

A valuable approach for creating synthetic polymers is the ring-opening metathesis polymerization (ROMP).[1,2] Well-defined metal carbene catalysts have been devised that afford control over the polymer chain length and architecture.[3–6] In addition, ruthenium carbene intiators have been developed with excellent air stability and functional group tolerance.[6,7] These catalysts enable the synthesis of polymers with a range of functionality,[8–15] thereby providing access to polymers for diverse applications.[16–19]
Extant polymers from ROMP, like the majority of synthetic polymers, are non-degradable. They therefore lead to refuse accumulation. A functional and degradable polymer from ROMP would allow the synthetically useful traits of ROMP reactions to be combined with the growing need for new degradable polymer scaffolds. To date, efforts to prepare degradable materials using ROMP have afforded polymers that are either functional or partially hydrolysable, but not both (Figure 1). For example, cargo can be attached via a linker such that it can be released from the polymer backbone by photolysis or hydrolysis in acid.[16,20–22] Still, the polymeric backbone persists. Alternatively, partially degradable polymers have been generated. A block copolymer can be generated from a modifiable monomer and a sacrificial dioxepine or dithiepine monomer.[8,23,24] In this scenario, one block is composed of a non-hydrolysable backbone and the degradable block contains acid-labile acetals or thioacetals that can be cleaved by hydrogenation. Polymers of this type only undergo partial degradation, as one block persists. The current state-of-the-art therefore demands a compromise between generating polymers that can be customized and polymers that can be easily degraded.
Figure 1.

Strategies to synthesize functionalizable and degradable ROMP polymers: a) ligand attachment via a cleavable linker, b) copolymerization with a sacrificial monomer, and c) homopolymerization of a functionalizable, heterocyclic oxazinone.
Applying ROMP to synthesize a modifiable homopolymer with a degradable backbone requires a monomer with three important attributes. First, it must be a strained cyclic or bicyclic olefin, so that it undergoes polymerization.[25] Second, it must contain core functionality that gives rise to a polymer that can be degraded. Third, a means to append desired functionality onto the monomer or polymer is needed to enable polymer diversification. Monomers with all of these attributes have been elusive. Many strained olefinic heterocycles spontaneously aromatize.[26,27] In addition, attempts to incorporate handles for diversification can further increase monomer instability.[28] Thus, traditional monomers used in ROMP cannot be simply modified to instil polymer degradability.
We sought a new class of monomers that would give rise to degradable materials. The generation of substrates with an 8-oxo-2-azabicylo[3.2.1]oct-6-en-3-one framework through a novel aza-[4+3] cycloaddition was recently reported (Scheme 1A).[29] We postulated that this bicyclic oxazinone would be a substrate for ROMP. Calculations suggest that this framework has a ring strain of 13.4 kcal/mol, which is comparable to that of trans-cyclooctene (a monomer that has favourable kinetics of polymerization using ROMP).[25,30] Reports of successful ring-opening cross metathesis on architecturally analogous 8-oxo-bicyclo[3.2.1.]oct-6-en-3-ones provided additional impetus.[31,32] Significantly, if the bicyclic oxazinone serves as a monomer in ROMP, the resulting framework should be both acid and base labile (Scheme 1B). Moreover, we postulated that we could modify this core monomer at a site distal to the polymerizable moieties and bridgehead carbons. Thus, without destabilizing the heterocycle, we could tailor the properties of the resulting materials.
Scheme 1.

Synthesis of bicyclic oxazinones that undergo ROMP to afford degradable polymers.
Despite our optimism that bicyclic oxazinones would serve as a monomer, we had concerns that its polymerization would yield highly oxygenated polymer products that would facilitate backbiting.[34,35] Our initial results confirmed our suspicions. The reaction of 3a using Grubbs’ second generation catalyst (GII)[33] in chloroform afforded polymer 4a (Table 1, entry 1), but we observed a concomitant increase in the PDI and decrease in the number averaged molecular weight (Mn) of the products as the polymerization progressed (Supporting Information, Table S1). One strategy to mitigate backbiting is to raise the reaction temperature to disrupt dative bonds.[36,37] However, in our system, an increase in temperature did not reduce backbiting (Table 1, entry 2). The problem of backbiting, nevertheless, was solved. We performed the polymerization reaction in tetrahydrofuran (THF), a solvent system that can compete with the polymer for catalyst coordination. We also employed catalyst 5, which has superior polymerization kinetics to GII.[7,36] These changes afforded dramatic improvements in the polymerization. We obtained stable Mn or PDI values even at long reaction times (Table 1, entry 5–6). In addition, the Mn increased proportionally to monomer to catalyst loading (Table 1, entry 3–7), and the degree of polyerization was found to be in good agreement with monomer conversion. Thus, structures such as compound 3a undergo ROMP to afford polymer scaffolds with a repeating oxazinane backbone.
Table 1.
Polymerization of monomer 3a using ROMP
| entry | [M]o/[I] | catalyst | solvent[a] | temp (°C) |
time (h) |
conversion (%)[b] |
Yield[c,d] | Mntheo (g/mol) |
MnNMR
[e] (g/mol) |
MnGPC
[f] (g/mol) |
PDI[f] (Mn/Mw) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 100/1 | GII | CHCl3 | 20 | 4 | 84 | 72 | 23600 | 103800 | 63200 | 2.8 |
| 2 | 100/1 | GII | CHCl3 | 45 | 18 | 81[g] | 49 | 22800 | 26000 | 24900 | 2.6 |
| 3 | 25/1 | 5 | THF | 20 | 1 | 87 | 57 | 6100 | 6500 | 9300 | 1.4 |
| 4 | 50/1 | 5 | THF | 20 | 1 | 85 | 71 | 12000 | 12500 | 16300 | 1.4 |
| 5 | 100/1 | 5 | THF | 20 | 1 | 81 | 80 | 23100 | 22000 | 21600 | 1.4 |
| 6 | 100/1 | 5 | THF | 20 | 18 | 81 | 85 | 23100 | 22100 | 22300 | 1.4 |
| 7 | 200/1 | 5 | THF | 20 | 1 | 73 | 83 | 41000 | 45200 | 50300 | 1.5 |
[M]o = 1M.
based off of 1H-NMR integrations of monomer olefin signals to polymer olefin signals.
isolated.
theoretical yield based off of monomer conversion.
based off 1H-NMR integrations of polymers olefin signals and polymers chain-end phenyl signals.
calibrated with polystyrene standards, eluted in THF,
additional 10–15% cyclic dimer (see Supporting Information for structural characterization of byproduct).
We next investigated the degradability of the new polyoxazinone scaffold. Small molecule oxazinones have been used to generate β-hydroxy carboxylic acids,[37,38] but the conditions used for ring opening have been harsh (i.e. extreme pH and high temperatures). We assessed more mild conditions. Utilizing gel permeation chromatography (GPC), we monitored the decomposition of polymer 4e under a range of acidic and basic conditions at room temperature (Figure 2). No appreciable breakdown occurred at pH values between 4.6 and 9.1 over 48 hours. This observation indicates that the oxazinane backbone is able to withstand exposure to a wide range of conditions, thereby facilitating polymer handling or modification. Still, the polymers are vulnerable under specific conditions. At pH values less than 1.0, degradation is fast with complete decomposition occurring in under an hour. The acid-catalyzed backbone decomposition also occurs readily at pH 2.5, with 66% of the polymer mass lost in 6 h. At pH values up to 4.5, slower degradation occurs. We also observed polymer backbone cleavage under basic conditions. Thus, either acid or base can promote degradation of polymers with an N-alkoxy-oxazinone backbone. These data indicate that these oxazinone polymers represent a unique class of backbone degradable ROMP polymer that is stable at neutral pH values but labile in either acidic or basic environments.
Figure 2.
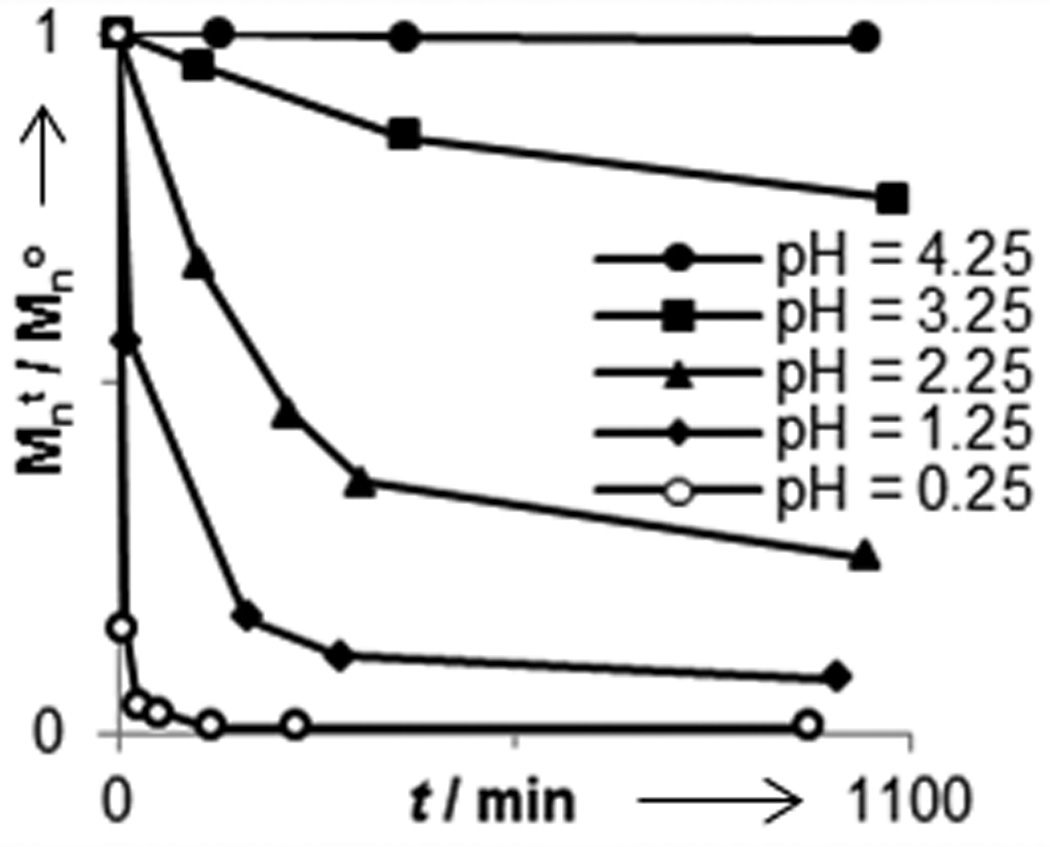
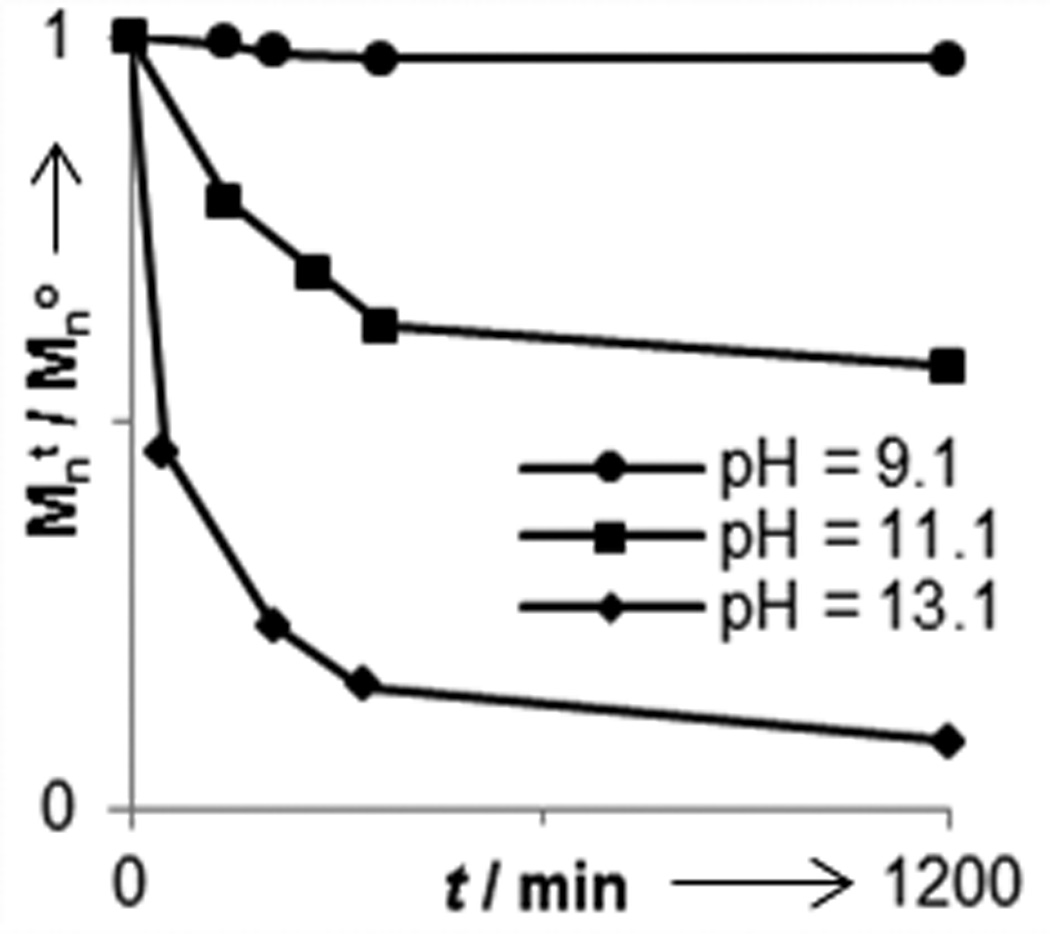
Degradation profile of polymer 4e under acidic and basic conditions. Mno = 18, 500 g/mol.
Further studies were conducted to characterize the degradation products. Because obtaining the necessary quantities of specific polymer degradation fragments for characterization is difficult, we employed a model. Specifically,, compound 3e was subjected to a ring-opening cross metathesis reaction with 1-hexene to yield heterocycle 9. When the product was exposed to an acidic methanol solution, ring-cleavage occurred to afford hydroxamic acid 10 (Scheme 2).[37] This mode of reactivity would promote fragmentation of the polyoxazinone backbone causing polymer degradation. It is expected that hydroxamic ester 10 undergoes further hydrolysis to a β-hydroxy carboxylic acid, however this species was not isolated.
Scheme 2.

Ring-opening cross metathesis of monomer 3a and subsequent hydrolysis of a 3-alkoxy-1, 3-oxazin-4-one under acid conditions.
Bicylic oxazinones can be used to access a new class of degradable polymers. However, the aforementioned monomers do not provide the means to imbue polymers with desired functional properties. To expand the utility of our strategy, we devised a monomer that can be further elaborated. We envisioned a facile and modular strategy to impart new functionality into the monomer is to use a bifunctional hydroxylamine. This approach facilitates the introduction of additional groups at a site distal from the monomer’s core polymerizable group. Moreover, no changes to the synthetic route are required (Scheme 1).
Starting with bromine functionalized hydroxylamine 1b, monomer 3b was accessed using the same synthetic strategy as monomer 3a.[29] Monomer 3b was also a substrate for ROMP, leading to polymer 4b. Additionally, monomer 3b can be elaborated through nucleophilic displacement of its alkyl bromide functionality. As a proof of concept, we exposed the bromine to sodium azide to yield compound 3c, which is a substrate for the azide-alkyne [3+2] cycloaddition (AAC). Because of the exquisite chemoselectivity of this Click chemistry reaction, it is widely used to introduce new functionality into polymers.[39–43] Azides, however, have been found to be incompatible with ruthenium catalysts.[22,44] Therefore, compound 3c was conjugated to 1-propargyl-α-D-mannose-2, 3, 4, 6-tetraacetate using a copper-catalyzed AAC to afford monomer 6 (Scheme 3). The functionalized monomer that resulted could undergo ROMP to afford polymer 7. The acetate groups were hydrolyzed to produce mannose-substituted polymer 8. The water solubility of this polymer facilitated analysis of its degradation in aqueous solution. At pH values similar to those that led to the loss of polymer 4e, we observed decomposition of 8.[45]
Scheme 3.

Synthesis of glycopolymer 8.
Substituents can be added to influence a polymer’s bioactivity or its mechanical or optical properties. To illustrate the generality of our strategy, we generated polymers from O-octyl hydroxylamine (1a), O-ethyl hydroxylamine (1d) or O-benzyl hydroxylamine (1e) starting materials. In this way, polymers 4a4d and 4e[46] with modified hydrocarbon functionality were generated. Additionally, pyrene conjugated hydroxylamine 1f was leveraged to assemble polymer 4f (Scheme 1B), which could serve as an optical sensor. This substance exhibited a red-shifted fluorescence spectrum indicative of pyrene exciplex emission (Supporting Information, Figure S1).[47] When polymer 4f undergoes hydrolytic degradation, a monomeric pyrene derivative is released from the backbone thereby diminishing exciplex emission. Changes in the ratio of fluorescence intensity of monomeric pyrene (λmax=377 nm) and polymeric exciplex (λmax=480 nm) over time report on the extent of backbone hydrolysis. We followed this spectral change to reveal that exposure to acidic conditions for 3 hours resulted in the degradation of approximately 80% of the polymer (Figure 3).
Figure 3.

Degradation of 4f was monitored by comparing the ratio of polymeric pyrene exiplex emission (λmax = 480 nm) to monomeric pyrene emission (λmax = 377 nm). λex = 250 nm, pH = 0.25 (3:1 THF:MeOH). Unfilled diamonds represent low emission ratios due to initial low polymer solubility; polymer completely dissolved in solution within 40 min.
In conclusion, ROMP can be used to synthesize a new class of degradable polymers. These polymers possess a backbone that is labile under either acidic or basic conditions. In addition, the polymers can be decorated through a modular monomer synthesis using bifunctional hydroxylamine building blocks. The process we describe affords functional and degradable polymers that can be used in specific applications. We envision that polymers of this type may yield new degradable plastics or resins. ROMP can be used on an industrial scale to generate new materials; therefore, the strategy described here could generate consumables with properties that complement those currently generated by ROMP.[2] In this way, ROMP can give rise to stable species or materials that can be broken down to simple building blocks. Furthermore, degradable and functional polymers can serve as scaffolds for directed drug delivery or regenerative medicine.[16,48,49] Therefore, we foresee that these polymers may find utility as novel biomaterials as well.
Supplementary Material
Acknowledgments
This research was supported by the NIH (R01 GM049975). The UW-Madison Chemistry NMR facility is supported by the NSF (CHE-9208463 and CHE-9629688) and NIH (1 s10 RRO 8389-01). The UW-Madison Soft Materials Laboratory is supported by the NSF UW Nanoscale Science and Engineering Center (NSEC, DMR-0425880) and UW Materials Research Science and Engineering Center (MRSEC, DMR-0520527).
Footnotes
Supporting information for this article, including experimental details and 1H and 13C NMR, is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Ivin KJ, Mol IC, editors. Olefin Metathesis and Metathesis Polymerization. Second Edition. Academic Press; 1996. [Google Scholar]
- 2.Grubbs, Ed RH, editor. Handbook of Metathesis. Vol. 1. Weinheim, Germany: Wiley-VCH; 2003. p. 3. [Google Scholar]
- 3.Gilliom LR, Grubbs RH. J. Am. Chem. Soc. 1986;108:733. [Google Scholar]
- 4.Wallace KC, Schrock RR. Macromolecules. 1987;20:448. [Google Scholar]
- 5.Schrock RR, Krouse SA, Knoll K, Feldman J, Murdzek JS, Yang DC. J. Mol. Catal. 1988;46:243. [Google Scholar]
- 6.Nguyen ST, Johnson LK, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1992;114:3974. [Google Scholar]
- 7.Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew. Chem. Int. Ed. 2002;41:4035. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 8.Hilf S, Kilbinger AFM. Nat. Chem. 2009;1:537. doi: 10.1038/nchem.347. [DOI] [PubMed] [Google Scholar]
- 9.Mortell KH, Weatherman RV, Kiessling LL. J. Am. Chem. Soc. 1996;118:2297. [Google Scholar]
- 10.Kiessling LL, Gestwicki JE, Strong LE. Curr. Opin. Chem. Biol. 2000;4:696. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 11.Puffer EB, Pontrello JK, Hollenbeck JJ, Kink JA, Kiessling LL. A.C.S. Chem. Biol. 2007;2:252. doi: 10.1021/cb600489g. [DOI] [PubMed] [Google Scholar]
- 12.Borrok MJ, Kolonko EM, Kiessling LL. A.C.S. Chem. Biol. 2008;3:101. doi: 10.1021/cb700211s. [DOI] [PubMed] [Google Scholar]
- 13.Courtney AH, Puffer EB, Pontrello JK, Yang Z-Q, Kiessling LL. Proc. Natl. Acad. Sci. 2009;106:2500. doi: 10.1073/pnas.0807207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baessler KA, Lee Y, Sampson NS. A.C.S. Chem. Biol. 2009;4:357. doi: 10.1021/cb900013d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S-G, Brown JM, Rogers CJ, Matson JB, Krishnamurthy C, Rawat M, Hsieh-Wilson LC. Chem. Sci. 2010;1:322. doi: 10.1039/C0SC00271B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith D, Pentzer EB, Nguyen ST. Polym. Rev. 2007;47:419. [Google Scholar]
- 17.Lienkamp K, Madkour AE, Musante A, Nelson CF, Nusslein K, Tew GN. J. Am. Chem. Soc. 2008;130:9836. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ambade AV, Yang SK, Weck M. Angew. Chem., Int. Ed. 2009;48:2894. doi: 10.1002/anie.200805116. [DOI] [PubMed] [Google Scholar]
- 19.Robertson NJ, Kostalik HAIV, Clark TJ, Mutolo PF, Abruna HD, Coates GW. J. Am. Chem. Soc. 2010;132:3400. doi: 10.1021/ja908638d. [DOI] [PubMed] [Google Scholar]
- 20.Carlise JR, Kriegel RM, Rees WS, Jr, Weck M. J. Org. Chem. 2005;70:5550. doi: 10.1021/jo050556a. [DOI] [PubMed] [Google Scholar]
- 21.Pichavant L, Bourget C, Durrieu M-C, Heroguez V. Macromolecules. 2011;44:7879. [Google Scholar]
- 22.Johnson JA, Lu Y-Y, Burts AO, Lim T-H, Finn MG, Koberstein JT, Turro NJ, Tirrell DA, Grubbs RH. J. Am. Chem. Soc. 2011;133:559. doi: 10.1021/ja108441d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fraser C, Hillmyer MA, Gutierrez E, Grubbs RH. Macromolecules. 1995;28:7256. [Google Scholar]
- 24.Hilf S, Kilbinger AFM. Macromolecules. 2009;42:4127. [Google Scholar]
- 25.Walker R, Conrad RM, Grubbs RH. Macromolecules. 2009;42:599. doi: 10.1021/ma801693q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boger DL, Coleman RS. J. Am. Chem. Soc. 1987;109:2717. [Google Scholar]
- 27.Afarinkia K, Vinader V, Nelson TD, Posner GH. Tetrahedron. 1992;48:9111. [Google Scholar]
- 28.Bandlish BK, Brown JN, Timberlake JW, Trefonas LM. J. Org. Chem. 1973;38:1102. [Google Scholar]
- 29.Jeffrey CS, Barnes KL, Eickhoff JA, Carson CR. J. Am. Chem. Soc. 2011;133:7688. doi: 10.1021/ja201901d. [DOI] [PubMed] [Google Scholar]
- 30.Howell J, Goddard JD, Tam W. Tetrahedron. 2009;65:4562. [Google Scholar]
- 31.Wright DL, Usher LC, Estrella-Jimenez M. Org. Lett. 2001;3:4275. doi: 10.1021/ol016936+. [DOI] [PubMed] [Google Scholar]
- 32.Mihovilovic MD, Groetzl B, Kandioller W, Snajdrova R, Muskotal A, Bianchi DA, Stanetty P. Adv. Synth. Catal. 2006;348:463. [Google Scholar]
- 33.Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 34.Buchowicz W, Holerca MN, Percec V. Macromolecules. 2001;34:3842. [Google Scholar]
- 35.Haigh DM, Kenwright AM, Khosravi E. Macromolecules. 2005;38:7571. [Google Scholar]
- 36.Kang E-H, Lee IS, Choi T-L. J. Am. Chem. Soc. 2011;133:11904. doi: 10.1021/ja204309d. [DOI] [PubMed] [Google Scholar]
- 37.Cardillo G, Hashem MA, Tomasini C. J. Chem. Soc., Perkin Trans. 1990;1:1487. [Google Scholar]
- 38.Bandini E, Martelli G, Spunta G, Bongini A, Panunzio M. Synlett. 1999;1999:1735. [Google Scholar]
- 39.Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Tornoe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 41.Binder WH, Kluger C. Macromolecules. 2004;37:9321. [Google Scholar]
- 42.Iha RK, Wooley KL, Nystrom AM, Burke DJ, Kade MJ, Hawker CJ. Chem. Rev. 2009;109:5620. doi: 10.1021/cr900138t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mansfeld U, Pietsch C, Hoogenboom R, Becer CR, Schubert US. Polym. Chem. 2010;1:1560. [Google Scholar]
- 44.Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao H, Lin Z, Jia G, Fokin VV. J. Am. Chem. Soc. 2008;130:8923. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- 45.Fishman JM, Kiessling LL. unpublished results. [Google Scholar]
- 46.Monomer 3e was a poorer substrate than the other monomers tested. See the Supporting Information for details.
- 47.Gorodetskaya IA, Gorodetsky AA, Vinogradova EV, Grubbs RH. Macromolecules. 2009;42:2895. [Google Scholar]
- 48.Shoichet MS. Macromolecules. 2010;43:581. [Google Scholar]
- 49.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Annu. Rev. Chem. Biomol. Eng. 2010;1:149. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.