

Abstract
Rationale
A number of studies have associated reduced Akt1 expression with vulnerability for schizophrenia. Although mice with deletion of a single copy of the Akt1 gene (Akt1+/−) show reduced Akt1 expression relative to wild-type (WT) animals, the extent to which these mice show schizophrenia-like phenotypic changes and/or increased susceptibility to epigenetic or non-genetic factors related to schizophrenia is unknown.
Objectives
Mutant mice were assessed on electroencephalographic/event related potential (EEG/ERP) and behavioral (acoustic startle and pre-pulse inhibition) measures relevant to schizophrenia. Mice were also assessed following exposure to the NMDA receptor antagonist ketamine, a potent psychotomimetic drug, in order to assess the role of reduced Akt1 expression as a vulnerability factor for schizophrenia.
Methods
Akt1+/−, Akt1−/− and WT mice received a series of paired-click, white noise stimuli, following ketamine (50 mg/kg) and saline injections. EEG was analyzed for ERPs and event-related power. Akt1+/− and WT mice were also assessed on PPI following ketamine (50 mg/kg) or saline injection.
Results
Akt1+/− and Akt1−/− mice displayed reduced amplitude of the P20 component of the ERP to the first click of a paired click stimulus, as well as reduced S1-S2 difference for P20 and N40 components, following ketamine. Mutant mice also showed increased reduction in gamma synchrony and theta suppression following ketamine. Akt1+/− mice displayed reduced pre-pulse inhibition.
Conclusions
Reduced genetic expression of Akt1 facilitated ketamine-induced changes of EEG and behavior in mice, suggesting that reduced Akt1 expression can serve as a vulnerability factor for schizophrenia.
Keywords: schizophrenia, ERSP, ITC, P50, N100, evoked-potential, Gamma, Theta, ketamine, drug abuse
Introduction
An association between the v-Akt murine thymoma viral oncogene homolog 1 gene (Akt1) and schizophrenia was originally suggested by Emamian, et al. in 2004, who identified a multi-single-nucleotide polymorphism (SNP) in patients with schizophrenia. Additionally, these researchers found reduced levels of Akt1 expression in patients with schizophrenia, a finding that has been confirmed in several subsequent studies (Balu et al. 2012; Blasi et al. 2012; Szamosi et al. 2012). Genetic variations in Akt1 have been linked to altered brain physiology, behavior and cognition. For example, two SNPs of the Akt1 gene (rs2494732 and rs1130233) have been linked to deficits in attention and reduced cortical gray matter (Ohi et al. 2011), working memory (Tan et al. 2008) and reduced hippocampal volume (Tan et al. 2011) in humans. Importantly, genetic variation in Akt1 has been associated with enhanced psychotomimetic response to amphetamine in rodents (Emamian et al. 2004) and psychotic response to cannabis in humans (Di Forti et al. 2012; van Winkel et al. 2011), suggesting that reduced Akt1 activity may confer greater susceptibility to the development of schizophrenia following exposure to non-genetic risk-factors for the disease.
Information about the role of Akt1 in behavior and psychopathology has been derived from the study of genetically engineered mice with a deletion of the Akt1 gene. These mice display a schizophrenia-like phenotype characterized by a larger reduction in PPI and working memory following dopamine challenge (Emamian et al. 2004; Lai et al. 2006), altered sensorimotor gating and impaired hippocampal learning and memory (Balu et al. 2012). Additionally, Akt1−/− mutant mice show schizophrenia-like neuropathological changes in dendritic morphology in the frontal cortex (Lai et al. 2006), and reduced hippocampal neurogenesis (Balu et al. 2012).
While these mice have been critical for supporting the hypothesis of a functional role for Akt1 in schizophrenia, deletion of the Akt1 gene completely abolishes production of the corresponding protein kinase, possibly leading to a more dramatic or different phenotype than that produced simply by reducing expression. For example, Akt1−/− mice display reduced body weight and altered metabolism (Wan et al. 2012), neither of which are seen in schizophrenia, and it is possible that such alterations could play a role in the behavioral changes observed in these mice independent of the relationship between reduced Akt1 expression and schizophrenia symptomology. In contrast to fully homozygous Akt1 null mice, heterozygous mice express Akt1 at approximately 20-40% the level observed in WT mice (Chen et al. 2012), suggesting that these hypomorphic mice could serve as a model of reduced, but not abolished, Akt1 expression, a condition more characteristic of the human disease state.
The present investigation sought to characterize the phenotype of Akt1+/− mice on several electroencephalographic (EEG) and event related potential (ERP) measures previously shown to have high translational validity for schizophrenia. Following the assessment of EEG, Akt1+/− and WT animals were assessed on PPI, a model of sensorimotor gating altered in schizophrenia (Braff et al. 1992). Given the potential importance of altered Akt1 as a vulnerability factor for the development of schizophrenia (Di Forti et al. 2012; Emamian et al. 2004; van Winkel et al. 2011), Akt1+/− mice were also assessed on each of these measures following administration of 50 mg/kg of ketamine. NMDA antagonists such as ketamine produce psychosis and cognitive impairment in human users (Krystal et al. 1994; Lahti et al. 2001; Malhotra et al. 1996; Newcomer et al. 1999), exacerbate psychotic symptoms (Lahti et al. 1995; Lahti et al. 2001; Malhotra et al. 1997) and recreate schizophrenia-like changes in the laboratory (Behrens et al. 2007; Ehrlichman et al. 2008; Lazarewicz et al. 2010; Moghaddam et al. 1997; Neill et al. 2010; Olney et al. 1999). It was predicted that Akt1+/− mice would show exaggerated EEG and behavioral responses to ketamine, suggestive of an enhanced vulnerability to evocation of behavioral changes associated with schizophrenia.
Materials and methods
Animals
Male Akt1−/−, Akt1+/− and WT mice were generated from a colony derived from founders provided by Dr. Morris Birnbaum backcrossed onto a C57BL/6 background over >10 generations (Cho et al. 2001). Genotyping was performed by PCR using the following primers: 5-AGCTCTTCTTCCAC CTGTCTC-3; 5-GCTCCATAAGCACACCTTCAGG-3; 5-GTGGATGTGGAATGTGTGCGAG-3. Mice were weaned at day 21 and housed 2–4 in standard sized cages and were singly housed following of surgery. Rooms were maintained at 22 (±2) °C and were kept on a 12:12 light/dark cycle (lights on at 08:00). All testing took place within the light phase of the light/dark cycle. Mice had standard laboratory mouse chow and water available ad libitum. Procedures were carried out according to the NIH Guide for the Care and Use of Laboratory Animals and the local Institutional Laboratory Animal Care and Use Committee (IACUC) guidelines. For EEG, a total of 28 mice were used (N= 11 WT, 8 Akt1+/− and 9 Akt1−/−), and 40 mice were used for PPI (N=10 WT saline, 10 WT ketamine, 10 Akt1+/− saline and 10 Akt1+/− ketamine). EEG mice were between 13 and 36 weeks of age (mean WT = 21.7 weeks; mean Akt1+/− = 20.1 weeks; mean Akt1−/− = 28.3 weeks). PPI mice were between 17 and 30 weeks of age (mean age WT mice = 23.4; mean age Akt1+/− = 23.8).
Electrode implantation
Mice were anesthetized under 1% isoflurane. The recording electrode was placed into the right hippocampal region at 1.8 mm posterior, 2.65 mm lateral, and 2.75 mm ventral to bregma. The positive electrode was referenced to the surface of the ipsilateral cortex (0.2 mm anterior, 2.65 mm lateral, and 0.75 mm ventral to bregma) and grounded cortex (0.8 mm posterior, 2.75 mm lateral, and 0.75 mm ventral to bregma). Electrodes were secured to the skull using Ethyl cyanoacrylate (Loctite) and dental cement (Ortho Jet). Animals were allowed seven days to recovery before EEG assessment. Given the distance between recording and reference electrodes this configuration records electrical activity beyond the localized field of the target region. By recording electrical activity across a widespread area, this configuration closely mimics EEG recording configurations used in humans. ERPs recorded using this configuration bear close resemblance to those obtained from the Cz scalp location in humans and show similar pharmacological and parametric responses to those obtained at the human Cz (Maxwell et al. 2006; Maxwell et al. 2004b; Rudnick et al. 2009; Siegel et al. 2003; Siegel et al. 2005).
EEG recording and analysis
Assessment of EEG took place in 8 standard mouse cages equipped with modified lids that allowed placement of a speaker on the top of the cage. Auditory stimuli were generated by Spike2, version 6.0, software and were delivered through a Power1401 interface (CED, UK). Mice were given 15 min to acclimate prior to the start of EEG recording. Each session consisted of a total of 300 click pairs (white noise, 10 msec duration, 85 dB). Each click pair was separated by a 500 msec interval presented every 8 sec. EEG was assessed using a within subjects design wherein each mouse was tested following both saline and ketamine (50 mg/kg) treatments in sequential sessions, with ketamine sessions following saline sessions. Each session occurred 40 min after administration of saline or ketamine.
Event-related potentials (ERP)
Spike2 software (CED, UK) was used to analyze and quantify ERPs. The amplitude of each component was quantified as the change in amplitude relative to the previous point of inflection within the relevant timeframe. These included the P20 (maximum value between 15 and 30 msec) and the N40 (minimum value occurring between 25 and 80 msec). Each component was quantified in response to the first (S1) and second (S2) stimulus of the click pair, thereby creating a P20 S1 and S2 and an N40 S1 and S2. Additionally, S1-S2 difference was calculated.
Baseline EEG power
Baseline power was assessed during a 60 sec period prior to auditory stimuli. Power decomposition was accomplished using the FFT function native to Spike2 (Hanning window, 0.81 Hz resolution). The resulting power spectra were analyzed for theta (4 to 12 Hz) and gamma (30 to 90 Hz) frequency bands.
Event-related Power
To analyze event-related power, EEG data were first exported from Spike2 into MATLAB. EEGLAB (Schwartz Center for Computational Neuroscience) was then used to process the data and create time-frequency measures. For each session, 300 single-trial epochs, ranging from −1 to 2 sec relative to tone onset, were extracted from the continuous EEG and further analyzed. Power was calculated within each epoch using Morlet wavelets in 116 logarithmically spaced frequency bins between 4 and 120 Hz, with wavelet cycle numbers ranging from 2 to 10 (Delorme and Makeig 2004). Theta was defined as the frequency band between 4 and 12 Hz and between 30 to 90 Hz. To create a single value for each measure, theta power was averaged between 0 and 400 msec, and gamma power was averaged between 0 to 60 msec. The ERSP is a method of event-related time-frequency power decomposition that measures changes in the power spectrum across time relative to a pre-event baseline period (Makeig 1993). The event period (−1 to 2 sec) is broken down into several overlapping family of waves with spectral changes averaged across numerous trials within each window. As such, the ERSP can be used to detect event-related changes in the power spectrum in response to auditory stimulation or other external stimuli. Intertrial coherence (ITC) is a measure of event-related phase synchronization of EEG across multiple trials. Both the ERSP and ITC were derived using EEGLAB.
Pre-pulse Inhibition (PPI)
PPI was assessed in four startle chambers (San Diego Instruments). The startle stimuli were white noise bursts (40 msec, 120 dB) presented through a speaker in the top of the chamber. Pre-pulse stimuli were white noise bursts (20 msec) presented at 69, 77 or 85 dB intensity 100 msec prior to the startle stimulus. Startle was measured with an accelerometer. Each session consisted of an initial 5 min acclimation period to background noise (60 dB), followed by five 120 dB startle pulses. Startle was then assessed in response to 0, 90, 95, 100, 105, 110, 115, and 120-db white noise bursts (40 msec) presented randomly with a mean interstimulus interval of 15 sec (10 sec min, 20 sec max). The last phase of each session assessed PPI of startle. For this, mice were exposed to both startle-alone stimuli and pre-pulse stimuli. PPI sessions consisted of 5 presentations of startle stimulus alone and five presentations of each pre-pulse. Each pre-pulse was also presented 5 times alone without the subsequent 120 dB startle pulse. PPI was calculated as [100−(startle+ pre-pulse/startle alone)×100] (%PPI). Ketamine and saline were given 40 min prior to assessment. Decibel levels for PPI stimuli were measured with a decibel meter (Radioshack Cat No. 33 2055) using A setting, fast response, max response.
Drugs
Ketamine (Ketalar, JHP Pharmaceuticals, MI) was diluted in 0.9% saline vehicle from a concentration of 100mg Ket per 1 ml saline. All injections were delivered into the intraperitoneal cavity (I.P.) in a volume of 0.1 ml 40 min prior to the start of testing. The dose of 50 mg/kg was chosen based on prior studies showing that this dose is minimally sufficient to produce reductions in EEG power that last throughout the timeframe of the testing period used here without inducing anesthesia or other severe side-effects.
Western Blot
Prefrontal cortex (PFC) was tissue dissected from WT (n=7) and Akt1+/− (n=7) mice. Proteins from left PFC were extracted in lysis buffer (25 mM Hepes pH=7.5, 300 mM NaCl, 0.2 mM EDTA, 1.5 mM MgCl2, 0.1% Triton, 0.5 mM DTT plus protease and phosphatase inhibitors). The protein concentration was determined using the Bradford method and 150 μg of proteins were loaded onto NuPage Novex 4-12% Bis-Tris precast gels (Invitrogen) and transferred onto PVDF membrane (Millipore). The membrane was incubated with the Akt1 antibody (rabbit monoclonal, #2938, cell signaling, 1/2000) followed by the secondary antibody (HRP conjugated Donkey anti Rabbit IgG (H+L), Jackson Immunoresearch, 1/10000). The blots were developed with chemoluminescence reagent ECL (Thermo Scientific). Films were scanned with a GS-800 Calibrated Densitometer and the signal was quantified using the Quantity One 4.6.3 software. Results are presented as measure of densitometric intensity after normalization to WT.
Statistics
Data were analyzed using repeated measures ANOVAs, with gene as a between subjects variable and drug, as well as stimulus (S1 versus S2) where applicable, as repeated within subjects variable. Significant interaction effects on the three-way ANOVAs were further analyzed using multiple two-way ANOVAs and Fisher’s LSD, while significant interaction effects on two-way ANOVAs were analyzed using Fisher’s LSD. Startle response was analyzed using a repeated measures ANOVA with gene and drug as between subject variables and pulse intensity as a repeated within subject variable. PPI sessions were analyzed in a separate repeated measures ANOVA with gene and drug as a between subject variable and intensity (69, 77 and 85 dB) as a within subjects variable. Western blot results were analyzed using an unpaired, two-tailed, t-test.
Results
Western blot analyses of tissue from the prefrontal cortex showed a significant decrease of AKT1 in heterozygous mice compare to the wild type mice (t (12) = 2.530, p <0.05) (Fig 1).
Fig 1.
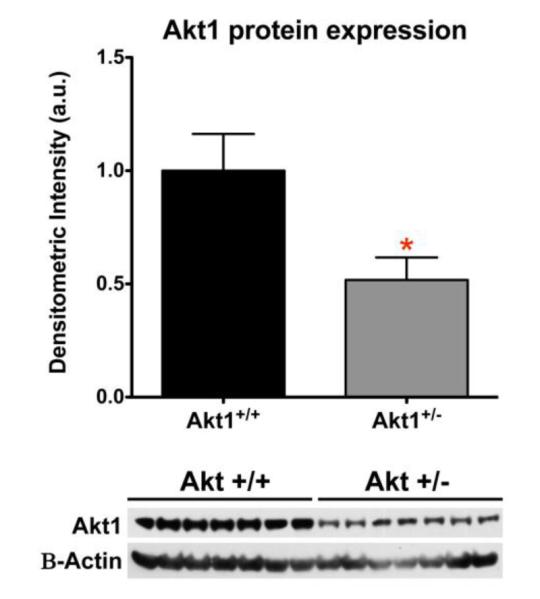
Western blot analysis for Akt1 in WT and Akt1+/− mice. A significant decrease in Akt1 expression was observed in Akt1+/− mice relative to WT controls (p<0.05).. Asterisk depicts significance at p<0.05
ERP measures
P20 amplitude
P20 amplitude response to the first stimulus (S1) did not differ as a function of genotype following either saline or ketamine treatment. However, a significant interaction was observed between gene × drug [F(2,25)=3.6, p<0.05] and for gene × drug × stimulus [F(2,25)=4.3, p<0.05]. Subsequent analyses showed a significant interaction between drug and gene on S1 [F(2, 25)=4.4, p<0.05], but not S2. Post hoc tests further revealed a significant decrease in P20 amplitude in Akt1−/− and Akt1+/− mice (p<0.01 for both) for P20 amplitude following ketamine treatment, relative to saline treatment. A similar change did not occur in WT animals, suggesting that ketamine significantly lowered P20 S1 amplitude in mutant, but not WT, mice (see Fig 4). Consistent with suppression or gating of the P20 following repetitive stimuli, amplitude of the S2 response was significantly lower than that of the S1 [F(1,25)=112.2, p<0.05]. Likewise, a significant interaction was seen for drug × stimulus [F(1,25)=38.7, p<0.05], with ketamine producing a more pronounced effect on the S1 response than S2 (see Fig 2).
Fig 4.

Event-Related Spectral Perturbation (ERSP) in WT, Akt1+/− and Akt1−/− mice. Figure A Top shows response following saline while Figure A Bottom shows response following 50 mg/kg ketamine. B. Quantification and analysis of ERSP response during saline treatment revealed a lasting post stimulus suppression of theta power, and this did not differ as a function of genotype. Ketamine significantly reduced post stimulus suppression in all groups but had a significantly greater effect in mutant mice (all p<0.05). C. A significant decrease in gamma power was observed following ketamine administration, but this did not differ as a function of genotype (p<0.05). Asterisk depicts significance at p<0.05
Fig 2.

Event-Related Potentials (ERP) in WT, Akt1+/− and Akt1−/− mice. A. Grand Average Waveform for the 200 msec period following presentation of the first white noise click of a paired-click stimulus (S1). WT, Akt1+/− and Akt1−/− mice are depicted in black, dark grey and light grey, respectively. B. Grand Average waveform following administration of 50 mg/kg of the NMDA antagonist ketamine for WT, Akt1+/− and Akt1−/− mice. C. Maximum positive deflection of the P20 response to the first click (S1) following saline (white) and 50 mg/kg ketamine (grey) treatment. Both Akt1+/− and Akt1−/− mice showed a significant decrease in P20 response following ketamine treatment (p<0.05). In contrast, a significant decrease was not observed following ketamine in WT mice. D. Maximum positive deflection of the P20 response to the second click (S2) of a paired-click stimulus following saline (white) and 50 mg/kg ketamine (grey) treatment. No differences were observed between groups on this measure. E. Maximum negative deflection of the N40 response to the first click (S1) following saline (white) and 50 mg/kg ketamine (grey) treatment. A significant main effect was observed for ketamine, but this did not differ as a function of Genotype. F. Maximum negative deflection of the N40 response to the second click (S2) following saline (white) and 50 mg/kg ketamine (grey) treatment. Asterisk depicts significance at p<0.05
In order to control for slight increases in baseline ERP response in mutant mice, a subset of mice from each group were matched according to P20 amplitude during saline treatment. Briefly, two mice that had P20 amplitudes above the range of the WT group were removed from the Akt1+/− group. Six mice were then selected from the WT group based on closeness of match to the remaining six Akt1+/− mice (scatterplots of these mice are shown in supplementary materials). Closeness of match was determined based on overall group average, such that the 6 WT mice selected proved mean P20 amplitude closest to mean of Akt1+/− mice. The same procedure was used to select Akt1−/− mice. An ANOVA conducted on P20 S1 amplitude following ketamine treatment revealed significant effect of genotype [F(2,15)=6.4, p<0.01]. Post hoc tests showed significantly lower P20 amplitude in Akt1+/− (p=0.006) and Akt1−/− mice (p=0.09) relative to WT. P20 S1 amplitude did not significantly differ between mutant mice (see figure 3). In contrast, no significance was observed for P20 S2 amplitude when subjects matched for baseline S1 response.
Fig 3.
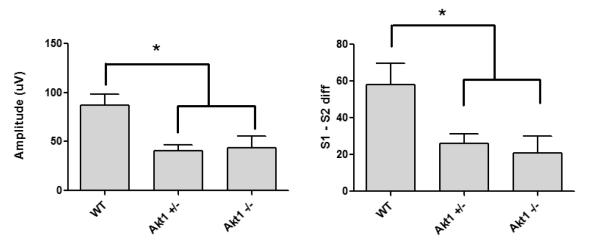
Effect of ketamine on P20 response in WT, Akt1+/− and Akt1−/− mice matched for P20 amplitude. A. The effect of ketamine on P20 amplitude was significantly greater in both Akt1+/− and Akt1−/− mice compared to WT (p<0.05). B. The effect of ketamine on P20 S1-S2 difference was significantly greater in both Akt1+/− and Akt1−/− mice compared to WT (p<0.05).
N40 amplitude
Ketamine reduced N40 amplitude relative to saline [F(1,25)=43.5, p<0.05], but this did not differ as a function of genotype. Likewise, N40 response was significantly lower following S2 than S1 [F(1,25)=68.3, p<0.05] and S1 was more significantly reduced by ketamine than S2 [F(1,25)=37.7, p<0.05]. Significance was observed for the three way interaction [F(2,25)=3.5, p<0.05]. Follow up ANOVAs showed significant reduction of both S1 and S2 amplitude following ketamine [F(1,25)=46.1 (S1) and 12.8 (S2)] and a trend towards significance for the interaction of drug × gene (p=0.1) (see Fig 2).
Analysis of N40 S1 response in the subset of mice created by matching P20 S1 amplitude (see above for details) failed to reveal a significant genotype difference in response to ketamine.
P20 (S1 – S2) Difference
Ketamine disproportionately affected S1-S2 difference across genotype conditions [F(2,25)=4.3,p<0.05]. Post hoc analyses revealed a significant decrease when comparing ketamine to saline periods in Akt1−/− and Akt1+/− (both p<0.01), but not in WT mice. Akt1+/− mice also showed increased S1-S2 difference during saline treatment relative to WT mice (p<0.001). Ketamine also produced a reduction in S1-S2 difference [F(1,25)=38.7, p<0.05] (Fig 2).
Since S1 – S2 difference is likely highly dependent upon the magnitude of initial baseline response (S1), a subset of mice were created in which subjects were matched based on similar P20 S1 amplitude response (see supplementary materials for scatterplots of these mice). A one way ANOVA conducted on P20 S1-S2 difference following ketamine treatment revealed a significant effect of genotype in this subset of mice [F(2,15)=5.0, p<0.02]. Post hoc tests showed a significant difference between WT mice and both mutant groups (Akt1+/− p=0.02; Akt1−/− p=0.011). Akt1+/− and Akt1−/− mice did not significantly differ from one another (see Fig 3).
N40 (S1-S2) Difference
Ketamine affected S1-S2 difference across genotype [F(2,25)=3.5, p<0.05], with Akt1−/− and Akt1+/− mice showing significant decreases in N40 difference when comparing post-ketamine to post-saline periods (p<0.01 for both). A similar change was not seen in WT mice. Ketamine also produced an overall reduction in S1-S2 difference [F(1,25)=37.7, p<0.05] (see Fig 2).
No differences were observed in the subset of mice matched for P20 S1 amplitude.
EEG Power
Baseline Power
No differences in baseline power were observed across gene conditions or for the gene by drug interaction, suggesting that baseline power did not differ as a function of genotype following either saline or ketamine treatment. Ketamine significantly increased baseline power for both gamma [F(2,25)=211.14, p<0.001] and theta [F(2,25)=14.2, p<0.001] across genotypes.
Event Related Power
No differences in event-related gamma power were observed as a function of genotype, either during saline or ketamine treatment. Ketamine significantly reduced event-related gamma power across all genotypes [F(2,25)=278.5, p<0.001] (see Fig 4).
For event-related theta, a significant main effect was found for gene [F(2,25)=3.9, p<0.05], with mutant mice showing higher theta power than WT mice. Likewise, ketamine significantly decreased theta power in both genotypes [F(2,25)=93.5, p<0.0001]. A significant interaction was observed between genotype and ketamine [F(2,25)=5.7, p<0.01]. Post hoc tests on these data failed to show an effect of gene during saline treatment. In contrast, significantly higher theta power was seen in Akt1+/− and Akt1−/− mice relative to WT mice (p=0.001 WT versus heterozygous; p=0.013 WT versus homozygous). Thus, increases in post stimulus theta power that normally occur following ketamine are greater in Akt1 mutant mice (see Fig 4).
Intertrial Coherence (ITC)
ITC within the gamma frequency band did not vary significantly across genotype, but was significantly reduced following ketamine treatment [F(2,25)=232.8, p<0.0001]. The ability for ketamine to reduce gamma ITC was significantly influenced by genotype, as suggested by a significant interaction between these two factors [F(2,25)=5.2, p<0.05]. Post hoc analyses failed to uncover a significant change in gamma ITC as a function of genotype following saline treatment, suggesting that there was no effect of Akt1 deletion or reduction on gamma ITC. In contrast, both Akt1−/− and Akt1+/− mice showed significant reductions in gamma ITC relative to WT mice following ketamine treatment (p<0.05 in both groups), suggesting that ketamine induced a greater reduction in gamma ITC in mutant mice versus WT mice (see Fig 5).
Fig 5.

Intertrial Coherence (ITC) in WT, Akt1+/− and Akt1−/− mice. Figure A Top shows ITC heat map following saline while Figure A bottom shows ITC heat map following 50 mg/kg ketamine. B. ITC for the theta frequency band was significantly reduced following ketamine treatment, but this did not differ as a function of genotype (p<0.05). C. A significant decrease in gamma power was observed following ketamine administration. Both Akt1+/− and Akt1−/− mice showed significantly reduced gamma ITC in response to ketamine relative to WT mice. Asterisk denotes significance at p<0.05
ITC within the theta range was not affected by gene nor was there a significant interaction between gene and drug, although there was a trend towards significance (p=0.056). Ketamine significantly reduced theta ITC [F(2,25)=90.8, p<0.0001] (see Fig 5).
Pre-pulse Inhibition (PPI)
A significant main effect was found for genotype, with Akt1+/− mice showing reduced PPI relative to WT mice [F(1,36)=4.9, p<0.05]. Additionally, a significant main effect was found for pre-pulse intensity [F(2,72)=46.99, p<0.05], with PPI increasing as a function as a function of pre-pulse intensity. Further, the disruptive effects of ketamine varied according to intensity of the pre-pulse, as suggested by a significant interaction between ketamine and pre-pulse intensity [F(2,72)=4.2, p<0.05]. Post hoc analysis of these data revealed a significant effect of ketamine only at the lowest pre-pulse intensity (69 dB, p<0.05). Additionally, this post hoc analysis revealed a significant difference between saline treated WT and ketamine treated Akt1+/− animals at the 69 dB intensity (p=0.006), with PPI being reduced in the latter group (see Fig 6).
Fig 6.

Pre-pulse inhibition (PPI) in WT and Akt1+/− mice following either saline or ketamine. A significant effect was observed for gene, with Akt1+/− animals displaying reduced PPI relative to WT mice. Additionally, 50 mg/kg ketamine produced a decrease in PPI in Akt1+/− mice relative to saline treated WT mice (p<0.05) on the lowest intensity pre-pulse. N=10 per group. Asterisk denotes significance at p<0.05, while # indicates significant difference from WT saline.
To assess basal startle amplitude, mice were exposed to pseudorandom presentations of pulse alone stimuli ranging from 90 to 120 dB, prior to PPI testing. Startle during this period failed to show significant differences for either drug treatment or genotype (see Supplementary materials). Assessment of startle response during no pulse trials also did not detect significant differences across genotype or drug (see Supplementary materials). Finally, analyses of pulse alone trials during PPI did not detect significant differences (see Supplementary materials). Thus, group on PPI were not likely due to differences in startle response.
Discussion
The current study is the first to examine event-related potentials and event-related oscillations in mice with altered Akt1 function. Alterations in the P50 and N100 components of the ERP are frequently observed in schizophrenic patients (Boutros et al. 2009; Boutros et al. 2004a; Boutros et al. 2004b; Freedman et al. 1983; Turetsky et al. 2009) and both measures show a relationship to positive symptoms as well as deficits in working memory and attention (Erwin et al. 1998; Lijffijt et al. 2009; Potter et al. 2006; Smith et al. 2010). Overlap between the mouse P20 and N40 and the human P50 and N100 is suggested by similarities in response to drugs, such as amphetamine (de Bruin et al. 1999; Light et al. 1999; Maxwell et al. 2004a) and nicotine (Adler et al. 1998; Phillips et al. 2007; Stevens et al. 1995), as well as responses to parametric manipulations (Ehrlichman et al. 2008; Siegel et al. 2003). As such, a paired-click auditory procedure was used to examine ERP responses in WT, Akt1−/− and Akt1+/− mice.
Neither Akt1−/− nor Akt1+/− mice showed reductions in P20 or N40 amplitude following exposure to saline vehicle, suggesting that these measures are not highly regulated by Akt1 function under normal conditions. Ketamine exposure also failed to produce a significant reduction in amplitude of either P20 or N40 component in WT mice, consistent with previous studies examining the effect of ketamine on P20 and N40 amplitude, in humans and rodents (Connolly et al. 2004; de Bruin et al. 1999; Oranje et al. 2002; van Berckel et al. 1998). In contrast, both Akt1−/− and Akt1+/− mice showed a significant reduction of the P20 response to the first click of the paired-click stimulus (S1) following ketamine. A significant reduction in S1-S2 difference was also observed in both groups of mutant mice following ketamine for the P20 component. This pattern of change is broadly consistent with that observed in schizophrenia patient populations and suggests that the reduction of Akt1 expression facilitated the ability for ketamine to induce electrophysiological changes similar to those observed in schizophrenia.
Separate cohorts of WT and Akt1+/− mice were examined for PPI. Akt1+/− mice showed reduced PPI relative to WT mice, independent of ketamine treatment. Analysis of baseline startle amplitude during pulse only trials did not differ across genotype, suggesting that the PPI reductions seen in Akt1+/− mice likely reflect impaired sensorimotor gating rather than more basic alterations in sensory reactivity. Consistent with previous reports, ketamine had little impact either on PPI or baseline startle response (de Bruin et al. 1999; Oranje et al. 2002; van Berckel et al. 1998). This stands in sharp contrast to the effect of ketamine reported here and elsewhere on ERP and EEG responses and strongly suggests that these two measures reflect very different neural and/or psychological mechanisms. It is interesting to note that ketamine-induced alterations in ERP response mostly involved reduction of the S1 response, with much less impact being observed on S2 response, suggesting a change in sensory registration or attention (“gating in”) rather than sensory gating or suppression, per se (“gating out”) (Brenner et al. 2009). Alternatively, the lack of effect on S2 could reflect a floor effect due to its relatively small baseline value.
Marked reductions in EEG event-related oscillatory response have been observed in schizophrenia (Doege et al. 2009; Johannesen et al. 2008; Kirihara et al. 2012; Kwon et al. 1999; Roach and Mathalon 2008), and these can be readily assessed and reproduced in animal models of schizophrenia (Behrens et al. 2007; Cao et al. 2012; Carlen et al. 2011; Ehrlichman et al. 2009; Featherstone et al. 2012; Lazarewicz et al. 2010). Here mice were assessed for event-related power and ITC. Consistent with previous studies examining the effects of NMDA hypofunction on EEG, ketamine significantly reduced both event-related gamma power and gamma ITC during the immediate post stimulus period (0 to 60 msec). The desynchronizing effect of ketamine on gamma ITC was significantly greater in Akt1+/− and Akt1−/− mice. A similar trend was observed for evoked-gamma power, although this did not reach statistical significance.
All mice showed a time-locked post-stimulus suppression of late theta power in response the first click of the stimulus pair and this was significantly reduced following ketamine. Importantly, Akt1 reduction enhanced this effect relative to WT mice. Theta suppression in response to sensory stimulation has been previously reported in rodents and in patients (Ford et al. 2008; Lazarewicz et al. 2010), but the importance of this phenomenon is unknown. Ford, et al, (2008) showed a decrease in theta power during a P300 task in healthy controls and an attenuation of this response in patients, suggesting that theta suppression could play a role in novelty detection. Suppression within the frequency range reported here may play a role in human long-term memory (Klimesch 1999). In the current study, theta suppression appeared to overlap strongly with enhanced gamma within the same time frame, suggesting that suppressed theta may play a role in maintaining gamma response following termination of the stimulus.
The present study demonstrates that genetic reduction in Akt1 expression can enhance the effect of ketamine on schizophrenia related measures in mice, consistent with the notion that reduced Akt1 expression may serve as a vulnerability factor for schizophrenia. While schizophrenia has a strong genetic basis, the fact that monozygotic twins can be discordant for the disease strongly implies a role for epigenetic or non-genetic factors. Evidence has suggested that the development of schizophrenia can be influenced by the use of illicit drugs, amongst other factors. Reduced Akt1 may act as a vulnerability factor for cannabis induced psychosis and cognitive impairment in humans (Di Forti et al. 2012; van Winkel et al. 2011). It is possible that Akt1 mediates the effect of other risk factors that impinge on glutamatergic signaling as a predisposition to disease emergence. Thus, further investigation of the role of Akt1 in mediating non-drug related risk factors in schizophrenia would be useful in elucidating physiological mechanisms that predispose emergence of symptoms of the disease.
Supplementary Material
Acknowledgements
We are grateful to Tiffany Hill-Smith for expert technical assistance. This study was supported by NIDA grant 5R01DA023210-02 to Steven J. Siegel and by USPHS grant MH 86599 to Irwin Lucki. None of the authors have a financial relationship with the organizations that sponsored the research outlined in the present manuscript.
Steven J. Siegel has received grants from Astellas, AstraZeneca, Abbott, NuPathe, Pfizer and GlaxoSmithKline that are unrelated to the content of this manuscript. Robert Featherstone has received grant support from Astellas that is unrelated to the content of this manuscript. Steven Siegel has served as a consultant to Abbott, NuPathe and Lundbeck.
The experiments described in this manuscript comply with the current laws of the USA.
References
- Adler LE, Olincy A, Waldo M, Harris JG, Griffith J, Stevens K, Flach K, Nagamoto H, Bickford P, Leonard S, Freedman R. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull. 1998;24:189–202. doi: 10.1093/oxfordjournals.schbul.a033320. [DOI] [PubMed] [Google Scholar]
- Balu DT, Carlson GC, Talbot K, Kazi H, Hill-Smith TE, Easton RM, Birnbaum MJ, Lucki I. Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus. 2012;22:30–40. doi: 10.1002/hipo.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–7. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Blasi G, Napolitano F, Ursini G, Taurisano P, Romano R, Caforio G, Fazio L, Gelao B, Di Giorgio A, Iacovelli L, Sinibaldi L, Popolizio T, Usiello A, Bertolino A. DRD2/AKT1 interaction on D2 c-AMP independent signaling, attentional processing, and response to olanzapine treatment in schizophrenia. Proc Natl Acad Sci U S A. 2012;108:1158–63. doi: 10.1073/pnas.1013535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros NN, Brockhaus-Dumke A, Gjini K, Vedeniapin A, Elfakhani M, Burroughs S, Keshavan M. Sensory-gating deficit of the N100 mid-latency auditory evoked potential in medicated schizophrenia patients. Schizophr Res. 2009;113:339–46. doi: 10.1016/j.schres.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros NN, Korzyuko O, Oliwa G, Feingold A, Campbell D, McClain-Furmanski D, Struve F, Jansen BH. Morphological and latency abnormalities of the mid-latency auditory evoked responses in schizophrenia: a preliminary report. Schizophr Res. 2004a;70:303–13. doi: 10.1016/j.schres.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Boutros NN, Korzyukov O, Jansen B, Feingold A, Bell M. Sensory gating deficits during the mid-latency phase of information processing in medicated schizophrenia patients. Psychiatry Res. 2004b;126:203–15. doi: 10.1016/j.psychres.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49:206–15. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- Brenner CA, Kieffaber PD, Clementz BA, Johannesen JK, Shekhar A, O’Donnell BF, Hetrick WP. Event-related potential abnormalities in schizophrenia: a failure to “gate in” salient information? Schizophr Res. 2009;113:332–8. doi: 10.1016/j.schres.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YA, Featherstone RE, Gandal MJ, Liang Y, Jutzeler C, Saunders J, Tatard-Leitman V, Chen J, Weinberger DR, Lerman C, Siegel SJ. Nicotine normalizes event related potentials in COMT-Val-tg mice and increases gamma and theta spectral density. Behav Neurosci. 2012;126:332–43. doi: 10.1037/a0027047. [DOI] [PubMed] [Google Scholar]
- Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Ruhlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2011;17:537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Chen YW, Hsu YF, Chang WT, Hsiao CK, Min MY, Lai WS. Akt1 deficiency modulates reward learning and reward prediction error in mice. Genes Brain Behav. 2012;11:157–69. doi: 10.1111/j.1601-183X.2011.00759.x. [DOI] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- Connolly PM, Maxwell C, Liang Y, Kahn JB, Kanes SJ, Abel T, Gur RE, Turetsky BI, Siegel SJ. The effects of ketamine vary among inbred mouse strains and mimic schizophrenia for the P80, but not P20 or N40 auditory ERP components. Neurochem Res. 2004;29:1179–88. doi: 10.1023/b:nere.0000023605.68408.fb. [DOI] [PubMed] [Google Scholar]
- de Bruin NM, Ellenbroek BA, Cools AR, Coenen AM, van Luijtelaar EL. Differential effects of ketamine on gating of auditory evoked potentials and prepulse inhibition in rats. Psychopharmacology (Berl) 1999;142:9–17. doi: 10.1007/s002130050856. [DOI] [PubMed] [Google Scholar]
- Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Methods. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Di Forti M, Iyegbe C, Sallis H, Kolliakou A, Falcone MA, Paparelli A, Sirianni M, La Cascia C, Stilo SA, Marques TR, Handley R, Mondelli V, Dazzan P, Pariante C, David AS, Morgan C, Powell J, Murray RM. Confirmation that the AKT1 (rs2494732) Genotype Influences the Risk of Psychosis in Cannabis Users. Biol Psychiatry. 2012;72:811–816. doi: 10.1016/j.biopsych.2012.06.020. [DOI] [PubMed] [Google Scholar]
- Doege K, Bates AT, White TP, Das D, Boks MP, Liddle PF. Reduced event-related low frequency EEG activity in schizophrenia during an auditory oddball task. Psychophysiology. 2009;46:566–77. doi: 10.1111/j.1469-8986.2009.00785.x. [DOI] [PubMed] [Google Scholar]
- Ehrlichman RS, Gandal MJ, Maxwell CR, Lazarewicz MT, Finkel LH, Contreras D, Turetsky BI, Siegel SJ. N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience. 2009;158:705–12. doi: 10.1016/j.neuroscience.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Ehrlichman RS, Maxwell CR, Majumdar S, Siegel SJ. Deviance-elicited changes in event-related potentials are attenuated by ketamine in mice. J Cogn Neurosci. 2008;20:1403–14. doi: 10.1162/jocn.2008.20097. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–7. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- Erwin RJ, Turetsky BI, Moberg P, Gur RC, Gur RE. P50 abnormalities in schizophrenia: relationship to clinical and neuropsychological indices of attention. Schizophr Res. 1998;33:157–67. doi: 10.1016/s0920-9964(98)00075-9. [DOI] [PubMed] [Google Scholar]
- Featherstone RE, Liang Y, Saunders JA, Tatard-Leitman VM, Ehrlichman RS, Siegel SJ. Subchronic ketamine treatment leads to permanent changes in EEG, cognition and the astrocytic glutamate transporter EAAT2 in mice. Neurobiol Dis. 2012;47:338–46. doi: 10.1016/j.nbd.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Ford JM, Roach BJ, Hoffman RS, Mathalon DH. The dependence of P300 amplitude on gamma synchrony breaks down in schizophrenia. Brain Res. 2008;1235:133–42. doi: 10.1016/j.brainres.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Adler LE, Waldo MC, Pachtman E, Franks RD. Neurophysiological evidence for a defect in inhibitory pathways in schizophrenia: comparison of medicated and drug-free patients. Biol Psychiatry. 1983;18:537–51. [PubMed] [Google Scholar]
- Johannesen JK, Bodkins M, O’Donnell BF, Shekhar A, Hetrick WP. Perceptual anomalies in schizophrenia co-occur with selective impairments in the gamma frequency component of midlatency auditory ERPs. J Abnorm Psychol. 2008;117:106–18. doi: 10.1037/0021-843X.117.1.106. [DOI] [PubMed] [Google Scholar]
- Kirihara K, Rissling AJ, Swerdlow NR, Braff DL, Light GA. Hierarchical organization of gamma and theta oscillatory dynamics in schizophrenia. Biol Psychiatry. 2012;71:873–80. doi: 10.1016/j.biopsych.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimesch W. EEG alpha and theta oscillations reflect cognitive and memory performance: a review and analysis. Brain Res Brain Res Rev. 1999;29:169–95. doi: 10.1016/s0165-0173(98)00056-3. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr., Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Kwon JS, O’Donnell BF, Wallenstein GV, Greene RW, Hirayasu Y, Nestor PG, Hasselmo ME, Potts GF, Shenton ME, McCarley RW. Gamma frequency-range abnormalities to auditory stimulation in schizophrenia. Arch Gen Psychiatry. 1999;56:1001–5. doi: 10.1001/archpsyc.56.11.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–67. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- Lai WS, Xu B, Westphal KG, Paterlini M, Olivier B, Pavlidis P, Karayiorgou M, Gogos JA. Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc Natl Acad Sci U S A. 2006;103:16906–11. doi: 10.1073/pnas.0604994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarewicz MT, Ehrlichman RS, Maxwell CR, Gandal MJ, Finkel LH, Siegel SJ. Ketamine modulates theta and gamma oscillations. J Cogn Neurosci. 2010;22:1452–64. doi: 10.1162/jocn.2009.21305. [DOI] [PubMed] [Google Scholar]
- Light GA, Malaspina D, Geyer MA, Luber BM, Coleman EA, Sackeim HA, Braff DL. Amphetamine disrupts P50 suppression in normal subjects. Biol Psychiatry. 1999;46:990–6. doi: 10.1016/s0006-3223(99)00034-7. [DOI] [PubMed] [Google Scholar]
- Lijffijt M, Lane SD, Meier SL, Boutros NN, Burroughs S, Steinberg JL, Moeller FG, Swann AC. P50, N100, and P200 sensory gating: relationships with behavioral inhibition, attention, and working memory. Psychophysiology. 2009;46:1059–68. doi: 10.1111/j.1469-8986.2009.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeig S. Auditory event-related dynamics of the EEG spectrum and effects of exposure to tones. Electroencephalogr Clin Neurophysiol. 1993;86:283–93. doi: 10.1016/0013-4694(93)90110-h. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17:141–50. doi: 10.1016/S0893-133X(97)00036-5. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, Breier A. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14:301–7. doi: 10.1016/0893-133X(95)00137-3. [DOI] [PubMed] [Google Scholar]
- Maxwell CR, Ehrlichman RS, Liang Y, Trief D, Kanes SJ, Karp J, Siegel SJ. Ketamine produces lasting disruptions in encoding of sensory stimuli. J Pharmacol Exp Ther. 2006;316:315–24. doi: 10.1124/jpet.105.091199. [DOI] [PubMed] [Google Scholar]
- Maxwell CR, Kanes SJ, Abel T, Siegel SJ. Phosphodiesterase inhibitors: a novel mechanism for receptor-independent antipsychotic medications. Neuroscience. 2004a;129:101–7. doi: 10.1016/j.neuroscience.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Maxwell CR, Liang Y, Weightman BD, Kanes SJ, Abel T, Gur RE, Turetsky BI, Bilker WB, Lenox RH, Siegel SJ. Effects of chronic olanzapine and haloperidol differ on the mouse N1 auditory evoked potential. Neuropsychopharmacology. 2004b;29:739–46. doi: 10.1038/sj.npp.1300376. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL, Snigdha S, Rajagopal L, Harte MK. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther. 2010;128:419–32. doi: 10.1016/j.pharmthera.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–18. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- Ohi K, Hashimoto R, Yasuda Y, Fukumoto M, Nemoto K, Ohnishi T, Yamamori H, Takahashi H, Iike N, Kamino K, Yoshida T, Azechi M, Ikezawa K, Tanimukai H, Tagami S, Morihara T, Okochi M, Tanaka T, Kudo T, Iwase M, Kazui H, Takeda M. The AKT1 gene is associated with attention and brain morphology in schizophrenia. World J Biol Psychiatry. 2011 doi: 10.3109/15622975.2011.591826. doi: 10.3109/15622975.2011.591826. [DOI] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–33. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Oranje B, Gispen-de Wied CC, Verbaten MN, Kahn RS. Modulating sensory gating in healthy volunteers: the effects of ketamine and haloperidol. Biol Psychiatry. 2002;52:887–95. doi: 10.1016/s0006-3223(02)01377-x. [DOI] [PubMed] [Google Scholar]
- Phillips JM, Ehrlichman RS, Siegel SJ. Mecamylamine blocks nicotine-induced enhancement of the P20 auditory event-related potential and evoked gamma. Neuroscience. 2007;144:1314–23. doi: 10.1016/j.neuroscience.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter D, Summerfelt A, Gold J, Buchanan RW. Review of clinical correlates of P50 sensory gating abnormalities in patients with schizophrenia. Schizophr Bull. 2006;32:692–700. doi: 10.1093/schbul/sbj050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach BJ, Mathalon DH. Event-related EEG time-frequency analysis: an overview of measures and an analysis of early gamma band phase locking in schizophrenia. Schizophr Bull. 2008;34:907–26. doi: 10.1093/schbul/sbn093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick ND, Koehler C, Picciotto MR, Siegel SJ. Role of beta2-containing nicotinic acetylcholine receptors in auditory event-related potentials. Psychopharmacology (Berl) 2009;202:745–51. doi: 10.1007/s00213-008-1358-6. [DOI] [PubMed] [Google Scholar]
- Siegel SJ, Connolly P, Liang Y, Lenox RH, Gur RE, Bilker WB, Kanes SJ, Turetsky BI. Effects of strain, novelty, and NMDA blockade on auditory-evoked potentials in mice. Neuropsychopharmacology. 2003;28:675–82. doi: 10.1038/sj.npp.1300087. [DOI] [PubMed] [Google Scholar]
- Siegel SJ, Maxwell CR, Majumdar S, Trief DF, Lerman C, Gur RE, Kanes SJ, Liang Y. Monoamine reuptake inhibition and nicotine receptor antagonism reduce amplitude and gating of auditory evoked potentials. Neuroscience. 2005;133:729–38. doi: 10.1016/j.neuroscience.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Smith AK, Edgar JC, Huang M, Lu BY, Thoma RJ, Hanlon FM, McHaffie G, Jones AP, Paz RD, Miller GA, Canive JM. Cognitive abilities and 50- and 100-msec paired-click processes in schizophrenia. Am J Psychiatry. 2010;167:1264–75. doi: 10.1176/appi.ajp.2010.09071059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens KE, Meltzer J, Rose GM. Nicotinic cholinergic normalization of amphetamine-induced loss of auditory gating in freely moving rats. Psychopharmacology (Berl) 1995;119:163–70. doi: 10.1007/BF02246157. [DOI] [PubMed] [Google Scholar]
- Szamosi A, Kelemen O, Keri S. Hippocampal volume and the AKT signaling system in first-episode schizophrenia. J Psychiatr Res. 2012;46:279–84. doi: 10.1016/j.jpsychires.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Tan HY, Chen AG, Chen Q, Browne LB, Verchinski B, Kolachana B, Zhang F, Apud J, Callicott JH, Mattay VS, Weinberger DR. Epistatic interactions of AKT1 on human medial temporal lobe biology and pharmacogenetic implications. Mol Psychiatry. 2011;17:1007–1016. doi: 10.1038/mp.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HY, Nicodemus KK, Chen Q, Li Z, Brooke JK, Honea R, Kolachana BS, Straub RE, Meyer-Lindenberg A, Sei Y, Mattay VS, Callicott JH, Weinberger DR. Genetic variation in AKT1 is linked to dopamine-associated prefrontal cortical structure and function in humans. J Clin Invest. 2008;118:2200–8. doi: 10.1172/JCI34725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turetsky BI, Bilker WB, Siegel SJ, Kohler CG, Gur RE. Profile of auditory information-processing deficits in schizophrenia. Psychiatry Res. 2009;165:27–37. doi: 10.1016/j.psychres.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berckel BN, Oranje B, van Ree JM, Verbaten MN, Kahn RS. The effects of low dose ketamine on sensory gating, neuroendocrine secretion and behavior in healthy human subjects. Psychopharmacology (Berl) 1998;137:271–81. doi: 10.1007/s002130050620. [DOI] [PubMed] [Google Scholar]
- van Winkel R, van Beveren NJ, Simons C. AKT1 moderation of cannabis-induced cognitive alterations in psychotic disorder. Neuropsychopharmacology. 2011;36:2529–37. doi: 10.1038/npp.2011.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M, Easton RM, Gleason CE, Monks BR, Ueki K, Kahn CR, Birnbaum MJ. Loss of Akt1 in mice increases energy expenditure and protects against diet-induced obesity. Mol Cell Biol. 2012;32:96–106. doi: 10.1128/MCB.05806-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.