

Abstract
Widespread bacterial resistance to antibiotics is a significant public health concern. To remain a step ahead of evolving bacteria, new methods to study resistance to antibacterials and to uncover novel antibiotics that evade resistance are urgently needed. Herein, microarray-based methods that have been developed to study aminoglycoside modification by resistance-causing enzymes are reviewed. These arrays can also be used to study the binding of aminoglycoside antibiotics to a mimic of their therapeutic target, the rRNA aminoacyl site (A-site), and how modification by resistance-causing enzymes affects their abilities to bind RNA.
Keywords: Antibiotics, Bacteria, Aminoglycosides, RNA, Antibiotic resistance, Glycoarray
1 INTRODUCTION
The therapeutic target of aminoglycosides is the bacterial ribosome (1–5). Different sites within the ribosome bind aminoglycosides, for example, streptomycin (6) binds the S12 protein and affects ribosomal assembly, while kanamycin A, neomycin B, and amikacin target the aminoacyl site (A-site) and interfere with decoding (7–10). A variety of structural and biochemical studies have investigated how aminoglycosides affect decoding and bind to the ribosome. Early work to identify aminoglycoside binding sites in rRNA was completed by Moazed and Noller (7). This work was extended by Purohit and Stern who showed that aminoglycosides bind the A-site as part of the entire ribosomal and as a small rRNA fragment similarly (11). High-resolution NMR structural analysis of the aminoglycoside–A-site complex was completed by the Puglisi group (9, 12–14). These early structural studies showed that local conformational changes within the RNA target occurred upon aminoglycoside binding. These NMR studies served as a catalyst to understand the molecular recognition of the codon–anticodon helix by the 16S rRNA using chemical modification. The results of that study showed that the 30S ribosome is critical for the decoding process by interacting with the codon–anticodon complex (15). Crystallographic studies completed on ribosomal subunits or entire ribosomes in the presence of aminoglycosides provided near atomic resolution information on codon–anticodon interactions and the complex’s interaction with aminoglycosides (16–18). These studies served as a springboard for the Pilch and Hermann groups to develop fluorescence-based methods to study the effect that aminoglycosides have on A-site dynamics (19–22). Subsequent crystallographic studies by the Cate group on whole intact Escherichia coli ribosomes revealed that, in addition to the A-site, aminoglycosides bind helix 69 in 70S ribosomes, helping to explain how aminoglycosides inhibit ribosomal recycling (8).
Bacterial resistance to aminoglycosides is not as complex as the mechanism involved in aminoglycoside inhibition of protein synthesis. The most common mechanism of resistance is due to enzymatic modification of the aminoglycoside. Modifications introduced into aminoglycosides that confer resistance include O-phosphorylation, O-nucleotidylation, and N-acetylation. Enzymes that confer these modifications are called aminoglycoside phosphotransferases (APH’s), aminoglycoside nucleotidyltransferase (ANT’s), and aminoglycoside acetyltransferases (AAC’s), respectively (3, 4, 23–25). Enzymatic modification is so extensive that many of the parent aminoglycoside antibacterials are no longer effective in a clinical setting. Figure 1 depicts some of the positions in the aminoglycoside ribostamycin that are modified by resistance-causing enzymes and the functional groups that are added by resistance-causing enzymes.
Fig. 1.
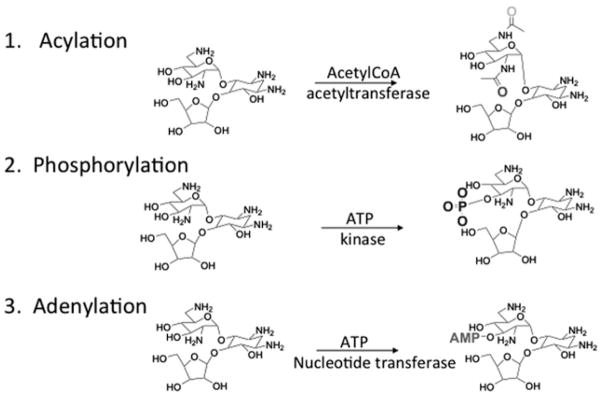
Types of enzymatic modification of aminoglycosides that confer resistance.
Modified aminoglycosides have a greatly diminished affinity (>10-fold, Fig. 2) toward their therapeutic RNA target (26). Investigation of the binding pockets of RNA–aminoglycoside complexes shows that diminished binding can be due to removing functional groups from forming direct contacts with RNA (e.g., acylation of amino groups) or introducing steric bulk or charge–charge repulsion (e.g., a phosphate group lying near the negatively charged backbone of RNA).
Fig. 2.
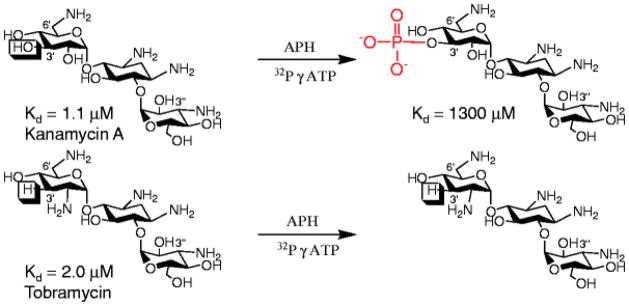
The structures of the aminoglycosides kanamycin A and tobramycin, their measured affinity to a mimic of the bacterial rRNA A-site (26) and the product of their modification by APH(3′)-IIIa. Note that kanamycin A is modified by APH(3′) because it contains a reactive hydroxyl group at the 3′ position, whereas tobramycin contains a hydrogen atom and is thus not susceptible to APH(3′) modification.
Based on these observations and the development of carbohydrate microarray technology (27–33), our group has developed a microarray approach that monitors aminoglycoside modification by resistance-causing enzymes and the effect of modification on rRNA A-site binding using site-specifically immobilized aminoglycoside substrates (34, 35). These studies, therefore, extend previous investigations that used nonspecifically immobilized aminoglycosides to only probe their binding to RNA (29) or resistance-causing enzymes (27). The methods outlined here focus on the use of radioactive labeling to detect enzymatic modification and the impacts of modification on binding RNA. Most recently (mid-2010), our group developed a fluorescence-based microarray approach that can be used to detect modification of carbohydrates by acetyltransferases (36). Please see that manuscript for a description of those studies.
The preparation of the agarose microarray surfaces is critical for these studies, as agarose provides a porous layer that allows for high ligand loading and supports high-density modification of sugars displayed on the array surface (as much as 80% of the total amount of immobilized aminoglycoside) (34, 35). Agarose array surfaces are also quite versatile in the number of chemically reactive handles that can be introduced onto the surface as reactions with amines, alkynes, and azides have all been reported (Fig. 3) (29, 36–41).
Fig. 3.
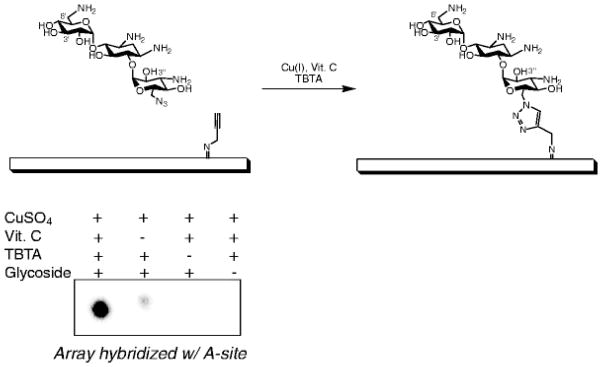
Using the Huisgen dipolar cycloaddition reaction to immobilize azide-functionalized kanamycin A onto an alkyne-functionalized microarray surface.
Portions of this document have been adapted from Aminova and Disney (42), primarily the construction of appropriately functionalized agarose microarray surfaces. Microarray fabrication is included such that two stand alone chapters would be available. That methods paper reviews our group’s work on the development of two-dimensional combinatorial screening (2DCS) (37, 38) and people interested in those methods should refer to that manuscript.
2 MATERIALS
2.1 Array Preparation and Spotting
This section is based on ref. 42, which describes the protocol for 2DCS.
31.8 mM solution of NaCNBH3: Dissolve 0.2 g of sodium cyanoborohydride in 100 mL of 4:1 1× PBS:ethanol. Prior to applying this solution to the array surface, ensure that all of the NaCNBH3 is dissolved by stirring the solution for 2–3 min. It is important to prepare a fresh solution each time.
10× Phosphate-buffered saline (PBS): Dissolve 14.2 g of Na2HPO4, 2.45 g of KH2PO4, 81.8 g of NaCl, 1.86 g of KCl in 900 mL of nanopure water. Adjust the pH to 7.5 using 1 M NaOH or 1 M H3PO4. Add nanopure water to bring volume to 1 L. Store the solution at room temperature.
0.2% Sodium dodecyl sulfate (SDS): Dissolve 2 g of SDS in 900 mL of nanopure water; add water to make 1 L of solution. Store the solution at room temperature.
-
10× Phosphate buffer: Prepare the following solutions:
1 M K2HPO4 (dissolve 1.74 g of K2HPO4 in 10 mL of nanopure water)
1 M KH2PO4 (dissolve 1.36 g of KH2PO4 in 10 mL of nanopure water)
1 M Na2HPO4 (dissolve 1.42 g of Na2HPO4 in 10 mL of nanopure water)
1 M NaH2PO4 (dissolve 1.2 g of NaH2PO4 in 10 mL of nanopure water)
Prepare a 100 mM potassium phosphate solution by mixing 940 μL of 1 M K2HPO4 60 μL of 1 M KH2PO4 and 9 mL of nanopure water.
Prepare a 100 mM sodium phosphate solution by mixing 932 μL of 1 M Na2HPO4, 68 μL of 1 M NaH2PO4, and 9 mL of nanopure water.
Finally, mix equal volumes of the two 100 mM solutions together; this solution should have a pH of ~8. Store the solution at room temperature.
1% Agarose solution: Dissolve 1 g of high melting agarose in nanopure water by heating the solution in a microwave or on a stir plate. Only use this solution once as repeated heating cycles will create a fragile microarray surface.
0.02 M NaIO4 solution. Dissolve 2.14 g of NaIO4 in 500 mL of nanopure water.
10% Ethylene glycol solution: Mix 20 mL of ethylene glycol with 180 mL of nanopure water.
0.1 M NaHCO3: Dissolve 4.2 g of NaHCO3 in 500 mL of nanopure water. Adjust the pH to 8.5 using NaOH.
10 mM 3-azidopropylamine: Prepare 3-azidopropylamine as previously described (37). Dissolve 0.1 g of 3-azidopropylamine in 100 mL of nanopure water.
10 mM propargylamine: Mix 64 μL of propargylamine in 100 mL of nanopure water.
Tris [(1-benzyl-1H-1,2,3-triazol-4-yl) methyl] amine (TBTA) solution (43): Stock solutions of TBTA are to be made in a 4:1 mixture of 2-butanol:dimethylsulfoxide and should be stored at 4°C. TBTA is now commercially available from Sigma-Aldrich (Milwaukee, WI), however, the synthesis is straightforward, high-yielding, and inexpensive (43).
5 mM (Tris (2-Carboxyethy) phosphine hydrochloride (TCEP) (10×): Dissolve 0.14 g of TCEP in 10 mL of nanopure water.
10× Spotting Solution A: Mix 121 mg of Tris–HCl, 25 mg of CuSO4·5H2O, 1.8 mg of ascorbic acid, 1 mL of glycerol, add nanopure water to 10 mL of total volume, add enough of the above TBTA solution to bring the concentration of TBTA to 1 mM, and pH to 8.5.
10× Spotting Solution B: Mix 1 mL of 10× phosphate buffer, 25 mg of CuSO4·5H2O, 1 mL of 5 mM TCEP, 1 mL of 1 mM TBTA, 1 mL of glycerol, and 6 mL of nanopure water.
Azide or Alkyne ligands: Prepare 1:5 serial dilutions of alkyne/azide-displaying ligands from 5 mM to 1 μM in 1× Spotting Solution A or B, respectively.
2.2 Preparation of Cellular Lysates
E. coli strains: JM109(DE3) and BL21(DE3) pLysS.
Plasmids encoding resistance-causing enzymes: pETSACG1 (encodes APH(3′)-IIIa); pET22b(+) (encodes ANT(2″)-Ia); pET22a (encodes AAC-(3)-IV); or plasmid containing a resistance-causing enzyme of interest.
LB medium: Dissolve 10 g of bacto tryptone, 5 g of bacto yeast extract, 10 g of NaCl, 1 mL of 1 M NaOH in 900 mL of deionized water. Adjust the pH to 7.0 with 1 M NaOH. Sterilize by autoclaving on the liquid cycle at 121°C for 20 min. Store the solution at room temperature.
1,000× Ampicilin solution: Dissolve 50 mg of ampicillin in 1 mL of water.
1,000× Kanamycin A solution: Dissolve 10 mg of kanamycin A in 1 mL of water.
1,000× Carbenicllin: Dissolve 50 mg of carbenicillin in 1 mL of water.
1 M (500 or 1,000×) isopropyl-β-D-1-thiogalactopyranoside (IPTG): Dissolve 2.83 g of IPTG in a total volume of 10 mL by the addition of nanopure water.
APH(3′)-IIIa (44) lysis buffer: Dissolve 606 mg of Tris–HCl, 1.1 g of NaCl, 1.6 mg of dithiolthreitol (DTT) to 100 mL. Adjust the pH to 8.0 and then add 17.4 mg of phenylmethylsulfonyl fluoride (PMSF; dissolved in ethanol).
APH(3′)-IIIa (44) dialysis buffer: Dissolve 24.2 g of Tris–HCl, 11.9 g of KCl, 8.13 g of MgCl2·6H2O to 4 L of nanopure water; adjust the pH to 7.5.
ANT(2″)-Ia (45) lysis buffer: Add 600 mg Tris–HCl, 101 mg of MgCl2·6H2O, and 35 μL of 2-mercaptoethanol, and nanaopure water to afford a solution with a total volume of 100 mL. Adjust the pH to 8.0.
ANT(2″)-Ia (45) dialysis buffer: Add 29 g of Tris–HCl, 32.5 g of MgCl2·6H2O, 85.6 g of NH4Cl, and 99 mg of DTT to 4 L of nanopure water and adjust the pH to 7.1.
AAC-(3)-IV (46) lysis buffer: Add 300 mg of triethanolamine and 17 mg of PMSF (dissolved in ethanol) to 100 mL of nanopure water. Adjust the pH of the solution to 7.8.
AAC-(3)-IV (46) dialysis buffer: Add 11.2 g of triethanolamine to 4 L of nanopure water and adjust the pH to 7.8.
BCA Protein Assay Kit (Pierce Biotechnologies/Thermo Fisher; catalog number 23227).
4× SDS-PAGE Stacking Buffer: Dissolve 30.4 g of Tris–HCl base and 2.0 g of SDS in 500 mL of nanopure water. Adjust the pH to 6.8 with 1 M HCl.
4× SDS-PAGE Resolving Buffer: Dissolve 91.0 g of Tris–HCl base and 2.0 g of SDS 500 mL of nanopure water. Adjust the pH to 8.8 with 1 M HCl.
5× SDS-PAGE Electrophoresis Buffer: Dissolve 15.1 g of Tris–HCl base, 72.0 g of Glycine, and 5.0 g of SDS in 1 L of nanopure water.
30% (w/v) Acrylamide/bis-acrylamide (19:1): Acrylamide solutions can be purchased from Sigma-Aldrich or prepared as follows: dissolve 28.5 g of acrylamide and 1.5 g of bis-acrylamide in 100 mL of nanopure water. (Caution: acrylamide is a known neurotoxin).
4× Protein gel sample loading buffer: Mix 2.0 mL of 1 M Tris–HCl, 0.8 g of SDS, 4.0 mL of 10% glycerol, 0.4 mL of 14.7 M β-mercaptoethanol, 1.0 mL of 0.5 M EDTA, and 8 mg of bromophenol blue.
2.3 Modification and Hybridization of Microarrays
1× ANT and APH assay buffer: Add 479 mg of HEPES, 22.3 mg of MgCl2·6H2O, and 16.4 mg of KCl to 10 mL of nanopure water. Adjust the pH to 7.5.
1× AAC-(3)-IV assay buffer: Add 1.916 g of HEPES to 40 mL of nanopure water and pH the solution to 7.5.
1× RNA hybridization buffer: Add 45 mg of Na2HPO4, 2 mg of EDTA, and 407 mg of NaCl to 40 mL of nanopure water. Adjust the pH of the solution to 7.1.
Fluorescently labeled A-site oligonucleotide mimic: A fluorescently labeled oligonucleotide mimic of the bacterial A-site can be purchased from Dharmacon or Integrated DNA Technologies, Inc.
Phosphorimager.
Microarray scanner.
3 METHODS
Although the microarrays are generally robust, we prefer to use them within approximately 1 month of their construction. This is due to surface cracking that can occur if the array dries out, producing sub-optimal images. When spotting microarrays, we suggest using a freshly prepared spotting solution. For the majority of our array work, dialyzed cellular lysates were applied to the array surface to monitor modification. Partially or totally purified proteins can also be used, however, their preparation can be time-consuming.
3.1 Preparation of Agarose Slides (42)
Prepare a 1% agarose solution (w/v) using nanopure water. Melt in a microwave on high for 2–3 min, swirling the solution every 20–30 s.
While the solution is hot, apply ~1.5 mL to the surface of a glass slide using a P-1000 pipette. Ensure that the solution is spread evenly over the slide surface. The solution can be spread over the surface by simply tracing the outside of the array with the pipette tip during application to the array surface. Allow the agarose to dry to a thin film overnight.
3.2 Functionalization of Agarose Slides (42)
Submerge the slides in 0.02 M NaIO4 for 30 min at room temperature, and then wash them with nanopure water for 30 min.
Submerge the slides in 10% (v/v) ethylene glycol for 1 h at room temperature to quench residual NaIO4. Wash with water for 1.5 h, changing the water every 20 min. Subheading 3.2, step 1 and 2 afford slides that display aldehydes.
-
To construct microarrays of amine-displaying ligands, complete the following:
Allow the slides to dry.
Prepare spotting solutions as follows: small molecule at desired concentration (typically serially diluted from 5 mM to 1 μM), 0.1 M NaHCO3, and 10% glycerol. Spot 0.4 μL of the solutions onto aldehyde-agarose slides in duplicate.
Incubate for 3 h at room temperature in a humidity chamber (box containing a saturated solution of NaCl). Wash the slides 3 × 10 min with 1× RNA hybridization buffer, followed by water, 2 × 10 min. Continue to Subheading 3.2, step 6.
For microarrays of alkyne-displaying ligands, submerge the slides in a solution of 0.1 M NaHCO3 and 10 mM 3-azidopropylamine for 3 h at room temperature. Continue to Subheading 3.2, step 6.
For microarrays of azide-displaying ligands, submerge the slides in a solution of 0.1 M NaHCO3 and 10 mM propargylamine. Continue to Subheading 3.2, step 6. For the series of aminoglycosides that we have tested in modification assays, all have been functionalized with an azide tag for surface immobilization (34, 36, 39).
-
Submerge slides in a solution of 31.8 mM NaCNBH3 for 3 min to reduce the imine formed on the microarray surface.
Wash the slides with 0.2% SDS, 3 × 15 min, and then with water, 2 × 15 min.
Dry the slides under a stream of air.
3.3 Immobilization of Azido- or Alkyne-Aminoglycosides on the Slide Surface (42)
In this section, spotting the aminoglycoside microarrays manually by delivering fixed volumes of solutions from a pipette is described. This method is feasible to produce microarrays with ≤100 features. The use of replicators or robotic arrayers can be used, if more features are desired. It should be noted that spot sizes should be ~500–1,000 μm in diameter and each spot should be separated by at least 2,000 μm for them to be observable using a phosphorimager. Therefore, ensure that the pin and spotting buffer combination produce features that are large enough to be observed.
-
Prepare spotting solutions as follows:
For azide-displaying small molecules: add the small molecule at the desired concentration (typically serially diluted from 5 mM to 1 μM) in 1× Spotting Solution A. Spot 0.4 μL of the solutions in duplicate onto alkyne-agarose slides.
For alkyne-displaying small molecules: add the small molecule at the desired concentration (typically serially diluted from 5 mM to 1 μM) in 1× Spotting Solution B. Spot 0.4 μL of the solutions in duplicate onto azide-agarose slides.
Incubate the slides for 3 h at room temperature in a humidity chamber (box containing a saturated solution of NaCl). Wash the slides 3 × 10 min with 1× RNA hybridization buffer, followed by water (2 × 10 min).
3.4 Preparation of Cellular Lysates
3.4.1 Preparation of APH(3′)-IIIa Lysate
Transform E. coli JM109(DE3) with the pETSACG1 plasmid containing the APH(3′)-IIIa resistance-causing gene (46) using standard protocols.
Grow a 1 L culture at 37°C in LB medium containing 100 mg/L of ampicillin (final concentration is 1× per Subheading 2.2, item 2). (As a negative control, grow a 1 L culture of JM109(DE3) cells that have not been transformed. Do not use ampicillin in the LB medium. Follow steps 3–7 and 9 in Subheading 3.4.1).
Once the culture reaches an OD600 of around 0.5, add IPTG to a final concentration of 1 mM (final concentration is 1× for 1,000× stock in Subheading 2.2, item 5). Incubate the culture for an additional 3 h at 37°C.
Pellet the cell suspension by centrifugation (10 min at 5,000 × g) and wash with ice cold lysis buffer (see Subheading 2.2, item 6).
Resuspended the pellet in a minimal volume of lysis buffer and lyse the cells by sonication.
Pellet cellular debris by centrifugation (20 min at 10,000 × g) and then dialyze the supernatant against 4 L of APH(3′) dialysis buffer (Subheading 2.2, item 7) for 24 h at 4°C.
Concentrate the lysate to 500 μL in using a centrifugal concentrator at 4°C.
Confirm the isolation of the resistance-causing enzyme by SDS-PAGE (47) and activity using a phosphocellulose capture assay (48).
Total protein content of the lysate can then be determined using a BCA Protein Assay Kit.
3.4.2 Preparation of ANT(2″)-Ia Lysate
Transform E. coli JM109(DE3) with pET 22b(+) plasmid containing the ANT(2″)-Ia resistance gene using standard protocols.
Grow a 1 L culture at 37°C in LB medium containing 50 mg/L of amplicillin (final concentration is 0.5× per Subheading 2.2, item 2) and 10 mg/L of kanamaycin A (final concentration is 1× per Subheading 2.2, item 3). (As a negative control, grow a 1 L culture of JM109(DE3) cells that have not been transformed. Do not use ampicillin or kanamycin A in the LB medium. Follow steps 3–7 and 9 in Subheading 3.4.2).
Once the culture reaches an OD600 of ~0.5, add IPTG to a final concentration of 0.5 mM (final concentration is 0.5× for 1,000× stock in Subheading 2.2, item 5). Incubate the culture for an additional 3 h at 37°C.
Pellet the cells by centrifugation (10 min at 5,000 × g) and wash with ice cold lysis buffer (see Subheading 2.2, item 8).
Resuspended the pellet in a minimal volume of lysis buffer and lyse the cells by sonication.
Pellet cellular debris by centrifugation (20 min at 10,000 × g) and dialyze the supernatant 4 L of ANT(2″) dialysis buffer (Subheading 2.2, item 9) for 24 h at 4°C.
Concentrate the lysate to 500 μL using a centrifugal concentrator.
Confirm the isolation of the resistance-causing enzyme by SDS-PAGE (47) and activity using a phosphocellulose capture assay (48).
Total protein content of the lysate can then be determined using a BCA Protein Assay Kit.
3.4.3 Preparation of AAC-(3)-IV Lysate
Transform E. coli BL21(DE3) pLysS with pET 22a plasmid containing the AAC-(3)-IV resistance-causing gene using standard protocols.
Grow a 1 L culture at 37°C in LB medium containing 50 mg/L of carbenicillin (final concentration is 1× per Subheading 2.2, item 4) for 24 h. (As a negative control, grow a 1 L culture of JM109(DE3) cells that have not been transformed. Do not use carbenicillin in the LB medium. Follow steps 3–7 and 9 in Subheading 3.4.3).
Pellet the cells by centrifugation (10 min at 5,000 × g).
Resuspend the pellet in 25 mL of AAC-(3)-IV lysis buffer (Subheading 2.2, item 10).
Lyse the cells by sonication. Add 50 U of DNase I and then stir the solution on ice for 30 min.
Pellet cellular debris by centrifugation (20 min at 10,000 × g) and dialyze the supernatant against 4 L of AAC-(3)-IV dialysis buffer (Subheading 2.2, items 9 and 11).
Concentrate the lysate to 500 μL using a centrifugal concentrator.
Confirm the isolation of the resistance-causing enzyme by SDS-PAGE (47) and activity using a phosphocellulose capture assay (48). Activity can also be confirmed by using a spectrophotometric assay as previously described (46).
Total protein content of the lysate can then be determined using a BCA Protein Assay Kit.
3.5 Modification of Array-Displayed Aminoglycosides by Resistance-Causing Enzymes
This section describes the mechanics of completing a modification experiment on an array surface. There are two ways in which these experiments can be completed: using a silicon gasket that produces microwells or hybridizing the entire microarray surface. Gaskets that afford 50 microwells are available from Grace Bio Labs (Bend, OR). Each well can hold approximately 12 μL of solvent. When hybridizing the entire slide surface with a solution containing the resistance-causing enzyme, a hybridization chamber or a hydrophobic marker can be used. It is important that hybridization chambers cover the entire array surface. [A variety of chambers can be purchased from Sigma-Aldrich (Milwaukee, WI).] A hydrophobic marker, or a PAP pen, can be used to draw a rectangle on the diameter of an array and can be purchased from Abchem (San Francisco, CA). PAP pens are much less expensive than hybridization chambers and allow customization of the array into various segments.
3.5.1 Modification of an Array Surface with 32P by Using ANT(2″)-Ia and APH(3′)-IIIa. Results Are Illustrated in Fig. 4
Fig. 4.
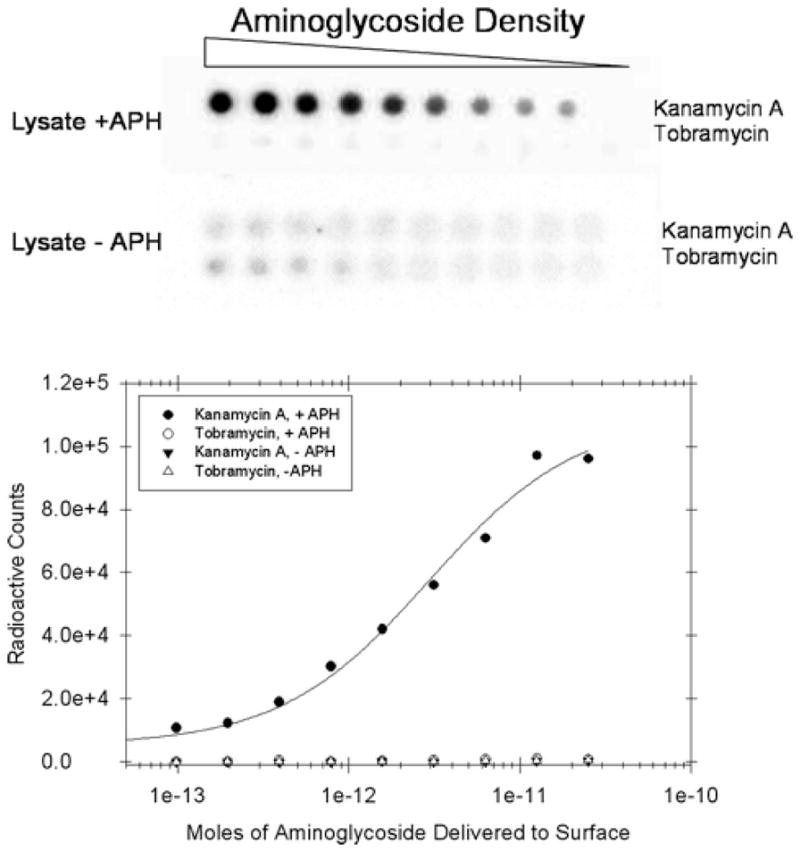
A microarray that is used to probe modification of array immobilized kanamaycin A and tobramycin by APH(3′)-IIIa. Radioactivity is only deposited where the reactive kanamcyin A is deposited on the array surface.
-
Pre-hybridize an air-dried microarray with 1× ANT and APH assay buffer. It is important to pre-hybridize arrays as application of a radioactive solution to a dry array leads to nonspecific deposition of radioactivity that is very difficult to remove.
For arrays with microwells, add 12 μL to each well, incubate for 5 min, and remove the buffer. Repeat four times.
For arrays that are affixed with a hybridization chamber or that use a PAP pen, add 1.5–2.0 mL of 1× ANT and APH assay buffer to the array surface and incubate for at least 5 min. Just prior to the addition of the resistance-causing enzyme solution, tip the array on its side to remove the buffer from the surface.
To assay aminoglycoside modification, add 2.3 nmol of phosphoenolpyruvate, 0.018 U of pyruvate kinase, 11.4 nmol of ATP to 12 μL of 1× ANT, or APH assay buffer. For APH modification, add 0.2 OD260 of cell lysate and 500,000 CPM of (γ-32P]ATP; for ANT modification, add 0.8 OD260 of ANT and 500,000 CPM of (α-32P]ATP. For control experiments, substitute the cell lysate with one that does not contain a resistance-causing enzyme.
Incubate the arrays for 12 h at 37°C and 100% humidity to prevent evaporation.
Remove the gasket from the surface and wash the slide by submerging it in a solution of 0.2% SDS (2× 15 min) and then in water for 30 min at 37°C.
Air dry the array surface, cover with plastic wrap, and expose in a phosphorimager cassette.
3.5.2 Modification of an Array Surface with 14C by Using AAC-(3)-IV
Pre-hybridize an air-dried microarray with 1× AAC-(3)-IV assay buffer for at least 5 min at room temperature. (These experiments were only completed using arrays constructed with a PAP pen.) Just prior to the addition of the resistance-causing enzyme solution, tip the array on its side to remove the buffer from the surface.
To assay aminoglycoside modification, prepare 0.13 nmol 14C-Acetyl coenzyme A (AcCoA), 5 nmol of unlabelled AcCoA, and 0.3 μg of total protein lysate in 240 μL of AAC-(3)-IV assay buffer. For control experiments, add 0.3 μg of total cell lysate that does not contain a resistance-causing enzyme.
Incubate the arrays for 20 h at 37°C and 100% humidity to prevent evaporation.
Remove the gasket from the surface. Wash the slide by submerging it in a solution of 0.2% SDS (2× 15 min) and then in water for 30 min at 37°C.
Air dry the array surface, cover with plastic wrap, and expose in a phosphorimager cassette for at least 48 h. If signal is low or background is high, see Notes 1 & 2.
3.6 Hybridization of the Arrays with a Fluorescently Labeled Mimic of the Bacterial rRNA A-Site. Results Are Illustrated in Fig. 5
Fig. 5.
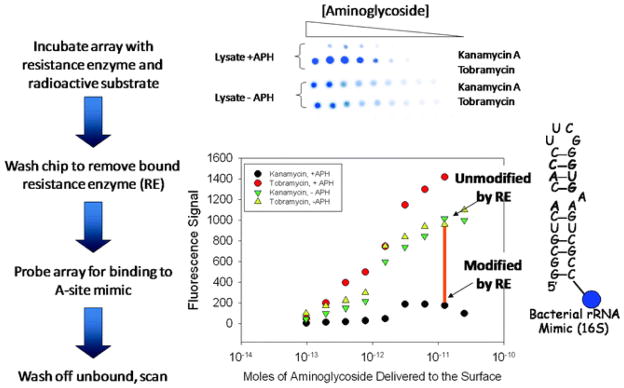
Overall approach to using microarrays to study antibacterial resistance that is conferred by aminoglycoside modification. The image illustrates results of binding of array-immobilized substrates that have been modified by APH(3′)-IIIa to a mimic of the bacterial rRNA A-site. The plot below the array image illustrates the decrease in affinity of the arrayed aminoglycoside upon modification by APH(3′)-IIIa; the orange bar shows the decreased signal due to the modification of kanamaycin A by APH(3′)-IIIa.
The activity of a variety of aminoglycoside resistance-causing enzymes has been probed on a microarray surface via these routes. In all but one case, the extent of array modification was high enough to observe a decrease in binding to an oligonucleotide mimic of the rRNA A-site. Interestingly, in the case in which decreased binding to the A-site was not observed, the aminoglycoside modifying enzyme used was not known to confer resistance to aminoglycosides in vivo (34). Thus, the extent of modification of arrayed ligands is critical to observe a decrease in ligand binding to biomolecules.
Pre-equilibrate the modified slide from Subheading 3.5.1 or 3.5.2 with 600 μL of 1× RNA hybridization buffer containing 200 μg/mL of BSA for at least 5 min at room temperature.
During the pre-hybridization, prepare a 600 μL solution of 1 μM of a fluorescently labeled RNA in 1× RNA hybridization buffer. Fold the RNA by heating at 95°C for 4 min and then cooling to room temperature. After cooling, add BSA to the sample to a final concentration of 100 μg/mL.
Pipette the solution containing the folded RNA onto the array surface and spread evenly using a custom-cut sheet of parafilm (same dimensions as the array surface).
Incubate the array for 40 min at room temperature in the dark.
Remove unbound RNA by delivering 10 × 1 mL aliquots of 1× RNA hybridization buffer containing 200 μg/mL of BSA to the surface. Remove any salt stuck to the surface by delivering 5 × 1 mL aliquots of water.
Image the slide using a microarray scanner. If signal is low or background is high, see Notes 1 & 2.
Acknowledgments
I thank Olivia Barrett, Alexei Pushechnikov, Meilan Wu, Pavel Tsitovich, and Jon French for their contributions to this project. Funding for this work was provided by the Research Corporation through a Cottrell Scholar award, the Camille and Henry Dreyfus Foundation through a New Investigator in the Chemical Sciences Award and a Teacher-Scholar Award, by NYSTAR through a J.D. Watson Young Investigator award, and by the National Institutes of Health (R01-GM079235)
Footnotes
- Check that the spotting solution was prepared properly.
- Check that the assay buffer was prepared properly.
- Use a new stock of radioactively labeled substrate ([α-] or [γ-32P] ATP or 14C-acetyl Coenzyme A).
- Ensure that the total concentration of substrate added is greater than the Km.
-
Ensure that the resistance-causing enzyme was overexpressed and is active.
- Compare the protein content of cellular lysates in which the resistance-causing enzyme was and was not expressed by SDS-PAGE. The resistance-causing enzyme should be a significant percentage of total protein (>30%). If this is not the case, ensure that the IPTG solution was prepared correctly. If so, either induce the cells with a higher concentration of IPTG (2 mM) or increase the induction time. Optimization of induction can be assessed by SDS-PAGE.
- Increase the amount of cellular lysate incubated with the slide surface. The amount of control cellular lysate should be increased to the same extent.
- If testing acetyltransferases, add a deacetylase inhibitor such as butyric acid.
- Reactive groups were not quenched after the immobilization of aminoglycosides. NaBH3CN is hygroscopic; obtain a new stock if clumps are present.
- Ensure that the slides were pre-hybridized prior to incubation with resistance-causing enzymes or fluorescently labeled RNA. Addition of 1% BSA may reduce the background.
- Wash the slides in 0.2% SDS for 5 min with gentle agitation.
References
- 1.Tenson T, Mankin A. Antibiotics and the ribosome. Mol Microbiol. 2006;59:1664–1677. doi: 10.1111/j.1365-2958.2006.05063.x. [DOI] [PubMed] [Google Scholar]
- 2.Gallego J, Varani G. Targeting RNA with small-molecule drugs: therapeutic promise and chemical challenges. Acc Chem Res. 2001;34:836–843. doi: 10.1021/ar000118k. [DOI] [PubMed] [Google Scholar]
- 3.Magnet S, Blanchard JS. Molecular insights into aminoglycoside action and resistance. Chem Rev. 2005;105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 4.Wright GD, Berghuis AM, Mobashery S. Aminoglycoside antibiotics. Structures, functions, and resistance. Adv Exp Med Biol. 1998;456:27–69. [PubMed] [Google Scholar]
- 5.Francois B, Russell RJ, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005;33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funatsu G, Wittmann HG. Ribosomal proteins. 33. Location of amino-acid replacements in protein S12 isolated from Escherichia coli mutants resistant to streptomycin. J Mol Biol. 1972;68:547–550. doi: 10.1016/0022-2836(72)90108-8. [DOI] [PubMed] [Google Scholar]
- 7.Moazed D, Noller HF. Interaction of Antibiotics With Functional Sites in 16 s Ribosomal-RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 8.Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol. 2007;14:727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- 9.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the A site of Escherichia coli 16 S ribosomal RNA complexed with an aminoglycoside antibiotic. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 10.Wong CH, Hendrix M, Priestley ES, Greenberg WA. Specificity of aminoglycoside antibiotics for the A-site of the decoding region of ribosomal RNA. Chemistry & Biology. 1998;5:397–406. doi: 10.1016/s1074-5521(98)90073-4. [DOI] [PubMed] [Google Scholar]
- 11.Purohit P, Stern S. Interactions of a small RNA with antibiotic and RNA ligands of the 30 S subunit. Nature. 1994;370:659–662. doi: 10.1038/370659a0. [DOI] [PubMed] [Google Scholar]
- 12.Fourmy D, Yoshizawa S, Puglisi JD. Paromomycin binding induces a local conformational change in the A-site of 16 S rRNA. J Mol Biol. 1998;277:333–345. doi: 10.1006/jmbi.1997.1551. [DOI] [PubMed] [Google Scholar]
- 13.Lynch SR, Gonzalez RL, Puglisi JD. Comparison of X-ray crystal structure of the 30 S subunit-antibiotic complex with NMR structure of decoding site oligonucleotide-paromomycin complex. Structure (Camb) 2003;11:43–53. doi: 10.1016/s0969-2126(02)00934-6. [DOI] [PubMed] [Google Scholar]
- 14.Recht MI, Fourmy D, Blanchard SC, Dahlquist KD, Puglisi JD. RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J Mol Biol. 1996;262:421–436. doi: 10.1006/jmbi.1996.0526. [DOI] [PubMed] [Google Scholar]
- 15.Yoshizawa S, Fourmy D, Puglisi JD. Recognition of the codon-anticodon helix by ribosomal RNA. Science. 1999;285:1722–1725. doi: 10.1126/science.285.5434.1722. [DOI] [PubMed] [Google Scholar]
- 16.Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 17.Yonath A. Structural insight into functional aspects of ribosomal RNA targeting. Chembiochem. 2003;4:1008–1017. doi: 10.1002/cbic.200300683. [DOI] [PubMed] [Google Scholar]
- 18.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30 S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 19.Barbieri CM, Kaul M, Pilch DS. Use of 2-aminopurine as a fluorescent tool for characterizing antibiotic recognition of the bacterial rRNA A-site. Tetrahedron. 2007;63:3567–6574. doi: 10.1016/j.tet.2006.08.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaul M, Barbieri CM, Pilch DS. Fluorescence-based approach for detecting and characterizing antibiotic-induced conformational changes in ribosomal RNA: comparing aminoglycoside binding to prokaryotic and eukaryotic ribosomal RNA sequences. J Am Chem Soc. 2004;126:3447–3453. doi: 10.1021/ja030568i. [DOI] [PubMed] [Google Scholar]
- 21.Kaul M, Barbieri CM, Pilch DS. Aminoglycoside-induced reduction in nucleotide mobility at the ribosomal RNA a-site as a potentially key determinant of antibacterial activity. J Am Chem Soc. 2006;128:1261–1271. doi: 10.1021/ja056159z. [DOI] [PubMed] [Google Scholar]
- 22.Shandrick S, Zhao Q, Han Q, Ayida BK, Takahashi M, Winters GC, Simonsen KB, Vourloumis D, Hermann T. Monitoring molecular recognition of the ribosomal decoding site. Angew Chem Int Ed Engl. 2004;43:3177–3182. doi: 10.1002/anie.200454217. [DOI] [PubMed] [Google Scholar]
- 23.Pfister P, Hobbie S, Vicens Q, Bottger EC, Westhof E, Vicens Q, Westhof E. The molecular basis for A-site mutations conferring aminoglycoside resistance: relationship between ribosomal susceptibility and X-ray crystal structures. Chembiochem. 2003;4:1078–1088. doi: 10.1002/cbic.200300657. [DOI] [PubMed] [Google Scholar]
- 24.Vetting M, Roderick SL, Hegde S, Magnet S, Blanchard JS, Vetting MW, Hegde SS, Javid-Majd F, Dam TK, Brewer CF. What can structure tell us about in vivo function? The case of aminoglycoside-resistance genes. Biochem Soc Trans. 2003;31:520–522. doi: 10.1042/bst0310520. [DOI] [PubMed] [Google Scholar]
- 25.Wright GD. Mechanisms of resistance to antibiotics. Curr Opin Chem Biol. 2003;7:563–569. doi: 10.1016/j.cbpa.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 26.Llano-Sotelo B, Azucena EF, Jr, Kotra LP, Mobashery S, Chow CS. Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem Biol. 2002;9:455–463. doi: 10.1016/s1074-5521(02)00125-4. [DOI] [PubMed] [Google Scholar]
- 27.Disney MD, Magnet S, Blanchard JS, Seeberger PH. Aminoglycoside Microarrays To Study Antibiotic Resistance. Angew Chem Int Ed Engl. 2004;43:1591–1594. doi: 10.1002/anie.200353236. [DOI] [PubMed] [Google Scholar]
- 28.Disney MD, Seeberger PH. The use of carbohydrate microarrays to study carbohydrate-cell interactions and to detect pathogens. Chem Biol. 2004;11:1701–1707. doi: 10.1016/j.chembiol.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 29.Disney MD, Seeberger PH. Aminoglycoside microarrays to explore interactions of antibiotics with RNAs and proteins. Chemistry. 2004;10:3308–3314. doi: 10.1002/chem.200306017. [DOI] [PubMed] [Google Scholar]
- 30.Ratner DM, Adams EW, Disney MD, Seeberger PH. Tools for glycomics: mapping interactions of carbohydrates in biological systems. Chembiochem. 2004;5:1375–1383. doi: 10.1002/cbic.200400106. [DOI] [PubMed] [Google Scholar]
- 31.Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH, Paulson JC. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci U S A. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukui S, Feizi T, Galustian C, Lawson AM, Chai W. Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nat Biotechnol. 2002;20:1011–1017. doi: 10.1038/nbt735. [DOI] [PubMed] [Google Scholar]
- 33.Park S, Lee MR, Shin I. Fabrication of carbohydrate chips and their use to probe protein-carbohydrate interactions. Nat Protoc. 2007;2:2747–2758. doi: 10.1038/nprot.2007.373. [DOI] [PubMed] [Google Scholar]
- 34.Barrett OJ, Pushechnikov A, Wu M, Disney MD. Studying aminoglycoside modification by the acetyltransferase class of resistance-causing enzymes via microarray. Carbohydr Res. 2008;343:2924–2931. doi: 10.1016/j.carres.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Disney MD, Barrett OJ. An aminoglycoside microarray platform for directly monitoring and studying antibiotic resistance. Biochemistry. 2007;46:11223–11230. doi: 10.1021/bi701071h. [DOI] [PubMed] [Google Scholar]
- 36.Tsitovich PB, Pushechnikov A, French JM, Disney MD. A chemoenzymatic route to diversify aminolgycosides enables a microarray-based method to probe acetyltransferase activity. Chembiochem. 2010;11:1656–1660. doi: 10.1002/cbic.201000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, Disney MD. A small molecule microarray platform to select RNA internal loop-ligand interactions. ACS Chem Biol. 2007;2:745–754. doi: 10.1021/cb700174r. [DOI] [PubMed] [Google Scholar]
- 38.Disney MD, Labuda LP, Paul DJ, Poplawski SG, Pushechnikov A, Tran T, Velagapudi SP, Wu M, Childs-Disney JL. Two-dimensional combinatorial screening identifies specific aminoglycoside-RNA internal loop partners. J. 2008 doi: 10.1021/ja803234t. [DOI] [PubMed] [Google Scholar]
- 39.Velagapudi SP, Seedhouse SJ, Disney MD. Structure-activity relationships through sequencing (StARTS) defines optimal and suboptimal RNA motif targets for small molecules. Angew Chem Int Ed Engl. 2010;49:3816–3818. doi: 10.1002/anie.200907257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran T, Disney MD. Two-dimensional combinatorial screening of a bacterial rRNA A-site-like motif library: defining privileged asymmetric internal loops that bind aminoglycosides. Biochemistry. 2010;49:1833–1842. doi: 10.1021/bi901998m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labuda LP, Pushechnikov A, Disney MD. Small molecule microarrays of RNA-focused peptoids help identify inhibitors of a pathogenic group I intron. ACS Chem Biol. 2009;4:299–307. doi: 10.1021/cb800313m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aminova O, Disney MD. A microarray-based method to perform nucleic acid selections. Methods Mol Biol. 2010;669:209–224. doi: 10.1007/978-1-60761-845-4_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 44.McKay GA, Thompson PR, Wright GD. Broad spectrum aminoglycoside phosphotransferase type III from Enterococcus: overexpression, purification, and substrate specificity. Biochemistry. 1994;33:6936–6944. doi: 10.1021/bi00188a024. [DOI] [PubMed] [Google Scholar]
- 45.Wright E, Serpersu EH. Isolation of aminoglycoside nucleotidyltransferase(2″)-Ia from inclusion bodies as active. monomeric enzyme Protein Express Purif. 2004;35:373–380. doi: 10.1016/j.pep.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Magalhaes ML, Blanchard JS. The kinetic mechanism of AAC3-IV aminoglycoside acetyltransferase from Escherichia coli. Biochemistry. 2005;44:16275–16283. doi: 10.1021/bi051777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 1, 2, and 3. Cold Spring Harbor Laboratory Press; NY: 1989. [Google Scholar]
- 48.Wybenga-Groot LE, Draker K, Wright GD, Berghuis AM. Crystal structure of an aminoglycoside 6′-N-acetyltransferase: defining the GCN5-related N-acetyltransferase superfamily fold. Structure. 1999;7:497–507. doi: 10.1016/s0969-2126(99)80066-5. [DOI] [PubMed] [Google Scholar]