

Abstract
Sjögren's syndrome (SjS) is chronic autoimmune disease manifested by the loss of saliva and/or tear secretion by salivary and/or lacrimal glands, respectively. The pathogenesis of the disease remains elusive, perhaps due to the multiple triggers of the disease. However, substantial advances have been made in attempting to resolve the complexity of SjS using both animal models and human subjects. The primary objectives of this review are to provide a better understanding of the disease processes with major emphasis on the use of mouse models, how genetic predisposition plays a role in the natural history of the disease, as well as a presentation of new findings pertaining to the role of TH1, TH2, and TH17 cells in the pathogenesis of SjS.
Keywords: Sjögren's syndrome, animal models, genetics, TH1 cells, TH2 cells, TH17 cells
Introduction
Maintenance of oral and ocular health depends heavily on the salivary and lacrimal gland systems to secrete saliva and tears, respectively. These fluids are rich in a variety of proteins and lipids produced by both acinar and ductal tissues. Saliva and tear fluids contain multiple enzymes that break down nucleic acids and proteins, anti-microbial substances such as secretory immunoglobulin (Ig), histatins, cystatins, and splunc molecules, and growth factors such as epidermal growth factor. Without these anti-bacterial and physiological mechanisms for moisturizing and buffering the oral and ocular surfaces, a general decline in oral and ocular hygiene can occur, leading to caries, loss of teeth, and/or reduced vision. Xerostomia sicca (dry mouth) and xerophthalmia (dry eye) result respectively from basic changes in the saliva and tear flow rates, the composition of saliva and tears, and/or combinations thereof. The underlying causes of xerostomia include the natural aging process, use of medications, asthma, mouth breathing, chemotherapy, radiation therapy, autoimmune processes, thyroid dysfunction, kidney dialysis, and/or stroke. Likewise, the fundamental causes of xerophthalmia include the natural aging process, physical injury, surgical procedures, meibomian gland dysfunction, and/or autoimmune attack against one or more of the multiple secretory tissues/glands of the eye (Hansen et al. 2003; Nguyen et al. 2007).
While there are multiple underlying causes for decreased secretions of saliva and tears, one of the causes of severe xerostomia sicca and xerophthalmia is the autoimmune disease Sjögren's syndrome (SjS). SjS is a chronic systemic autoimmune disease in which the immune system targets the salivary and/or lacrimal glands and leads to the loss of secretory function (Fox and Kang 1992; Nguyen et al. 2007). SjS is classified as either primary SjS (pSjS) or secondary SjS (sSjS). pSjS is a chronic autoimmune attack involving both lacrimal and salivary glands in the absence of other autoimmune diseases, whereas sSjS is an autoimmune attack against the lacrimal and/or salivary glands in the presence of other autoimmune diseases, most often rheumatoid arthritis, systemic lupus erythmatosus (SLE), or scleroderma (Fox and Kang 1992; Fox and Michelson 2000). In addition to the salivary and lacrimal glands, other organs often affected include the entire GI tract, skin, lungs, vasculature, kidneys, bladder, and the vagina. Involvement of the musculature can lead to fibromyalgia-like symptoms and chronic fatigue. The clinical presentation of Sjögren's syndrome is presented in Table 1. Like other autoimmune connective tissue diseases, SjS shows a sexual dimorphism, with women affected 10–20 times more frequently than men, suggesting hormonal involvement (Carroll 1998; Sullivan 1997; Sullivan 2004; Toda et al. 1998). While not considered a lethal disease, between 4–10% of patients with SjS will develop non-Hodgkin's B--cell lymphomas, a portion of which will advance to high--grade malignancies (Ambrosetti et al. 2004; De Vita et al. 1997; Ramos-Casals et al. 2004; Voulgarelis and Moutsopoulos 2003). Although there are no prevalence studies in the US, the National Arthritis Data Workgroup recently estimated the number of individuals with pSjS in the USA is 1.3 million with a range of 0.4–3.1 million, placing SjS as one of the more common rheumatic and autoimmune diseases (Helmick et al. 2008).
Table 1. Clinical manifestation of Sjögren's syndrome in human patients.
| Organs | Manifestations | |
|---|---|---|
| Primary target organs | Mouth | Xerostomia: oral dryness, manifested by inability to swallow dry food with out aid of liquid, unclear pronunciation of words and/or chalky feeling in mouth |
| Eyes | Xerophtalmia: dry eyes, manifested by burning, sandy and itchy feeling, conjunctiva redness and increased photosensitivity | |
|
| ||
| Secondary target organs | General | Low-grade fever, weight loss, vasculitis, lymphadenopathy, lymphoma |
| Respiratory tract | Cough, pharynx irritation | |
| GI tract | Parotid enlargement | |
| Genitourinary tract | Nephritis, dyspareunia, decreased sexual activity, splenomegaly | |
| Skin | Ulcerations, dryness and itchiness, urticaria, increased risk of epidermal abrasion and skin infection | |
| Muscle and joint | Muscle and joint pain, arthritis, myositis, fibromyalgia | |
| Neurological | Hearing loss, peripheral neuropathy | |
Despite on-going efforts to define the autoimmune basis of SjS, the underlying etiology of this disease remains elusive. Over the past 20 years, in addition to extensive data gathered from studies of SjS patients, a diverse number of mouse strains have been developed to study the immuno-pathophysiological nature of SjS. In this review we will provide an in-depth analysis of predominant mouse models of SjS as well as present recent insights into the pathogenesis of SjS, including the genetic contribution and interconnection of various effector T-cell populations, specifically CD4+ TH17 memory cells.
Mouse Models of Sjs
In an effort to identify the underlying pathogenesis leading to SjS in humans, numerous spontaneous and experimentally induced mouse models exhibiting various aspects of SjS are being intensively investigated (Nguyen et al. 2007). Typically, these mouse models show lymphocyte infiltration of the exocrine glands, increased expressions of pro-inflammatory cytokines, generation of unique autoantibodies (especially anti-nuclear antigens (ANAs), anti-α-fodrin, and anti-muscarinic type II receptor (M3R)), and eventually impairment of secretory function. Strains studied extensively include MRL/lpr, NZB/NZW F1-hybrids, NOD (non-obese diabetic), and NFS/sld. Recently, several new strains have been generated, including the Id3 gene knock-out (KO) mouse, the aromatase gene KO mouse, the Baff gene knock-in mouse, as well as the IQI/Jic mouse and the C57BL/6.NOD-Aec1Aec2 congenic line. Each of these models along with their general disease profiles are listed in Table 2. Detailed description of three well-characterized mouse models, MRL/lpr, NOD/NOD.B10-H2b congenic, and C57BL/6.NOD--Aec1Aec2, are discussed below.
Table 2. Mouse models of Sjögren's syndrome-like disease.
| Mouse strains | Characteristics | Disease manifestation/phenotypes |
|---|---|---|
| MRL/lpr | Mutation of lpr gene that encodes Fas protein |
|
|
| ||
| NOD | Spontaneous insulitis and diabetes | SjS-like disease phenotype with loss of secretion, anti-M3R antibodies, and focal lymphocytic infiltration |
|
| ||
| NOD.B10-H2b | NOD with H2b from C57BL/10 | SjS-like disease phenotypes without type I diabetes |
|
| ||
| C57BL/6.NOD-Aec1Aec2 | C57BL/6 mouse carrying Aec1 (Idd3) genetic region on Chr. 3 and Aec2 (Idd5) genetic region on Chr. 1 from NOD mouse |
|
|
| ||
| (NZB/NZW) F1 | Naturally occurring mouse model by crossing NZB and NZW | Lacrimal gland involvement that shows greater percentage of B cells compared with MRL/lpr mouse |
|
| ||
| NFS/sld | Autosomal recessive gene with sublingual gland differentiation arrest | Anti-α-fodrin antibodies |
|
| ||
| Id3 gene knock-out (KO) | No Id3 (basic helix-loop-helix transcription factor) production, a dominant negative inhibitor of gene expression | Impaired TCR-mediated T-cell selection, loss of secretion and presence of anti-Ro and anti-La antibodies |
|
| ||
| Aromatase gene KO | Estrogen deficient due to absence of enzyme catalyzing the conversion of testosterone to estradiol | B cell hyperplasia in the BM and spleen and anti-α-fodrin antibodies |
|
| ||
| Baff gene knock-in | Over-expression of the B-cell survival factor, BAFF (BLys) | Lymphocytic infiltration with a majority of B cells, leading to loss of secretion by 15–17 months of age |
|
| ||
| IQI/Jic | Inbred strain originating from ICR with SjS-like disease in the absence of diabetes | Anti-kallikrein-1 and -13 antibodies |
The MRL/lpr mouse
MRL/lpr mice were originally developed as a model for the study of SLE carrying a mutated lpr gene (Andrews et al. 1978). The mutated lpr gene encodes a defective TNFSF6 (Fas) protein that results in failure of apoptosis and clonal deletion of lymphocytes in peripheral lymphoid organs (Hoffman et al. 1984; Singer and Abbas 1994; Watson et al. 1992). MRL/lpr mice develop dacryoadenitis of the lacrimal glands and sialadenitis of the submandibular, parotid, and sublingual glands (Jonsson et al. 1987). In addition, they manifest an autoantibody pattern resembling SjS patients, including anti-dsDNA, anti-ssDNA, ANA, and rheumatoid factor (Theofilopoulos and Dixon 1985). Although an early report suggested that MRL/lpr mice failed to synthesize anti-SSA/Ro and anti-SSB/La auto-antibodies (Hoffman et al. 1984), a recent study indicated that nearly 30% of mice developed anti-52 KDa SSA/Ro antibodies, 6% developed anti-60-kDa SSA/Ro antibodies, and 6% developed anti-SSB/La antibodies (Wahren et al. 1994). Although the animals manifested extensive lymphocytic infiltrations of the exocrine glands, anti-M3R antibodies, which could explain abnormal salivary secretion, were not observed (Hoffman et al. 1984). One interesting observation exemplifying the dichotomy between the disease process of lacrimal and salivary glands in this mouse model is the distinctive presence of a TH1 immune profile in the spleen, lymph node, and salivary glands (Mustafa et al. 1998; Takahashi et al 1996), whereas a TH2 profile predominated in the lacrimal glands (Jabs et al. 2000). In the absence of IL-4, the TH2 autoimmune profile converted to the TH1 profile (Jabs et al. 2007).
The NOD mouse and its NOD.B10-H2b congenic line
The inbred NOD mouse strain has been widely used as an animal model for insulin-dependent diabetes mellitus, which is also called type I diabetes (T1D) (Makino et al. 1980). During the past decade, the NOD mouse has also become a popular animal model for SjS-like disease, in part due to its many similarities to human SjS, ranging from decreased secretory function to the presence of lymphocytic infiltrates in the exocrine glands (Hu et al. 1992; Humphreys-Beher et al. 1993; Humphreys-Beher et al. 1994; Robinson et al. 1996). These SjS-like phenotypes, along with the detection of autoantibodies, make the NOD mice an ideal model for secondary SjS. Interestingly, the genetic predisposition for SjS-like disease in NOD mice is independent of or only weakly dependent on major histocompatibility (MHC)-associated genes, mimicking SjS in humans. The first indication of a non-MHC association involved the studies of the congenic strain NOD.B10-H2b, in which the NOD MHC I-Ag7 T1D susceptibility locus is replaced by the T1D non-susceptibility MHCI-Ab locus from C57BL/10 mice (Kong et al. 1998). The NOD.B10--H2b mice failed to exhibit insulitis and the development of T1D, but continued to show complete SjS-like disease manifestations, including salivary and lacrimal gland dysfunction. Thus NOD.B10-H2b mice represent a standard model for primary SjS (Kong et al. 1998).
The C57BL/6.NOD-Aec1Aec2 mouse
One major drawback of the NOD and NOD-derived mouse models is the lack of appropriate normal control mice to perform disease comparative analyses; it was therefore critical to develop a mouse model to allow separation of age- versus disease-dependent associations. While more than 19 insulin-dependent diabetes (Idd) loci have been identified that contribute to diabetes susceptibility in the NOD mouse model, only two of these loci (Idd3 and Idd5) appear to be essential for the development of sialadenitis and loss of secretory functions (Brayer et al. 2000). Introducing both the Idd3 and Idd5 genetic regions derived from NOD mice into SjS non-susceptible C57BL/6 mice resulted in the appearance of SjS-like disease, confirming the contributions of these two genetic loci to the development and onset of disease (Argueso et al. 2002; Brayer et al. 2000). In follow-up studies we have now narrowed the genetic region of Idd3 to a centromeric segment approximately 20 cM from 48.5 cM in size and Idd5 from 73.3 cM to approximately 10 cM located at 79±5 cM distal to the centromere (Nguyen et al. 2006; Nguyen et al. 2008). Extensive analysis of this mouse model revealed that the pre-clinical non-immune aspects of the disease (e.g. increased expression of caspases, matrix metalloproteinases, PSP proteolytic enzymes, increased apoptosis, and altered protein secretions) are associated with the Idd5 locus of chromosome 1. In addition, the immunological aspects of the disease (e.g. appearance of activated T and B cells, increased expression of pro-inflammatory cytokines, appearance of autoantibodies, and loss of secretory function) are associated with Idd3 of chromosome 3. However, recapitulation of the full disease profile requires genes within both these genetic loci. This SjS-susceptible C57BL/6 congenic mouse, currently carrying our designation C57BL/6.NOD--Aec1Aec2 (where Aec1 corresponds to Idd3 of the NOD mouse and Aec2 corresponds to Idd5 of the NOD mouse), offers many genetic advantages over the NOD mouse model while maintaining the NOD SjS-like disease profile in the absence of T1D susceptibility.
Etiologies of Sjs and Sjs-Like Disease
In 1933, Dr. Henrik Sjögren (1899–1986) presented data describing a cluster of women over 40 years of age in a small Swedish town presenting with keratoconjunctivitis, lymphoid infiltrations of the conjunctiva, cornea, lacrimal glands, and parotid glands, a history of arthritis, swelling of the salivary glands, and dryness of the oronasopharynx. Two years later, this observation was connected with Mikulicz's disease, and together formed the general basis for SjS. Since that time, extensive research has focused on identifying specific etiologies that may be directly involved in SjS development. A number of hypotheses have been postulated in attempting to resolve the complexity of SjS-related dry mouth and eyes, including: (a) the role of epithelial cells apoptosis, (b) the function of autoreactive T lymphocytes, (c) the role of autoreactive B cells, (d) the role of neurological dysfunction, and (e) the role of viral infection (Crouse et al. 1990; Fox et al 1992; Pflugfelder et al 1990). Other factors such as androgen deficiency (Sullivan et al. 1999a; Sullivan et al. 1999b; Sullivan 2004) and meibomian gland function (Shimazaki et al. 1998; Sullivan et al. 2002) have expanded the search for answers. New findings associated with genetic contributions to SjS and the possible involvement of the CD4+ TH17 memory T--cell system with the TH1 and TH2 system open new insights into the genetic predisposition and autoimmune aspect of SjS, and critical roles are upheld for this cell population, offering new and novel approaches for interventions.
Genetic predisposition for SjS
Two elements considered essential for the development and onset of autoimmune diseases such as SjS are an underlying genetic susceptibility and an environmental trigger for activating the immune system. Although the environmental triggers responsible for initiating SjS remain elusive, it is apparent that genes of the MHC are involved because the MHC genes dictate the basic development of the immune system and ultimately the immune responses per se. At the same time, non-MHC genes and their products function to regulate most aspects of immune responses. Not surprisingly, therefore, studies using animal models of SjS indicate that disease susceptibility is multi-genic, encompassing many critical causal elements. Whether linked to MHC or non-MHC genes, these elements remain mostly speculative (Bolstad and Jonsson 2002). This is even less clear in humans, due in part to the relatively weak tendency toward familial aggregation, perhaps due to the lack of large twin and/or large cohort studies. Nevertheless, observations reported by Harley et al. (Harley et al. 1986) suggest that SjS susceptibility is linked to HLA--DQ genes, specifically DQ1 and DQ2, when associated with the presence of anti-SS-A/Ro and anti-SS-B/La autoantibodies. Gottenberg et al. (Gottenberg et al. 2003) likewise reported that the frequency of the HLA--DRβ1*15 allele in Caucasians with SjS was 13% higher than in healthy non-diseased individuals or patients with anti-SSA/Ro antibody-positive sera. In addition, patients with both anti-SSA/Ro and anti-SSB/La serum antibodies displayed a 44% frequency of HLA-DRβ1*3 compared with 12% in patients negative for anti-La and anti-Ro, 19% in anti-Ro-positive patients, and 10% in healthy controls. In a third report there was a direct correlation between anti-La antibodies and HLA-A1, B8, DR3 (Ricchiuti et al. 1994). Considered as a whole, then, different MHC genetic linkages should be expected in different ethnic groups, a point clearly illustrated by the fact that Northern and Western European Caucasians and North Americans show a higher frequency of B8, DRw52, and DR3 genes whereas Scandinavians and Greeks show linkages predominantly to the DR2 and DR5 genes, respectively (Bolstad and Jonsson 2002). These various linkages, while of interest, may result merely from the relative homogeneity of the populations studied.
Perhaps more interesting is the increasing number of reports identifying polymorphism and expression level of non-MHC genes linked to SjS-susceptibility or a specific aspect of its pathogenesis. A few of these genes are Apoe (apolipoprotein-E) (Pertovaara et al. 2004), Car2 (carbonic anhydrase-2) (Hu et al. 2007), Clu (clusterin) (Tsubota et al. 2007), Il7r (interleukin 7 receptor) (Teutsch et al. 2003), Il10 (interleukin 10) (Nguyen et al. 2009), Lyzs (lysozyme) (Kumagai et al. 1997), Mbl (mannose-binding protein) (Wang et al. 2004), Tnfsf6 (tumor necrosis factor, soluble factor 6 or Fas) (Bolstad et al. 2000), Tnfa (tumor necrosis factor-α) (Gottenberg et al. 2004), and Gstm1 (glutathione S-transferase-1). GSTM1 has been reported to contribute to anti-SS--A autoantibody production (Morinobu et al. 1999). An additional linkage involves the Tap2 gene. TAP2 is critical for antigen processing and presentation by macrophages and dendritic cells. In an examination of 108 SjS patients in Japan, Kumagai et al. (Kumagai et al. 1997) identified a number of polymorphisms in the Tap1 and Tap2 genes, with one unique base-pair substitution at codon 577 of Tap2 showing a higher prevalence in SjS patients manifesting elevated anti-Ro autoantibody. With several large-scale SNP analysis studies just underway, genetic polymorphisms within specific genes linked to SjS-susceptibility, like that observed in Tap2, should begin to define those genes that alter a biological process that subsequently identifies the autoimmune genotype/phenotype. Additional genes, recently identified in microarray studies involving the C57BL/6.NOD--Aec1Aec2 mouse model for pSjS and hypothesized to be involved in the underlying pathology, include several chemokines, cathepsins, cystatins, complement components, proteins involved in lipid and fatty acid homeostasis, genes regulating apoptosis, and lymphotoxin-β and its receptor. These genes represent potential candidate genes whose polymorphisms should be determined in the equivalent human genes.
The role of TH1, TH2, and TH17 cells in the pathogenesis of SjS
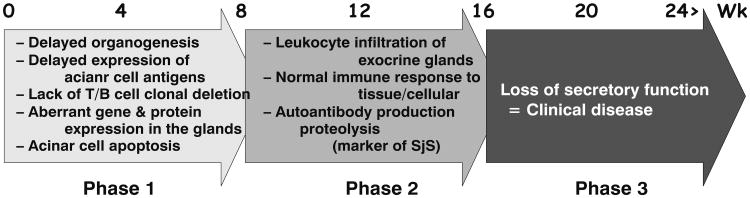
The conventional approach to simplify the complexity of many autoimmune diseases is to compartmentalize their etiologies into specific effector T-cell subsets. Despite the extensive data from the animal models mentioned above and from human subjects, it is still premature to resolve the complexity of SjS based on specific T--cell functions. Recently we proposed that SjS-like disease in our NOD and C57BL/6. NOD-Aec1Aec2 mouse models progresses through three distinct but continuous phases. In phase 1 (initiation of glandular pathology), a number of aberrant genetic, physiological, and biochemical activities associated with retarded salivary gland organogenesis and increased acinar cell apoptosis occur sequentially prior to and independent of detectable autoimmunity (Berditchevski 2001). In phase 2 (onset of autoimmunity believed to result from the acinar cell apoptosis), leukocytes expressing pro-inflammatory cytokines infiltrate the exocrine glands, thereafter establishing lymphocytic foci, first of T-cell clusters, followed by recruitment of B lymphocytes (Kong et al. 1998). In phase 3 (onset of clinical disease), loss of salivary and lacrimal gland secretory functions occur, most likely the result of antagonistic (auto)-antibodies reactive with the M3Rs (Abdul Ajees et al. 2006; Brayer et al. 2001; Kong et al. 1998; Nguyen et al. 2000). These three phases are illustrated in Fig. 1.
Fig. 1. Three phases of SjS--like disease development.
Previous studies carried out in both NOD.IFNγ−/− and NOD.IL4−/− mice revealed that both IFN-γ from TH1 cells and IL-4 from TH2 cells are critical cytokines for the development of SjS-like disease in these animal models (Brayer et al. 2001; Cha et al. 2004). TH1 cells release IFN-γ and regulate cell-mediated immunity through activation of macrophages, NK cells, and CD8+ T cells, and this process is driven by IL-12 through activating the transcription factor signal transducer and activator of transcription 4, resulting in the expression of the transcription factor T-bet. An interesting aspect of IFN-γ in SjS-like disease is that it affects organogenesis of the salivary glands by retarding cellular growth. Examination of NOD.Ifnγ−/− and NOD.IfnyR−/− mice revealed normal acinar cell proliferation and maturation as well as normal development of their salivary glands (Batten et al. 2004). Both NOD. Ifnγ−/− and NOD.IfnγR−/− mice failed to develop any aspect of SjS--like disease of the salivary glands, including acinar cell apoptosis around 8–10 weeks of age or the subsequent leukocyte infiltration of the salivary glands normally observed 10 weeks and beyond. This delay in acinar cell maturation may therefore prevent the expression of cellular antigens at the critical time of self-tolerance, resulting in the lack of proper clonal deletion. In addition to its early involvement in SjS, IFN-γ appears to be critical for the early phase of the adaptive immune response, as well. Progressive lymphocyte infiltration is often seen in salivary lacrimal glands during the development of SjS. Cells present in these infiltrates are mostly T cells, especially during the early stages of disease and again during the very late stages, with the highest numbers being CD4+ T helper cells and a lesser number of B cells. One of the critical functions of IFN--γ is the induction of glandular adhesion molecules such as vascular cell adhesion molecule-1, α4β1 integrin, peripheral node addressin (PNAd), L-selectin, and LFA-1. Recently we found that mRNA transcripts encoding CCL8, CCL19, CCL5, CXCL16 (an IFN-γ--regulated chemokine that attracts NK and memory T cells), CXCL9, and CXCL13 were all up-regulated in the salivary glands of C57BL/6.NOD-Aec1Aec2 mice at the time of disease onset (unpublished data), correlating with dendritic cell, T-cell, and B-cell infiltrations. As anticipated, elimination of IFN-γ in the disease-susceptible mice prevents the influx of lymphocytes into the glands, thereby eliminating the autoimmune response. Other TH1 cytokines such as IL-18 might also play an important role in the development of SjS. IL-18 is detected in CD68+ macrophages and ductal and acinar cells of SjS salivary glands (Bombardieri et al. 2004; Manoussakis et al. 2007; Sakai et al. 2008) and is also secreted at a significantly higher level in sera and saliva of SjS patients and NOD mice (Bombardieri et al. 2004; Delaleu et al. 2008). Therefore it is possible that IL-18 produced by activated macrophages and T cells can stimulate the production of other inflammatory cytokines, chemokines, and adhesion molecules to attract inflammatory cells to the glands.
IL-4, a product of TH2 cells, appears to be an essential factor during the early period of the adaptive immune response in SjS. Earlier studies using the NOD.IL4−/− and NOD.B10-H2b.IL4−/− mice indicated that secretory function of salivary glands was restored to normal levels when the Il4 gene was genetically knocked-out, despite the fact that these mice continued to exhibit the expected pathophysiological abnormalities and leukocyte infiltrations in the exocrine glands concomitantly with production of ANAs (Brayer et al. 2001; Gao et al. 2006). These earlier data suggested that IL-4 plays an important role during the clinical phase, while having little or no effect on the pathology associated with the preclinical disease state. IL-4 intracellular signaling can follow either the STAT-6 or IRS pathway and it appears capable of regulating physiological functions that subsequently modulate the secretory activities of exocrine glands. Interestingly, Il4 gene KO mice fail to produce IgG1 isotypic autoantibodies against M3R, yet produce normal levels of M3R of other isotypes, for example IgG2a, IgG2b, IgG3, IgM, and IgA, pointing to a possible critical role for IgG1 isotype switching (Gao 2004). This observation was further supported by studies on Stat6 gene knock-out NOD.B10-H2b mice, which renders the animal unable to produce IgG1 immunoglobulin. As the result of this specific gene elimination, the mice failed to make IgG1 isotype against M3R. More importantly, purified IgG fractions isolated from the sera of NOD.B10-H2b.Stat6−/− mice were not able to temporarily inhibit saliva flow rates when infused into naive C57BL/6 mice, while IgG fractions from the wild-type parental mice were able to reduce saliva secretion in normal C57BL/6 mice (Nguyen et al. 2007). Thus during the development of SjS, IL-4 is a critical effector cytokine, not only intimately involved in the proliferation and differentiation of B/T lymphocytes, but also actively affecting the isotypic switching mechanism to produce pathogenic IgG1 autoantibody channeling through the IL-4/STAT6 pathway.
While the above observations suggest a requirement for both TH1 and TH2 cell-associated functions for the onset of clinical disease, our recent identification of CD4+ TH17 memory cells within the lymphocytic foci present in the salivary and lacrimal glands of SjS-susceptible C57BL/6.NOD-Aec1Aec2 mice and human SjS patients indicates a much greater complexity (Nguyen et al. 2008). The TH17 cell population is a subset of CD4+ memory effector T cells that appears to be distinct and unrelated to either the TH1 or TH2 cell lineages (Bettelli et al. 2006; Harrington et al. 2005; Mangan et al. 2006; Park et al. 2005; Veldhoen et al. 2006). TH17 effector cells have the unique ability to secrete one or more cytokines belonging to the IL-17 family, a function under the control of the master transcription regulator RORγt (T cell-specific orphan nuclear receptor-γ) (Ivanov et al. 2006). Although the IL-17 family of cytokines consists of six members: IL-17A (IL-17), IL--17B, IL-17C, IL-17D, IL-17E (IL-25), and IL-17F, most attention has focused on IL-17A, especially in autoimmunity. These molecules are potent pro-inflammatory cytokines which are actively involved in tissue inflammation via induction of pro-inflammatory cytokines and chemokine expressions, such as IL-6, TNF-α, MIP-2 (macrophage inflammatory protein-2), G-CSF (granulocyte colony-stimulating factor), CXCL1, CXCL2, CXCL5, IL-21, IL-22, NO (nitric oxide), PGE2 (prostaglandin-E2), MMP-3 (matrix metalloproteinase-3), and MMP-13 (Weaver et al. 2007). As part of the local inflammatory response, IL-17 is involved in the mobilization, maturation, and migration of neutrophils via the release of IL-8 at the site of injury (Kastelein et al. 2007). Most importantly, the TH17 cell population per se has been shown to be a tissue-seeking T-cell population intimately involved in autoimmune diseases, for example Crohn's disease (Duerr et al. 2006; Hue et al. 2006), experimental autoimmune encephalomyelitis (Cua et al. 2003), collagen-induced arthritis (Cua et al. 2003), and others. It will be interesting to determine if the TH17 cell population is attracted to the exocrine glands by the chemokine CXCL16. The temporal interaction of TH1, TH2, and TH17 cells is presented in Fig. 2.
Fig. 2. A model of temporal interaction of TH1, TH2, and TH17 cells for the progression of SjS-like disease.

As stated above, our recent studies indicate the presence of the TH17 cell population within the lymphocytic infiltrations appearing in exocrine glands of both SjS patients and C57BL/6.NOD-Aec1Aec2 mice, correlating with higher levels of IL-6 and IL-17 found in the saliva and sera of a few SjS patients. We postulate that these TH17 cells contribute to tissue destruction, in part from their up-regulation of MMP activity, the latter known to be highly expressed in inflamed lacrimal and salivary glands during development of SjS and SjS-like disease. This concept is further supported by our data using a newly generated set of C57BL/6.NOD-Aec1Aec2R(n) recombinant inbred lines in which the Aec2 genetic region of C57BL/6.NOD-Aec1Aec2 mice is redefined, identifying a shortened region essential for disease. Residing within this newly defined Aec2-susceptibility locus is the gene encoding Tnfsf4 (commonly known as OX40L). Although a number of distinct cell populations express OX40L, including activated dendritic cells (Baum et al. 1994), OX40L can function as a regulatory factor for the maturation of Treg1 cells (Ito et al. 2006), a cell population that produces IL-10 and IFN-γ. The Treg1 cells, in conjunction with IL-27-producing dendritic cells, inhibit effector CD4+ TH17 cells (Bettelli et al. 2008). Since Tnfsf4 expression is down-regulated during the development of SjS-like disease, we suggest that during the development of the autoimmune attack against the exocrine gland tissues there is reduced Treg1 cell function and IL-27-producing dendritic cell function that would normally regulate CD4+ TH17 cell function, leading to the activation of effector CD4+ TH17. If this concept proves accurate, and an imbalance in the TH17:Treg1/DCIL27 ratio that favors the TH17 population(s) in concert with activities of TH1 and TH2 cell populations is identified as an important element in disease development, then there is a new area to target intervention therapies.
Conclusion
SjS is an multifaceted autoimmune disease. The complexity is compounded by the elusive genetic contribution, unexplained changes in glandular homeostasis, and an interconnection among different effector T-cell populations, especially the newly described TH17 cells, and an important involvement of B lymphocytes and antigen-presenting cells. The development and application of various animal models concomitantly with human studies will provide better insights into the pathogenesis of the disease and eventually lead to better diagnostic approaches and novel therapies for the disease.
Acknowledgments
This work was supported in part by: C.Q. Nguyen is supported by a post-doctoral fellowship from PHS grant 5T32 AR007603. We would like to thank Dr. Ammon Peck for his insights and critical reading of the manuscript.
References
- Abdul Ajees A, Gunasekaran K, Volanakis JE, et al. The structure of complement C3b provides insights into complement activation and regulation. Nature. 2006;444:221–225. doi: 10.1038/nature05258. [DOI] [PubMed] [Google Scholar]
- Ambrosetti A, Zanotti R, Pattaro C, et al. Most cases of primary salivary mucosa-associated lymphoid tissue lymphoma are associated either with Sjoegren syndrome or hepatitis C virus infection. Br J Haematol. 2004;126:43–49. doi: 10.1111/j.1365-2141.2004.04993.x. [DOI] [PubMed] [Google Scholar]
- Andrews BS, Eisenberg RA, Theofilopoulos AN, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueso P, Balaram M, Spurr-Michaud S, et al. Decreased levels of the goblet cell mucin MUC5AC in tears of patients with Sjogren syndrome. Invest Ophthalmol Vis Sci. 2002;43:1004–1011. [PubMed] [Google Scholar]
- Batten M, Fletcher C, Ng LG, et al. TNF deficiency fails to protect BAFF transgenic mice against autoimmunity and reveals a predisposition to B cell lymphoma. J Immunol. 2004;172:812–822. doi: 10.4049/jimmunol.172.2.812. [DOI] [PubMed] [Google Scholar]
- Baum PR, Gayle RB, 3rd, Ramsdell F, et al. Molecular characterization of murine and human OX40/OX40 ligand systems: identification of a human OX40 ligand as the HTLV-1--regulated protein gp34. EMBO J. 1994;13:3992–4001. doi: 10.1002/j.1460-2075.1994.tb06715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berditchevski F. Complexes of tetraspanins with integrins: more than meets the eye. J Cell Sci. 2001;114:4143–4151. doi: 10.1242/jcs.114.23.4143. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Korn T, Oukka M, et al. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad AI, Jonsson R. Genetic aspects of Sjogren's syndrome. Arthritis Res. 2002;4:353–359. doi: 10.1186/ar599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad AI, Wargelius A, Nakken B, et al. Fas and Fas ligand gene polymorphisms in primary Sjogren's syndrome. J Rheumatol. 2000;27:2397–2405. [PubMed] [Google Scholar]
- Bombardieri M, Barone F, Pittoni V, et al. Increased circulating levels and salivary gland expression of interleukin-18 in patients with Sjogren's syndrome: relationship with autoan-tibody production and lymphoid organization of the periductal inflammatory infiltrate. Arthritis Res Ther. 2004;6:R447–456. doi: 10.1186/ar1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayer JB, Cha S, Nagashima H, et al. IL-4-dependent effector phase in autoimmune exocrinopathy as defined by the NOD.IL-4-gene knockout mouse model of Sjogren's syndrome. Scand J Immunol. 2001;54:133–140. doi: 10.1046/j.1365-3083.2001.00958.x. [DOI] [PubMed] [Google Scholar]
- Brayer J, Lowry J, Cha S, et al. Alleles from chromosomes 1 and 3 of NOD mice combine to influence Sjo-gren's syndrome-like autoimmune exocrinopathy. J Rheumatol. 2000;27:1896–1904. [PubMed] [Google Scholar]
- Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol. 1998;16:545–568. doi: 10.1146/annurev.immunol.16.1.545. [DOI] [PubMed] [Google Scholar]
- Cha S, Brayer J, Gao J, et al. A dual role for interferon-gamma in the pathogenesis of Sjogren's syndrome-like autoimmune exocrinopathy in the nonobese diabetic mouse. Scand J Immunol. 2004;60:552–565. doi: 10.1111/j.0300-9475.2004.01508.x. [DOI] [PubMed] [Google Scholar]
- Crouse CA, Pflugfelder SC, Cleary T, et al. Detection of Epstein-Barr virus genomes in normal human lacrimal glands. J Clin Microbiol. 1990;28:1026–1032. doi: 10.1128/jcm.28.5.1026-1032.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- De Vita S, Boiocchi M, Sorrentino D, et al. Characterization of prelymphomatous stages of B cell lymphopro-liferation in Sjogren's syndrome. Arthritis Rheum. 1997;40:318–331. doi: 10.1002/art.1780400217. [DOI] [PubMed] [Google Scholar]
- Delaleu N, Immervoll H, Cornelius J, et al. Biomarker profiles in serum and saliva of experimental Sjogren's syndrome: associations with specific autoimmune manifestations. Arthritis Res Ther. 2008;10:R22. doi: 10.1186/ar2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RI, Kang HI. Pathogenesis of Sjogren's syndrome. Rheum Dis Clin North Am. 1992;18:517–538. [PubMed] [Google Scholar]
- Fox RI, Luppi M, Pisa P, et al. Potential role of Epstein--Barr virus in Sjogren's syndrome and rheumatoid arthritis. J Rheumatol Suppl. 1992;32:18–24. [PubMed] [Google Scholar]
- Fox RI, Michelson P. Approaches to the treatment of Sjogren's syndrome. J Rheumatol Suppl. 2000;61:15–21. [PubMed] [Google Scholar]
- Gao J. Biological functions and molecular mechanisms of the interleukin 4 signaling pathways in autoimmune exocrinopathy using the NOD.B10.H2b mouse model of Sjögren's syndrome. Department of Pathology, Immunology and Laboratory Medicine, University of Florida; Gainesville: 2004. [Google Scholar]
- Gao J, Killedar S, Cornelius JG, et al. Sjogren's syndrome in the NOD mouse model is an interleukin-4 time-dependent, antibody isotype-specific autoimmune disease. J Autoimmun. 2006;26:90–103. doi: 10.1016/j.jaut.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Gottenberg JE, Busson M, Loiseau P, et al. In primary Sjogren's syndrome, HLA class II is associated exclusively with autoantibody production and spreading of the autoimmune response. Arthritis Rheum. 2003;48:2240–2245. doi: 10.1002/art.11103. [DOI] [PubMed] [Google Scholar]
- Gottenberg JE, Busson M, Loiseau P, et al. Association of transforming growth factor beta1 and tumor necrosis factor alpha polymorphisms with anti-SSB/La antibody secretion in patients with primary Sjogren's syndrome. Arthritis Rheum. 2004;50:570–580. doi: 10.1002/art.20060. [DOI] [PubMed] [Google Scholar]
- Hansen A, Lipsky PE, Dorner T. New concepts in the pathogenesis of Sjogren syndrome: many questions, fewer answers. Curr Opin Rheumatol. 2003;15:563–570. doi: 10.1097/00002281-200309000-00007. [DOI] [PubMed] [Google Scholar]
- Harley JB, Alexander EL, Bias WB, et al. Anti-Ro (SS--A) and anti-La (SS-B) in patients with Sjogren's syndrome. Arthritis Rheum. 1986;29:196–206. doi: 10.1002/art.1780290207. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Helmick CG, Felson DT, Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- Hoffman RW, Alspaugh MA, Waggie KS, et al. Sjogren's syndrome in MRL/l and MRL/n mice. Arthritis Rheum. 1984;27:157–165. doi: 10.1002/art.1780270206. [DOI] [PubMed] [Google Scholar]
- Hu S, Wang J, Meijer J, et al. Salivary proteomic and genomic biomarkers for primary Sjogren's syndrome. Arthritis Rheum. 2007;56:3588–3600. doi: 10.1002/art.22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Nakagawa Y, Purushotham KR, et al. Functional changes in salivary glands of autoimmune disease-prone NOD mice. Am J Physiol. 1992;263:E607–614. doi: 10.1152/ajpendo.1992.263.4.E607. [DOI] [PubMed] [Google Scholar]
- Hue S, Ahern P, Buonocore S, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys-Beher MG, Brinkley L, Purushotham KR, et al. Characterization of antinuclear autoantibodies present in the serum from nonobese diabetic (NOD) mice. Clin Immunol Immunopathol. 1993;68:350–356. doi: 10.1006/clin.1993.1137. [DOI] [PubMed] [Google Scholar]
- Humphreys-Beher MG, Hu Y, Nakagawa Y, et al. Utilization of the non-obese diabetic (NOD) mouse as an animal model for the study of secondary Sjogren's syndrome. Adv Exp Med Biol. 1994;350:631–636. doi: 10.1007/978-1-4615-2417-5_105. [DOI] [PubMed] [Google Scholar]
- Ito T, Wang YH, Duramad O, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci USA. 2006;103:13138–13143. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Jabs DA, Lee B, Whittum-Hudson JA, et al. Th1 versus Th2 immune responses in autoimmune lacrimal gland disease in MRL/Mp mice. Invest Ophthalmol Vis Sci. 2000;41:826–831. [PubMed] [Google Scholar]
- Jabs DA, Prendergast RA, Campbell AL, et al. Autoimmune Th2-mediated dacryoadenitis in MRL/MpJ mice becomes Th1-mediated in IL-4 deficient MRL/MpJ mice. Invest Ophthalmol Vis Sci. 2007;48:5624–5629. doi: 10.1167/iovs.07-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson R, Tarkowski A, Backman K, et al. Sialadenitis in the MRL-l mouse: morphological and immunohistochemi-cal characterization of resident and infiltrating cells. Immunology. 1987;60:611–616. [PMC free article] [PubMed] [Google Scholar]
- Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- Kong L, Robinson CP, Peck AB, et al. Inappropriate apoptosis of salivary and lacrimal gland epithelium of immun-odeficient NOD-scid mice. Clin Exp Rheumatol. 1998;16:675–681. [PubMed] [Google Scholar]
- Kumagai S, Kanagawa S, Morinobu A. Association of a new allele of the TAP2 gene, TAP2*Bky2 (Val577), with susceptibility to Sjogren's syndrome. Arthritis Rheum. 1997;40:1685–1692. doi: 10.1002/art.1780400919. [DOI] [PubMed] [Google Scholar]
- Makino S, Kunimoto K, Muraoka Y, et al. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Manoussakis MN, Boiu S, Korkolopoulou P, et al. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren's syndrome: correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum. 2007;56:3977–3988. doi: 10.1002/art.23073. [DOI] [PubMed] [Google Scholar]
- Morinobu A, Kanagawa S, Koshiba M, et al. Association of the glutathione S-transferase M1 homozygous null genotype with susceptibility to Sjogren's syndrome in Japanese individuals. Arthritis Rheum. 1999;42:2612–2615. doi: 10.1002/1529-0131(199912)42:12<2612::AID-ANR15>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Mustafa W, Zhu J, Deng G, et al. Augmented levels of macrophage and Th1 cell-related cytokine mRNA in sub-mandibular glands of MRL/lpr mice with autoimmune siaload-enitis. Clin Exp Immunol. 1998;112:389–396. doi: 10.1046/j.1365-2249.1998.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen CQ, Cha SR, Peck AB. Sjögren's syndrome (SjS)-like disease of mice: the importance of B lymphocytes and autoantibodies. Front Biosci. 2007;12:1767–1789. doi: 10.2741/2187. [DOI] [PubMed] [Google Scholar]
- Nguyen CQ, Cornelius JG, Cooper L, et al. Identification of possible candidate genes regulating Sjögren's syndrome-associated autoimmunity: a potential role for Tnfsf4 in autoimmune exocrinopathy. Arthritis Res Ther. 2008;10:R137. doi: 10.1186/ar2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen CQ, Gao JH, Kim H, et al. IL-4-STAT6 signal transduction-dependent induction of the clinical phase of Sjogren's syndrome-like disease of the nonobese diabetic mouse. J Immunol. 2007;179:382–390. doi: 10.4049/jimmunol.179.1.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen CQ, Hu MN, Li Y, et al. Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjögren's syndrome: findings in humans and mice. Arthritis Rheum. 2008;58:734–743. doi: 10.1002/art.23214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C, Singson E, Kim JY, et al. Sjogren's syndrome-like disease of C57BL/6.NOD-Aec1 Aec2 mice: gender differences in keratoconjunctivitis sicca defined by a cross-over in the chromosome 3 Aec1 locus. Scand J Immunol. 2006;64:295–307. doi: 10.1111/j.1365-3083.2006.01828.x. [DOI] [PubMed] [Google Scholar]
- Nguyen CQ, Sharma SA, She JX, et al. differential gene expressions in the lacrimal gland during development and onset of keratoconjunctivitis sicca in Sjogren's Syndrome (SjS)-like disease of the C57BL/6.NOD-Aec1Aec2 mouse. Exp Eye Res. 2009 doi: 10.1016/j.exer.2008.10.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KH, Brayer J, Cha S, et al. Evidence for anti-muscarinic acetylcholine receptor antibody-mediated secretory dysfunction in nod mice. Arthritis Rheum. 2000;43:2297–2306. doi: 10.1002/1529-0131(200010)43:10<2297::AID-ANR18>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertovaara M, Lehtimaki T, Rontu R, et al. Presence of apolipoprotein E epsilon4 allele predisposes to early onset of primary Sjogren's syndrome. Rheumatology. 2004;43:1484–1487. doi: 10.1093/rheumatology/keh383. [DOI] [PubMed] [Google Scholar]
- Pflugfelder SC, Tseng SC, Pepose JS, et al. Epstein-Barr virus infection and immunologic dysfunction in patients with aqueous tear deficiency. Ophthalmology. 1990;97:313–323. doi: 10.1016/s0161-6420(90)32595-2. [DOI] [PubMed] [Google Scholar]
- Ramos-Casals M, Trejo O, Garcia-Carrasco M, et al. Triple association between hepatitis C virus infection, systemic autoimmune diseases, and B cell lymphoma. J Rheumatol. 2004;31:495–499. [PubMed] [Google Scholar]
- Ricchiuti V, Isenberg D, Muller S. HLA association of anti-Ro60 and anti-Ro52 antibodies in Sjogren's syndrome. J Autoimmun. 1994;7:611–621. doi: 10.1006/jaut.1994.1045. [DOI] [PubMed] [Google Scholar]
- Robinson CP, Yamamoto H, Peck AB, et al. Genetically programmed development of salivary gland abnormalities in the NOD (nonobese diabetic)-scid mouse in the absence of detectable lymphocytic infiltration: a potential trigger for sialoadenitis of NOD mice. Clin Immunol Immunopathol. 1996;79:50–59. doi: 10.1006/clin.1996.0050. [DOI] [PubMed] [Google Scholar]
- Sakai A, Sugawara Y, Kuroishi T, et al. Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren's syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J Immunol. 2008;181:2898–2906. doi: 10.4049/jimmunol.181.4.2898. [DOI] [PubMed] [Google Scholar]
- Shimazaki J, Goto E, Ono M, et al. Meibomian gland dysfunction in patients with Sjogren syndrome. Ophthalmology. 1998;105:1485–1488. doi: 10.1016/S0161-6420(98)98033-2. [DOI] [PubMed] [Google Scholar]
- Singer GG, Abbas AK. The fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1:365–371. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Sullivan DA. Sex hormones and Sjogren's syndrome. J Rheumatol Suppl. 1997;50:17–32. [PubMed] [Google Scholar]
- Sullivan DA. Androgen deficiency and dry eye syndromes. Arch Soc Esp Oftalmol. 2004;79:49–50. [PubMed] [Google Scholar]
- Sullivan DA, Krenzer KL, Sullivan BD, et al. Does androgen insufficiency cause lacrimal gland inflammation and aqueous tear deficiency? Invest Ophthalmol Vis Sci. 1999;40:1261–1265. [PubMed] [Google Scholar]
- Sullivan DA, Sullivan BD, Evans JE, et al. Androgen deficiency, Meibomian gland dysfunction, and evaporative dry eye. Ann N Y Acad Sci. 2002;966:211–222. doi: 10.1111/j.1749-6632.2002.tb04217.x. [DOI] [PubMed] [Google Scholar]
- Sullivan DA, Wickham LA, Rocha EM, et al. Androgens and dry eye in Sjogren's syndrome. Ann N Y Acad Sci. 1999;876:312–324. doi: 10.1111/j.1749-6632.1999.tb07656.x. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Fossati L, Iwamoto M, et al. Imbalance towards Th1 predominance is associated with acceleration of lupus-like autoimmune syndrome in MRL mice. J Clin Invest. 1996;97:1597–1604. doi: 10.1172/JCI118584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teutsch SM, Booth DR, Bennetts BH, et al. Identification of 11 novel and common single nucleotide polymorphisms in the interleukin-7 receptor-alpha gene and their associations with multiple sclerosis. Eur J Hum Genet. 2003;11:509–515. doi: 10.1038/sj.ejhg.5200994. [DOI] [PubMed] [Google Scholar]
- Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–239. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- Toda I, Wickham LA, Sullivan DA. Gender and androgen treatment influence the expression of proto-oncogenes and apoptotic factors in lacrimal and salivary tissues of MRL/lpr mice. Clin Immunol Immunopathol. 1998;86:59–71. doi: 10.1006/clin.1997.4466. [DOI] [PubMed] [Google Scholar]
- Tsubota K, Mishima K, Obara K, et al. Tear Film and Ocular Surface 2007. Taormina, Italy: 2007. Reactive oxygen species can be controlled by the secretory glycoprotein, clus-terin, from side population cells in the lacrimal gland: a new intervention for age-related dry eye disorders. [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Voulgarelis M, Moutsopoulos HM. Lymphoproliferation in autoimmunity and Sjogren's syndrome. Curr Rheumatol Rep. 2003;5:317–323. doi: 10.1007/s11926-003-0011-y. [DOI] [PubMed] [Google Scholar]
- Wahren M, Skarstein K, Blange I, et al. MRL/lpr mice produce anti-Ro 52,000 MW antibodies: detection, analysis of specificity and site of production. Immunology. 1994;83:9–15. [PMC free article] [PubMed] [Google Scholar]
- Wang H, Nakamura K, Inoue T, et al. Mannose-binding lectin polymorphisms in patients with Behcet's disease. J Dermatol Sci. 2004;36:115–117. doi: 10.1016/j.jdermsci.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Watson ML, Rao JK, Gilkeson GS, et al. Genetic analysis of MRL-lpr mice: relationship of the Fas apoptosis gene to disease manifestations and renal disease-modifying loci. J Exp Med. 1992;176:1645–1656. doi: 10.1084/jem.176.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, et al. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]