

Abstract
Adenylation or adenylate-forming enzymes (AEs) are widely found in nature and are responsible for the activation of carboxylic acids to intermediate acyladenylates, which are mixed anhydrides of AMP. In a second reaction, AEs catalyze the transfer of the acyl group of the acyladenylate onto a nucleophilic amino, alcohol, or thiol group of an acceptor molecule leading to amide, ester, and thioester products, respectively. Mycobacterium tuberculosis encodes for more than 60 adenylating enzymes, many of which represent potential drug targets due to their confirmed essentiality or requirement for virulence. Several strategies have been used to develop potent and selective AE inhibitors including high-throughput screening, fragment-based screening, and the rationale design of bisubstrate inhibitors that mimic the acyladenylate. In this review, a comprehensive analysis of the mycobacterial adenylating enzymes will be presented with a focus on the identification of small molecule inhibitors. Specifically, this review will cover the aminoacyl tRNA-synthetases (aaRSs), MenE required for menaquinone synthesis, the FadD family of enzymes including the fatty acyl-AMP ligases (FAAL) and the fatty acyl-CoA ligases (FACLs) involved in lipid metabolism, and the nonribosomal peptide synthetase adenylation enzyme MbtA that is necessary for mycobactin synthesis. Additionally, the enzymes NadE, GuaA, PanC, and MshC involved in the respective synthesis of NAD, guanine, pantothenate, and mycothiol will be discussed as well as BirA that is responsible for biotinylation of the acyl CoA-carboxylases.
Keywords: Adenylation, adenylate-forming, tuberculosis, bisubstrate inhibitor
INTRODUCTION
Tuberculosis (TB), caused by the acid-fast Gram positive bacillus Mycobacterium tuberculosis, remains the leading source of bacterial infectious disease mortality [1–3]. TB is readily transmitted through the air by aerosolized droplets produced during coughing of an actively infected individual. Following inhalation, M. tuberculosis establishes an infection through invasion of alveolar macrophages. Both innate and adaptive immune responses serve to physically contain the infection through the formation of a granuloma. In the majority of individuals, M. tuberculosis switches its metabolism to a non-replicating state and these individuals are considered to have latent TB that is noninfectious. Approximately 10% of latently-infected individuals will develop active TB during their lifetime, which can be precipitated by malnutrition, immunosupression, or other dietary factors such as iron overload. The advent of chemotherapy in the middle of the 20th century led to a dramatic reduction in TB mortality in the industrialized world, but TB has continued to rage in the developing world.
Current treatment of TB employs four first-line drugs (isoniazid, rifampin, pyrazinamide, and ethambutol) that must be taken daily for a two month intensive phase followed by four to six months of a continuation phase with isoniazid and rifampin [4]. For susceptible TB strains, this therapy is 95% effective, but the emergence of multidrug resistant (MDR) strains, defined as resistant to isoniazid and rifampin, requires the use of less effective and more toxic second-line TB drugs such as one of the injectable antibiotics including amakacin, kanamyin, or capreomycin in conjunction with a fluoroquinolone that must be given daily for up to 18 months. In the last few years, extensively drug resistant (XDR) TB strains have been increasingly recognized that display the MDR-phenotype and are additionally resistant to a fluoroquinolone and one of the three aforementioned injectable antibiotics. Treatment of XDR-TB is particularly challenging as one is left with the most toxic and least effective antitubercular drugs including para-aminosalicylic acid, ethionamide, cycloserine, and thiacetazone [5]. The challenges of TB drug development are thus multifold and require the identification of novel targets as well as drugs that are effective against drug resistant strains, active against latent nonreplicating bacilli in order to reduce the duration of treatment, and compatible with antiretroviral drugs (i.e. do not induce CYP3A4).
ADENYLATING ENZYMES AS DRUG TARGETS
The sequencing of the genome of M. tuberculosis H37Rv, the virulent laboratory strain, in 1998 created much excitement and provided the first insight into the unique metabolic capabilities of this organism [6]. Over the last decade, tremendous effort has been expended to further genetically validate and biochemically characterize many of the corresponding ‘essential’ gene products [7–9]. M. tuberculosis putatively encodes more than 60 adenylating enzymes (see Table 1) that catalyze a multitude of essential biochemical processes in protein synthesis, glycolysis, lipid metabolism, and cofactor biosynthesis (biotin, coenzyme A, and nicotine adenine dinucleotide) as well as synthesis of small molecule metabolites including the mycobactins (siderophores for iron acquisition) and mycothiols (a thiol to protect against oxidative stress). We believe adenylating enzymes represent attractive targets for the development of new anti-tubercular agents due to their druggability (vide infra) and confirmed essentiality. In this short review we will first provide an overview of the adenylating enzymes (AEs) in M. tuberculosis based on their function. Next, we will discuss strategies for inhibitor design. Finally, we will review the literature of the mycobacterial adenylating enzyme with an emphasis on reported small molecule inhibitors and the potential of AEs as drug targets.
Table 1.
Genes Encoding Adenylate-Forming Enzymes
| Entry | Gene(s) | Accession Number | Annotated Function | Essential (E) or Virulence Factor (V) |
|---|---|---|---|---|
| 1 | alaS, argG, argS, aspS, cysS1, gltS, glyS, hisS, ileS, leuS, lysS, lysX, metS, pheS, pheT, proS, serS, thrS, trpS, tyrS, valS | Rv2555c, Rv1658, Rv1292, Rv2572c, Rv3580c, Rv2992, Rv2357c, Rv2580c, Rv1536, Rv0041, Rv3598c, Rv1640c, Rv1007c, Rv1649, Rv1650, Rv2845c, Rv3834c, Rv2614c, Rv3336c, Rv1689, Rv2448c | Aminoacyl tRNA synthetase (Protein biosynthesis) | yes (E) |
| 2 | acs | Rv3667 | Acetyl coenzyme synthetase (glycolysis) | no |
| 3 | menE | Rv0542c | O-Succinylbenzoic acid CoA synthetase (menaquinone biosynthesis) | yes (E) |
| 4 | fadD1, fadD2, fadD3, fadD4, fadD5, fadD6, fadD7, fadD8, fadD9, fadD11, fadD12, fadD13, fadD14, fadD15, fadD16, fadD17, fadD18, fadD19, fadD35, fadD36 | Rv1750c, Rv0270, Rv3561, Rv0214, Rv0166, Rv1206, Rv0119, Rv0551c, Rv2590, Rv1550, Rv1427c, Rv3089, Rv1058, Rv2187, Rv0852, Rv3506, Rv3513c, Rv3515c, Rv2505c, Rv1193 | Fatty acyl CoA synthetase (fatty acid catabolism, cholesterol catabolism) | no (except fadD36) |
| 5 | fadD10 | Rv0099 | Possibly involved in synthesis of the nonribosomal peptide encoded by nrp (adjacent to nrp) | no |
| 5 | fadD21 | Rv1185c | Possibly involved in mycolipanoic, mycolipenic, mycolipodienic acid synthesis (adjacent to pks3/4) | no |
| 6 | fadD22 | Rv2948c | Phenolglycolipid synthesis (loading of 4-hydroxybenzoic acid onto Pks15/1) | no |
| 7 | fadD23 | Rv3826 | Phthioceronic and hydroxyphthioceronic acid portion of the sulfolipids (adjacent to pks2) | no |
| 8 | fadD24 | Rv1529 | Possibly involved in lipooligosaccharides synthesis (adjacent to pks5) | no |
| 9 | fadD26 | Rv2930 | Synthesis of the phthiocerol portion of the dimycocerosate esters (loading of fatty acid onto PpsA) | no |
| fadD28 | Rv2941 | Synthesis of the mycocerosic acid portion of the dimycocerosate esters (loading of fatty acid onto Mas) | no | |
| 10 | fadD30 | Rv0404 | Synthesis of an unknown polar lipid (loading of fatty acid onto Pks6) | yes (E) |
| 11 | fadD32 | Rv3801c | Mycolic acid synthesis (loading Pks13) | yes (E) |
| 12 | mbtM (fadD33) | Rv1345 | Mycobactin synthesis (loading of MbtL) | no (mutant slow growing) |
| 13 | fadD25, fadD29, fadD31, fadD34 | Rv1521, Rv2950c, Rv1925, Rv0035 | Unknown (fatty acyl-AMP ligases, FAALs) | no |
| 14 | nrp | Rv0101 | Multifunctional NRPS (unknown function) | no |
| 15 | mbtA, mbtB, mbtE, mbtF | Rv2384, Rv2383c, Rv2380c, Rv2379c | NRPS Adenylation domains (mycobactin biosynthesis) | yes (V) |
| 16 | nadE | Rv2438c | Glutamine dependent NAD synthetase | yes (E) |
| 17 | guaA | Rv3396c | GMP synthetase | yes (E) |
| 18 | panC | Rv3602c | Pantothenate synthetase (Coenzyme A synthesis) | yes (V) |
| 19 | mshC | Rv2130c | Cysteine:1-D-myoinosityl 2-amino-2-deoxy-D-glucopyranose ligase (mycothiol biosynthesis) | yes (E) |
| 20 | birA | Rv3279c | Biotin ligase (biotinylation of AccA1, AccD4, Pcc) | yes (E) |
ADENYLATION ENZYME MECHANISM
Adenylating or adenylate-forming enzymes catalyze a two-step reaction as shown in Fig. (1) [10, 11]. In the first reaction, binding of a carboxylic acid substrate 1 and ATP is followed by nucleophilic attack of the substrate carboxylate on the α-phosphate of ATP to generate acyladenylate 2 and the release of pyrophosphate Fig. (1, part a). In the second reaction, the enzyme binds an acceptor molecule and transfers the acyladenylate 2 to a nucleophilic oxygen, sulfur or nitrogen atom of the acceptor leading to products 3–6 in M. tuberculosis Fig. (1, part b). The acceptor moiety is the 2′ or 3′ alcohol from the ribose sugar of the terminal adenosine residue of a cognate tRNA molecule for the 21 aminoacyl tRNA synthetases (see 3, Fig. (1 and entry 1, Table 1). The acceptor moiety is the terminal sulfur atom of the coenzyme A molecule for the 26 CoA ligases (see 4, Fig. (1 and entries 2–4, Table 1) and terminal sulfur atom of the phosphopantetheinyl (ppant) cofactor arm of a cognate polyketide synthase (PKS) carrier domain for the 12 long chain fatty acyl AMP ligases (see 5, Fig. (1 and entries 5–13, Table 1) and 6 nonribosomal peptide synthetase (NRPS) adenylation domains (see 5, Fig. (1 and entries 14–15, Table 1). The acceptor molecule is ammonia for NadE and GuaA (see 6a, Fig. (1 and entries 16–17, Table 1), the amino group of β-alanine for PanC (see 6b, Fig. (1 and entry 18, Table 1), the 2-amino group in 1-D-myo-inosityl-2-amino-2-deoxy-α-D-glucopyranoside for MshC (see 6c, Fig. (1 and entry 19, Table 1), and the conserved lysine residue of a biotin carrier domain for BirA (see 6d, Fig. (1 and entry 20, Table 1).
Fig. 1.

Adenylation enzyme mechanism.
APPROACHES FOR INHIBITOR DESIGN
Bisubstrate Inhibitors
One of the most straightforward strategies to prepare an inhibitor of adenylating enzymes is to design a mimic of the acyladenylate. The rationale is based on the findings that the intermediate acyladenylates bind several orders of magnitude more tightly than the substrate acids since they simultaneously occupy both substrate-binding pockets (acid and ATP). Thus acyladenylate analogues that incorporate a stabile bioisostere of the labile acylphosphate linkage function can potentially provide potent small molecule bisubstrate inhibitors [12–14]. Bisubstrate inhibitors (A-B) can realize substantial enhancement in binding energy compared to the sum of the Gibbs binding energies of the respective fragments (A and B) as first articulated by Jencks due to a smaller entropy barrier to binding of A-B as compared to A + B [15]. Fig. (2C) shows several isosteric replacements of the native acylphosphate linkage of the acyladenylate 2 Fig. (2C, entry 1) that have been investigated including alkyl phosphates [16] Fig. (2C, entry 2), β-ketophosphonates [17] Fig. (2C, entry 3), acylsulfamates [18] Fig. (2C, entry 4), and sulfamates [19] Fig. (2C, entry 5). As first demonstrated by Ishida and coworkers, the acylsulfamate linkage (see 7, Fig. (2A)) inspired from the natural product ascamycin 8 Fig. (2B) has been extensively employed as a stable bioisostere of the labile acylphosphate linkage in 2, since this most closely resembles the overall molecular geometry and charge distribution of the acylphosphate [18, 20]. Additionally, the acylsulfamate moiety pKa is higher (+2 to +4) than a typical phosphonate or phosphate (−2), so that a substantially greater fraction will exist as the neutral conjugate acid at physiological pH, providing better membrane penetration, which is critical for whole-cell antibacterial activity. Simple bisubstrate inhibitors of the generic structure 7 typically possess potent nanomolar dissociation constants and display exquisite biochemical selectivity toward other adenylating enzymes, but lack selectivity against related human orthologs.
Fig. 2.

Bisubstrate inhibitors. (A) Replacement of the acylphosphate linkage of an acyl-adenylate with an acylsulfamate provides bisubstrate inhibitor 7. (B) Structure of ascamycin 8, a nucleoside natural product. (C) Table of different linkers that have been used to generate bisubstrate inhibitors, entry 1 is the native unstable acyl-phosphate while entries 2–5 represent chemically stabile isosteres.
High-Throughput Screening
An alternate approach to identify inhibitors of adenylating enzymes is by high-throughput screening (HTS). These efforts focus on screening large chemical libraries against the biological target (protein or whole-cell) in order to identify hit compounds which affect that target. These libraries are composed of thousands to millions of compounds and have a large amount of structural diversity. Due to the recent advances in liquid handling and robotic instrumentation, these sizable libraries may be screened in a timely manner (days). The output from HTS campaigns are not final drugs, but are starting points, or hit compounds, which require further medicinal chemistry optimization to identify lead compounds [21].
For the purpose of this review, HTS technologies for screening inhibitors that disrupt the kinetic activity of pure enzyme targets (and not whole-cell HTS assays) will be discussed. Several biophysical methods suitable for HTS implementation are available to measure enzyme activity (and inhibition), including time-resolved fluorescence resonance energy transfer (tr-FRET) [22], scintillation proximity assay (SPA) [23], fluorescence intensity [24], and fluorescence polarization [25]. Once a detection method has been chosen, careful attention must be given to designing the enzyme assay parameters so that initial velocity is maintained throughout the assay window. This requirement is crucial in HTS kinetic-based assays since the enzyme activity is most sensitive during the initial velocity phase [21]. Furthermore, when screening an enzyme target, it is useful to identify hits with varying inhibition modalities (competitive, uncompetitive, noncompetitive) and to accomplish this, Copeland recommends maintaining substrate concentrations equal to their KM [21].
Following optimization of enzyme concentration, reaction time, and substrate concentrations, the assay must be deemed robust enough to enter the library screening phase. A powerful gauge of assay robustness is referred to as the Z′ factor, which takes into account both the assay’s signal to noise as well as the standard deviation [26]. An assay possessing a Z′ from 0.5–1.0 is considered an excellent assay suitable for HTS. After the screen is performed, the hits are rank ordered based on their percent inhibition of the enzyme target. Hits are then validated using a secondary assay that is orthogonal to the primary assay used during the high-throughput screen. Lastly, confirmed hits are ordered from commercial sources, and their structure and purity are confirmed by NMR and LC-MS. A full dose-response curve is then obtained to yield both the IC50 and the Hill slope of the compound. Compounds with Hill slopes that vary greatly from unity may be discarded, as this suggests nonspecific enzyme inhibition [21].
Fragment-Based Screening
Fragment-based screening (FBS) has developed into a promising alternative to HTS. Whereas HTS libraries consist of hundreds of thousands of larger molecules (typically >250 Da), FBS libraries have fewer compounds (1000–5000) that are much smaller (typically <250 Da), highly diverse and have good solubility [27]. Due to their small size and decreased functionality, these fragments often bind to the protein target with low affinities (μM - mM), but also possess higher binding energy per heavy atom in order to overcome a large entropic barrier upon binding to protein [27,28]. Those fragments which display target binding are then elaborated through rational design to provide potent inhibitors. The resulting ligands obtained via FBS should have higher ‘ligand efficiencies’ than inhibitors discovered through HTS campaigns, where ligand efficiency is defined as the ratio of the free energy of ligand binding to the number of non-hydrogen atoms [29,30].
The overall flow of FBS is shown in Fig. (3). First, the target enzyme is screened against the fragment library using one of several biophysical techniques, including differential scanning fluorimetry [31], surface plasmon resonance (SPR) [32], NMR spectroscopy [33], and X-ray crystallography [34]. During this primary screen, fragment compounds are added at high concentrations (1–10 mM) to the protein due to their intrinsic low affinity. After initial hit fragments are identified from the primary screen, secondary assays must be run in order to confirm and characterize binding. If NMR was not used as the primary screen, saturation transfer difference NMR (STD NMR) may be used as a secondary screen. STD-NMR detects ligand binding to proteins and does not require [15N]-labeled protein [35]. Whereas STD-NMR can only provide confirmation of fragment binding, isothermal titration calorimetry (ITC) and SPR can provide both confirmation and the binding affinity of the fragment with the protein [36]. Therefore, ITC and SPR represent powerful validation assays in FBS.
Fig. 3.

Schematic flow of fragment-based drug development.
Following confirmation of fragment binding via secondary assays, X-ray crystallography, arguably the most powerful tool in FBS [27], is employed to determine the binding mode of the fragment [34]. Fragments are either soaked into the protein crystal or co-crystallized with the protein at high (mM) concentrations. The resulting fragment bound protein structures provide key insight into fragment binding sites as well as the orientation of the fragments in those sites [37]. The resulting structural data allows one to design larger, more potent molecules that incorporate the original fragments [27]. These more elaborate inhibitors are built by either ‘linking’ fragments that are in close proximity to each other or by ‘extending’ the original fragment into neighboring pockets. The proposed molecules are then synthesized and their affinities, kinetic inhibition constants, and binding modes are determined by ITC, kinetic enzyme assays, and X-ray crystallography, respectively.
ADENYLATING ENZYME BY FUNCTION
In the following section a brief background on the biochemical pathway of the mycobacterial adenylating enzymes will be presented followed by available information on its respective kinetic mechanism, structure, and reported inhibitor(s).
Aminoacyl tRNA Synthetases
In ribosomal protein biosynthesis, aminoacyl-tRNA synthetases (aaRSs) serve as the enzymatic link between the genetic information encoded in DNA and the functional protein realm [14]. Thus, it is not surprising that aaRSs are essential enzymes found in all living organisms [13]. M. tuberculosis encodes for all 21 aaRSs, which are responsible for activating and loading each amino acid onto its cognate tRNA molecule. In the first half reaction, the aaRS forms an acyl adenylate 10 Fig. (4) between the amino acid 9 and ATP, where in the second half reaction, the amino acid is transferred onto the 3′-OH of the cognate tRNA 11 to form the charged, acylated tRNA 12 [14].
Fig. 4.

Adenylation-Ligation reaction catalyzed by aminoacyl-tRNA synthetases.
Inhibitors of aaRSs have been reviewed elsewhere, thus only a limited discussion will be presented here [13,14, 38]. Historically, aaRSs were the first adenylating enzymes for which inhibitors were developed. Notably, the natural product mupirocin 13 is a clinically approved topical antibiotic that inhibits isoleucine-tRNA synthetase [39]. Mupirocin is effective for many Gram-positive bacteria, but has no antimycobacterial activity [40]. Several research groups have pursued the development of aaRS inhibitors for antibiotic development by designing small molecule mimics of the acyl-adenylate intermediate. Prolyl-AMS 14 Fig. (5) is a representative bisubstrate inhibitor and displayed potent activity against the proline tRNA synthetase (ProRS) from E. coli (IC50 = 4.3 nM); however, 14 inhibited the human ortholog with even greater potency (IC50 = 0.6 nM) [41]. Microorganism selective aaRS inhibitors exemplified by 15 were successfully developed by Cubist Pharmaceuticals [12–13, 38]. Compound 15 selectively inhibited the isoleucine-tRNA synthetase (IleRS) from E. coli (IC50 1 nM) versus the human ortholog (IC50 = 570 nM) and importantly 15 displayed in vivo activity against several pathogens, including S. aureus (MIC = 10 μg/mL), S. pyogenes (MIC = 0.5 μg/mL) and E. coli (MIC = 10 μg/mL); however, activity against M. tuberculosis was not reported [13, 38]. Bisubstrate inhibitor 16 is the only inhibitor that has been specifically evaluated against a mycobacterial enzyme and it showed moderate in vitro activity towards methionyl-tRNA synthetase (Met-RS) from M. tuberculosis, but did not possess antitubercular activity [42,43]. Several additional aaRS inhibitors have been discovered through high throughput screening efforts and have been reviewed elsewhere [38]. One compound in particular, REP8839 17, is currently under clinical investigation as a topical agent for superficial skin infections caused by S. aureus [44]. Although these HTS-derived compounds possess whole-cell antibacterial activity towards several devastating pathogens, no activity towards M. tuberculosis has been disclosed.
Fig. 5.

Aminoacyl-tRNA synthetase inhibitors.
MenE
M. tuberculosis uses menaquinone (a napthoquinone) in the electron transport chain for ATP synthesis, which functions analogously to ubiquinone (a benzoquinone) used in humans. Menaquinone (vitamin K2) has been proposed as an attractive target since humans do not utilize menaquinone in their electron transport chain, ATP synthesis is essential for the viability of hypoxic nonreplicating M. tuberculosis, and the transposon mutagenesis studies by Sassetti and coworkers demonstrated the essentiality of several genes in the pathway [45–47]. In analogy to the pathway in E. coli, menaquinone biosynthesis begins from chorismate 18 that is converted to ortho-succinylbenzoate (OSB) 19 through the activities of MenF, MenD, and MenC Fig. (6) [48]. The acyl-CoA synthetase MenE then converts 19 into the corresponding CoA ester 20, which is cyclized to dihydronaphthoquinone 21 by MenB. Methylation and prenylation of 21 is carried out by MenA and MenH, respectively to afford 22 and 23. The prenyl side chain of menaquinone in M. tuberculosis consists of nine isoprenoid units, represented by structure 22 and the most abundant menaquinone in M. tuberculosis is the dihydro derivative 23, due to reduction of the second isoprene unit [49]. Additionally, the novel sulfated menaquinone 24 was recently isolated [50].
Fig. 6.

Biosynthesis of menaquinone.
The adenylating enzyme MenE from M. tuberculosis is a 37 kDa CoA ligase responsible for the fourth step in menaquinone biosynthesis and catalyzes the ATP-dependent ligation of ortho-succinylbenzoate (OSB) 19 to CoA ester 20 Fig. (8A), but has not been structurally or functionally characterized. However, detailed kinetic characterization of MenE from Bacillus anthracis (BaMenE) showed it catalyzes the reaction via a bi-uni-uni-bi ping-pong kinetic mechanism with sequential ordered binding of ATP followed by OSB and sequential ordered release of OSB-CoA followed by AMP Fig. (8B) [51]. The apparent KM values for ATP, OSB, and CoASH were determined as 27, 22, and 304 μM, respectively. Pre-steady state kinetic analysis in the absence of CoASH showed the adenylation half-reaction proceeded with an initial burst of 4 min−1 corresponding to a single turnover (formation of OSB-AMP) and release of PPi. The observed kcat of 155 min−1, which is approximately 39-times faster than the initial burst rate in the adenylation half-reaction, demonstrates that addition of CoASH accelerates the first half-reaction. Based on the kinetic and structural studies of 4-chlorobenzoate CoA ligase by Gulick and Dunaway-Mariano, the kinetic enhancement induced by CoASH is likely caused by a large conformation movement of the C-terminal domain of MenE upon binding CoASH that may permit dissociation of the product PPi [52,53]. More limited characterization of MenE from E. coli (EcMenE) confirmed it was a Mg2+ dependent enzyme that exists as a homotetramer in solution with similar KM values for ATP, OSB, and CoASH of 74, 16, and 360 μM, respectively [54]. Recently, the crystal structure of MenE from Staphylococcus aureus has been deposited in the Protein Data Bank (PDB code 3IPL) and shares approximately 40% similarity and 26% identity to the M. tuberculosis ortholog Fig. (7). Together these kinetic and structural studies of MenE orthologs from other organisms provides a reasonable foundation for inhibitor design and discovery.
Fig. 8.

(A) Adenylation–ligation reaction catalyzed by MenE. (B) Bi-uni-uni-bi ping-pong kinetic mechanism for MenE.
Fig. 7.

Structure of MenE from S. aureus (PDB 3IPL). The C-terminal subdomain (orange) rotates nearly 30 ° towards the N-terminal subdomain (yellow) upon ligand binding. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
Because of the spontaneous spirolactonization [55] of OSB-CoA 20 to afford spirodilactone 28 Fig. (9), the bisubstrate inhibitors 29–33 (Table 2) were synthesized by protection of the aryl carboxylic acid as a methyl ester (29–32) or bioisosteric replacement with a trifluoromethyl group (33). The IC50 values for 29–33 were obtained against EcMenE while the reported Kii value of 33 was obtained with BaMenE. Compounds 29–31 contain a sulfamate, sulfamide, and vinylsulfonamide moieties as isosteres of the native acylphosphate linkage. Interestingly, vinylsulfonamde 31 was the most potent with an IC50 value of 5.7 μM. The bi-substrate inhibitor 32 was designed to avoid spirolactonization through replacement of the ketone group with the isosteric terminal olefin; however, this compound was inactive. Detailed kinetic investigation was only reported for 33 and this analysis showed it exhibited noncompetitive inhibition with respect to both ATP (Kii = 5.2 μM, Kis = 108 μM) and OSB (Ki = 5.6 μM, Kis = 25 μM) and uncompetitive inhibition with respect to CoASH (Ki = 8.9 μM) where the Kii is the dissociation constant for the binary E·I complex and Kis is the dissociation constant for the E·S·I ternary complex. Surprisingly, the Kii values of 33 are only 4–5 lower than the corresponding Km values for the substrates. In summary, none of the reported compounds exhibited very potent activity against MenE that would be expected for a bisubstrate inhibitor and none exhibited antibacterial activity.
Fig. 9.

Degradation of OSB-CoA.
Table 2.
MenE Inhibitors
| compound | IC50 (Kii), μM |
|---|---|
29

|
38 ± 3 |
30

|
34 ± 3 |
31

|
5.7 ± 0.7 |
32

|
> 200 |
33
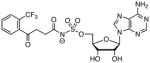
|
(5.4 ± 0.8)a |
Kii value: Kii(ATP) = 5.2 ± 0.8 μM, Kii(OSB) = 5.6 ± 0.8 μM
FadDs
Mycobacterial Lipid Metabolism
Mycobacteria produce a tremendously diverse array of lipophilic molecules ranging from simple short chain fatty acids to the very complex long-chained mycolic acids [56–58]. Approximately 5% of the coding genome of M. tuberculosis is dedicated to fatty acid metabolism [6]. Many of the unique lipids including mycolic acids are essential components of the mycobacterial cell envelope and several antitubercular drugs target mycolic acid biosynthesis. Other lipids such as phthiocerol dimycocerosates (PDIMs) are associated with hypervirulent TB-strains [59]. Fatty acid degradation is also believed to be important and several lines of evidence support the hypothesis that mycobacteria are primarily lipolytic in vivo, deriving their nutrients by breakdown of host fatty acids [60,61]. Sassetti and co-workers demonstrated that cholesterol catabolism was essential for persistence and that the breakdown products of cholesterol were efficiency incorporated into mycobacterial lipids [62].
FadD’s are Fatty acyl-AMP/CoA Ligases
Gokhale and co-workers recently characterized the fadD family of genes involved in both lipid biosynthesis as well as catabolism [63,64]. M. tuberculosis encodes for an astonishing 34 fadD’s. By comparison, E. coli encodes a single fadD, whose gene product is an acyl-CoA ligase that converts free fatty acids into acyl-coenzyme A (CoA) thioesters, the first step of fatty acid degradation. The fadD genes can be grouped into two classes: fatty acyl-CoA ligases (FACLs) involved in lipid catabolism and fatty acyl-AMP ligases (FAALs) involved in lipid biosynthesis Fig. (10) [63]. Gokhale has shown that the mechanistic divergence between the two classes of FadD’s is due to an approximately 20 amino acid insertion sequence between the N- and C-terminal subdomains, which is found in FAALs but not in FACLS Fig. (11). This insertion sequence modulates the C-terminal domain movement by stabilizing the adenylation conformation thereby preventing binding of CoA. Gokhale has proposed to rename the FadDs by their functional classification, thus FadD6, a fatty acyl-CoA ligase, is now annotated as FACL6 and FadD28, a fatty acyl-AMP ligase, is now FAAL28. We will use this nomenclature through the following section.
Fig. 10.

FadD enzyme mechanism.
Fig. 11.
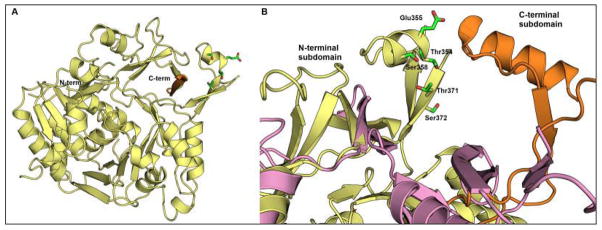
Structure of FAAL28 from M. tuberculosis (PDB 3E53). (A) N-terminal domain of the FAAL28 protein. The C-terminal subdomain is too disordered to be defined in this apo structure. It extends from the orange C-terminus position and has been hypothesized to rotate towards the N-terminal subdomain (yellow), similar to other adenylation enzymes. (B) Active site structure of FAAL28 as modeled from superimposed structures of PheA (PDB 1AMU, orange), acetyl-CoA synthetase (PDB 1PG4, pink), and FAAL28 (yellow). The N-terminal subdomain residues Thr354, Glu355, Ser358, Thr371, and Ser372, shown with green carbon atoms, and red oxygen atoms, may interfere with the C-terminal domain when it adopts one of the catalytic conformations. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
Fatty acyl-CoA Ligases (FACLs)
The precise biochemical roles of the 20 FACLs are largely unknown. FACL2 is required for efficient M. avium invasion of mucosal epithelial cells [65]. FACL17 and FACL19 have been implicated in cholesterol degradation, but this proposed activity has not yet been confirmed [66,67]. FACL6, FACL13, FACL15, FACL17, and FACL19 have been biochemically confirmed as CoA ligases, but the native substrates for these enzymes remain unknown [63,64, 68]. The lack of essentiality of the FACLS, based on the transposon mutagenesis studies of Sassetti, may be due to their functional redundancy [47]. Thus, FACL6, FACL15, and FACL19 were all shown to possess a remarkably broad substrate specificity and were able to synthesize CoA esters from medium to long-chain fatty acids with modifications at the α-, β-, ω-, and (ω-υ)-positions [64].
Fatty acyl-AMP Ligases (FAALs)
The FAALs activate and transfer fatty acids to polyketide synthase (PKS) for further chain extension to afford most of the highly functionalized lipids found in M. tuberculosis. Polyketide synthases are complex multienzyme systems that are characterized by a modular architecture and sequentially condense simple carboxylic acids such as malonyl-CoA or (n-alkyl)malonyl CoA derivatives. Unlike the functionally related fatty acid synthases (FASs), PKSs utilize methyl-malonyl CoA, which leads to methyl branching in the lipid. Additionally, PKSs often lack catalytic domains involved in reductive processing resulting in the introduction of unsaturation and hydroxyl groups in the lipid chain Fig. (12). FAALs thereby link fatty acid and polyketide synthesis. In fact, 10 of the 14 FAALs are found adjacent to pks or the related nrps genes in the M. tuberculosis genome and this genetic link allows one to infer function in the absence of any biochemical characterization.
Fig. 12.

Unique lipids found in cell envelope of Mycobacterium tuberculosis. All of the molecules shown exist as a suite of related isomers that vary in the lipid chain length. If reported, the major isomer is shown otherwise a represetative molecule is depicted. Specific FadD’s are responsible for installation of the lipid chains highlighted in red. (A) Phthiocerol dimycocerosate A (PDIM A, 38) biosynthesis requires FadD26 and FadD28 for synthesis of phthiocerol and mycocerosic acid moieties, respectively. (B) Phenolic glycolipids (PGLs, 39) require FadD22 and FadD29 for assembly of the phenolphthiocerol lipid as well as FadD28 for the two mycocerosic acids. (C) The mycobactins (MBTs, 40) employ FadD33 for installation of the C-20 lipid residue on the central lysine moiety. (D) Sulfolipids represented by SL-1 41 require FadD23 for biosynthesis of the phthioceranic acid and two hydroxyphthioceranic acid groups. (E) The mycolic acids, represented by the most abundant α-mycolic acid (α-MA, 42) employ FadD32 for introduction of the meromycolic acid subunit. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
Several of the FAALs have been biochemically characterized and shown to activate acid substrates and load these onto cognate PKS proteins. Quadri and co-workers demonstrated FAAL22 is essential for synthesis of the virulence-conferring phenolic glycolipids (PGLs, see 39, Fig. (12)) through genetic inactivation and also biochemically confirmed FAAL22 is responsible for the activation and transfer of 4-hydroxybenzoic acid onto PKS15/1 [69,70]. FAAL26 and FAAL28 are essential for synthesis of the dimycocerosate esters (DIMs), which are important virulence factors comprised of phthiocerol esterified with two mycocerosic acid moieties [71–73]. Gokhale and co-workers showed FAAL26 initiates phthiocerol synthesis by loading the polyketide synthase PpsA with long-chained fatty acids while FAAL28 initiates mycocerosic acid synthesis by loading the PKS protein mycocerosic acid synthase (Mas) [63, 74]. FAAL32 has been the most extensively studied of the FAALs since it is required for synthesis of the mycolic acids (42, Fig. (12)) and catalyzes the transfer of meromycolic acid onto PKS13 [63, 75, 76]. Targeted disruption of faal32 confirmed its essentiality while knockdown studies showed faal32 is a highly vulnerable target and depletion of FAAL32 was bactericidal [77, 78]. Accordingly FAAL32 represents an extremely attractive target. FAAL33 (reannotated as MbtM) is part of an operon involved in activation and attachment of the lipid moiety to the mycobactins (40, Fig. (12)), which are small-molecule iron chelators produced by M. tuberculosis for iron acquisition [79]. Biochemical studies with MbtM demonstrated that it activated dodecanoic acid (a model substrate) and loaded this onto the acyl carrier protein MbtL. Genetic disruption and complementation of mbtM determined its requirement for growth of M. tuberculosis in a hepatocyte cell line and in the liver of BALB/c mice [80, 81].
While all FACLs utilize CoA as a common acceptor substrate, each FAAL has a unique acceptor substrate, which is not a small molecule, but rather an acyl carrier protein domain of a modular PKS protein. The fidelity and efficiency of acyl chain transfer between the FAAL-PKS proteins is likely governed by specific protein-protein interactions between the FAAL and PKS proteins in analogy to PKS systems [82]. This additional level of specificity is expected to disfavor functional overlap between 14 FAALs and indeed genetic inactivation of FAALs as discussed above results in strains unable to synthesize specific lipids, demonstrating that the other 13 FAALs or 20 FACLs cannot rescue function.
Inhibition of FACLs and FAALs
5′-O-[N-(Dodecanoyl)sulfamoyl]adenosine 43 (Table 3) was recently described as a multitarget FadD inhibitor and shown to inhibit both FAAL28 and FACL19 with apparent Ki values of 1.5 μM and 4.9 μM respectively and possessed very modest antitubercular activity with a MIC of approximately 100 μM [83]. Treatment of M. tuberculosis with 43 led to a dose-dependent decrease in mycolic acid synthesis and a concomitant increase in fatty acid methyl esters indicating 43 does not inhibit simple fatty acid biosynthesis. The corresponding dodecylphosphate-AMP analogue 44 was also described as a FadD inhibitor [75]. The authors demonstrated 44 inhibited FAAL32, inhibited mycolic acid biosynthesis, and growth of M. smegmatis. 5′-O-[N-(4-Hydroxybenzoyl)sulfamoyl]adenosine 45 was designed as an inhibitor of FAAL22 employing the acylsulfamate isostere of the acylphosphate [69]. Compound 45 was a modest inhibitor of FAAL22 with an apparent Ki of 6 μM and comparable inhibition of PGL biosynthesis in M. tuberculosis and other mycobacterial species with an IC50 value of 9 μM. Since PGL biosynthesis is dispensable for growth in vitro, 45 was not expected to exhibit antitubercular activity and indeed no inhibition of growth was observed up to 800 μM 45. Interestingly, inhibition of PGL synthesis correlated with a modest increase in PDIM production, likely due to increased metabolic flux of precursor building blocks due to blockage of the PGL pathway. Given the likely promiscuous activity and poor physicochemical attributes of these bisubstrate inhibitors they can only be considered tool compounds. A fluorescence polarization high-throughput assay has been described for FAAL28 that hopefully can be used to identify nonnucleoside-based small molecule inhibitors of the FAAL proteins [84].
Table 3.
FadD Inhibitors
| compound | IC50, μM (FadD) | MIC, μM |
|---|---|---|
43

|
1.5 (FAAL28) 4.9 (FACL19) |
100 |
44

|
< 1 (FAAL32) | < 20 |
45

|
6 ± 1 (FAAL22) | > 800 |
MbtA
M. tuberculosis requires iron for survival in mammalian hosts; however, the concentration of iron in human serum and body fluids is too low to support bacterial growth. To circumvent this limitation, mycobacteria and other pathogenic microorganisms have evolved elegant mechanisms for acquiring this essential nutrient from their host. The most common mechanism employed by bacteria for iron acquisition is the synthesis, secretion, and re-internalization of small molecule iron chelators referred to as siderophores [85, 86]. M. tuberculosis produce a pair of structurally related siderophores, the water soluble mycobactins (also known as the carboxymycobactins) 46 [87] and the lipophilic mycobactins 47 [88], that vary by the nature of the lipid residue Fig. (13A) [89, 90]. The mycobactins are biosynthesized via a multi-enzyme complex (MbtA-F) that constitutes a mixed nonribosomal peptide synthetase-polyketide synthase (NRPS-PKS) assembly line Fig. (13B) [79, 91, 92]. Mycobactin biosynthesis begins with a stand alone adenylating enzyme, MbtA, which activates salicylic acid as an adenylate and transfers salicylate onto the carrier protein, MbtB. Following MbtB-catalyzed addition of salicylate onto the tethered serine, MbtE installs a lysine residue onto the growing chain. MbtC and MbtD are dedicated to installing the β-hydroxybutyrate moiety, while MbtF appends on the final lysine residue. The thioesterase (TE) domain of MbtF catalyzes the release of premycobactin via lactamization. Subsequent N-hydroxylation of the two lysine residues mediated by MbtG and acylation of the central N-lysine by MbtK affords the mycobactins [79, 93]. The mycobactin assembly thus contains one stand alone adenylating enzyme, MbtA, and three additional adenylation domain embedded in the multifunctional nonribosomal peptide synthetase (NRPS) domains of MbtB, MbtE, and MbtF. MbtA that activates salicylic acid has been the most investigated among the four NRPS adenylating enzymes since it catalyzes the first step of mycobactin biosynthesis and is functionally distinct from the other three NRPS adenylation domains, which activate amino acids.
Fig. 13.

The NRPS-PKS pathway for mycobactin biosynthesis.
In a key experiment, mycobactins were shown to be critical for the virulence of M. tuberculosis [94]. In this study, Barry and coworkers genetically disrupted the mbtB gene and the resulting strain was unable to produce mycobactins, restricted for growth in iron-replete media, and impaired for growth in macrophage THP-1 cells. In vivo gene expression profiles of M. tuberculosis showed the iron-responsive gene mbtB was highly upregulated [61]. The finding that mycobactins directly acquire intracellular iron through lipid trafficking provided the first evidence that these siderophores are important in vivo [95]. In addition several other observations have indirectly provided evidence for the importance of mycobacterial iron metabolism. Thus, iron overload and active TB infection are inversely correlated and M. tuberculosis containing a constitutively expressed IdeR iron repressor homologue exhibited attenuated virulence in a BALB/c mouse model [96,97]. Collectively, these results indicate that targeting the biosynthesis of mycobactins may provide selective therapeutics to combat TB.
MbtA is a 59 kDa aryl-acid adenylating enzyme responsible for priming the initial NRPS carrier protein domain (MbtB) of the mycobactin assembly line with the salicylic acid starter unit. This reaction is catalyzed in a two-step reaction; in the first-half reaction, MbtA catalyzes the condensation of salicylic acid (48, Fig. (14A) with ATP to form the aryl-adenylate 49 (Sal-AMP). In the second-half reaction, MbtA ligates salicylic acid onto the phosphopantetheinyl cofactor that is tethered to the carrier protein (CP) domain of MbtB to afford 50. Although MbtA has only been superficially biochemically characterized [91] the E. coli homologue, EntE, that bears 40% amino acid identity has been extensively studied [98]. EntE adenylates 2,3-dihydroxybenzoic acid (DHB) as opposed to salicylic acid (SAL). The steady-state kinetic mechanism for EntE is a bi-uni-uni-bi ping-pong kinetic mechanism with sequential ordered binding of DHB and ATP Fig. (14B) [98]. Initial velocity studies provided a kcat value of 2.8 sec−1 and KM values of 2.5, 430, and 2.9 μM for DHB, ATP, and holo-EntB (analogous to MbtB), respectively. The crystal structures of DhbE and BasE were solved with bound intermediate acyladenylate intermediates Fig. (15) providing a solid structural foundation for this class of aryl acid adenylating enzymes [99,100]. DhbE the aryl-acid adenylating enzyme from Bacillus subtilis and BasE from Acinetobacter baumannii share 45% and 37% sequence identity with MbtA, respectively.
Fig. 14.

(A) Adenylation-ligation reaction catalyzed by MbtA. (B) Bi-uni-uni-bi ping-pong kinetic mechanism for EntE.
Fig. 15.

2,3-DHB-AMS co-crystallized with BasE from A. baumannii (3O82). (A) N-terminal subdomain with inhibitor bound. C-terminal subdomain is beyond the hinge residue Lys437 as indicated. Enzyme residues are shown with green carbon atoms, red oxygen atoms, and blue nitrogen atoms. Ligand is shown with cyan carbon atoms, blue nitrogen atoms, red oxygen atoms, and yellow sulfur atoms. Hydrogen bonds are indicated by red dashed lines. (B) Residues Asp420 and Val344 recognize the adenosine sugar, while Glu341 and His241 bind the sulfamoyl linker analogous to the native phosphonate. 2,3-DHB is held in a planar conformation due to hydrogen bonding between the 2-hydroxyl and the sulfamoyl nitrogen (pKa ~ 2). Ser247 hydrogen bonds to the 3-hydroxyl, while Phe243 and Val344 form a stacking and hydrophobic interaction respectively with the phenyl ring. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
The bisubstrate inhibitor 5′-O-[N-(salicyl)sulfamoyl] adenosine (Sal-AMS, 51, Fig. (16)) wherein the acylphosphate linkage of the Sal-AMP aryl-adenylate intermediate 49 was replaced with the isosteric acylsulfamate linkage was shown to be a potent inhibitor of MbtA and also exhibited impressive antitubercular activity with an MIC of 0.39 μM under iron-limiting conditions (Table 4) [101–103]. Isothermal titration calorimetry (ITC) experiments employing a large excess of salicylic acid as a competitive ligand revealed the true dissociation constant of 51 as approximately one picomolar [104]. Detailed kinetic studies with EntE from E. coli, a homolog of MbtA, demonstrated this bisubstrate inhibitor exhibits slow-onset time-dependent inhibition via a one-step slow association mechanism [98]. Based on the promising activity of 51, extensive structure activity relationship (SAR) studies were performed that systematically explored the importance of every atom of this modular inhibitor scaffold, which can be dissected into four domains as depicted in Fig. (16) [102, 104–106].
Fig. 16.

Modular scaffold of Sal-AMS 51.
Table 4.
MbtA Inhibitors: Linker Domain SAR

| |||
|---|---|---|---|
| Compound, R = | Kiapp (μM) | MIC99 (μM) (iron-poor) | MIC99 (μM) (iron-rich) |
 51
51
|
0.005 ± 0.001 | 0.39 | 1.56 |
 52
52
|
0.004 ± 0.001 | 0.19 | 0.39 |
 53
53
|
0.41 ± 0.06 | > 100 | > 100 |
 54
54
|
3.3 ± 0.6 | 25 | > 100 |
 55
55
|
> 100 | > 100 | > 100 |
 56
56
|
1.25 ± 0.05 | 200 | > 200 |
 57
57
|
143 ± 9 | 100 | > 100 |
Nature of the Linker [102, 107]
The most crucial consideration of the inhibitors is the linker since this must be metabolically stabile and appropriately position both the nucleoside and the aryl domains to obtain optimal interactions with their respective binding sites. The initial lead compound, Sal-AMS (51, Table 4), contained an acylsulfamate linkage [102]. Replacement of the 5′-oxygen atom with a nitrogen atom furnished acylsulfamide 52 that possessed an apparent Ki of 4 nM [102]. Deletion of the linker carbonyl moiety provided sulfamate 53 and an 80-fold loss in potency. Substitution of the central nitrogen atom with a carbon atom yielded β-ketosulfonamide 54 and led to a profound 870-loss in potency while the correspondingα,α-difluoro analogue 55 was completely inactive [107]. This last result was contrary to expectations since CF2 moieties are typically considered excellent isosteres of an oxygen atom (the native acylphosphate contains an ‘oxygen’ atom at this position). Acylurea analogue 56 explored the importance of the tetrahedral geometry of the sulfonyl moiety for activity and displayed modest activity (1.25 μM), which represents a 329-fold loss in binding affinity relative to acylsulfamide 52 [105]. Vinylsulfonamide 57 realized an approximately 100,000 loss in potency concordant with removal of the linker carbonyl (100-fold loss) and linker nitrogen atom (1000-fold loss) [108].
Based on the observed structure-activity relationships, molecular modeling results, and X-ray co-crystal structural data, the key interactions between the ligand and protein are through Lys542 that simultaneously interacts with the linker carbonyl and the sulfonyl oxo group [100, 109]. The sulfonyl moiety is additionally predicted to interact with His257 and Ala356 as shown in Table 4. Significantly, the ligand forms an internal hydrogen bond with the phenol and sulfamate nitrogen atom, which is deprotonated (pKa = 2.0), that enforces a coplanar arrangement of the salicyl group. Molecular mechanics simulations revealed that the required coplanar conformation for 54 and 55 were greater than 5 kcal/mol over the lowest energy conformation explaining their lack of activity [107]. The SAR data underscores how exquisitely sensitive the linker region is toward modification. Overall, the in vitro enzyme inhibition and whole-cell activity were well correlated. Analogues 51 and 52 containing the acylsulfamate and acylsulfamide linkages displayed the highest activity with MIC99’s of 0.39 and 0.19 μM respectively under iron limiting conditions. However, these compounds also exhibited activity under iron-rich conditions suggesting that the inhibitors are operating by siderophore-independent mechanisms. The ratio of MIC99 (iron-poor)/MIC99 (iron-rich) is referred herein to as the selectivity ratio and is a measure of the inhibitor specificity.
Importance of the Aryl Ring [105]
To Whom It May Concern: explore the significance of the ortho-hydroxy group, a systematic series of analogues were prepared bearing substitution at the 2-position [105]. Deletion of the hydroxyl group afforded benzoyl analogue 58 and resulted in a 14-fold loss of binding affinity (Table 5). Substitution of the ortho-hydroxy group reduced potency in all cases but by widely varying amounts: by 6-fold for 2-fluoro 59, 117-fold for 2-amino 60, 1100-fold for 2-nitro 61, and 2700-fold for 2-chloro 62. Analogues 63–65 were prepared to define the steric requirements of the shallow binding pocket and to identify potential sites for further modification [105]. The SAR from this chloroscan showed that substitution at the 4-position of the aryl ring was most tolerated resulting in a modest 2-fold decrease in potency relative to 51 whereas substitution at the 3- and 5-positions reduced inhibitor potency 3- and 9-fold, respectively. Since the optimal position was found to be the 4-position, several additional substituents (F, Br, CH3, CF3, and NH2) were examined at this position [105]. None of the analogues in this series (66–70) exceeded the potency of 4-Cl 64 and strongly electron-withdrawing substituents were strongly disfavored. Thus, 4-CF3 69 exhibited a remarkable 313-fold loss of potency relative to the 4-CH3 68. Additionally, many other analogues were prepared containing heterocyclic, alkyl, cycloalkyl, benzyl, and aminoacyl groups (not shown); however these were all inactive [105].
Table 5.
MbtA Inhibitors: Aryl Domain SAR

| ||||
|---|---|---|---|---|
| Compound, R = | KIapp (μM) | MIC99 (μM) (iron-deficient) | MIC99 (μM) (iron-rich) | |

|
51 OH | 0.0066 ± 0.00015d | 0.39 | 1.56 |
| 58 H | 0.092 ± 0.007 | 12.5 | 50 | |
| 59 F | 0.038 ± 0.008 | 12.5 | 50 | |
| 60 NH2 | 0.77 ± 0.12 | > 200 | > 200 | |
| 61 NO2 | 7.32 ± 0.9 | > 200 | > 200 | |
| 62 Cl | 18.1 ± 1.7 | > 200 | > 200 | |

|
63 3-Cl | 0.061 ± 0.003 | 50 | > 200 |
| 64 4-Cl | 0.012 ± 0.0006 | 12.5 | 50 | |
| 65 5-Cl | 0.020 ± 0.0014 | 12.5 | 50 | |
| 66 4-F | 0.012 ± 0.0016 | 0.098 | 0.39 | |
| 67 4-Br | 0.021 ± 0.001 | 6.25–12.5 | 50 | |
| 68 4-CH3 | 0.014 ± 0.0007 | 1.56 | 25 | |
| 69 4-CF3 | 4.38 ± 0.44 | > 200 | > 200 | |
| 70 4-NH2 | 0.040 ± 0.004 | 1.56 | 25 | |
| 71 6-F | 0.0073 ± 0.0006 | 0.78 | 3.13 | |
|
72 | 0.137 ± 0.012 | > 200 | > 200 |

|
73 | 3.7 ± 0.5 | > 200 | > 200 |
The MIC99 values against M. tuberculosis for the aryl-modified analogues paralleled the enzyme inhibition data. The p-aminosalicyl derivative 70 displayed improved selectivity showing a 16-fold difference in MIC under iron-deficient conditions relative to iron-rich conditions, suggesting that this modification reduced off-target binding. The collective SAR from this series of compounds demonstrated that the aryl domain is poorly tolerant to modification in accord with the strict specificity of NRPS adenylation enzymes. Although, the 2-hydroxy group is required for optimal activity, the findings that this can be replaced with a fluoro may be useful to increase the metabolic stability as phenols are well known to undergo glucuronidation.
Role of the Ribose
Modification of the ribose subunit was explored to determine the importance of each atom towards binding affinity [110]. Carbocyclic analogue 74 (Table 6) displayed a 3-fold increase in potency demonstrating that the ribofuranose ring-oxygen is dispensable for activity. Deletion of either the 2′-hydroxyl in 75 or the 3′-hydroxyl in 76 resulted in an approximately 2-fold increase in potency. Although Asp435 is positioned to form a bidendate hydrogen bond with both the 2′- and 3′-hydroxyl groups and thus fix the sugar in the adenosine binding pocket, the observed SAR shows that only one of these interactions is required to maintain potency. Dideoxy dehydro carbocyclic analogue 77 exhibited a pronounced 126-fold loss of potency while the saturated dideoxy carbocycle 78 resulted in a mere 9-fold loss in binding affinity despite removal of the 2′ and 3′ alcohols and the ribofuranose ring oxygen. Formation of a salt bridge between Arg451 and Asp435 may compensate for the loss of hydrogen bonds between 78 and MbtA. The attenuated activity of 77 is most likely a result of the rigidity of the cyclopentene moiety that is unable to adopt the required C3′ endo conformation. Acyclo analogue 79 was approximately 2500-fold less active illustrating the importance of the conformational rigidity of parent ribose moiety.
Table 6.
MbtA Inhibitors: Glycosyl Domain SAR

| |||
|---|---|---|---|
| Compound, R = | Kiapp (μM)a | MIC99 (μM) (iron-deficient) | MIC99 (μM) (iron-rich) |
 51
51
|
0.0066 ± 0.00015 | 0.39 | 1.56 |
 74
74
|
0.0023 ± 0.0004 | 1.56 | 6.25 |
 75
75
|
0.0035 ± 0.0004 | 25 | 100 |
 76
76
|
0.0032 ± 0.0005 | 1.56 | 12.5 |
 77
77
|
0.830 ± 0.078 | > 200 | > 200 |
 78
78
|
0.061 ± 0.005 | > 200 | > 200 |
 79
79
|
16.7 ± 0.5 | > 200 | > 200 |
The biological activity of 74-79 paralleled the enzyme inhibition except for compounds 75 and 78. The poor antibacterial activity of 75 is likely due to the known chemical instability of 2′-deoxynucleosides, thus 75 may decompose over the approximately 1–2 weeks of the whole-cell M. tuberculosis assay. A rationale for the observed discrepancy between enzyme inhibition and whole-cell biological activity of 78 is that the sugar is important for recognition by a putative transporter. M. tuberculosis encodes 37 ABC (ATP-dependent binding cassette) transporters of which 16 have been unambiguously assigned as importers responsible for assimilation of amino acids, nucleotides, and other essential cofactors [111]. Additionally, M. tuberculosis transports many hydrophilic solutes via a class of proteins known as the porins (the major porin is known as MspA), which have also been shown to play an important role in the transport of antibiotics such as the β-lactams and aminoglycosides [112].
Impact of the Nucleobase [104]
A systematic series of analogues was prepared to examine the impact of modification to the nucleobase domain of 51 (Table 7) [104]. Deletion of N-7 in 80 led to a 4-fold decrease in potency while removal of N-1, N-3, and N-7 in indole analogue 81 did not further impact binding affinity relative to 80 (Table 7). These data were consistent with a homology model that showed no electrostatic interactions for N-1 and N-3 and a poorly aligned hydrogen bond between Gly329 and N-7 [104]. At least one hydrogen bond donor at N-6 was found to be essential for activity of this class of compounds as inosine 82 and dimethyl 83 analogues exhibited pronounced reductions in potency. Substitution of N-6 with C1-C3 chains in 84-87 and 89 was beneficial leading to incremental increases in potency and reaching a maximum with cyclopropyl 89. The potency followed a logarithmic decline as the chain length increased to C4 and C5, then leveled off at C5 (see 88 and 90-92). The C-8 modified analogues 93-95 exhibited drastically reduced potencies, illustrating the exquisite sensitivity to modification at this position. Substitution at C-2 was initially explored with 2-iodo 96, which showed a 2-fold increase in affinity. Encouraged by this result, a small series of analogues were synthesized by various cross-coupling reactions to provide 2-phenylamino 97, phenylethynyl 98, and 2-phenyl 99 Fig. (17). These compounds were 7, 17, and 24-times more potent than 51. Modeling studies predicted that additional room at this binding site was available at either the meta- or para-positions of the phenyl ring in 99. Accordingly, biphenyl derivatives 100-102 were prepared and found to exhibit the following trend: meta~para > ortho. The 174-fold loss in potency of ortho-biphenyl 100 compared to 99 is consistent with a steric clash of the biphenyl with backbone resi-residues in the N-terminal domain, while the meta and para phenyl groups successfully access the interdomain pocket. The ability to tolerate these bulky nonpolar substituents at C-2 was remarkable, but consistent with molecular dynamics simulations, which showed a high degree of flexibility in this region [104, 109]. The antitubercular activity of these compounds was evaluated against whole-cell M. tuberculosis H37Rv and 97-99 exhibited the most potent whole-cell antitubercular activity yet reported for this new class of antibacterial agents. Even more significantly, cyclopropyl analogue 89 displayed enhanced selectivity with an MIC99 of 98 nM under iron-deficient conditions, but 6.25 μM under iron-rich conditions, representing a 64-fold difference in activity, suggesting that this modification reduced off-target binding.
Table 7.
MbtA Inhibitors: Nucleobase Domain SAR

| ||||
|---|---|---|---|---|
| Compound, R = | KIapp (μM) | MIC99(μM) (iron-deficient) | MIC99 (μM) (iron-rich) | |

|
51, X = Y = N | 0.0066 ± 0.0015 | 0.39 | 1.56 |
| 80, X = N; Y = CH | 0.0243 ± 0.0070 | 6.25 | 50 | |
| 81, X = Y= CH | 0.0201 ± 0.0023 | 6.25 | 50 | |
|
| ||||

|
82, =O | 0.80 ± 0.05 | >100 | >100 |
| 83, –NMe2 | 0.38 ± 0.03 | 50 | n.d. | |
| 84, –NHMe | 0.0016 ± 0.0002 | 0.39 | 1.56 | |
| 85, –NHEt | 0.0044 ± 0.0008 | 0.39 | 1.56 | |
| 86, –NHn-Pr | 0.0035 ± 0.0002 | 0.39 | 1.56 | |
| 87, –NHi-Pr | 0.0070 ± 0.0009 | 25 | >100 | |
| 88, –NHi-Bu | 1.14 ± 0.23 | >100 | >100 | |
| 89, –NHcyclopropy | 0.0019 ± 0.0001 | 0.098 | 6.25 | |
| 90, –NHcyclobutyl | 0.124 ± 0.012 | >100 | >100 | |
| 91, –NHcyclopenty | 9.4 ± 5.2 | >100 | >100 | |
| 92, –NHBn | 8.3 ± 0.8 | >100 | >100 | |
|
| ||||

|
93, –Br | 2.05 ± 0.30 | >50 | >50 |
| 94, –N3 | 42.5 ± 6.2 | >50 | >50 | |
| 95, –NH2 | 183 ± 19 | >50 | >50 | |
|
| ||||

|
96, –I | 0.0030 ± 0.0003 | 0.19 | 3.12 |
| 97, –NHPh | 0.0009 ± 0.0002 | 0.049 | 0.39 | |
| 98, –CCPh | 0.0004 ± 0.0001 | 0.049 | 0.39 | |
| 99, –Ph | 0.0003 ± 0.0001 | 0.049 | 0.39 | |
| 100, biphen-2-yl | 0.047 ± 0.002 | >50 | >50 | |
| 101, biphen-3-yl | 0.0014 ± 0.0002 | 3.13–6.25 | 12.5–25 | |
| 102, biphen-4-y | 0.0010 ± 0.00002 | 3.13–6.25 | 12.5–25 | |
103

|
0.0033 ± 0.0003 | 3.13 | >50 | |
|
104
|
0.0027 ± 0.0004 | >25 | >50 | |
Fig. 17.

Model of 99 bound to MbtA. Ligand 99 is illustrated in a predicted binding mode (QM/MM/6-31G(d)). The conformations of Lys332 and Val448 in the original homology model with Sal-AMS (51) are shown in green, indicating the conformational shift necessary to permit binding of ligands with large C2-substituents. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
High-Throughput Screening
Neres and co-workers developed a fluorescence polarization HTS displacement assay using ligand 105 Fig. (18), a fluorescently labeled analogue of the parent ligand 51 [113]. More than 85,000 compounds were screened in duplicate against the aryl acid adenylating enzyme BasE from Acinetobacter baumannii, which shares 37% sequence identity to MbtA. A total of 92 compounds were found to result in greater than 25% displacement of the probe after performing secondary assays to remove false positives. Pyrazolo[3,4-b]pyridine 106 Fig. (18) emerged as the most potent scaffold with a KD of 78 nM against BasE and 3.7 μM against MbtA [113,114]. Subsequent SAR studies have identified compounds with low nanomolar affinity for MbtA [114]. Biochemical and structural characterization of 106 with BasE showed it exhibits competitive inhibition against both ATP and 2,3-dihydroxybenzoic acid (DHB, the aryl acid substrate for BasE) [100]. Based on the similarity of 106 to adenine, it was expected that it might bind in the adenylate binding pocket with the phenyl group analogously occupying the position of the C-2 phenyl substituent of ligand 99 and the 4-pyridylmethyl group occupying the ribose binding pocket [113]. However, 106 bound to BasE in an unexpected binding mode Fig. (19) and did not superimpose with the adenylate, but rather projected the phenyl group into the pantetheine tunnel. Additionally, the 4-pyridylmethyl group occupied the DHB binding pocket with the pyridyl nitrogen accepting a hydrogen bond from the side chain of Asn242, in a manner similar to the 2- and 3-hydroxy groups of DHB Fig. (19) [100]. The binding mode of 106 was consistent with the biochemical results, thus 106 can also be considered a novel multisubstrate inhibitor of adenylating enzymes, and unlike the substrate-based inhibitors, 106 is expected to have superior physicochemical properties.
Fig. 18.

Structures of ligand 105 used for the fluorescence polarization assay and pyrazolo[3,4-b]pyridine 106 identified by high-throughput screening.
Fig. 19.

Pyrazolo[3,4-b]pyridine 106 co-crystallized with BasE from A. baumannii (PDB 3O84). (A) N-terminal subdomain with 106 bound. The C-terminal subdomain is beyond the hinge residue Lys437 as indicated. The C-terminal domain is disordered in the crystal lattice and could not be modeled. Enzyme residues are shown with green carbon atoms, red oxygen atoms, and blue nitrogen atoms. Ligand is shown with cyan carbon atoms, red oxygen atoms, and blue nitrogen atoms. Hydrogen bonds are indicated by red dashed lines. (B) Binding mode of 106 differs greatly from 2,3-DHB-AMS. The N2 nitrogen of the pyrazole ring forms a hydrogen bond with the amide nitrogen of Gly338, and the pyridine ring accepts a hydrogen bond from the side chain of Asn242. The phenyl ring of 106 occupies the DHB subsite by stacking with His241 on one side and hydrophobic Val286 on the other. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
NadE
Nicotinamide adenine dinucleotide (NAD) is an essential cofactor used in reduction-oxidation reactions that is required for energy metabolism [115]. Energy metabolism has been shown to be a critical process for the viability of hypoxic nonreplicating M. tuberculosis suggesting NAD biosynthesis is an ideal pathway for the development of new antitubercular agents [46]. Several important nonredox functions of NAD have also been reported including protein deacetylation catalyzed by the sirtuins, DNA repair performed by the NAD-dependent prokaryotic DNA ligase, and ADP-ribosylation mediated by ADP-ribosyltransferases [116]. NAD can be synthesized de novo from dihydroxyacetone phosphate 108 and aspartate 109, or it can be recycled via the Preiss-Handler salvage pathway from nicotinamide 107 Fig. (20). Both pathways are utilized in vivo by M. tuberculosis [117]. The NAD de novo and salvage pathways converge at nicotinate mononucleotide (NaMN, 110), which is then converted to nicotinic acid adenine dinucleotide (NaAD, 111) by NadD, a nicotinic acid mononucleotide adenylyltransferase, and then to nicotinamide adenine dinucleotide (NAD, 112) by NadE, a NAD synthetase. Based on the redundancy of both NAD de novo synthesis and salvage pathways, only the final two steps catalyzed by NadD and NadE are valid drug targets. The adenylating enzyme NadE shares 23% identity to the human ortholog and the protein from M. tuberculosis has been functionally and structurally characterized providing the foundation for the development of small molecule inhibitors [118].
Fig. 20.

NAD salvage and de novo biosynthetic pathways.
NadE from M. tuberculosis is an approximately 75 kDa protein containing an N-terminal glutamine amidotransferase (GAT) domain that generates ammonia through hydrolysis of glutamine and a C-terminal adenylation domain, which catalyzes the two-step amidation of nicotinic acid adenine dinucleotide (NaAD) [118]. The active sites are tightly coupled and steady-state kinetic analysis suggests that adenylation of NaAD 111 to afford NaAD-AMP 113 occurs initially Fig. (21). Formation of the acyladenylate 113 then triggers a putative conformational change that activates the GAT domain, which subsequently catalyzes glutamine hydrolysis to afford ammonia that is channeled through an approximately 40 Å ammonia tunnel to the adenylation domain [118]. Nucleophilic attack of ammonia on the intermediate acyladenylate 113 then provides NAD and AMP. The co-crystal structure of NaAD and mtNadE from M. tuberculosis has been solved Fig. (22) [118]. Additionally, the co-crystal structure of bsNadE from Bacillus subtilus with the acyladenylate 113 has been reported [119].
Fig. 21.

NAD synthetase enzyme mechanism.
Fig. 22.

NAD synthetase from M. tuberculosis co-crystallized with NaAD+ (PDB 3DLA). (A) The N-terminal subdomain from chain A (yellow) and the C-terminal subdomain from chain D (purple) come together to bind NaAD+ (cyan carbon atoms, blue nitrogen atoms, red oxygen atoms, and orange phosphate atoms). Enzyme residues are shown with green carbon atoms, blue nitrogen atoms, and red oxygen atoms. Hydrogen bonds are indicated by red dashed lines. (B) Active site binding shows hydrogen bond stabilization of the adenosine by side chain of Arg354 and the backbone of Gly475, as well as key π-stacking with Phe634. Hydrogen bond contacts with the phosphates are expected and observed with the side chains of Asn471 and Lys635. Less electron density stabilization of the nicotinic acid ring, indicating flexibility at this site, perhaps allowing for the introduction of ATP into the active site. Key stabilizing interactions include the Asp497 side chain hydrogen bonding with the carboxylic acid, as well as Trp490 hydrogen bonding to the positively charge nitrogen on the nicotinic acid ring. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
Brouillette and co-workers described the synthesis of a series of simplified NaAD substrate mimics termed ‘tethered dimers’ containing two heteroaryl moieties separated by a spacer where an indole was used to target the adenosine binding pocket and a N-methypyridinium or N,N,N-trimethylanilinium ion was used to target the nicotinamide binding pocket [120,121]. Initially these compounds were evaluated against bsNadE and the SAR showed that the optimal linker comprised a simple hydrophobic alkyl chain between 6–9 atoms while the optimal heteroaryl head groups were 5-benzyloxyindole and either N-methylpyridinium or N,N,N-trimethylanilinium ions. Removal of the positively charged aromatic groups resulted in complete loss of activity. Barry and co-workers independently synthesized several of the most potent compounds and evaluated them against NadE from M. tuberculosis as well as for antimycobacterial activity under replicating and non-replicating conditions and these results are shown in Table 8 [117]. Although it is not possible to delineate rigorous structure-activity relationships from this small series of five compounds, the biochemical activity and MIC values did exhibit reasonable correlation. Furthermore, the most active compounds 118 and 120 were bactericidal with MBC (minimum bactericidal activity) values of 10 μM and 40 μM, respectively. Additionally they exhibited nearly equipotent activity under non-replicating anaerobic conditions as measured by their MAC (minimum anaerobic activity) values. Interestingly, the tethered dimers were designed as NadD substrate mimics, but the modality of inhibition has never been reported. The IC50 values determined by Barry and co-workers [117] were measured at 4 mM NaAD, which is approximately 30-fold higher than its respective Michaelis-Menten constant (KM = 0.13 mM [118]). More recently, Brouillette and co-workers utilized virtual screening to identify lead inhibitors of bsNadE [122]. The most promising compound was sulfonamide-diarylurea 122, which possessed an IC50 value of 6.4 μM against bsNadE. Synthesis of a 76-membered library of analogues failed to identify a more potent compound [123].
Table 8.
Activities of NadE Inhibitorsa
| compound | IC50 (μM) | MIC (μM) | MBC (μM) | MAC (μM) |
|---|---|---|---|---|
117

|
insol.b | 42 | n.d.c | n.d. |
118

|
25 | 15 | 10 | 19 |
119

|
63 | 155 | n.d. | n.d. |
120

|
22 | 20 | 40 | 42 |
121

|
58 | 40 | n.d. | n.d. |
122

|
10d | n.d. | n.d. | n.d. |
Summary of in vitro activity against NadE (IC50), MIC against M. tuberculosis H37Rv, minimum bactericidal concentration (MBC), minimum anaerobicidal concentration (MAC) defined as the concentration causing a 10-fold kill of bacteria in nonreplicating anaerobic culture after 7 days exposure.
insol., insoluble.
n.d., not determined.
IC50 against NadE from Bacillus subtilus.
GuaA
Nucleotide biosynthesis is a ubiquitous and essential process found throughout all forms of life. Nucleotides play crucial roles in numerous cellular functions such as DNA replication and transcription, chemical energy production, enzymatic catalysis, and intracellular secondary messaging [124]. The genes responsible for the de novo biosynthesis of purine nucleotides in M. tuberculosis have been putatively assigned, although many of the experimental details necessary to confirm enzymatic function are lacking [125]. Purine biosynthesis proceeds through a 10-step enzymatic process starting from 5′-phosphoribosylpyrophosphate (PRRP, 123, Fig. (23)) to yield inosine monophosphate (IMP, 124). IMP represents a divergence point in purine biosynthesis, since it can be further elaborated via separate pathways to form either adenosine monophosphate (AMP, 126) or guanosine monophosphate (GMP, 128). In the penultimate step in GMP biosynthesis, the enzyme IMP dehydrogenase converts IMP into xanthosine monophosphate (XMP, 127). During the final step, GMP synthetase catalyzes the conversion of XMP into GMP.
Fig. 23.

Purine biosynthesis.
GMP synthetase from M. tuberculosis, encoded by guaA, is a 56 kDa amidotransferase that has not been functionally or structurally characterized. The M. tuberculosis GMP synthetase shares 49% sequence identity to the E. coli ortholog, which has been extensively characterized [126–129]. The E. coli enzyme exists as a homodimer of 58 kDa subunits and catalyzes the formation of an O2-adenylate (129, XMP-AMP, Fig. (24)) between XMP 127 and ATP [130]. The adenylate formation activates the C2 carbon of the XMP portion of 129 for attack by ammonia to form GMP 128 and release of AMP [129, 131]. GMP synthetase is organized in a modular function and consists of two separate functional domains; the “amidotransferase” domain which abstracts the amide nitrogen of the glutamine substrate, and the “synthetase” domain which is responsible for coupling the abstracted ammonia to the activated XMP-adenylate 129 [131]. The apparent KM values for XMP, ATP, and glutamine were found to be 65, 320, and 13 mM, respectively [129]. Lastly, Smith and coworkers solved the crystal structure of GMP synthetase from E. coli (PDB code 1GPM) at a resolution of 2.2 Å Fig. (25) [131].
Fig. 24.

Adenylation–ligation reaction catalyzed by GMP synthetase.
Fig. 25.
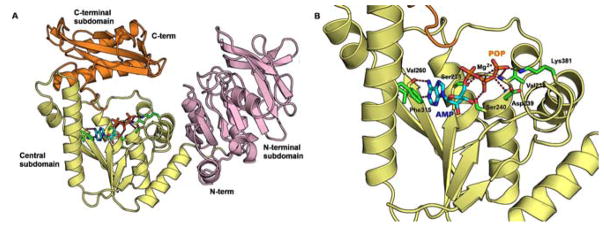
AMP and pyrophosphate co-crystallized with GMP synthetase from E. coli (PDB 1GPM). (A) Full length GMP synthetase. The N- and C-terminal subdomains (purple and orange, respectively) are not involved in binding of AMP and pyrophosphate. Only the Central subdomain (yellow) forms important contacts with side chains shown as green carbon atoms, blue nitrogen atoms, and red carbon atoms. A critical magnesium atom is shown as a gray sphere. Ligands are shown with cyan carbon atoms, blue nitrogen atoms, red oxygen atoms, and orange phosphate atoms. (B) Active site binding of AMP (cyan carbon atoms, blue nitrogen atoms, red oxygen atoms, and orange phosphate atoms) and POP (red oxygen atoms and orange phosphate atoms) was observed upon soaking crystals with XMP and ATP, likely through spontaneous hydrolysis [131]. Key binding interactions at the nucleoside base are π-stacking of Phe315 and hydrogen bonding from the backbone of Val260. Electrostatic stabilization of POP with Mg2+ as well as side chain hydrogen bonds with Ser235, Val238, Asp239, Ser240, and Lys381 are expected to stabilize ATP binding. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
Several inhibitors of GMP synthetase from E. coli have been developed. The majority of these inhibitors including acivicin, 5-oxo-L-norleucine, and azaserine (130-132, Fig. (26)), are glutamine analogues that target the amidotransferase domain of E. coli GMP synthetase [132]. As an orthogonal strategy, Davisson and coworkers designed inhibitor 133, which targets the XMP binding pocket of GMP synthetase [133]. The authors note that early studies of GMPS suggested that the enzyme was inhibited by a nucleotide derivative that was likely the product of hydroxylamine attack on adenylate 129 [133–135]. Therefore, the researchers synthesized N2-OH GMP 133 and showed it to be a time-dependent inhibitor that is competitive with respect to XMP with a Ki = 92 nM.
Fig. 26.

E. coli GMP synthetase inhibitors.
PanC
Pantothenate also known as vitamin B5 is the core of coenzyme A, an essential cofactor in the central pathways of respiration and lipid metabolism [136]. The phosphopantetheine moiety of coenzyme A is also the prosthetic group in more than thirty carrier proteins found in the fatty acid synthases, polyketide synthases, and non-ribosomal peptide synthetases in M. tuberculosis Fig. (27). Accordingly, pantothenate biosynthesis has been considered an attractive target in the development of new antimicrobial agents since this pathway is absent in mammals, but essential in microbes. A gene knockout of panC, which encodes for the fourth enzyme in the pantothenate biosynthetic pathway in M. tuberculosis, exhibited highly attenuated virulence in two immunocompetent mouse models serving to validate pantothenate as a potential target [137, 138].
Fig. 27.

Pantothenate 134 is transformed to coenzyme A 135, which is converted to CoA thioesters 136 and holo-acyl carrier proteins 137 by CoA ligases and phosphopantetheinyl transferases, respectively.
Pantothenate biosynthesis is accomplished in four steps employing the enzymes PanB and PanE to synthesize pantoic acid and PanD, an aspartate α-decarboxylase, to produce β-alanine. The adenylating enzyme PanC then uses ATP to catalyze the condensation of pantoic acid and β-alanine to afford pantothenate Fig. (28A). Blanchard and Zheng demonstrated that PanC from M. tuberculosis follows a bi-uni-uni-bi ping pong kinetic mechanism with ATP binding preceding binding of pantoic acid Fig. (28B) [139,140]. Eisenberg and Wang solved the three-dimensional structures of PanC from M. tuberculosis in complex with AMPCPP (a non-hydrolyzable ATP analogue), pantoic acid, a ternary complex of AMPCPP and pantoic acid, the pantoyl-adenylate, and a ternary complex of AMP and β-alanine providing a step-by-step view of the PanC catalyzed reaction Fig. (29) [141,142]. Overall, PanC is the best biochemically and structurally characterized adenylating enzyme in M. tuberculosis.
Fig. 28.

A. Adenylation–ligation reaction catalyzed by PanC. B. Bi-uni-uni-bi ping-pong kinetic mechanism for PanC.
Fig. 29.

Structure of pantoyl-adenylate co-crystal with PanC from M. tuberculosis (PDB 1N2I). (A) Full-length PanC. The N-terminal subdomain (yellow) contains the majority of the key interactions for adenylate binding, with the C-terminal subdomain (orange) includes conserved nucleobase interactions. Ligand is shown with cyan carbon atoms, blue nitrogen atoms, red oxygen atoms, and orange phosphate atoms. Enzyme residues are shown with green carbon atoms, blue nitrogen atoms, red oxygen atoms and yellow sulfur atoms. (B) Active site binding shows pantoate specificity derived from multiple hydrogen bond interactions with Gln72 and Gln164. Adenylate specificity is conserved by hydrogen bond interactions with backbone atoms, highlighted by Val187. The sugar ring and phosphate linkage are stabilized by hydrogen bond interactions of Asp161, His47, and Met40. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
High-throughput screening of PanC against a 4080-membered library using the coupled myokinase, pyruvate kinase and lactate dehydrogenase assay system for detection of released AMP led to the identification of nafronyl oxalate 141, which was a weak inhibitor with an IC50 value of 75 μM (Table 9) [143]. Crystal soaking experiments showed that nafronyl bound in the active site, although the 2.9 Å co-crystal structure did not enable determination of the precise binding mode [143]. Further screening at the Southern Research Institute (SRI) against a 65,422-membered library led to the identification of a pyrazol-4-yl-tetrahyrdro[d]isoxazole 142 that was a potent inhibitor with an IC50 value of 120 nM against PanC [144]. Limited SAR was performed, which showed substitution of the benzyl subunit attached to the pyrazole ring was tolerated in analogues 143-146 and 2-napthyl derivative 148 without any deleterious effect on binding affinity (Table 9). However, removal of the benzyl group in analogue 147 led to a 500-fold loss in activity [145]. Despite the fairly potent biochemical activity, none of the pyrazol-4-yl-tetrahydro[d]isoxazole analogues demonstrated appreciable antimycobacterial activity (MIC ≥ 128 μM).
Table 9.
HTS and Bisubstrate Inhibitors of PanC
| compound | IC50 (μM) | ||
|---|---|---|---|
| 141 |

|
75.0 | |
|
| |||
| 142 |

|
||
| 143 | |||
| 144 | |||
| 145 | |||
| 146 | |||
| 147 | |||
| 148 | |||
|
| |||
| 149 |

|
0.22 | |
| 150 |

|

|
4.0 |
| 151 | 18.0 | ||
Abell and co-workers have described the synthesis of a series of bisubstrate inhibitors that mimic the acyladenylate PAN-AMP 139 using the sulfamate as an isostere of the phosphate moiety [146]. Due to the known instability of the adenylate presumably caused by lactonization to generate pantolactone 152 Fig. (30) [139], the C-4 alcohol in the pantoate portion of bisubstrate inhibitors 149-151 (Table 9) was removed [146]. Compound 149 (PAN-AMS) resembles the acyladenylate most closely and was evaluated as a di-astereomeric mixture at the C-2 hydroxy position of the pantoate moiety and shown to exhibit competitive inhibition with respect to ATP. The Ki value for 149 was 0.22 μM, which is approximately 1,500-fold lower than the KM value for ATP. Replacement of the C-2 hydroxy group of the pan toate fragment with an amino group provided analogue 150 and concomitant deletion of the 3-methyl group furnished 151. These analogues were 18- and 82-fold less active than 149, respectively. Isothermal titration calorimetry showed the true dissociation constant for 149 was 125 nM, which is several orders of magnitude higher than other related bisubstrate inhibitors described in this review. This is likely attributed to the deletion of the terminal hydroxyl group of the pantoate fragment and the intrinsically weak binding affinity of the pantoate fragment as evidenced by the high KM value (0.13 mM) of pantoate for PanC. Unfortunately, the antimycobacterial activity of 149-151 has not been reported. Abell and co-workers also described several other related inhibitors involving other phosphate isosteres that were evaluated against PanC from E. coli; however, none of these displayed appreciable activity demonstrating the strict requirement for the sulfamate moiety [147].
Fig. 30.

Degradation of PAN-AMP.
In a third strategy to identify PanC inhibitors, Abell and co-workers employed a fragment-based approach [36]. A library of approximately 1,300 fragments was screened by WaterLOGSY NMR spectroscopy, which led to the identification of 5-methoxyindole 153 that possessed a KD of 1 mM as measured by isothermal titration calorimetry (Table 10). The binding mode of 153 was determined by soaking crystals of PanC and X-ray crystallographic analysis provided the structure at 1.6 Å resolution. 5-Methoxyindole 153 possessed fairly narrow structure activity relationships; however, attachment of a carboxylic acid at C-2 or a carboxymethyl group at N-1 of the indole led to fragments 154 and 155 respectively, with incremental increases in potency. Elaboration of fragment 154 provided acylsulfonamide 156 with a KD of 210 μM, whose binding mode was confirmed by X-ray crystallography. The co-crystal structure of 156 and PanC suggested further opportunities to enhance binding affinity were available by growing from the sulfonyl moiety. Attachment of a 4-methylpyridine to the sulfonyl moiety provided 157 with a KD of 29 μM, representing a nearly 10-fold increase in binding affinity. Finally, attachment of a carboxymethyl group at N-1 of indole 157 yielded 158 with a KD of 1.5 μM, which was shown to be an ATP-competitive inhibitor in a coupled continuous kinetic assay. Thus, starting from 5-methoxyindole 153 the authors were able to increase the binding affinity by more than 730-fold in only four iterations requiring the synthesis of less than ten analogues. In a parallel approach, benzofuran-2-carboxylic acid was identified from a fluorescence-based thermal-shift assay and X-ray studies showed it bound in an adjacent binding pocket to 5-methoxyindole [36]. A fragment linking approach was used ultimately leading to the identification of 159 with a KD of 1.8 μM. These complimentary fragment growing and linking strategies highlight the power of fragment-based drug discovery and suggest this may be a fruitful approach for the identification of novel inhibitors for adenylating enzymes.
Table 10.
Fragment-based PanC Inhibitors
| compound | KD (μM) |
|---|---|
153

|
1100 |
154

|
500 |
155

|
500 |
156

|
210 |
157

|
29.0 |
158

|
1.5 |
159

|
1.8 |
MshC
Mycobacteria and most members of the Actinobacteria use a low molecular weight thiol known as mycothiol (1-D-myo-inosityl-2-(N-acetyl-L-cysteinyl)amido-2-deoxy-α-D-glucopyranoside, 164, Fig. (31)) that functions analogously to glutathione in eukaryotes in detoxification by forming S-conjugates with drugs [148, 149]. Mycothiol biosynthesis is carried out in five steps from uridine diphosphate N-acetylglucosamine 160 and myo-inositol-1-phosphate 161 catalyzed by MshA, an unknown phosphatase, MshB, MshC, and MshD, whose corresponding genes are scattered throughout the genome Fig. (31). Targeted genetic disruption studies have shown that mshA and mshC are essential while mshB and mshD are dispensable for growth of M. tuberculosis [148]. Although mycothiol synthesis is essential for M. tuberculosis viability, mycothiol is also required for bioactivation of ethionamide. Thus, inhibition of mycothiol synthesis could lead to decreased susceptibility to this important second-line antitubercular agent [150,151]. Nevertheless, the mycothiol biosynthetic pathway has been considered attractive for the development of potential antitubercular agents since mycothiol is unique to mycobacteria, the pathway has been confirmed as essential through targeted genetic disruption studies, and detailed kinetic analysis as well as the 1.6 Å crystal structure of MshC have been reported [152, 153].
Fig. 31.

Biosynthesis of mycothiol.
The adenylating enzyme MshC is a 47 kDa cysteine ligase responsible for the penultimate step in mycothiol biosynthesis that catalyzes the ATP-dependent ligation of cysteine 165 to GlcN-Ins 162 to afford desacetyl mycothiol 163 Fig. (32A). Blanchard, Fan and co-workers demonstrated that MshC follows a bi uni uni bi ping pong kinetic mechanism analogous to the adenylating enzyme PanC discussed earlier in this review with ordered sequential binding of ATP followed by cysteine Fig. (32B). To provide a baseline for comparing inhibition constants for reversible inhibitors, the reported KM values for ATP, cysteine, and GlcN-INs were found to be 1.8, 0.1, and 0.16 mM, respectively [152]. Blanchard, Tremblay and co-workers determined the co-crystal structure of MshC with the acyladenylate mimic 5′-O-[N-(cysteinyl)sulfamoyl]adenosine (Cys-AMS, 167, Fig. (34)) and the key interactions are shown in Fig. (33).
Fig. 32.

(A) Adenylation–ligation reaction catalyzed by MshC. (B) Bi-uni-uni-bi ping-pong kinetic mechanism for PanC.
Fig. 34.

MshC inhibitors.
Fig. 33.

Cys-AMS co-crystallized with MshC from M. smegmatis (PDB 3C8Z). (A) The N-terminal chain of MshC with Cys-AMS bound. Enzyme residues are shown with green carbon atoms, blue nitrogen atoms, and red oxygen atoms. Ligand is shown with cyan carbon atoms, blue nitrogen atoms, red oxygen atoms, and orange sulfur atoms. Zn cofactor is a gray sphere. (B) The AMS moiety is stabilized by hydrogen bond interactions with Asp251, the backbone nitrogen of Gly249 and Ile, Thr58, and His55. The cysteine moiety of the ligand is bound by hydrogen bond interactions with side chains Thr46, Thr83, and Trp227, backbone interactions with Gly44, and direction interaction with Zn+.(For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
The bisubstrate inhibitor 5′-O-[N-(cysteinyl)sulfamoyl] adenosine (Cys-AMS, 167, Fig. (34)) was shown to exhibit competitive inhibition toward ATP with a Ki value of 304 nM, which is 500-fold lower than the KM value for ATP [152, 154]. Cys-AMS 167 also displayed noncompetitive inhibition toward cysteine with Kii (dissociation constant for the binary E·I complex) and Kis (dissociation constant for the E·S·I ternary complex) values of 290 nM and 2.4 μM, respectively, that is consistent with the kinetic mechanism presented in Fig. (32). While Cys-AMS 167 serves as useful tool compound for mechanistic analysis, this compound is not considered attractive since it inhibits the corresponding mammalian cysteinyl-tRNA synthetase. Furthermore, the presence of the free thiol renders Cys-AMS 167 metabolically unstable and it’s zwitterionic nature renders it highly polar, thus limiting membrane permeability and antibacterial activity.
In a complementary approach to identify inhibitors of MshC, Fahey, Newton and co-workers developed a high-throughput screening (HTS) spectrophotometric coupled assay detecting the released pyrophosphate using pyrophosphatase and malachite green [155, 156]. The authors screened a focused library of 2024 compounds that target ATP binding enzymes and after counterscreening, dibenzothiazepinone 168 Fig. (34) was the only compound identified that exhibited a well-behaved dose response curve with an IC50 of 100 μM and an MIC of 16 μM against M. smegmatis. Additionally, 168 inhibited mycothiol production and led to an accumulation of the MshC substrate GlcN-Ins. Over 500 additional analogues were evaluated, yet none were more potent than the initial hit compound. Bewley, Boshoff and co-workers at the NIH identified dequalinium chloride 169 Fig. (34) from 3100 compounds using a luceriferase reporter HTS assay that detects the consumption of ATP [157]. Dequalinium chloride 169 was shown to be an ATP-competitive inhibitor of MshC with a KD of 0.22 μM and inhibited M. tuberculosis under both aerobic and anaerobic growth conditions with MIC values of 2.3 and 0.6 μM, respectively. Despite the small library size employed for both of the aforementioned screens, useful tool compounds were identified, which suggests a larger screening effort is warranted.
BirA
M. tuberculosis produces a wide variety of complex fatty acids that contribute to the pathogen’s virulence and viability. Therefore, it is no surprise that the enzymes found within the biosynthetic pathways of these lipids represent promising and validated targets for antitubercular drug discovery [158]. Fatty acids are assembled in a modular fashion from simple malonyl-CoA building blocks and the enzymes responsible for synthesizing these malonyl-CoA monomers are the acyl coenzyme A carboxylases (ACCs) [77, 159]. ACCs are only functionally active after a biotin group has been posttranslationally installed onto the biotin carboxylase carrier protein (BCCP) domain of the ACC. A single adenylating enzyme in M. tuberculosis, biotin protein ligase encoded by birA, is responsible for the biotinylation, and thus activation, of all ACCs [160]. Since BirA globally regulates lipid metabolism via a single posttranslational event, this adenylating enzyme represents a promising target for antitubercular development.
Similar to the previous adenylating enzymes described in this review, BirA catalyzes the installment of biotin onto ACCs in a two-step reaction. During the first-half reaction, BirA binds biotin (170, Fig. (35)) and ATP and catalyzes the formation of a biotinyl adenylate (171, Bio-AMP). In the second-half reaction, BirA transfers the biotin group onto a conserved lysine residue within ACC 172 to form holo, functional ACC 173. The apparent KM values for biotin, ATP and BCCP with BirA were found to be 0.42, 21.1, and 5.2 μM, respectively, while the average kcat value was 0.031 s−1 [160]. Recently, the crystal structure of BirA from M. tuberculosis was determined and deposited into the Protein Data Bank (PDB code 3L2Z).
Fig. 35.

Adenylation-ligation reaction catalyzed by BirA.
Beckett and coworkers described acylsulfamate inhibitor 174 Fig. (36) in which the labile acyl-phosphate linkage of Bio-AMP was replaced with a sulfamate linkage [161,162]. They found that bisubstrate inhibitor 174 bound 1.4-fold more weakly than biotin to EcBirA from E. coli. This finding was surprising since, as described with several other adenylating enzymes in this review, replacement of the labile phosphate linkage of the adenylate intermediate with the more stabile sulfamate linkage provided potent bisubstrate inhibitors. Sulfamate 174 was independently prepared and found to rapidly decomposes via cyclonucleoside formation to yield N-biotinylsulfamic acid 176 and 3,5′-cyclo-5′-deoxyadenosine 177 [163]. Replacement of the 5′-oxygen atom in sulfamate 174 with a nitrogen atom afforded sulfamide 175 (Bio-AMS) that was chemically stable. Bio-AMS 175 was evaluated against BirA from M. tuberculosis using isothermal titration calorimetry (ITC) and the dissociation constant (KD) was 530 pM as compared to a KD of only 0.94 μM for biotin [160]. The binding is solely enthalpically driven (ΔH = −15.4 kcal/mol), which agrees well with the numerous hydrogen bond and electrostatic interactions observed in the co-crystal structure Fig. (37). Bio-AMS 175 was active against M. tuberculosis H37Rv as well as several multidrug- (MDR) and extensively drug-resistant (XDR) TB strains. The minimum inhibitor concentration that inhibited 99% of cell growth (MIC99) for all strains ranged from 0.16–0.625 μM. Furthermore, Bio-AMS 175 was found to be selective towards TB as the compound was inactive against a panel of Gram-negative bacteria (A. baumanii, E. coli, K. pneumoniae, P. aeruginosa), Gram-positive bacteria (E. faecalis and S. aureus) two fungal strains (C. neoformans, C. albicans) and two mammalian cell lines. Taken together, these data suggest that BirA represents a promising target for further antitubercular therapeutic development.
Fig. 36.

Bisubstrate inhibitors of BirA.
Fig. 37.

Bio-AMS co-crystallized with BPL from M. tuberculosis (PDB 3RUX, to be released). (A) The N-terminal chain of MtBPL with Bio-AMS bound. Enzyme residues are shown with green carbon atoms, blue nitrogen atoms, and red oxygen atoms. Ligand is shown with cyan carbon atoms, blue nitrogen atoms, red oxygen atoms, and yellow sulfur atoms. (B) Stabilization of the AMS moiety supported by backbone hydrogen bonding of Arg69, a unique interaction of the 2′-OH with Asp167, and hydrogen bonding with Lys138; thought to be the critical residue for adenylation. The biotin moiety shows extensive hydrogen bonding with the backbone of Arg67, and the side chains for Ser38, Thr39, and Gln63. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper).
CONCLUSION
Adenylation or adenylate-forming enzymes (AEs) in M. tuberculosis are involved in a myriad of biological processes and many are essential for survival or virulence. The abundance of structural, biochemical, and genetic data on the mycobacterial adenylating enzymes provides a solid foundation to develop new rationally designed antitubercular agents. Several strategies exist to identify AE inhibitors including high-throughput screening, fragment-based screening, and synthesis of bisubstrate inhibitors that mimic the intermediate acyladenylate. All of these strategies have been successfully employed as detailed in this short review with varying levels of success. Fragment-based screening is gaining popularity as a means to rationally design inhibitors; however, only a single example of this approach has been reported for a mycobacterial enzyme. Clearly, further application of this powerful method is warranted. The preparation of bisubstrate inhibitors of the acyladenylate wherein the adenylate phosphate moiety is replaced with a sulfamate represents the most straightforward method to prepare potent inhibitors of AEs. In some cases, the acylsulfamate inhibitors are not stable and decompose via lactonization, spirolactonization, or cyclonucleoside formation. The bisubstrate inhibitors developed for MbtA serve to highlight the power of this method and illustrate how one can systematically and methodically modify the inhibitor scaffold to modulate the antimycobacterial activity and physicochemical properties. Interest in target-based approaches for antitubercular drug discovery is waning with most new efforts emphasizing whole-cell phenotypic assays. Nevertheless, we believe many of the AEs represent excellent drug targets, given their confirmed essentiality and vulnerability, coupled with the ease of preparing potent and selective inhibitors by the strategies outlined in this review.
Acknowledgments
This was supported in part by NIH grant AI-070219.
ABBREVIATIONS
- ACC
Acyl-CoA carboxylases
- AE
Adenylating or adenylate-forming enzymes
- BPL
Biotin protein ligase
- BCCP
Biotin carboxylase carrier protein
- CP
Carrier protein
- DHB
2,3-dihydroxybenzoic acid
- DIM
Dimycocerosate esters
- FAAL
Fatty acyl-AMP ligase
- FACL
Fatty acyl-CoA ligase
- FAS
Fatty acid synthase
- FBS
Fragment-based screening
- HTS
High-throughput screening
- ITC
Isothermal titration calorimetry
- LC-MS
High-performance liquid chromatography-mass spectrometry
- MDR
Multidrug resistant
- NMR
Nuclear magnetic resonance spectroscopy
- NRPS
Nonribosomal peptide synthetase
- PKS
Polyketide synthase
- PGL
Phenolic glycolipid
- SAL
Salicylic acid
- SAR
Structure-activity relationship
- SPR
Surface plasmon resonance
- TB
Tuberculosis
- XDR
Extensively drug resistant
Footnotes
CONFLICT OF INTEREST
None declared.
References
- 1.World Health Organization. Global tuberculosis control. 2010 http://www.who.int/tb/publications/global_report/2010/en/index.html.
- 2.Barry CE, 3rd, Blanchard JS. The chemical biology of new drugs in the development for tuberculosis. Curr Opin Chem Biol. 2010;14:456–466. doi: 10.1016/j.cbpa.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. The challenge of new drug discovery for tuberculosis. Nature. 2011;469:483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 4.Aldrich CC, Boshoff HI, Remmel RP. Antitubercular Agents. In: Rotella D, Abraham DJ, editors. Burgers Medicinal Chemistry, Drug Discovery, and Development. 7. Vol. 7. Wiley; Hoboken, N. J: 2010. pp. 713–812. [Google Scholar]
- 5.Jacobson KR, Tierney DB, Jeon CY, Mitnick CD, Murray MB. Treatment outcomes among patients with extensively drug-resistant tuberculosis: systematic review and meta-analysis. Clin Infect Dis. 2010;51:6–14. doi: 10.1086/653115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 7.Galagan JE, Sisk P, Stolte C, Weiner B, Koehrsen M, Wymore F, Reddy TB, Zucker JD, Engels R, Gellesch M, Hubble J, Jin H, Larson L, Mao M, Nitzberg M, White J, Zachariah ZK, Sherlock G, Ball CA, Schoolnik GK. TB database 2010: overview and update. Tuberculosis (Edinb) 2010;90:225–235. doi: 10.1016/j.tube.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 8.Chim N, Habel JE, Johnston JM, Krieger I, Miallau L, Sankaranarayanan R, Morse RP, Bruning J, Swanson S, Kim H, Kim CY, Li H, Bulloch EM, Payne RJ, Manos-Turvey A, Hung LW, Baker EN, Lott JS, James MN, Terwilliger TC, Eisenberg DS, Sacchettini JC, Goulding CW. The TB structural genomics consortium: a decade of progress. Tuberculosis (Edinb) 2011;91:155–172. doi: 10.1016/j.tube.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList-10 years after. Tuberculosis (Edinb) 2011;91:1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Gulick AM. Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmelz S, Naismith JH. Adenylate-forming enzymes. Curr Opin Struct Biol. 2009;19:666–671. doi: 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill JM, Yu G, Shue Y-K, Zydowsky TM, Rebek J. Aminoacyl adenylate mimics as novel antimicrobial and antiparasitic agents. 5,726,195, US Patent. 1998 Mar 10;
- 13.Schimmel P, Tao J, Hill J. Aminoacyl tRNA synthetases as targets for new anti-infectives. FASEB J. 1998;12:1599–1609. [PubMed] [Google Scholar]
- 14.Kim S, Lee SW, Choi EC, Choi SY. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl Microbiol Biotechnol. 2003;61:278–288. doi: 10.1007/s00253-003-1243-5. [DOI] [PubMed] [Google Scholar]
- 15.Jencks WP. On the attribution and additivity of binding energies. Proc Natl Acad Sci USA. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pope AJ, Lapointe J, Mensah L, Benson N, Brown MJ, Moore KJ. Characterization of isoleucyl-tRNA synthetase from Staphylococcus aureus. I: Kinetic mechanism of the substrate activation reaction studied by transient and steady-state techniques. J Biol Chem. 1998;273:31680–31690. doi: 10.1074/jbc.273.48.31680. [DOI] [PubMed] [Google Scholar]
- 17.Southgate CC, Dixon HB. Phosphonate analogues of aminoacyl adenylates. Biochem J. 1978;175:461–465. doi: 10.1042/bj1750461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueda HSY, Hayashi N, Mitsunaga JY, Doi M, Inoue M, Ishida M. X-ray crystallographic conformational study of 5′-O-[N-(L-alanyl)-sulfamoyl]adenosine: A substrate analogue for alanyl-tRNA synthetase. Biochim Biophys Acta. 1991;1080:126–134. doi: 10.1016/0167-4838(91)90138-p. [DOI] [PubMed] [Google Scholar]
- 19.Brown MJ, Mensah LM, Doyle ML, Broom NJ, Osbourne N, Forrest AK, Richardson CM, O’Hanlon PJ, Pope AJ. Rational design of femtomolar inhibitors of isoleucyl tRNA synthetase from a binding model for pseudomonic acid-A. Biochemistry. 2000;39:6003–6011. doi: 10.1021/bi000148v. [DOI] [PubMed] [Google Scholar]
- 20.Isono K, Uramoto M, Kusakabe H, Miyata N, Koyama H, Ubukata M, Sethi SK, McCloskey JA. Ascamycin and dealanylascamycin, nucleoside antibiotics from Streptomyces sp. J Antibiot. 1984;37:670–672. doi: 10.7164/antibiotics.37.670. [DOI] [PubMed] [Google Scholar]
- 21.Copeland RA. Mechanism, and Data Analysis. 2. Wiley-VCH; New York: 2000. Enzymes, A Practical Introduction to Structure. [Google Scholar]
- 22.Moshinsky DJ, Ruslim L, Blake RA, Tang F. A widely applicable, high-throughput TR-FRET assay for the measurement of kinase autophosphorylation: VEGFR-2 as a prototype. J Biomol Screen. 2003;8:447–452. doi: 10.1177/1087057103255282. [DOI] [PubMed] [Google Scholar]
- 23.Bays NW, Hill AD, Kariv I. A simplified scintillation proximity assay for fatty acid synthase activity: development and comparison with other FAS activity assays. J Biomol Screen. 2009;14:636–642. doi: 10.1177/1087057109335746. [DOI] [PubMed] [Google Scholar]
- 24.Vasan M, Neres J, Williams J, Wilson DJ, Teitelbaum AM, Remmel RP, Aldrich CC. Inhibitors of the salicylate synthase (MbtI) from Mycobacterium tuberculosis discovered by high-throughput screening. Chem Med Chem. 2010;5:2079–2087. doi: 10.1002/cmdc.201000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaudet EA, Huang KS, Zhang Y, Huang W, Mark D, Sportsman JR. A homogeneous fluorescence polarization assay adaptable for a range of protein serine/threonine and tyrosine kinases. J Biom Screen. 2003;8:164–175. doi: 10.1177/1087057103252309. [DOI] [PubMed] [Google Scholar]
- 26.Zhang JH, Chung TDY, Oldenbug KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 27.Ciulli A, Abell C. Fragment-based approaches to enzyme inhibition. Curr Opin Biotechnol. 2007;18:489–496. doi: 10.1016/j.copbio.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray CW, Verdonk ML. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J Comput Aided Mol Des. 2002;16:741–753. doi: 10.1023/a:1022446720849. [DOI] [PubMed] [Google Scholar]
- 29.Erlanson DA. Fragment-based lead discovery: a chemical update. Curr Opin Biotechnol. 2006;17:643–652. doi: 10.1016/j.copbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 31.Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 32.Neumann T, Junker HD, Schmidt K, Sekul R. SPR-based fragment screening: advantages and applications. Curr Top Med Chem. 2007;7:1630–1642. doi: 10.2174/156802607782341073. [DOI] [PubMed] [Google Scholar]
- 33.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 34.Blundell TL, Jhoti H, Abell C. High-throughput crystallography for lead discovery in drug design. Nat Rev Drug Discov. 2002;1:45–54. doi: 10.1038/nrd706. [DOI] [PubMed] [Google Scholar]
- 35.Mayer M, Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed. 1999;38:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 36.Hung AW, Silvestre HL, Wen S, Ciulli A, Blundell TL, Abell C. Application of fragment growing and fragment linking to the discovery of inhibitors of Mycobacterium tuberculosis pantothenate synthetase. Angew Chem Int Ed. 2009;48:8452–8456. doi: 10.1002/anie.200903821. [DOI] [PubMed] [Google Scholar]
- 37.Carr R, Jhoti H. Structure-based screening of low-affinity compounds. Drug Discov Today. 2002;7:522–527. doi: 10.1016/s1359-6446(02)02245-6. [DOI] [PubMed] [Google Scholar]
- 38.Hurdle JG, O’Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob Agents Chemother. 2005;49:4821–4833. doi: 10.1128/AAC.49.12.4821-4833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chain EB, Mellows G. Pseudomonic acid. Part 1. The structure of pseudomonic acid A, a novel antibiotic produced by Pseudomonas fluorescens. J Chem Soc, Perkin Trans. 1977;1:294–309. [PubMed] [Google Scholar]
- 40.Sassanfar M, Kranz JE, Gallant P, Schimmel P, Shiba K. A eubacterial Mycobacterium tuberculosis tRNA synthetase is eukaryote-like and resistant to a eubacterial-specific antisynthetase drug. Biochemistry. 1996;35:9995–10003. doi: 10.1021/bi9603027. [DOI] [PubMed] [Google Scholar]
- 41.Heacock D, Forsyth CJ, Shiba K, Musier-Forsyth K. Synthesis and aminoacyl-tRNA synthetase inhibitory activity of prolyl adenylate analogs. Bioorg Chem. 1996;24:273–289. [Google Scholar]
- 42.Lee J, Kang SU, Kang MK, Chun MW, Jo YJ, Kwak JH, Kim S. Methionyl adenylate analogues as inhibitors of methionyl-tRNA synthetase. Bioorg Med Chem Lett. 1999;9:1365–1370. doi: 10.1016/s0960-894x(99)00206-1. [DOI] [PubMed] [Google Scholar]
- 43.Ingvarsson H, Jones TA, Unge T. Crystallization of Mycobacterium smegmatis methionyl-tRNA synthetase in the presence of methionine and adenosine. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009;65:618–620. doi: 10.1107/S1744309109016704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Critchley IA, Ochsner UA. Recent advances in the preclinical evaluation of the topical antibacterial agent REP8839. Curr Opin Chem Biol. 2008;12:409–417. doi: 10.1016/j.cbpa.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 45.Dhiman RK, Mahapatra S, Slayden RA, Boyne ME, Lenaerts A, Hinshaw JC, Angala SK, Chatterjee D, Biswas K, Narayanasamy P, Kurosu M, Crick DC. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol Microbiol. 2009;72:85–97. doi: 10.1111/j.1365-2958.2009.06625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao SP, Alonso S, Rand L, Dick T, Pethe K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2008;105:11945–11950. doi: 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 48.Meganathan R. Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q): a perspective on enzymatic mechanisms. Vitam Horm. 2001;61:173–218. doi: 10.1016/s0083-6729(01)61006-9. [DOI] [PubMed] [Google Scholar]
- 49.Collins MD, Pirouz T, Goodfellow M, Minnikin DE. Distribution of menaquinones in actinomycetes and corynebacteria. J Gen Microbiol. 1977;100:221–230. doi: 10.1099/00221287-100-2-221. [DOI] [PubMed] [Google Scholar]
- 50.Holsclaw CM, Sogi KM, Gilmore SA, Schelle MW, Leavell MD, Bertozzi CR, Leary JA. Structural characterization of a novel sulfated menaquinone produced by stf3 from Mycobacterium tuberculosis. ACS Chem Biol. 2008;3:619–624. doi: 10.1021/cb800145r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tian Y, Suk DH, Cai F, Crich D, Mesecar AD. Bacillus anthracis o-succinylbenzoyl-CoA synthetase: reaction kinetics and a novel inhibitor mimicking its reaction intermediate. Biochemistry. 2008;47:12434–12447. doi: 10.1021/bi801311d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reger AS, Wu R, Dunaway-Mariano D, Gulick AM. Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry. 2008;47:8016–8025. doi: 10.1021/bi800696y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu R, Cao J, Lu X, Reger AS, Gulick AM, Dunaway-Mariano D. Mechanism of 4-chlorobenzoate:coenzyme a ligase catalysis. Biochemistry. 2008;47:8026–8039. doi: 10.1021/bi800698m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwon O, Bhattacharyya DK, Meganathan R. Menaquinone (vitamin K2) biosynthesis: overexpression, purification, and properties of o-succinylbenzoyl-coenzyme A synthetase from Escherichia coli. J Bacteriol. 1996;178:6778–6781. doi: 10.1128/jb.178.23.6778-6781.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meganathan R, Bentley R. Menaquinone (vitamin K2) biosynthesis: conversion of o-succinylbenzoic acid to 1,4-dihydroxy-2-naphthoic acid by Mycobacterium phlei enzymes. J Bacteriol. 1979;140:92–98. doi: 10.1128/jb.140.1.92-98.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gokhale RS, Saxena P, Chopra T, Mohanty D. Versatile polyketide enzymatic machinery for the biosynthesis of complex mycobacterial lipids. Nat Prod Rep. 2007;24:267–277. doi: 10.1039/b616817p. [DOI] [PubMed] [Google Scholar]
- 57.Onwueme KC, Vos CJ, Zurita J, Ferreras JA, Quadri LE. The dimycocerosate ester polyketide virulence factors of mycobacteria. Prog Lipid Res. 2005;44:259–302. doi: 10.1016/j.plipres.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, Kaplan G, Barry CE., 3rd A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–87. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 60.McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr, Russell DG. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 61.Timm J, Post FA, Bekker LG, Walther GB, Wainwright HC, Manganelli R, Chan WT, Tsenova L, Gold B, Smith I, Kaplan G, McKinney JD. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc Natl Acad Sci USA. 2003;100:14321–14326. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trivedi OA, Arora P, Sridharan V, Tickoo R, Mohanty D, Gokhale RS. Enzymatic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature. 2004;428:441–445. doi: 10.1038/nature02384. [DOI] [PubMed] [Google Scholar]
- 64.Arora P, Vats A, Saxena P, Mohanty D, Gokhale RS. Promiscuous fatty acyl CoA ligases produce acyl-CoA and acyl-SNAC precursors for polyketide biosynthesis. J Am Chem Soc. 2005;127:9388–9389. doi: 10.1021/ja052991s. [DOI] [PubMed] [Google Scholar]
- 65.Dam T, Danelishvili L, Wu M, Bermudez LE. The fadD2 gene is required for efficient Mycobacterium avium invasion of mucosal epithelial cells. J Infect Dis. 2006;193:1135–1142. doi: 10.1086/501469. [DOI] [PubMed] [Google Scholar]
- 66.Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci USA. 2007;104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilbrink MH, Petrusma M, Dijkhuizen L, van der Geize R. FadD19 of Rhodococcus rhodochrous DSM43269, a steroid-coenzyme A ligase essential for degradation of C-24 branched sterol side chains. Appl Environ Microbiol. 2011;77:4455–4464. doi: 10.1128/AEM.00380-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khare G, Gupta V, Gupta RK, Gupta R, Bhat R, Tyagi AK. Dissecting the role of critical residues and substrate preference of a fatty acyl-CoA synthetase (FadD13) of Mycobacterium tuberculosis. PLoS One. 2009;4:e8387. doi: 10.1371/journal.pone.0008387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferreras JA, Stirrett KL, Lu X, Ryu JS, Soll CE, Tan DS, Quadri LE. Mycobacterial phenolic glycolipid virulence factor biosynthesis: mechanism and small-molecule inhibition of polyketide chain initiation. Chem Biol. 2008;15:51–61. doi: 10.1016/j.chembiol.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He W, Soll CE, Chavadi SS, Zhang G, Warren JD, Quadri LE. Cooperation between a coenzyme A-independent stand-alone initiation module and an iterative type I polyketide synthase during synthesis of mycobacterial phenolic glycolipids. J Am Chem Soc. 2009;131:16744–16750. doi: 10.1021/ja904792q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Camacho LR, Constant P, Raynaud C, Laneelle MA, Triccas JA, Gicquel B, Daffe M, Guilhot C. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J Biol Chem. 2001;276:19845–19854. doi: 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- 72.Infante E, Aguilar LD, Gicquel B, Pando RH. Immunogenicity and protective efficacy of the Mycobacterium tuberculosis fadD26 mutant. Clin Exp Immunol. 2005;141:21–28. doi: 10.1111/j.1365-2249.2005.02832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sirakova TD, Fitzmaurice AM, Kolattukudy P. Regulation of expression of mas and fadD28, two genes involved in production of dimycocerosyl phthiocerol, a virulence factor of Mycobacterium tuberculosis. J Bacteriol. 2002;184:6796–6802. doi: 10.1128/JB.184.24.6796-6802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trivedi OA, Arora P, Vats A, Ansari MZ, Tickoo R, Sridharan V, Mohanty D, Gokhale RS. Dissecting the mechanism and assembly of a complex virulence mycobacterial lipid. Mol Cell. 2005;17:631–643. doi: 10.1016/j.molcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 75.Leger M, Gavalda S, Guillet V, van der Rest B, Slama N, Montrozier H, Mourey L, Quemard A, Daffe M, Marrakchi H. The dual function of the Mycobacterium tuberculosis FadD32 required for mycolic acid biosynthesis. Chem Biol. 2009;16:510–519. doi: 10.1016/j.chembiol.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 76.Gavalda S, Leger M, van der Rest B, Stella A, Bardou F, Montrozier H, Chalut C, Burlet-Schiltz O, Marrakchi H, Daffe M, Quemard A. The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J Biol Chem. 2009;284:19255–19264. doi: 10.1074/jbc.M109.006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Portevin D, de Sousa-D’Auria C, Montrozier H, Houssin C, Stella A, Laneelle MA, Bardou F, Guilhot C, Daffe M. The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth: identification of the carboxylation product and determination of the acyl-CoA carboxylase components. J Biol Chem. 2005;280:8862–8874. doi: 10.1074/jbc.M408578200. [DOI] [PubMed] [Google Scholar]
- 78.Carroll P, Faray-Kele MC, Parish T. Identifying vulnerable pathways in Mycobacterium tuberculosis by using a knockdown approach. Appl Environ Microbiol. 2011;77:5040–5043. doi: 10.1128/AEM.02880-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krithika R, Marathe U, Saxena P, Ansari MZ, Mohanty D, Gokhale RS. A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:2069–2074. doi: 10.1073/pnas.0507924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rindi L, Fattorini L, Bonanni D, Iona E, Freer G, Tan D, Deho G, Orefici G, Garzelli C. Involvement of the fadD33 gene in the growth of Mycobacterium tuberculosis in the liver of BALB/c mice. Microbiology. 2002;148:3873–3880. doi: 10.1099/00221287-148-12-3873. [DOI] [PubMed] [Google Scholar]
- 81.Rindi L, Bonanni D, Lari N, Garzelli C. Requirement of gene fadD33 for the growth of Mycobacterium tuberculosis in a hepatocyte cell line. New Microbiol. 2004;27:125–131. [PubMed] [Google Scholar]
- 82.Buchholz TJ, Geders TW, Bartley FE, 3rd, Reynolds KA, Smith JL, Sherman DH. Structural basis for binding specificity between subclasses of modular polyketide synthase docking domains. ACS Chem Biol. 2009;4:41–52. doi: 10.1021/cb8002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arora P, Goyal A, Natarajan VT, Rajakumara E, Verma P, Gupta R, Yousuf M, Trivedi OA, Mohanty D, Tyagi A, Sankaranarayanan R, Gokhale RS. Mechanistic and functional insights into fatty acid activation in Mycobacterium tuberculosis. Nat Chem Biol. 2009;5:166–173. doi: 10.1038/nchembio.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grimes KD, Aldrich CC. A high-throughput screening fluorescence polarization assay for fatty acid adenylating enzymes in Mycobacterium tuberculosis. Anal Biochem. 2011;417:264–273. doi: 10.1016/j.ab.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 86.Quadri LE. Assembly of aryl-capped siderophores by modular peptide synthetases and polyketide synthases. Mol Microbiol. 2000;37:1–12. doi: 10.1046/j.1365-2958.2000.01941.x. [DOI] [PubMed] [Google Scholar]
- 87.Gobin J, Moore CH, Reeve JR, Jr, Wong DK, Gibson BW, Horwitz MA. Iron acquisition by Mycobacterium tuberculosis: isolation characterization of a family of iron-binding exochelins. Proc Natl Acad Sci USA. 1995;92:5189–5193. doi: 10.1073/pnas.92.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Snow GA. Isolation and structure of mycobactin-T, a growth factor of Mycobacterium tuberculosis. Biochem J. 1965;97:166–175. doi: 10.1042/bj0970166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Snow GA. Mycobactins: iron-chelating growth factors from mycobacteria. Bacteriol Rev. 1970;34:99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vergne AF, Walz AJ, Miller MJ. Iron chelators from mycobacteria (1954–1999) and potential therapeutic applications. Nat Prod Rep. 2000;17:99–116. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]
- 91.Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 92.Chavadi SS, Stirrett KL, Edupuganti UR, Vergnolle O, Sadhanandan G, Marchiano E, Martin C, Qiu WG, Soll CE, Quadri LE. Mutational and phylogenetic analyses of the mycobacterial mbt gene cluster. J Bacteriol. 2011;193:5905–5913. doi: 10.1128/JB.05811-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moody DB, Young DC, Cheng TY, Rosat JP, Roura-Mir C, O’Connor PB, Zajonc DM, Walz A, Miller MJ, Levery SB, Wilson IA, Costello CE, Brenner MB. T cell activation by lipopeptide antigens. Science. 2004;303:527–531. doi: 10.1126/science.1089353. [DOI] [PubMed] [Google Scholar]
- 94.De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., 3rd The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc Natl Acad Sci USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luo M, Fadeev EA, Groves JT. Mycobactin-mediated iron acquisition within macrophages. Nat Chem Biol. 2005;1:149–153. doi: 10.1038/nchembio717. [DOI] [PubMed] [Google Scholar]
- 96.Murray MJ, Murray AB, Murray MB, Murray CJ. The adverse effect of iron repletion on the course of certain infections. Br Med J. 1978;2:1113–1115. doi: 10.1136/bmj.2.6145.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manabe YC, Saviola BJ, Sun L, Murphy JR, Bishai WR. Attenuation of virulence in Mycobacterium tuberculosis expressing a constitutively active iron repressor. Proc Natl Acad Sci USA. 1999;96:12844–12848. doi: 10.1073/pnas.96.22.12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sikora AL, Wilson DJ, Aldrich CC, Blanchard JS. Kinetic and inhibition studies of dihydroxybenzoate-AMP ligase from Escherichia coli. Biochemistry. 2010;49:3648–3657. doi: 10.1021/bi100350c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.May JJ, Kessler N, Marahiel MA, Stubbs MT. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc Natl Acad Sci USA. 2002;99:12120–12125. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Drake EJ, Duckworth BP, Neres J, Aldrich CC, Gulick AM. Biochemical and structural characterization of bisubstrate inhibitors of BasE, the self-standing nonribosomal peptide synthetase adenylate-forming enzyme of acinetobactin synthesis. Biochemistry. 2010;49:9292–9305. doi: 10.1021/bi101226n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- 102.Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J Med Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 103.Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 2006;273:409–419. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]
- 104.Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, 3rd, Bennett EM, Aldrich CC. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J Med Chem. 2008;51:5349–5370. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qiao CH, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, 3rd, Aldrich CC. 5′-O-[(N-Acyl) sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: An adenylation enzyme required for siderophore biosynthesis of the mycobactins. J Med Chem. 2007;50:6080–6094. doi: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gupte A, Boshoff HI, Wilson DJ, Neres J, Labello NP, Somu RV, Xing C, Barry CE, 3rd, Aldrich CC. Inhibition of siderophore biosynthesis by 2-triazole substituted analogues of 5′-O-[N-(salicyl)sulfamoyl]adenosine: antibacterial nucleosides effective against Mycobacterium tuberculosis. J Med Chem. 2008;51:7495–7507. doi: 10.1021/jm8008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vannada J, Bennett EM, Wilson DJ, Boshoff HI, Barry CE, 3rd, Aldrich CC. Design, synthesis, and biological evaluation of b-ketosulfonamide adenylation inhibitors as potential antitubercular agents. Org Lett. 2006;8:4707–4710. doi: 10.1021/ol0617289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qiao CH, Wilson DJ, Bennett EM, Aldrich CC. A mechanism-based aryl carrier protein/thiolation domain affinity probe. J Am Chem Soc. 2007;129:6350–6351. doi: 10.1021/ja069201e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Labello NP, Bennett EM, Ferguson DM, Aldrich CC. Quantitative three dimensional structure linear interaction energy model of 5′-O-[N-(salicyl)sulfamoyl]adenosine and the aryl acid adenylating enzyme MbtA. J Med Chem. 2008;51:7154–7160. doi: 10.1021/jm800668u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, 3rd, Aldrich CC. Antitubercular nucleosides that inhibit siderophore biosynthesis: SAR of the glycosyl domain. J Med Chem. 2006;49:7623–7635. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Braibant M, Gilot P, Content J. The ATP-binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol Rev. 2000;24:449–467. doi: 10.1111/j.1574-6976.2000.tb00550.x. [DOI] [PubMed] [Google Scholar]
- 112.Niederweis M. Mycobacterial porins - new channel proteins in unique outer membranes. Mol Microbiol. 2003;49:1167–1177. doi: 10.1046/j.1365-2958.2003.03662.x. [DOI] [PubMed] [Google Scholar]
- 113.Neres J, Wilson DJ, Celia L, Beck BJ, Aldrich CC. Aryl acid adenylating enzymes involved in siderophore biosynthesis: fluorescence polarization assay, ligand specificity, and discovery of non-nucleoside inhibitors via high-throughput screening. Biochemistry. 2008;47:11735–11749. doi: 10.1021/bi801625b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.unpublished results, João Neres, Courtney Aldrich.
- 115.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 116.Lin H. Nicotinamide adenine dinucleotide: beyond a redox coenzyme. Org Biomol Chem. 2007;5:2541–2554. doi: 10.1039/b706887e. [DOI] [PubMed] [Google Scholar]
- 117.Boshoff HI, Xu X, Tahlan K, Dowd CS, Pethe K, Camacho LR, Park TH, Yun CS, Schnappinger D, Ehrt S, Williams KJ, Barry CE., 3rd Biosynthesis recycling of nicotinamide cofactors in Mycobacterium tuberculosis. An essential role for NAD in nonreplicating bacilli. J Biol Chem. 2008;283:19329–19341. doi: 10.1074/jbc.M800694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.LaRonde-LeBlanc N, Resto M, Gerratana B. Regulation of active site coupling in glutamine-dependent NAD(+) synthetase. Nat Struct Mol Biol. 2009;16:421–429. doi: 10.1038/nsmb.1567. [DOI] [PubMed] [Google Scholar]
- 119.Rizzi M, Bolognesi M, Coda A. A novel deamido-NAD+-binding site revealed by the trapped NAD-adenylate intermediate in the NAD+ synthetase structure. Structure. 1998;6:1129–1140. doi: 10.1016/s0969-2126(98)00114-2. [DOI] [PubMed] [Google Scholar]
- 120.Velu SE, Cristofoli WA, Garcia GJ, Brouillette CG, Pierson MC, Luan CH, DeLucas LJ, Brouillette WJ. Tethered dimers as NAD synthetase inhibitors with antibacterial activity. J Med Chem. 2003;46:3371–3381. doi: 10.1021/jm030003x. [DOI] [PubMed] [Google Scholar]
- 121.Velu SE, Luan CH, Delucas LJ, Brouillette CG, Brouillette WJ. Tethered dimer inhibitors of NAD synthetase: parallel synthesis of an aryl-substituted SAR library. J Comb Chem. 2005;7:898–904. doi: 10.1021/cc050063j. [DOI] [PubMed] [Google Scholar]
- 122.Moro WB, Yang Z, Kane TA, Brouillette CG, Brouillette WJ. Virtual screening to identify lead inhibitors for bacterial NAD synthetase (NADs) Bioorg Med Chem Lett. 2009;19:2001–2005. doi: 10.1016/j.bmcl.2009.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Moro WB, Yang Z, Kane TA, Zhou Q, Harville S, Brouillette CG, Brouillette WJ. SAR Studies for a New Class of Antibacterial NAD Biosynthesis Inhibitors. J Comb Chem. 2009;11(4):617–625. doi: 10.1021/cc9000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hyde JE. Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr Drug Targets. 2007;8:31–47. doi: 10.2174/138945007779315524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hatfull G, Jacobs WR., Jr . Molecular Genetics of Mycobacteria. American Society for Microbiology; Washington D. C: 2000. [Google Scholar]
- 126.Fukuyama TT. Formation of an adenyl xanthosine monophosphate intermediate by xanthosine 5′-phosphate aminase and its inhibition by psicofuranine. J Biol Chem. 1966;241:4745–4749. [PubMed] [Google Scholar]
- 127.Zyk N, Citri N, Moyed HS. Conformative response of xanthosine 5′-phosphate aminase. Biochemistry. 1969;8:2787–2794. doi: 10.1021/bi00835a015. [DOI] [PubMed] [Google Scholar]
- 128.Patel N, Moyed HS, Kane JF. Xanthosine-5′-phosphate amidotransferase from Escherichia coli. J Biol Chem. 1975;250:2609–2613. [PubMed] [Google Scholar]
- 129.von der Saal W, Crysler CS, Villafranca JJ. Positional isotope exchange and kinetic experiments with Escherichia coli guanosine-5′-monophosphate synthetase. Biochemistry. 1985;24:5343–5350. doi: 10.1021/bi00341a011. [DOI] [PubMed] [Google Scholar]
- 130.Sakamoto N, Hatfield GW, Moyed HS. Physical properties and subunit structure of xanthosine 5′-phosphate aminase. J Biol Chem. 1972;247:5880–5887. [PubMed] [Google Scholar]
- 131.Tesmer JJ, Klem TJ, Deras ML, Davisson VJ, Smith JL. The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nat Struct Biol. 1996;3:74–86. doi: 10.1038/nsb0196-74. [DOI] [PubMed] [Google Scholar]
- 132.Chittur SV, Klem TJ, Shafer CM, Davisson VJ. Mechanism for acivicin inactivation of triad glutamine amidotransferases. Biochemistry. 2001;40:876–887. doi: 10.1021/bi0014047. [DOI] [PubMed] [Google Scholar]
- 133.Deras ML, Chittur SV, Davisson VJ. N2-hydroxyguanosine 5′-monophosphate is a time-dependent inhibitor of Escherichia coli guanosine monophosphate synthetase. Biochemistry. 1999;38:303–310. doi: 10.1021/bi981980r. [DOI] [PubMed] [Google Scholar]
- 134.Moyed HS, Magasanik B. Enzymes essential for the biosynthesis of nucleic acid guanine; xanthosine 5′-phosphate aminase of Aerobacter aerogenes. J Biol Chem. 1957;226:351–363. [PubMed] [Google Scholar]
- 135.Fukuyama TT, Donovan KL. Mechanism of hydroxylamine inhibition of xanthosine 5′-phosphate aminase. J Biol Chem. 1968;243:5798–5801. [PubMed] [Google Scholar]
- 136.Webb ME, Smith AG, Abell C. Biosynthesis of pantothenate. Nat Prod Rep. 2004;21:695–721. doi: 10.1039/b316419p. [DOI] [PubMed] [Google Scholar]
- 137.Sambandamurthy VK, Wang X, Chen B, Russell RG, Derrick S, Collins FM, Morris SL, Jacobs WR., Jr A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat Med. 2002;8:1171–1174. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- 138.Sambandamurthy VK, Derrick SC, Jalapathy KV, Chen B, Russell RG, Morris SL, Jacobs WR., Jr Long-term protection against tuberculosis following vaccination with a severely attenuated double lysine and pantothenate auxotroph of Mycobacterium tuberculosis. Infect Immun. 2005;73:1196–1203. doi: 10.1128/IAI.73.2.1196-1203.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zheng R, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry. 2001;40:12904–12912. doi: 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- 140.Zheng R, Dam TK, Brewer CF, Blanchard JS. Active site residues in Mycobacterium tuberculosis pantothenate synthetase required in the formation and stabilization of the adenylate intermediate. Biochemistry. 2004;43:7171–7178. doi: 10.1021/bi049676n. [DOI] [PubMed] [Google Scholar]
- 141.Wang S, Eisenberg D. Crystal structures of a pantothenate synthetase from M. tuberculosis and its complexes with substrates and a reaction intermediate. Protein Sci. 2003;12:1097–1108. doi: 10.1110/ps.0241803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang S, Eisenberg D. Crystal structure of the pantothenate synthetase from Mycobacterium tuberculosis, snapshots of the enzyme in action. Biochemistry. 2006;45:1554–1561. doi: 10.1021/bi051873e. [DOI] [PubMed] [Google Scholar]
- 143.White EL, Southworth K, Ross L, Cooley S, Gill RB, Sosa MI, Manouvakhova A, Rasmussen L, Goulding C, Eisenberg D, Fletcher TM., 3rd A novel inhibitor of Mycobacterium tuberculosis pantothenate synthetase. J Biomol Screen. 2007;12:100–105. doi: 10.1177/1087057106296484. [DOI] [PubMed] [Google Scholar]
- 144.Mycobacterium tuberculosis Pantothenate Synthetase Assay. NIH BioAssay. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=375.
- 145.Velaparthi S, Brunsteiner M, Uddin R, Wan B, Franzblau SG, Petukhov PA. 5-tert-butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide derivatives as novel potent inhibitors of Mycobacterium tuberculosis pantothenate synthetase: initiating a quest for new antitubercular drugs. J Med Chem. 2008;51:1999–2002. doi: 10.1021/jm701372r. [DOI] [PubMed] [Google Scholar]
- 146.Ciulli A, Scott DE, Ando M, Reyes F, Saldanha SA, Tuck KL, Chirgadze DY, Blundell TL, Abell C. Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. Chembiochem. 2008;9:2606–2611. doi: 10.1002/cbic.200800437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tuck KL, Saldanha SA, Birch LM, Smith AG, Abell C. The design and synthesis of inhibitors of pantothenate synthetase. Org Biomol Chem. 2006;4:3598–3610. doi: 10.1039/b609482a. [DOI] [PubMed] [Google Scholar]
- 148.Newton GL, Buchmeier N, Fahey RC. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol Mol Biol Rev. 2008;72:471–494. doi: 10.1128/MMBR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fan F, Vetting MW, Frantom PA, Blanchard JS. Structures and mechanisms of the mycothiol biosynthetic enzymes. Curr Opin Chem Biol. 2009;13:451–459. doi: 10.1016/j.cbpa.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sareen D, Newton GL, Fahey RC, Buchmeier NA. Mycothiol is essential for growth of Mycobacterium tuberculosis Erdman. J Bacteriol. 2003;185:6736–6740. doi: 10.1128/JB.185.22.6736-6740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vilcheze C, Av-Gay Y, Attarian R, Liu Z, Hazbon MH, Colangeli R, Chen B, Liu W, Alland D, Sacchettini JC, Jacobs WR., Jr Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol. 2008;69:1316–1329. doi: 10.1111/j.1365-2958.2008.06365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fan F, Luxenburger A, Painter GF, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium smegmatis cysteine ligase (MshC) Biochemistry. 2007;46:11421–11429. doi: 10.1021/bi7011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tremblay LW, Fan F, Vetting MW, Blanchard JS. The 1. 6 A crystal structure of Mycobacterium smegmatis MshC: the penultimate enzyme in the mycothiol biosynthetic pathway. Biochemistry. 2008;47:13326–13335. doi: 10.1021/bi801708f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gutierrez-Lugo MT, Bewley CA. Susceptibility and mode of binding of the Mycobacterium tuberculosis cysteinyl transferase mycothiol ligase to tRNA synthetase inhibitors. Bioorg Med Chem Lett. 2011;21:2480–2483. doi: 10.1016/j.bmcl.2011.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fahey RC, Newton GL, Ta PV, Buchmeier N. Inhibitors of MshC and homologs thereof, and methods of identifying same. 2008/036139. WO. 2008 Mar 27;
- 156.Newton GL, Buchmeier N, La Clair JJ, Fahey RC. Evaluation of NTF1836 as an inhibitor of the mycothiol biosynthetic enzyme MshC in growing and non-replicating Mycobacterium tuberculosis. Bioorg Med Chem. 2011;19:3956–3964. doi: 10.1016/j.bmc.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gutierrez-Lugo MT, Baker H, Shiloach J, Boshoff H, Bewley CA. Dequalinium, a new inhibitor of Mycobacterium tuberculosis mycothiol ligase identified by high-throughput screening. J Biomol Screen. 2009;14:643–652. doi: 10.1177/1087057109335743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Slayden RA, Lee RE, Barry CE., 3rd Isoniazid affects multiple components of the type II fatty acid synthase system of Mycobacterium tuberculosis. Mol Microbiol. 2000;38:514–525. doi: 10.1046/j.1365-2958.2000.02145.x. [DOI] [PubMed] [Google Scholar]
- 159.Gago G, Kurth D, Diacovich L, Tsai SC, Gramajo H. Biochemical and structural characterization of an essential acyl coenzyme A carboxylase from Mycobacterium tuberculosis. J Bacteriol. 2006;188:477–486. doi: 10.1128/JB.188.2.477-486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Purushothaman S, Gupta G, Srivastava R, Ramu VG, Surolia A. Ligand specificity of group I biotin protein ligase of Mycobacterium tuberculosis. PLoS One. 2008;3:e2320. doi: 10.1371/journal.pone.0002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Brown PH, Cronan JE, Grotli M, Beckett D. The biotin repressor: modulation of allostery by corepressor analogs. J Mol Biol. 2004;337:857–869. doi: 10.1016/j.jmb.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 162.Brown PH, Beckett D. Use of binding enthalpy to drive an allosteric transition. Biochemistry. 2005;44:3112–3121. doi: 10.1021/bi047792k. [DOI] [PubMed] [Google Scholar]
- 163.Duckworth BP, Geders TW, Tiwari D, Boshoff HI, Sibbald PA, Barry CE, 3rd, Schnappinger D, Finzel BC, Aldrich CC. Inhibition of biotin protein ligase from Mycobacterium tuberculosis. Chem Biol. 2011;18:1432–1441. doi: 10.1016/j.chembiol.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]