. 1997;19(4):849–861. doi: 10.1016/s0896-6273(00)80966-6. [DOI] [PubMed] [Google Scholar]
- 19.Michael GJ, Averill S, Nitkunan A, et al. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. Journal of Neuroscience. 1997;17(21):8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silverman JD, Kruger L. Selective neuronal glycoconjugate expression in sensory and autonomic ganglia: relation of lectin reactivity to peptide and enzyme markers. Journal of Neurocytology. 1990;19(5):789–801. doi: 10.1007/BF01188046. [DOI] [PubMed] [Google Scholar]
- 21.Zylka MJ, Rice FL, Anderson DJ. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron. 2005;45(1):17–25. doi: 10.1016/j.neuron.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 22.Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neliropeptides, the P2X3 purinoceptor and IB4 binding sites. European Journal of Neuroscience. 1999;11(3):946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- 23.Gerke MB, Plenderleith MB. Binding sites for the plant lectin Bandeiraea simplicifolia I-isolectin B4 are expressed by nociceptive primary sensory neurones. Brain Research. 2001;911(1):101–104. doi: 10.1016/s0006-8993(01)02750-0. [DOI] [PubMed] [Google Scholar]
- 24.Cavanaugh DJ, Lee H, Lo L, et al. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(22):9075–9080. doi: 10.1073/pnas.0901507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L, Rutlin M, Abraira VE, et al. The functional organization of cutaneous low-threshold mechanosensory neurons. Cell. 2011;147(7):1615–1627. doi: 10.1016/j.cell.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150(3699):971–979. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- 27.Zeilhofer HU, Wildner H, Yévenes GE. Fast synaptic inhibition in spinal sensory processing and pain control. Physiological Reviews. 2012;92(1):193–235. doi: 10.1152/physrev.00043.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui L, Kim YR, Kim HY, et al. Modulation of synaptic transmission from primary afferents to spinal substantia gelatinosa neurons by group III mGluRs in GAD65-EGFP transgenic mice. Journal of Neurophysiology. 2011;105(3):1102–1111. doi: 10.1152/jn.00108.2010. [DOI] [PubMed] [Google Scholar]
- 29.Heinke B, Ruscheweyh R, Forsthuber L, Wunderbaldinger G, Sandkühler J. Physiological, neurochemical and morphological properties of a subgroup of GABAergic spinal lamina II neurones identified by expression of green fluorescent protein in mice. Journal of Physiology. 2004;560(1):249–266. doi: 10.1113/jphysiol.2004.070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Todd AJ, Sullivan AC. Light microscope study of the coexistence of GABA-like and glycine-like immunoreactivities in the spinal cord of the rat. Journal of Comparative Neurology. 1990;296(3):496–505. doi: 10.1002/cne.902960312. [DOI] [PubMed] [Google Scholar]
- 31.Grudt TJ, Perl ER. Correlations between neuronal morphology and electro-physiological features in the rodent superficial dorsal horn. Journal of Physiology. 2002;540(1):189–207. doi: 10.1113/jphysiol.2001.012890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hantman AW, van den Pol AN, Perl ER. Morphological and physiological features of a set of spinal substantia gelatinosa neurons defined by green fluorescent protein expression. Journal of Neuroscience. 2004;24(4):836–842. doi: 10.1523/JNEUROSCI.4221-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Y, Perl ER. A specific inhibitory pathway between substantia gelatinosa neurons receiving direct C-fiber input. Journal of Neuroscience. 2003;23(25):8752–8758. doi: 10.1523/JNEUROSCI.23-25-08752.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, Perl ER. Modular organization of excitatory circuits between neurons of the spinal superficial dorsal horn (laminae I and II) Journal of Neuroscience. 2005;25(15):3900–3907. doi: 10.1523/JNEUROSCI.0102-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng J, Lu Y, Perl ER. Inhibitory neurones of the spinal substantia gelatinosa mediate interaction of signals from primary afferents. Journal of Physiology. 2010;588(12):2065–2075. doi: 10.1113/jphysiol.2010.188052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasaka T, Kato G, Furue H, et al. Cell-type-specific excitatory and inhibitory circuits involving primary afferents in the substantia gelatinosa of the rat spinal dorsal horn in vitro. Journal of Physiology. 2007;581(part 2):603–618. doi: 10.1113/jphysiol.2006.123919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Todd AJ, McKenzie J. GABA-immunoreactive neurons in the dorsal horn of the rat spinal cord. Neuroscience. 1989;31(3):799–806. doi: 10.1016/0306-4522(89)90442-9. [DOI] [PubMed] [Google Scholar]
- 38.Maxwell DJ, Belle MD, Cheunsuang O, Stewart A, Morris R. Morphology of inhibitory and excitatory interneurons in superficial laminae of the rat dorsal horn. Journal of Physiology. 2007;584(part 2):521–533. doi: 10.1113/jphysiol.2007.140996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polgár E, Shehab SAS, Watt C, Todd AJ. GABAergic neurons that contain neuropeptide Y selectively target cells with the neurokinin I receptor in laminae III and IV of the rat spinal cord. Journal of Neuroscience. 1999;19(7):2637–2646. doi: 10.1523/JNEUROSCI.19-07-02637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polgár E, Sardella TCP, Watanabe M, Todd AJ. Quantitative study of NPY-expressing GABAergic neurons and axons in rat spinal dorsal horn. Journal of Comparative Neurology. 2011;519(6):1007–1023. doi: 10.1002/cne.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puskár Z, Polgár E, Todd AJ. A population of large lamina I projection neurons with selective inhibitory input in rat spinal cord. Neuroscience. 2001;102(1):167–176. doi: 10.1016/s0306-4522(00)00445-0. [DOI] [PubMed] [Google Scholar]
- 42.Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological Reviews. 2010;62(3):405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jahr CE, Jessell TM. Synaptic transmission between dorsal root ganglion and dorsal horn neurons in culture: antagonism of monosynaptic excitatory postsynaptic potentials and glutamate excitation by kynurenate. Journal of Neuroscience. 1985;5(8):2281–2289. doi: 10.1523/JNEUROSCI.05-08-02281.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kangrga I, Randić M. Outflow of endogenous aspartate and glutamate from the rat spinal dorsal horn in vitro by activation of low- and high-threshold primary afferent fibers. Modulation by μ-opioids. Brain Research. 1991;553(2):347–352. doi: 10.1016/0006-8993(91)90848-p. [DOI] [PubMed] [Google Scholar]
- 45.Gerber G, Randić M. Excitatory amino acid-mediated components of synaptically evoked input from dorsal roots to deep dorsal horn neurons in the rat spinal cord slice. Neuroscience Letters. 1989;106(1-2):211–219. doi: 10.1016/0304-3940(89)90228-0. [DOI] [PubMed] [Google Scholar]
- 46.Yoshimura M, Jessell T. Amino acid-mediated EPSPs at primary afferent synapses with substantia gelatinosa neurones in the rat spinal cord. Journal of Physiology. 1990;430:315–335. doi: 10.1113/jphysiol.1990.sp018293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li P, Wilding TJ, Kim SJ, Calejesan AA, Huettner JE, Zhuo M. Kainate-receptor-mediated sensory synaptic transmission in mammalian spinal cord. Nature. 1999;397(6715):161–164. doi: 10.1038/16469. [DOI] [PubMed] [Google Scholar]
- 48.Youn DH, Voitenko N, Gerber G, Park YK, Galik J, Randić M. Altered long-term synaptic plasticity and kainate-induced Ca2+ transients in the substantia gelatinosa neurons in GLUK6-deficient mice. Molecular Brain Research. 2005;142(1):9–18. doi: 10.1016/j.molbrainres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Li P, Zhou M. Silent glutamatergic synapses and nociception in mammalian spinal cord. Nature. 1998;393(6686):695–698. doi: 10.1038/31496. [DOI] [PubMed] [Google Scholar]
- 50.Baba H, Doubell TP, Moore KA, Woolf CJ. Silent NMDA receptor-mediated synapses are developmentally regulated in the dorsal horn of the rat spinal cord. Journal of Neurophysiology. 2000;83(2):955–962. doi: 10.1152/jn.2000.83.2.955. [DOI] [PubMed] [Google Scholar]
- 51.Todd AJ, Watt C, Spike RC, Sieghart W. Colocalization of GABA, glycine, and their receptors at synapses in the rat spinal cord. Journal of Neuroscience. 1996;16(3):974–982. doi: 10.1523/JNEUROSCI.16-03-00974.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chéry N, de Koninck Y. Junctional versus extrajunctional glycine and GABA(A) receptor-mediated IPSCs in identified lamina I neurons of the adult rat spinal cord. Journal of Neuroscience. 1999;19(17):7342–7355. doi: 10.1523/JNEUROSCI.19-17-07342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baccei ML, Fitzgerald M. Development of GABAergic and glycinergic transmission in the neonatal rat dorsal horn. Journal of Neuroscience. 2004;24(20):4749–4757. doi: 10.1523/JNEUROSCI.5211-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshimura M, Nishi S. Primary afferent-evoked glycine- and GABA-mediated IPSPs in substantia gelatinosa neurones in the rat spinal cord in vitro. Journal of Physiology. 1995;482(part 1):29–38. doi: 10.1113/jphysiol.1995.sp020497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lisman J, Malenka RC, Nicoll RA, Malinow R. Learning mechanisms: the case for CaM-KII. Science. 1997;276(5321):2001–2002. doi: 10.1126/science.276.5321.2001. [DOI] [PubMed] [Google Scholar]
- 56.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276(5321):2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 57.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405(6789):955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 58.Lee HK, Takamiya K, Han JS, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112(5):631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 59.Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nature Reviews Neuroscience. 2012;13(3):169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Banke TG, Bowie D, Lee HK, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. Journal of Neuroscience. 2000;20(1):89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benke TA, Lüthi A, Isaac JTR, Collingridge GL. Modulation of ampa receptor unitary conductance by synaptic activity. Nature. 1998;393(6687):793–797. doi: 10.1038/31709. [DOI] [PubMed] [Google Scholar]
- 62.Shi SH, Hayashi Y, Petralia RS, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284(5412):1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 63.Liu SQJ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405(6785):454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- 64.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annual Review of Neuroscience. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 65.Schulz PE. Long-term potentiation involves increases in the probability of neurotransmitter release. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(11):5888–5893. doi: 10.1073/pnas.94.11.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kullmann DM, Nicoll RA. Long-term potentiation is associated with increases in quantal content and quantal amplitude. Nature. 1992;357(6375):240–244. doi: 10.1038/357240a0. [DOI] [PubMed] [Google Scholar]
- 67.Lisman J, Raghavachari S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Science’s STKE. 2006;2006(356):p. re11. doi: 10.1126/stke.3562006re11. [DOI] [PubMed] [Google Scholar]
- 68.Bliss TV, Collingridge GL. Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Molecular Brain. 2013;6(article 5) doi: 10.1186/1756-6606-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 70.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 71.Lynch MA. Long-Term potentiation and memory. Physiological Reviews. 2004;84(1):87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 72.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313(5790):1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 73.Liu XG, Sandkühler J. Long-term potentiation of C-fiber-evoked potentials in the rat spinal dorsal horn is prevented by spinal N-methyl-D-aspartic acid receptor blockage. Neuroscience Letters. 1995;191(1-2):43–46. doi: 10.1016/0304-3940(95)11553-0. [DOI] [PubMed] [Google Scholar]
- 74.Randić M. Plasticity of excitatory synaptic transmission in the spinal cord dorsal horn. Progress in Brain Research. 1996;113:463–506. doi: 10.1016/s0079-6123(08)61104-8. [DOI] [PubMed] [Google Scholar]
- 75.Liu XG, Sandkühler J. Activation of spinal N-methyl-D-aspartate or neurokinin receptors induces long-term potentiation of spinal C-fibre-evoked potentials. Neuroscience. 1998;86(4):1209–1216. doi: 10.1016/s0306-4522(98)00107-9. [DOI] [PubMed] [Google Scholar]
- 76.Liu XG, Sandkühler J. Characterization of long-term potentiation of C-fiber-evoked potentials in spinal dorsal horn of adult rat: essential role of NK1 and NK2 receptors. Journal of Neurophysiology. 1997;78(4):1973–1982. doi: 10.1152/jn.1997.78.4.1973. [DOI] [PubMed] [Google Scholar]
- 77.Azkue JJ, Liu XG, Zimmermann M, Sandkühler J. Induction of long-term potentiation of C fibre-evoked spinal field potentials requires recruitment of group I, but not group II/III metabotropic glutamate receptors. Pain. 2003;106(3):373–379. doi: 10.1016/j.pain.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 78.Terman GW, Eastman CL, Chavkin C. Mu opiates inhibit long-term potentiation induction in the spinal cord slice. Journal of Neurophysiology. 2001;85(2):485–494. doi: 10.1152/jn.2001.85.2.485. [DOI] [PubMed] [Google Scholar]
- 79.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(10):4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kemp A, Manahan-Vaughan D. Hippocampal long-term depression: master or minion in declarative memory processes? Trends in Neurosciences. 2007;30(3):111–118. doi: 10.1016/j.tins.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 81.Massey PV, Bashir ZI. Long-term depression: multiple forms and implications for brain function. Trends in Neurosciences. 2007;30(4):176–184. doi: 10.1016/j.tins.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 82.Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiological Reviews. 2001;81(3):1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- 83.Cheng G, Randić M. Involvement of intracellular calcium and protein phosphatases in long-term depression of A-fiber-mediated primary afferent neurotransmission. Developmental Brain Research. 2003;144(1):73–82. doi: 10.1016/s0165-3806(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 84.Sandkühler J, Chen JG, Cheng G, Randić M. Low-frequency stimulation of afferent aδ-fibers induces long-term depression at primary afferent synapses with substantia gelatinosa neurons in the rat. Journal of Neuroscience. 1997;17(16):6483–6491. doi: 10.1523/JNEUROSCI.17-16-06483.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiological Reviews. 2009;89(2):707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- 86.Ruscheweyh R, Wilder-Smith O, Drdla R, Liu XG, Sandkühler J. Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Molecular Pain. 2011;7(article 20) doi: 10.1186/1744-8069-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ikeda H, Stark J, Fischer H, et al. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312(5780):1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- 88.Drdla-Schutting R, Benrath J, Wunderbaldinger G, Sandkühler J. Erasure of a spinal memory trace of pain by a brief, high-dose opioid administration. Science. 2012;335(6065):235–238. doi: 10.1126/science.1211726. [DOI] [PubMed] [Google Scholar]
- 89.Sandkühler J, Liu X. Induction of long-term potentiation at spinal synapses by noxious stimulation or nerve injury. European Journal of Neuroscience. 1998;10(7):2476–2480. doi: 10.1046/j.1460-9568.1998.00278.x. [DOI] [PubMed] [Google Scholar]
- 90.Ohnami S, Tanabe M, Shinohara S, Takasu K, Kato A, Ono H. Role of voltage-dependent calcium channel subtypes in spinal long-term potentiation of C-fiber-evoked field potentials. Pain. 2011;152(3):623–631. doi: 10.1016/j.pain.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 91.Park CK, Lü N, Xu ZZ, Liu T, Serhan CN, Ji RR. Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. Journal of Neuroscience. 2011;31(42):15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu WT, Li HC, Song XS, Huang ZJ, Song XJ. EphB receptor signaling in mouse spinal cord contributes to physical dependence on morphine. FASEB Journal. 2009;23(1):90–98. doi: 10.1096/fj.08-114462. [DOI] [PubMed] [Google Scholar]
- 93.Luo C, Gangadharan V, Bali KK, et al. Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PLoS Biology. 2012;10(3) doi: 10.1371/journal.pbio.1001283.e1001283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cheng LZ, Lü N, Zhang YQ, Zhao ZQ. Ryanodine receptors contribute to the induction of nociceptive input-evoked long-term potentiation in the rat spinal cord slice. Molecular Pain. 2010;6(article 1) doi: 10.1186/1744-8069-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang XC, Zhang YQ, Zhao ZQ. Involvement of nitric oxide in long-term potentiation of spinal nociceptive responses in rats. NeuroReport. 2005;16(11):1197–1201. doi: 10.1097/00001756-200508010-00013. [DOI] [PubMed] [Google Scholar]
- 96.Zhang XC, Zhang YQ, Zhao ZQ. Different roles of two nitric oxide activated pathways in spinal long-term potentiation of C-fiber-evoked field potentials. Neuropharmacology. 2006;50(6):748–754. doi: 10.1016/j.neuropharm.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 97.Pedersen LM, Jacobsen LM, Mollerup S, Gjerstad J. Spinal cord long-term potentiation (LTP) is associated with increased dorsal horn gene expression of IL-1β, GDNF and iNOS. European Journal of Pain. 2010;14(3):255–260. doi: 10.1016/j.ejpain.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 98.Youn DH, Royle G, Kolaj M, Vissel B, Randić M. Enhanced LTP of primary afferent neurotransmission in AMPA receptor GluR2-deficient mice. Pain. 2008;136(1-2):158–167. doi: 10.1016/j.pain.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 99.Ikeda H, Heinke B, Ruscheweyh R, Sandkühler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299(5610):1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- 100.Zhou HY, Chen SR, Chen H, Pan HL. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. Journal of Neuroscience. 2010;30(12):4460–4466. doi: 10.1523/JNEUROSCI.5857-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. Journal of Neuroscience. 1998;18(24):10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Montgomery JM, Pavlidis P, Madison DV. Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron. 2001;29(3):691–701. doi: 10.1016/s0896-6273(01)00244-6. [DOI] [PubMed] [Google Scholar]
- 103.Jung SJ, Kim SJ, Park YK, Oh SB, Cho K, Kim J. Group I mGluR regulates the polarity of spike-timing dependent plasticity in substantia gelatinosa neurons. Biochemical and Biophysical Research Communications. 2006;347(2):509–516. doi: 10.1016/j.bbrc.2006.06.134. [DOI] [PubMed] [Google Scholar]
- 104.Wei F, Vadakkan KI, Toyoda H, et al. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. Journal of Neuroscience. 2006;26(3):851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacological Reviews. 1999;51(1):7–61. [PubMed] [Google Scholar]
- 106.Hollmann M, Heinemann S. Cloned glutamate receptors. Annual Review of Neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 107.Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. The Journal of Biological Chemistry. 1997;272(51):32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 108.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 109.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287(5461):2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 110.Shi SH, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105(3):331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 111.Zamanillo D, Sprengel R, Hvalby O, et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284(5421):1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- 112.Plant K, Pelkey KA, Bortolotto ZA, et al. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nature Neuroscience. 2006;9(5):602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- 113.Jia Z, Agopyan N, Miu P, et al. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17(5):945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 114.Meng Y, Zhang Y, Jia Z. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 2003;39(1):163–176. doi: 10.1016/s0896-6273(03)00368-4. [DOI] [PubMed] [Google Scholar]
- 115.Tomita S, Adesnik H, Sekiguchi M, et al. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature. 2005;435(7045):1052–1058. doi: 10.1038/nature03624. [DOI] [PubMed] [Google Scholar]
- 116.Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45(2):269–277. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 117.Tomita S, Chen L, Kawasaki Y, et al. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. Journal of Cell Biology. 2003;161(4):805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Milstein AD, Nicoll RA. TARP modulation of synaptic AMPA receptor trafficking and gating depends on multiple intracellular domains. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(27):11348–11351. doi: 10.1073/pnas.0905570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature. 2013;493(7433):495–500. doi: 10.1038/nature11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Spike RC, Maxwell DJ, Todd AJ. GluR1 and GluR2/3 subunits of the AMPA-type glutamate receptor are associated with particular types of neurone in laminae I–III of the spinal dorsal horn of the rat. European Journal of Neuroscience. 1998;10(1):324–333. doi: 10.1046/j.1460-9568.1998.00048.x. [DOI] [PubMed] [Google Scholar]
- 121.Engelman HS, Allen TB, MacDermott AB. The distribution of neurons expressing calcium-permeable AMPA receptors in the superficial laminae of the spinal cord dorsal horn. Journal of Neuroscience. 1999;19(6):2081–2089. doi: 10.1523/JNEUROSCI.19-06-02081.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gu JG, Albuquerque C, Lee CJ, MacDermott AB. Synaptic strengthening through activation of Ca2+-permeable AMPA receptors. Nature. 1996;381(6585):793–796. doi: 10.1038/381793a0. [DOI] [PubMed] [Google Scholar]
- 123.Popratiloff A, Weinberg RJ, Rustioni A. AMPA receptor subunits underlying terminals of fine-caliber primary afferent fibers. Journal of Neuroscience. 1996;16(10):3363–3372. doi: 10.1523/JNEUROSCI.16-10-03363.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438(7065):185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 125.Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. The EMBO Journal. 2003;22(12):2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yao Y, Harrison CB, Freddolino PL, Schulten K, Mayer ML. Molecular mechanism of ligand recognition by NR3 subtype glutamate receptors. The EMBO Journal. 2008;27(15):2158–2170. doi: 10.1038/emboj.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321(6069):519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 128.Mayer ML, Westbrook GL, Buthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309(5965):261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 129.Lester RAJ, Clements JD, Westbrook GL, Jahr CE. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 1990;346(6284):565–567. doi: 10.1038/346565a0. [DOI] [PubMed] [Google Scholar]
- 130.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Current Opinion in Neurobiology. 2001;11(3):327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 131.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12(3):529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 132.Wyllie DJA, Béhé P, Colquhoun D. Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. Journal of Physiology. 1998;510(part 1):1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vicini S, Wang JF, Li JH, et al. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. Journal of Neurophysiology. 1998;79(2):555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- 134.Das S, Sasaki YF, Rothe T, et al. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature. 1998;393(6683):377–381. doi: 10.1038/30748. [DOI] [PubMed] [Google Scholar]
- 135.Pérez-Otaño I, Schulteis CT, Contractor A, et al. Assembly with the NR1 subunit is required for surface expression of NR3A-containing NMDA receptors. Journal of Neuroscience. 2001;21(4):1228–1237. doi: 10.1523/JNEUROSCI.21-04-01228.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chatterton JE, Awobuluyi M, Premkumar LS, et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature. 2002;415(6873):793–798. doi: 10.1038/nature715. [DOI] [PubMed] [Google Scholar]
- 137.MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Critical Reviews in Neurobiology. 2006;18(1-2):71–84. doi: 10.1615/critrevneurobiol.v18.i1-2.80. [DOI] [PubMed] [Google Scholar]
- 138.Köhr G, Jensen V, Koester HJ, et al. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. Journal of Neuroscience. 2003;23(34):10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bartlett TE, Bannister NJ, Collett VJ, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 2007;52(1):60–70. doi: 10.1016/j.neuropharm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 140.Tang YP, Shimizu E, Dube GR, et al. Genetic enhancement of learning and memory in mice. Nature. 1999;401(6748):63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 141.Erreger K, Dravid SM, Banke TG, Wyllie DJA, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. Journal of Physiology. 2005;563(2):345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cheung HH, Gurd JW. Tyrosine phosphorylation of the N-methyl-D-aspartate receptor by exogenous and postsynaptic density-associated Src-family kinases. Journal of Neurochemistry. 2001;78(3):524–534. doi: 10.1046/j.1471-4159.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 143.Nakazawa T, Komai S, Tezuka T, et al. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluRε2 (NR2B) subunit of the N-methyl-D-aspartate receptor. The Journal of Biological Chemistry. 2001;276(1):693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- 144.Ng D, Pitcher GM, Szilard RK, et al. Neto1 is a novel CUB-domain NMDA receptor-interacting protein required for synaptic plasticity and learning. PLoS Biology. 2009;7(2, article e41) doi: 10.1371/journal.pbio.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48(2):289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 146.Scheetz AJ, Nairn AC, Constantine-Paton M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nature Neuroscience. 2000;3(3):211–216. doi: 10.1038/72915. [DOI] [PubMed] [Google Scholar]
- 147.Gong R, Chang SP, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. The Journal of Biological Chemistry. 2006;281(27):18802–18815. doi: 10.1074/jbc.M512524200. [DOI] [PubMed] [Google Scholar]
- 148.Tran DH, Gong R, Tang SJ. Differential roles of NR2A and NR2B subtypes in NMDA receptor-dependent protein synthesis in dendrites. Neuropharmacology. 2007;53(2):252–256. doi: 10.1016/j.neuropharm.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 149.Bradshaw KD, Emptage NJ, Bliss TVP. A role for dendritic protein synthesis in hippocampal late LTP. European Journal of Neuroscience. 2003;18(11):3150–3152. doi: 10.1111/j.1460-9568.2003.03054.x. [DOI] [PubMed] [Google Scholar]
- 150.Tölle TR, Berthele A, Zieglgänsberger W, Seeburg PH, Wisden W. The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and in periaqueductal gray. Journal of Neuroscience. 1993;13(12):5009–5028. doi: 10.1523/JNEUROSCI.13-12-05009.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Luque JM, Bleuel Z, Malherbe P, Richards JG. Alternatively spliced isoforms of the N-methyl-D-aspartate receptor subunit 1 are differentially distributed within the rat spinal cord. Neuroscience. 1994;63(3):629–635. doi: 10.1016/0306-4522(94)90510-x. [DOI] [PubMed] [Google Scholar]
- 152.Boyce S, Wyatt A, Webb JK, et al. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology. 1999;38(5):611–623. doi: 10.1016/s0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- 153.Nagy GG, Watanabe M, Fukaya M, Todd AJ. Synaptic distribution of the NR1, NR2A and NR2B subunits of the N-methyl-D-aspartate receptor in the rat lumbar spinal cord revealed with an antigen-unmasking technique. European Journal of Neuroscience. 2004;20(12):3301–3312. doi: 10.1111/j.1460-9568.2004.03798.x. [DOI] [PubMed] [Google Scholar]
- 154.Shiokawa H, Kaftan EJ, MacDermott AB, Tong CK. NR2 subunits and NMDA receptors on lamina II inhibitory and excitatory interneurons of the mouse dorsal horn. Molecular Pain. 2010;6(article 26) doi: 10.1186/1744-8069-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Momiyama A. Distinct synaptic and extrasynaptic NMDA receptors identified in dorsal horn neurones of the adult rat spinal cord. Journal of Physiology. 2000;523(part 3):621–628. doi: 10.1111/j.1469-7793.2000.t01-1-00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sandkühler J. Understanding LTP in pain pathways. Molecular Pain. 2007;3(article 9) doi: 10.1186/1744-8069-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Georgiev SK, Furue H, Baba H, Kohno T. Xenon inhibits excitatory but not inhibitory transmission in rat spinal cord dorsal horn neurons. Molecular Pain. 2010;6(article 25) doi: 10.1186/1744-8069-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Benrath J, Kempf C, Georgieff M, Sandkühler J. Xenon blocks the induction of synaptic long-term potentiation in pain pathways in the rat spinal cord in vivo. Anesthesia and Analgesia. 2007;104(1):106–111. doi: 10.1213/01.ane.0000250368.27822.31. [DOI] [PubMed] [Google Scholar]
- 159.Kim HY, Lee KY, Lu Y, et al. Mitochondrial Ca2+ uptake is essential for synaptic plasticity in pain. Journal of Neuroscience. 2011;31(36):12982–12991. doi: 10.1523/JNEUROSCI.3093-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lerma J, Paternain AV, Rodríguez-Moreno A, López-Garciáa JC. Molecular physiology of kainate receptors. Physiological Reviews. 2001;81(3):971–998. doi: 10.1152/physrev.2001.81.3.971. [DOI] [PubMed] [Google Scholar]
- 161.Bettler B, Egebjerg J, Sharma G, et al. Cloning of a putative glutamate receptor: a low affinity kainate-binding subunit. Neuron. 1992;8(2):257–265. doi: 10.1016/0896-6273(92)90292-l. [DOI] [PubMed] [Google Scholar]
- 162.Werner P, Voigt M, Keinanen K, Wisden W, Seeburg PH. Cloning of a putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells. Nature. 1991;351(6329):742–744. doi: 10.1038/351742a0. [DOI] [PubMed] [Google Scholar]
- 163.Herb A, Burnashev N, Werner P, Sakmann B, Wisden W, Seeburg PH. The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron. 1992;8(4):775–785. doi: 10.1016/0896-6273(92)90098-x. [DOI] [PubMed] [Google Scholar]
- 164.Sommer B, Burnashev N, Verdoorn TA, Keinanen K, Sakmann B, Seeburg PH. A glutamate receptor channel with high affinity for domoate and kainate. The EMBO Journal. 1992;11(4):1651–1656. doi: 10.1002/j.1460-2075.1992.tb05211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schiffer HH, Swanson GT, Heinemann SF. Rat GluR7 and a carboxy-terminal splice variant, GluR7b, are functional kainate receptor subunits with a low sensitivity to glutamate. Neuron. 1997;19(5):1141–1146. doi: 10.1016/s0896-6273(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 166.Gregor P, O’Hara BF, Yang X, Uhl GR. Expression and novel subunit isoforms of glutamate receptor genes GluR5 and GluR6. NeuroReport. 1993;4(12):1343–1346. doi: 10.1097/00001756-199309150-00014. [DOI] [PubMed] [Google Scholar]
- 167.Burnashev N, Zhou Z, Neher E, Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. Journal of Physiology. 1995;485(part 2):403–418. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Egebjerg J, Heinemann SF. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(2):755–759. doi: 10.1073/pnas.90.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Burnashev N, Villarroel A, Sakmann B. Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. Journal of Physiology. 1996;496(part 1):165–173. doi: 10.1113/jphysiol.1996.sp021674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sailer A, Swanson GT, Pérez-Otaño I, et al. Generation and analysis of GluR5(Q636R) kainate receptor mutant mice. Journal of Neuroscience. 1999;19(20):8757–8764. doi: 10.1523/JNEUROSCI.19-20-08757.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kohler M, Burnashev N, Sakmann B, Seeburg PH. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron. 1993;10(3):491–500. doi: 10.1016/0896-6273(93)90336-p. [DOI] [PubMed] [Google Scholar]
- 172.Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nature Reviews Neuroscience. 2003;4(6):481–495. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- 173.Bortolotto ZA, Clarke VRJ, Delany CM, et al. Kainate receptors are involved in synaptic plasticity. Nature. 1999;402(6759):297–301. doi: 10.1038/46290. [DOI] [PubMed] [Google Scholar]
- 174.Lauri SE, Bortolotto ZA, Nistico R, et al. A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synapses in the hippocampus. Neuron. 2003;39(2):327–341. doi: 10.1016/s0896-6273(03)00369-6. [DOI] [PubMed] [Google Scholar]
- 175.Weisskopf MG, Nicoll RA. Presynaptic changes during mossy fibre LTP revealed by NMDA receptor-mediated synaptic responses. Nature. 1995;376(6537):256–259. doi: 10.1038/376256a0. [DOI] [PubMed] [Google Scholar]
- 176.Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nature Neuroscience. 1999;2(7):625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29(1):209–216. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- 178.Hwang SJ, Pagliardini S, Rustioni A, Valtschanoff JG. Presynaptic kainate receptors in primary afferents to the superficial laminae of the rat spinal cord. Journal of Comparative Neurology. 2001;436(3):275–289. [PubMed] [Google Scholar]
- 179.Kerchner GA, Wilding TJ, Huettner JE, Zhuo M. Kainate receptor subunits underlying presynaptic regulation of transmitter release in the dorsal horn. Journal of Neuroscience. 2002;22(18):8010–8017. doi: 10.1523/JNEUROSCI.22-18-08010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kerchner GA, Wilding TJ, Li P, Zhuo M, Huettner JE. Presynaptic kainate receptors regulate spinal sensory transmission. Journal of Neuroscience. 2001;21(1):59–66. doi: 10.1523/JNEUROSCI.21-01-00059.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11(6):1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- 182.Petralia RS, Wang YX, Wenthold RJ. Histological and ultrastructural localization of the kainate receptor subunits, KA2 and GluR6/7, in the rat nervous system using selective antipeptide antibodies. Journal of Comparative Neurology. 1994;349(1):85–110. doi: 10.1002/cne.903490107. [DOI] [PubMed] [Google Scholar]
- 183.Huettner JE. Glutamate receptor channels in rat DRG neurons: activation by kainate and quisqualate and blockade of desensitization by Con A. Neuron. 1990;5(3):255–266. doi: 10.1016/0896-6273(90)90163-a. [DOI] [PubMed] [Google Scholar]
- 184.Youn DH, Randić M. Modulation of excitatory synaptic transmission in the spinal substantia gelatinosa of mice deficient in the kainate receptor GluR5 and/or GluR6 subunit. Journal of Physiology. 2004;555(part 3):683–698. doi: 10.1113/jphysiol.2003.057570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annual Review of Cell and Developmental Biology. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 186.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacological Reviews. 2005;57(4):411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 187.Buraei Z, Yang J. The β subunit of voltage-gated Ca2+ channels. Physiological Reviews. 2010;90(4):1461–1506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Snutch TP, Tomlinson WJ, Leonard JP, Gilbert MM. Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron. 1991;7(1):45–57. doi: 10.1016/0896-6273(91)90073-9. [DOI] [PubMed] [Google Scholar]
- 189.Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347(6290):281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 190.Lee SJR, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458(7236):299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347(6292):477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 192.Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. Journal of Neurophysiology. 1996;76(5):3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- 193.Morgan SL, Teyler TJ. Electrical stimuli patterned after the theta-rhythm induce multiple forms of LTP. Journal of Neurophysiology. 2001;86(3):1289–1296. doi: 10.1152/jn.2001.86.3.1289. [DOI] [PubMed] [Google Scholar]
- 194.Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349(6304):67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- 195.Huang YY, Malenka RC. Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the induction of LTP. Journal of Neuroscience. 1993;13(2):568–576. doi: 10.1523/JNEUROSCI.13-02-00568.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kochlamazashvili G, Henneberger C, Bukalo O, et al. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca2+ channels. Neuron. 2010;67(1):116–128. doi: 10.1016/j.neuron.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Evers MR, Salmen B, Bukalo O, et al. Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. Journal of Neuroscience. 2002;22(16):7177–7194. doi: 10.1523/JNEUROSCI.22-16-07177.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Harris EW, Cotman CW. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl D-aspartate antagonists. Neuroscience Letters. 1986;70(1):132–137. doi: 10.1016/0304-3940(86)90451-9. [DOI] [PubMed] [Google Scholar]
- 199.Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. Journal of Neuroscience. 1999;19(23):10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kapur A, Yeckel MF, Gray R, Johnston D. L-type calcium channels are required for one form of hippocampal mossy fiber LTP. Journal of Neurophysiology. 1998;79(4):2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12(2):261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 202.Remy S, Spruston N. Dendritic spikes induce single-burst long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(43):17192–17197. doi: 10.1073/pnas.0707919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. Journal of Neuroscience. 1998;18(16):6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Carlin KP, Jones KE, Jiang Z, Jordan LM, Brownstone RM. Dendritic L-type calcium currents in mouse spinal motoneurons: implications for bistability. European Journal of Neuroscience. 2000;12(5):1635–1646. doi: 10.1046/j.1460-9568.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- 205.Naka A, Gruber-Schoffnegger D, Sandkühler J. Non-Hebbian plasticity at C-fiber synapses in rat spinal cord lamina I neurons. Pain. 2013;154(8):1333–1342. doi: 10.1016/j.pain.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tanabe M, Murakami H, Honda M, Ono H. Gabapentin depresses C-fiber-evoked field potentials in rat spinal dorsal horn only after induction of long-term potentiation. Experimental Neurology. 2006;202(2):280–286. doi: 10.1016/j.expneurol.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 207.Hara K, Saito Y, Kirihara Y, Sakura S, Kosaka Y. Antinociceptive effects of intrathecal L-type calcium channel blockers on visceral and somatic stimuli in the rat. Anesthesia and Analgesia. 1998;87(2):382–387. doi: 10.1097/00000539-199808000-00027. [DOI] [PubMed] [Google Scholar]
- 208.Neugebauer V, Vanegas H, Nebe J, Rümenapp P, Schaible HG. Effects of N- and L-type calcium channel antagonists on the responses of nociceptive spinal cord neurons to mechanical stimulation of the normal and the inflamed knee joint. Journal of Neurophysiology. 1996;76(6):3740–3749. doi: 10.1152/jn.1996.76.6.3740. [DOI] [PubMed] [Google Scholar]
- 209.Malmberg AB, Yaksh TL. Voltage-sensitive calcium channels in spinal nociceptive processing: blockade of N- and P-type channels inhibits formalin-induced nociception. Journal of Neuroscience. 1994;14(8):4882–4890. doi: 10.1523/JNEUROSCI.14-08-04882.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sluka KA. Blockade of calcium channels can prevent the onset of secondary hyperalgesia and allodynia induced by intradermal injection of capsaicin in rats. Pain. 1997;71(2):157–164. doi: 10.1016/s0304-3959(97)03354-x. [DOI] [PubMed] [Google Scholar]
- 211.Sluka KA. Blockade of N- and P/Q-type calcium channels reduces the secondary heat hyperalgesia induced by acute inflammation. Journal of Pharmacology and Experimental Therapeutics. 1998;287(1):232–237. [PubMed] [Google Scholar]
- 212.Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. Journal of Pharmacology and Experimental Therapeutics. 1994;269(3):1117–1123. [PubMed] [Google Scholar]
- 213.Fossat P, Dobremez E, Bouali-Benazzouz R, et al. Knockdown of L calcium channel subtypes: differential effects in neuropathic pain. Journal of Neuroscience. 2010;30(3):1073–1085. doi: 10.1523/JNEUROSCI.3145-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Favereaux A, Thoumine O, Bouali-Benazzouz R, et al. Bidirectional integrative regulation of Cav1.2 calcium channel by microRNA miR-103: role in pain. The EMBO Journal. 2011;30(18):3830–3841. doi: 10.1038/emboj.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hatakeyama S, Wakamori M, Ino M, et al. Differential nociceptive responses in mice lacking the α 1B subunit of N-type Ca2+ channels. NeuroReport. 2001;12(11):2423–2427. doi: 10.1097/00001756-200108080-00027. [DOI] [PubMed] [Google Scholar]
- 216.Kim DS, Yoon CH, Lee SJ, Park SY, Yoo HJ, Cho HJ. Changes in voltage-gated calcium channel α 1 gene expression in rat dorsal root ganglia following peripheral nerve injury. Molecular Brain Research. 2001;96(1-2):151–156. doi: 10.1016/s0169-328x(01)00285-6. [DOI] [PubMed] [Google Scholar]
- 217.Murakami M, Nakagawasai O, Suzuki T, et al. Antinociceptive effect of different types of calcium channel inhibitors and the distribution of various calcium channel α 1 subunits in the dorsal horn of spinal cord in mice. Brain Research. 2004;1024(1-2):122–129. doi: 10.1016/j.brainres.2004.07.066. [DOI] [PubMed] [Google Scholar]
- 218.Umeda M, Ohkubo T, Ono J, Fukuizumi T, Kitamura K. Molecular and immunohistochemical studies in expression of voltage-dependent Ca2+ channels in dorsal root ganglia from streptozotocin-induced diabetic mice. Life Sciences. 2006;79(21):1995–2000. doi: 10.1016/j.lfs.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 219.Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366(6451):156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- 220.Ogasawara M, Kurihara T, Hu Q, Tanabe T. Characterization of acute somatosensory pain transmission in P/Q-type Ca2+ channel mutant mice, leaner. FEBS Letters. 2001;508(2):181–186. doi: 10.1016/s0014-5793(01)03052-6. [DOI] [PubMed] [Google Scholar]
- 221.Pelkey KA, Topolnik L, Lacaille JC, McBain CJ. Compartmentalized Ca2+ channel regulation at divergent mossy-fiber release sites underlies target cell-dependent plasticity. Neuron. 2006;52(3):497–510. doi: 10.1016/j.neuron.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 222.Sarihi A, Mirnajafi-Zadeh J, Jiang B, et al. Cell type-specific, presynaptic LTP of inhibitory synapses on fast-spiking GABAergic neurons in the mouse visual cortex. Journal of Neuroscience. 2012;32(38):13189–13199. doi: 10.1523/JNEUROSCI.1386-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Heinke B, Balzer E, Sandkühler J. Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. European Journal of Neuroscience. 2004;19(1):103–111. doi: 10.1046/j.1460-9568.2003.03083.x. [DOI] [PubMed] [Google Scholar]
- 224.Yamamoto T, Sakashita Y. Differential effects of intrathecally administered N- and P-type voltage-sensitive calcium channel blockers upon two models of experimental mononeuropathy in the rat. Brain Research. 1998;794(2):329–332. doi: 10.1016/s0006-8993(98)00306-0. [DOI] [PubMed] [Google Scholar]
- 225.Luvisetto S, Marinelli S, Panasiti MS, et al. Pain sensitivity in mice lacking the Cav2.1 α 1 subunit of P/Q-type Ca2+ channels. Neuroscience. 2006;142(3):823–832. doi: 10.1016/j.neuroscience.2006.06.049. [DOI] [PubMed] [Google Scholar]
- 226.Fukumoto N, Obama Y, Kitamura N, et al. Hypoalgesic behaviors of P/Q-type voltage-gated Ca2+ channel mutant mouse, rolling mouse Nagoya. Neuroscience. 2009;160(1):165–173. doi: 10.1016/j.neuroscience.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 227.Gruner W, Silva LR. ω-conotoxin sensitivity and presynaptic inhibition of glutamatergic sensory neurotransmission in vitro. Journal of Neuroscience. 1994;14(5, part 1):2800–2808. doi: 10.1523/JNEUROSCI.14-05-02800.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Holz GGT, Dunlap K, Kream RM. Characterization of the electrically evoked release of substance P from dorsal root ganglion neurons: methods and dihydropyridine sensitivity. Journal of Neuroscience. 1988;8(2):463–471. doi: 10.1523/JNEUROSCI.08-02-00463.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Evans AR, Nicol GD, Vasko MR. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Research. 1996;712(2):265–273. doi: 10.1016/0006-8993(95)01447-0. [DOI] [PubMed] [Google Scholar]
- 230.Jeon D, Kim C, Yang YM, et al. Impaired long-term memory and long-term potentiation in N-type Ca2+ channel-deficient mice. Genes, Brain and Behavior. 2007;6(4):375–388. doi: 10.1111/j.1601-183X.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- 231.Ahmed MS, Siegelbaum SA. Recruitment of N-type Ca2+ channels during LTP enhances low release efficacy of hippocampal CA1 perforant path synapses. Neuron. 2009;63(3):372–385. doi: 10.1016/j.neuron.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Santicioli P, Del Bianco E, Tramontana M, Geppetti P, Maggi CA. Release of calcitonin gene-related peptide like-immunoreactivity induced by electrical field stimulation from rat spinal afferents is mediated by conotoxin-sensitive calcium channels. Neuroscience Letters. 1992;136(2):161–164. doi: 10.1016/0304-3940(92)90039-a. [DOI] [PubMed] [Google Scholar]
- 233.Maggi CA, Tramontana M, Cecconi R, Santicioli P. Neurochemical evidence for the involvement of N-type calcium channels in transmitter secretion from peripheral endings of sensory nerves in guinea pigs. Neuroscience Letters. 1990;114(2):203–206. doi: 10.1016/0304-3940(90)90072-h. [DOI] [PubMed] [Google Scholar]
- 234.Vanegas H, Schaible HG. Effects of antagonists to high-threshold calcium channels upon spinal mechanisms of pain, hyperalgesia and allodynia. Pain. 2000;85(1-2):9–18. doi: 10.1016/s0304-3959(99)00241-9. [DOI] [PubMed] [Google Scholar]
- 235.Winquist RJ, Pan JQ, Gribkoff VK. Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuropathic pain. Biochemical Pharmacology. 2005;70(4):489–499. doi: 10.1016/j.bcp.2005.04.035. [DOI] [PubMed] [Google Scholar]
- 236.Saegusa H, Kurihara T, Zong S, et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. The EMBO Journal. 2001;20(10):2349–2356. doi: 10.1093/emboj/20.10.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Fukuizumi T, Ohkubo T, Kitamura K. Spinal sensitization mechanism in vincristine-induced hyperalgesia in mice. Neuroscience Letters. 2003;343(2):89–92. doi: 10.1016/s0304-3940(03)00332-x. [DOI] [PubMed] [Google Scholar]
- 238.Matthews EA, Dickenson AH. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92(1-2):235–246. doi: 10.1016/s0304-3959(01)00255-x. [DOI] [PubMed] [Google Scholar]
- 239.Cizkova D, Marsala J, Lukacova N, et al. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Experimental Brain Research. 2002;147(4):456–463. doi: 10.1007/s00221-002-1217-3. [DOI] [PubMed] [Google Scholar]
- 240.Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41(1):127–138. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 241.Andrade A, Denome S, Jiang YQ, Marangoudakis S, Lipscombe D. Opioid inhibition of N-type Ca2+ channels and spinal analgesia couple to alternative splicing. Nature Neuroscience. 2010;13(10):1249–1256. doi: 10.1038/nn.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Brittain JM, Duarte DB, Wilson SM, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nature Medicine. 2011;17(7):822–829. doi: 10.1038/nm.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chen CC, Shen JW, Chung NC, Min MY, Cheng SJ, Liu IY. Retrieval of context-associated memory is dependent on the CaV3.2 T-type calcium channel. PLoS ONE. 2012;7(1) doi: 10.1371/journal.pone.0029384.e29384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wang Y, Rowan MJ, Anwyl R. LTP induction dependent on activation of Ni2+-sensitive voltage-gated calcium channels, but not NMDA receptors, in the rat dentate gyrus in vitro. Journal of Neurophysiology. 1997;78(5):2574–2581. doi: 10.1152/jn.1997.78.5.2574. [DOI] [PubMed] [Google Scholar]
- 245.Isomura Y, Fujiwara-Tsukamoto Y, Imanishi M, Nambu A, Takada M. Distance-dependent Ni2+-sensitivity of synaptic plasticity in apical dendrites of hippocampal CA1 pyramidal cells. Journal of Neurophysiology. 2002;87(2):1169–1174. doi: 10.1152/jn.00536.2001. [DOI] [PubMed] [Google Scholar]
- 246.Song D, Wang Z, Berger TW. Contribution of T-Type VDCC to TEA-induced long-term synaptic modification in hippocampal CA1 and dentate gyrus. Hippocampus. 2002;12(5):689–697. doi: 10.1002/hipo.10105. [DOI] [PubMed] [Google Scholar]
- 247.Suzuki E, Okada T. Stratum oriens stimulation-evoked modulation of hippocampal long-term potentiation involves the activation of muscarinic acetylcholine receptors and the inhibition of Kv7/M potassium ion channels. European Journal of Neuroscience. 2012;36(1):1984–1992. doi: 10.1111/j.1460-9568.2012.08127.x. [DOI] [PubMed] [Google Scholar]
- 248.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. Journal of Neuroscience. 1999;19(6):1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Huang LYM. Calcium channels in isolated rat dorsal horn neurones, including labelled spinothalamic and trigeminothalamic cells. Journal of Physiology. 1989;411:161–177. doi: 10.1113/jphysiol.1989.sp017566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ryu PD, Randić M. Low- and high-voltage-activated calcium currents in rat spinal dorsal horn neurons. Journal of Neurophysiology. 1990;63(2):273–285. doi: 10.1152/jn.1990.63.2.273. [DOI] [PubMed] [Google Scholar]
- 251.Walsh MA, Graham BA, Brichta AM, Callister RJ. Evidence for a critical period in the development of excitability and potassium currents in mouse lumbar superficial dorsal horn neurons. Journal of Neurophysiology. 2009;101(4):1800–1812. doi: 10.1152/jn.90755.2008. [DOI] [PubMed] [Google Scholar]
- 252.Ku WH, Schneider SP. Multiple T-type Ca2+ current subtypes in electrophysiologically characterized hamster dorsal horn neurons: possible role in spinal sensory integration. Journal of Neurophysiology. 2011;106(5):2486–2498. doi: 10.1152/jn.01083.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Matthews EA, Dickenson AH. Effects of ethosuximide, a T-type Ca2+ channel blocker, on dorsal horn neuronal responses in rats. European Journal of Pharmacology. 2001;415(2-3):141–149. doi: 10.1016/s0014-2999(01)00812-3. [DOI] [PubMed] [Google Scholar]
- 254.Drdla R, Sandkühler J. Long-term potentiation at C-fibre synapses by low-level presynaptic activity in vivo. Molecular Pain. 2008;4(article 18) doi: 10.1186/1744-8069-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Bourinet E, Alloui A, Monteil A, et al. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. The EMBO Journal. 2005;24(2):315–324. doi: 10.1038/sj.emboj.7600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Scroggs RS, Fox AP. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. Journal of Physiology. 1992;445:639–658. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Shin J-B, Martinez-Salgado C, Heppenstall PA, Lewin GR. A T-type calcium channel required for normal function of a mammalian mechanoreceptor. Nature Neuroscience. 2003;6(7):724–730. doi: 10.1038/nn1076. [DOI] [PubMed] [Google Scholar]
- 258.Choi S, Na HS, Kim J, et al. Attenuated pain responses in mice lacking CaV3.2 T-type channels. Genes, Brain and Behavior. 2007;6(5):425–431. doi: 10.1111/j.1601-183X.2006.00268.x. [DOI] [PubMed] [Google Scholar]
- 259.Kim D, Park D, Choi S, et al. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science. 2003;302(5642):117–119. doi: 10.1126/science.1088886. [DOI] [PubMed] [Google Scholar]
- 260.Dogrul A, Gardell LR, Ossipov MH, Tulunay FC, Lai J, Porreca F. Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain. 2003;105(1-2):159–168. doi: 10.1016/s0304-3959(03)00177-5. [DOI] [PubMed] [Google Scholar]
- 261.Wen XJ, Xu SY, Chen Z-X, Yang CX, Liang H, Li H. The roles of T-type calcium channel in the development of neuropathic pain following chronic compression of rat dorsal root ganglia. Pharmacology. 2010;85(5):295–300. doi: 10.1159/000276981. [DOI] [PubMed] [Google Scholar]
- 262.Todorovic SM, Jevtovic-Todorovic V, Meyenburg A, et al. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron. 2001;31(1):75–85. doi: 10.1016/s0896-6273(01)00338-5. [DOI] [PubMed] [Google Scholar]
- 263.Todorovic SM, Meyenburg A, Jevtovic-Todorovic V. Redox modulation of peripheral T-type Ca2+ channels in vivo: alteration of nerve injury-induced thermal hyperalgesia. Pain. 2004;109(3):328–339. doi: 10.1016/j.pain.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 264.Maeda Y, Aoki Y, Sekiguchi F, et al. Hyperalgesia induced by spinal and peripheral hydrogen sulfide: evidence for involvement of Cav3.2 T-type calcium channels. Pain. 2009;142(1-2):127–132. doi: 10.1016/j.pain.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 265.Takahashi T, Aoki Y, Okubo K, et al. Upregulation of Cav3.2 T-type calcium channels targeted by endogenous hydrogen sulfide contributes to maintenance of neuropathic pain. Pain. 2010;150(1):183–191. doi: 10.1016/j.pain.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 266.Myoga MH, Regehr WG. Calcium microdomains near R-type calcium channels control the induction of presynaptic long-term potentiation at parallel fiber to purkinje cell synapses. Journal of Neuroscience. 2011;31(14):5235–5243. doi: 10.1523/JNEUROSCI.5252-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Yasuda R, Sabatini BL, Svoboda K. Plasticity of calcium channels in dendritic spines. Nature Neuroscience. 2003;6(9):948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]
- 268.Takahashi H, Magee JC. Pathway interactions and synaptic plasticity in the dendritic tuft regions of CA1 pyramidal neurons. Neuron. 2009;62(1):102–111. doi: 10.1016/j.neuron.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 269.Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. Journal of Physiology. 1995;487(part 1):67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Fang Z, Hwang JH, Kim JS, Jung SJ, Oh SB. R-type calcium channel isoform in rat dorsal root ganglion neurons. Korean Journal of Physiology and Pharmacology. 2010;14(1):45–49. doi: 10.4196/kjpp.2010.14.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Bourinet E, Stotz SC, Spaetgens RL, et al. Interaction of SNX482 with domains III and IV inhibits activation gating of α 1E (CaV2.3) calcium channels. Biophysical Journal. 2001;81(1):79–88. doi: 10.1016/S0006-3495(01)75681-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Matthews EA, Bee LA, Stephens GJ, Dickenson AH. The Cav2.3 calcium channel antagonist SNX-482 reduces dorsal horn neuronal responses in a rat model of chronic neuropathic pain. European Journal of Neuroscience. 2007;25(12):3561–3569. doi: 10.1111/j.1460-9568.2007.05605.x. [DOI] [PubMed] [Google Scholar]
- 273.Saegusa H, Kurihara T, Zong S, et al. Altered pain responses in mice lacking α 1E subunit of the voltage-dependent Ca2+ channel. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(11):6132–6137. doi: 10.1073/pnas.100124197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Ji R-R, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends in Neurosciences. 2003;26(12):696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 275.Deisseroth K. Optogenetics. Nature Methods. 2011;8(1):26–29. doi: 10.1038/nmeth.f.324. [DOI] [PMC free article] [PubMed] [Google Scholar]