

Abstract
Neuroblastoma continues to be a devastating childhood solid tumor and is responsible for over 15% of all childhood cancer related deaths. Focal adhesion kinase (FAK) and vascular endothelial growth factor receptor-3 (VEGFR-3) are protein tyrosine kinases that are overexpressed in a number of human cancers, including neuroblastoma. These two kinases can directly interact and provide survival signals to cancer cells. In this study, we utilized siRNA to VEGFR-3 to demonstrate the biologic importance of this kinase in neuroblastoma cell survival. We also used confocal microscopy and immunoprecipitation to show that FAK and VEGFR-3 bind in neuroblastoma. Finally, employing a 12-amino-acid peptide (AV3) specific to VEGFR-3, we showed that the colocalization between FAK and VEGFR-3 could be disrupted, and that disruption resulted in decreased neuroblastoma cell survival. These studies provide insight to the FAK - VEGFR-3 interaction in neuroblastoma and demonstrate its importance in this tumor type. Focusing upon the FAK - VEGFR-3 interaction may provide a novel therapeutic target for the development of new strategies for treatment of neuroblastoma.
Keywords: AV3, FLT-4, Pediatric, PTK-2, neuroblastoma
Introduction
Neuroblastoma, a tumor of neural crest origin, is the most common extracranial solid tumor of childhood. This tumor carries a dismal prognosis in children presenting with advanced stage disease, with a survival of less than 30% [1]. Some progress has been made with neuroblastoma towards improved outcomes, but successful treatment of this disease will require the development of novel therapies.
Focal adhesion kinase (FAK) is a nonreceptor cytoplasmic 125kDa protein tyrosine kinase. FAK has been shown to be important in the tumorigenesis of a number of human tumors, controlling cell signaling pathways involved in cellular proliferation, viability, motility, and survival [2,3,4,5]. The inhibition of FAK with antisense oligonucleotides or a dominant-negative FAK protein (FAK-CD) has been shown to cause decreased growth in human breast cancer cells and melanoma cells [6,7,8,9]. Silencing FAK expression with small interfering RNAs resulted in decreased migration of lung cancer cells [10] and glioblastoma cells [11]. Finally, FAK inhibition has been shown to lead to apoptosis in human fibroblasts [12] and ovarian cancer cells [13]. Our initial studies have revealed that both the abundance of FAK mRNA and the expression of FAK protein are significantly increased in aggressive human neuroblastoma tumors [14] and human neuroblastoma cell lines [15], and inhibition of FAK results in decreased neuroblastoma survival [15,16,17].
Vascular endothelial growth factor receptor-3 (VEGFR-3) is a receptor tyrosine kinase that is important in normal lymphangiogenesis and in the development of the cardiovascular system [18,19]. VEGFR-3 is modulated by its ligands vascular endothelial growth factors C and D [20]. Recent evidence shows that VEGFR-3 plays a key role in tumor lymphangiogenesis and promotes the lymphatic spread of breast [21], non-small cell lung [22], bladder [23], and prostate cancer [24] and is involved in tumor angiogenesis [25]. Inhibition of VEGFR-3 function with competing antibodies has been shown to decrease the growth of several human tumor xenografts [26]. Previously, we noted that VEGFR-3 mRNA is differentially expressed in human neuroblastoma cells in vitro [27,28]. Recently, Lagodny and colleagues demonstrated that lymphangiogenesis was involved in neuroblastoma tumors and that the ligands VEGF-C and VEGF-D were expressed in some neuroblastoma cell lines [29]. In addition, the work of Rossler and colleagues summarized data regarding the importance of angiogenesis in neuroblastoma, especially in advanced stage and aggressive disease [30]. Since neuroblastoma has been shown to express VEGFR-3, we wished to determine if abrogation of VEGFR-3 would result in decreased cell survival in vitro.
Recent data suggested that FAK and VEGFR-3 bind and promote tumor cell survival [31,32], and we hypothesized that they would also bind in neuroblastoma. Furthermore, we believed that the disruption of the FAK and VEGFR-3 interaction in neuroblastoma would lead to decreased tumor cell survival. The data from the current study showed that inhibition of VEGFR-3 with siRNA resulted in decreased neuroblastoma cell survival, and that FAK and VEGFR-3 interact in select human neuroblastoma cell lines. The current study also demonstrated that targeting the FAK and VEGFR-3 interaction with the peptide, AV3, derived from the binding site of VEGFR-3, resulted in neuroblastoma cell detachment and apoptosis. The results of this study demonstrated that interfering with the interaction between FAK and VEGFR-3 may be a potential target for novel therapeutics for neuroblastoma.
Materials and Methods
Cells and cell culture
Human neuroblastoma cells lines, SK-N-AS (CRL-2137, American Type Culture Collection, ATCC, Manassas, VA) and SK-N-BE(2) (CRL-2271, ATCC) were maintained as follows: SK-N-AS in Dulbecco's modified Eagle's medium with 4 mM L-glutamine, 0.1 mM nonessential amino acids, 1 µg/mL penicillin and 1 µg/mL streptomycin, and 10% fetal bovine serum (FBS),and SK-N-BE(2) in a 1:1 mixture of MEM and F12 medium with 1 µg/mL penicillin, 1 µg/mL streptomycin, and 10% FBS.
Antibodies and reagents
Antibodies used for Western blotting were as follows. Phospho-specific VEGFR-3 antibody (pc460, rabbit polyclonal) was from Calbiochem (EMD Millipore, Billerica, MA). Antibodies for VEGFR-3 were either from Millipore (MAB3757, clone 9D9F9) or Santa Cruz (sc-321, rabbit polyclonal, Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Polyclonal rabbit antibody for cleaved PARP (9542s) was from Cell Signaling (Cell Signaling Technology, Inc., Danvers, MA). Monoclonal anti-FAK (4.47) and rabbit polyclonal anti-phospho-FAK (Y397) antibodies were obtained from Millipore (05–537), and Invitrogen (4624G, Invitrogen Corp., Carlsbad, CA), respectively. GAPDH and β-actin antibodies were from Santa Cruz.
Antibodies used for immunofluorescence were as listed: primary antibody to VEGFR-3 was a rabbit polyclonal (sc-321, 1:1000 dilution, Santa Cruz), and to FAK (4.47) was a mouse monoclonal (05–537, 1:1000 dilution, Millipore). Secondary antibodies for immunofluorescence were from Invitrogen and included goat anti-rabbit Alexa Fluor 488 (A-11008, 1:200 dilution) and goat anti-mouse Alexa Fluor 594 (A-21044, 1:200 dilution).
Recombinant human VEGF-C was from R&D Systems (Cys156Ser, Minneapolis, MN).
AV3 peptide and control have been described previously [32]. AV3 is a 12 amino-acid peptide with a sequence (WHWRPWTPCKMF) that is homologous to VEGFR-3. This peptide was identified through phage display. The association between the NH(2) terminus of VEGFR-3, containing AV3 sequence, and the COOH terminus of FAK was noted by both in vitro and in vivo binding studies, confirming its specificity [32]. The AV3 peptide was coupled to a TAT sequence (YGRKKRRQRRR) to allow for cellular penetration. The TAT sequence alone was utilized as a control.
Immunoprecipitation and Western blotting
For immunoprecipitation, cells were lysed with NP40 lysis buffer, and total protein was precleared at 4 °C for 1 hour with A/G-agarose beads (Calbiochem) followed by incubation with 5µg of antibody overnight and a 2 hour incubation with A/G-agarose beads at 4 °C. Precipitates were washed and the beads were resuspended in SDS-PAGE sample loading buffer, boiled for 5 minutes, and resolved by SDS-PAGE. Membranes were probed with the appropriate antibody and detected with chemiluminescence using Amersham ECL Western blotting detection reagents (GE Healthcare Life Sciences, Piscataway, NJ).
Western blots were performed as previously described [15,33]. Briefly, cells were treated with the agent under study, then lysed on ice for 30 min in a buffer containing 50mM Tris-HCL, (pH 7.5), 150 mM NaCl, 1% Triton-X, 0.5% NaDOC, 0.1% SDS, 5mM EDTA, 50mM NaF, 1 mM NaVO3, 10% glycerol, and protease inhibitors: 10 µg/mL leupeptin, 10 µg/mL PMSF and 1 µg/mL aprotinin. The lysates were cleared by centrifugation at 10 000 rpm for 30 min at 4 °C. Protein concentrations were determined using a Bio-Rad kit (Bio-Rad, Hercules, CA) and proteins were separated by electrophoresis on SDS-PAGE gels. Antibodies were used according to manufacturer's recommended conditions. Molecular weight markers were used to confirm the expected size of the target proteins. Immunoblots were developed with chemiluminescence Amersham ECL Western blotting detection reagents (GE Healthcare Life Sciences). Blots were stripped with stripping solution (Bio-Rad) at 37 °C for 15 minutes and then reprobed with selected antibodies. Immunoblotting with antibody to β-actin or GAPDH provided an internal control for equal protein loading.
siRNA transfection
Small interfering RNA’s (siRNA) were obtained from Dharmacon (Thermo Fisher Scientific, Lafayette, CO) and used as previously described [15]. Briefly, cells were plated and allowed to attach for 24 hours. The cells were transfected with Lipofectamine 2000 (Invitrogen) alone, Lipofectamine 2000 (Invitrogen) plus control GAPDH siRNA (ON-TARGET plus siRNA GAPDH, 0.14 µM, Dharmacon), or Lipofectamine 2000 (Invitrogen) plus VEGFR-3 siRNA (SMARTpool FLT4, 0.14 µM, Dharmacon) according to manufacturer's protocol. Cells were incubated 48 hours after transfection and then used for experiments.
Cell viability assays
Equal numbers of cells were plated and allowed to attach for 24 hours. Cells were treated with siRNA, AV3, or control peptide as described. Cellular viability was measured using trypan blue exclusion and cell counting with a hemacytometer. Viability was further measured with MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assays [15]. In brief, 5 × 103 cells/well were plated and allowed to attach for 24 hours. Following treatment with siRNA or peptides (48 hours), 100 µg of MTT was added to each well and the absorbance at 590 nm was measured using a kinetic microplate reader (Vmax, Molecular Devices, LLC, Sunnyvale, CA).
Cellular detachment assay
Equal numbers of cells were plated and allowed to attach for 24 hours. The cells were then treated with AV3 or control peptide as described. After 48 hours of treatment, detached and adherent cells were collected separately and counted with a hemacytometer. The percentage of detached cells was determined (detached / attached + detached × 100) and reported as change in percent detached cells.
Apoptosis assays
Apoptosis was determined by two methods. Following treatment with peptides for 48 hours, cells were stained with Hoechst 33258 as previously described [33,34]. All cells, both floating and adherent were harvested, fixed to a glass slide and stained with Hoechst 33258. Hoechst positive cells were counted with fluorescence microscopy and the percentage of apoptotic cells was calculated in three independent fields with 100 nuclei per field.
Apoptosis was also detected by immunoblotting for cleaved PARP expression. Cells were treated as described, lysates were collected, and immunoblotting for cleaved PARP was performed. Bands were detected by chemiluminescence and β-actin or GAPDH served as an internal control.
Immunofluorescence
Cells were plated on glass chamber slides and allowed to attach for 24 hours, treated for 24 hours, then fixed with 3% paraformaldehyde. Cells were permeablized with 0.15% Triton X-100 and the first primary antibody (anti-VEGFR-3, C-20, Santa Cruz) was added and incubated at room temperature (RT) for 1 hour followed by the addition of the second primary antibody (anti-FAK 4.47, Millipore) that was also incubated for 1 hour at RT. The Alexa Fluor 488 secondary antibody was added for 45 minutes at RT. After washing, the second secondary antibody, Alexa 594, was added and incubated as above. Prolong Gold antifade reagent with DAPI (P36931, Invitrogen) was used for mounting. Imaging was performed with a Zeiss LSM 710 Confocal Scanning Microscope with Zen 2008 software (Carl Zeiss MicroImaging, LLC, Thornwood, NY) using a 63X objective with a zoom of 0.9.
Data analysis
Densitometry of immunoblots was performed utilizing Scion Image Program (http://www.nist.gov/lispix/imlab/prelim/dnld.html). Determination of confocal colocalization was completed using Meta Imaging Series Software (Version 7.6, Analytical Technologies, Molecular Devices). Experiments were repeated at least in triplicate, and data were reported as mean ± standard error of the mean. An ANOVA or student’s t-test was used as appropriate to compare data between groups. Statistical significance was determined at P<0.05.
Results
Vascular endothelial growth factor receptor-3 (VEGFR-3) was present in human neuroblastoma cell lines
The expression of VEGFR-3 and its ligand VEGF-C by human neuroblastoma tumors or cell lines has been reported in the literature [27,29,35,36]. Since we were planning on examining the FAK - VEGFR-3 interaction, our choice of cell lines was based upon our previous data in neuroblastoma. Previously we have shown that neuroblastoma cell lines with MYCN amplification have greater FAK expression and are more sensitive to FAK inhibition than MYCN non-amplified cell lines [15]. Therefore, for our investigations we chose the SK-N-AS cell line that has lower FAK expression [15,17,33] (Fig. 1A) and was MYCN non-amplified [37] (Fig. 1B) and the SK-N-BE(2) cell line that has higher FAK expression [15,17,33] (Fig. 1A) and was MYCN amplified [38] (Fig. 1B). We confirmed the expression of VEGFR-3 in these two human neuroblastoma cell lines with immunoblotting (Fig. 1C). We noted that these cell lines had differing expression of VEGFR-3, that is, less in the SK-N-AS cells and more in the SK-N-BE(2) cells (Fig. 1C). Variations in VEGFR-3 expression at the mRNA (Supplemental Data Fig. 1A) and protein level (Supplemental Data Fig. 1B) were also seen in other neuroblastoma cell lines. However, studies in isogenic MYCN +/− cell lines did not show a relation between MYCN expression and VEGFR-3 (Supplemental Data Fig. 1C). Immunoblotting also demonstrated the presence of phosphorylated VEGFR-3 protein in the SK-N-AS and SK-N-BE(2) neuroblastoma cell lines (Fig. 1C), which may point to the physiological activity and significance of VEGFR-3 in neuroblastoma cells.
Figure 1. Expression of vascular endothelial growth factor receptor-3 (VEGFR-3) in human neuroblastoma cell lines.
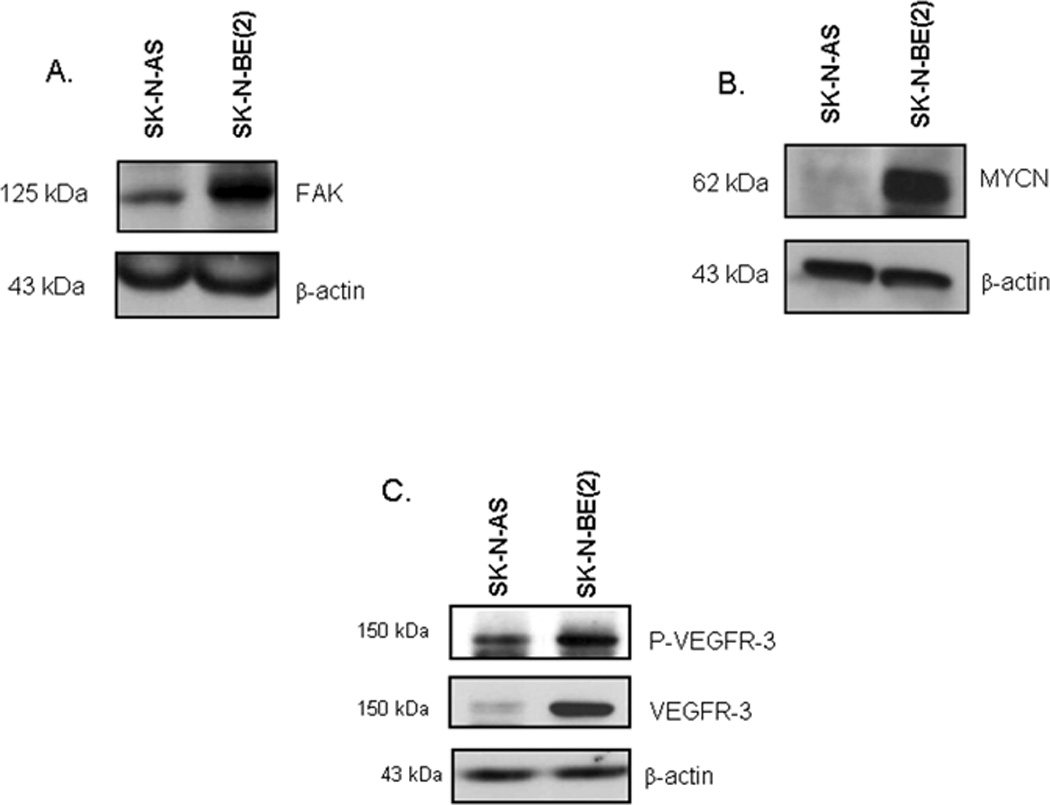
Two neuroblastoma cell lines were chosen for study, SK-N-AS (non-amplified MYCN) and SK-N-BE(2) (amplified MYCN). A. Immunoblotting for FAK revealed that these two cell lines had differing FAK expression, with the SK-N-BE(2) cells having stronger FAK expression than the SK-N-AS cells. B. Western blotting for MYCN showed more MYCN protein in the MYCN amplified SK-N-BE(2) cell line compared to the non-amplified SK-N-AS cells. C. Immunoblotting was performed and revealed the presence of VEGFR-3 protein and phosphorylation in these two cell lines, and that the two cell lines had differing VEGFR-3 expression.
Ligand induced phosphorylation of VEGFR-3 in neuroblastoma cell lines
VEGF-C, and specifically VEGF-C156S, is the ligand associated with VEGFR-3, and has been shown to activate VEGFR-3 [39]. Both the abundance of VEGF-C mRNA and expression of VEGF-C by immunohistochemistry have been shown to correlate with higher clinical stage in neuroblastoma [35,36,40]. Therefore, we wanted to investigate whether stimulation with exogenous VEGF-C would have an effect upon the phosphorylation of VEGFR-3 in the SK-N-AS and SK-N-BE(2) neuroblastoma cell lines. These two cell lines were treated with recombinant human VEGF-C156S (1 nM) for 0, 5, 15, 30 and 60 minutes. The SK-N-AS cell line showed an increase in VEGFR-3 phosphorylation after 15 minutes of exposure to VEGF-C156S which was not sustained over time (Fig. 2A). These findings were confirmed by densitometry evaluation of immunoblots (Fig. 2B). In contrast, the SK-N-BE(2) cells had an early and sustained increase in VEGFR-3 phosphorylation noted after 5 minutes of treatment with VEGF-C156S (Fig. 3A). Again, densitometry of the immunoblots confirmed this evaluation (Fig. 3B). The VEGF-C156S ligand did not affect the expression of FAK or its phosphorylation in either cell line (Supplemental Data, Fig. 2). These data showed that VEGF-C156S (ligand) led to receptor (VEGFR-3) phosphorylation in these two neuroblastoma cell lines, and strengthened our hypothesis that VEGFR-3 receptor had physiologic significance for neuroblastoma cells.
Figure 2. Activation of VEGFR-3 in SK-N-AS neuroblastoma cells by VEGF-C156S.
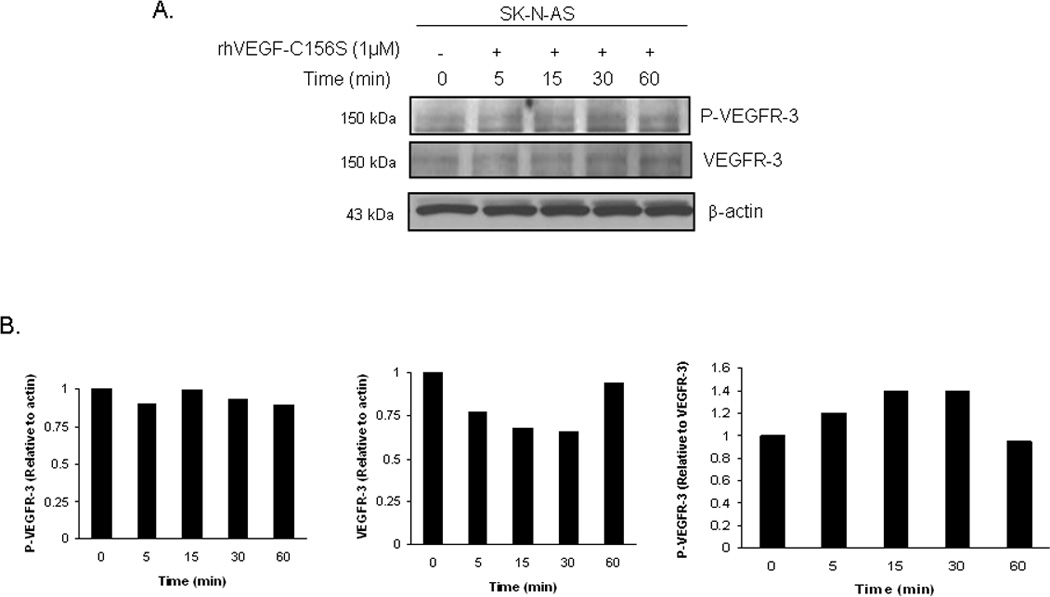
A. SK-N-AS cells were treated with VEGF-C156S (1 nM), a specific ligand for VEGFR-3, for increasing time periods. Phosphorylation of VEGFR-3 was detected by immunoblotting. There was an increase in phosphorylation of VEGFR-3 seen after 15 minutes. B. Densitometry was done on immunoblots comparing the bands for phosphorylated VEGFR-3 to total VEGFR-3, after both were normalized to β-actin bands. These data further illustrated the findings of ligand-induced phosphorylation.
Figure 3. Activation of VEGFR-3 in SK-N-BE(2) neuroblastoma cells by VEGF-C156S.
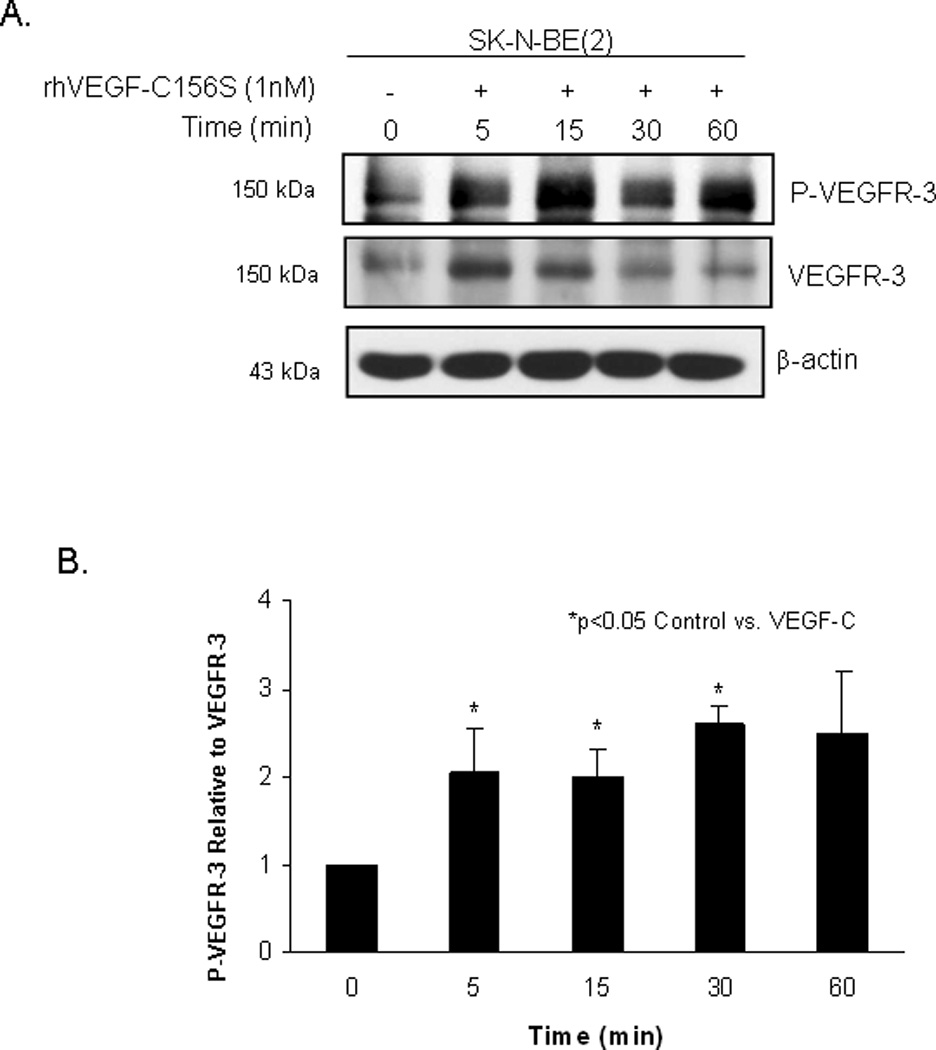
A. SK-N-BE(2) cells were treated with VEGF-C156S (1 nM) for increasing periods of time. Phosphorylation of VEGFR-3 and VEGFR-3 were detected by immunoblotting. There was an increase in VEGFR-3 phosphorylation seen after 5 minutes. B. Densitometry was done on the immunoblots to compare the bands for phosphorylated VEGFR-3 to total VEGFR-3, after both were normalized to the bands for β-actin. These data were presented to further illustrate the findings of ligand-induced phosphorylation in the SK-N-BE(2) cell line.
Downregulation of VEGFR-3 with siRNA led to decreased viability of neuroblastoma cells
Previous studies have shown that inhibition of VEGFR-3 resulted in decreased tumor progression in a variety of tumors [41,42,43,44]. Since VEGFR-3 was present in our cell lines, we wanted to determine if its inhibition would have a biologic effect upon the cells. Transfection of the SK-N-AS and SK-N-BE(2) cell lines with VEGFR-3 siRNA resulted in decreased expression of VEGFR-3 by both cell lines (Fig. 4A, B), with the control siRNA having nominal effects upon VEGFR-3 expression in either cell line (Fig. 4A, B). Inhibition of VEGFR-3 expression resulted in a decrease in the relative viability of the SK-N-AS cells that was statistically significant (1 vs. 0.84 ± 0.02, p ≤ 0.01, control siRNA vs. VEGFR-3 siRNA) (Fig. 4C, left panel). Similarly, RNAi inhibition of VEGFR-3 in the SK-N-BE(2) cell line also led to a small, but statistically significant decrease in cell viability (1 vs. 0.92 ± 0.002, p ≤ 0.01, control siRNA vs. VEGFR-3 siRNA) (Fig. 4C, right panel). These results, did however, demonstrate that VEGFR-3 has a biologic significance for these neuroblastoma cell lines.
Figure 4. Decreased neuroblastoma cell viability with siRNA inhibition of vascular endothelial growth factor receptor-3 (VEGFR-3).
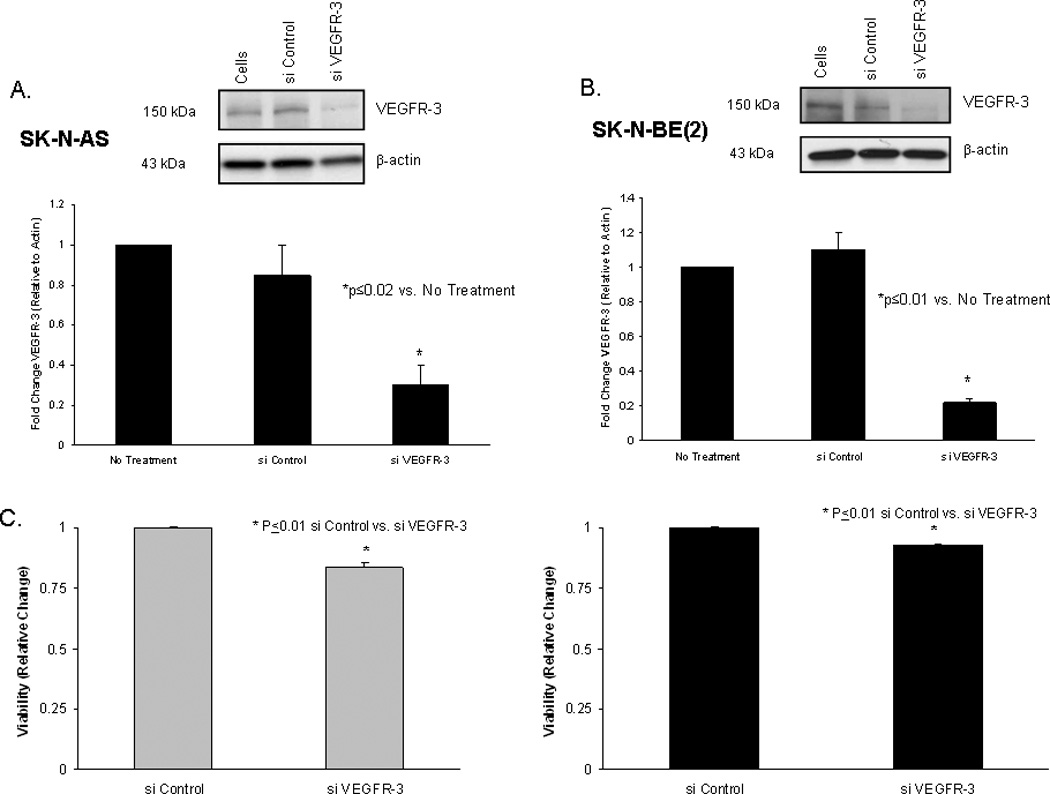
A. VEGFR-3 siRNA (0.14 µM) transfection for 48 hours resulted in decreased VEGFR-3 expression in SK-N-AS neuroblastoma cells compared to control siRNA (top panel). Densitometry was used to further show the differences (bottom panel) with VEGFR-3 expression normalized to β-actin and reported as a fold change from cells with no treatment. B. The same RNAi treatment of the SK-N-BE(2) cell line resulted in a decrease in VEGFR-3 expression compared to control siRNA (top panel). Densitometry was used to further show the differences (bottom panel) with VEGFR-3 expression normalized to β-actin and reported as a fold change from cells with no treatment. C. Cell viability was detected with MTT assay. Viability of SK-N-AS cells was significantly decreased with 48 hours of siRNA inhibition of VEGFR-3 (left panel). Treatment of SK-N-BE(2) cells with VEGFR-3 siRNA for 48 hours also significantly decreased viability in these cells (right panel).
Association of FAK and VEGFR-3 in neuroblastoma cell lines
Previous publications have documented that FAK and VEGFR-3 interact, leading to a survival advantage for cancer cells [31,32]. We have previously shown that FAK was differentially expressed and provided a survival advantage in neuroblastoma cell lines [14,15]. Since both FAK and VEGFR-3 were present in our cell lines (Fig. 1), we hypothesized that they may also interact in neuroblastoma cell lines. Immunoprecipitation in both directions (FAK and VEGFR-3 antibodies) was used to determine whether FAK and VEGFR-3 interaction was present in these cell lines. Immunoprecipitation with FAK antibody followed by immunoblotting for VEGFR-3 showed co-precipitation of VEGFR-3 protein with FAK, so an association between these two proteins was present in both cell lines (Fig. 5A). This association was also detected by immunoprecipitation with VEGFR-3 antibody followed by immunoblotting for FAK (Fig. 5C). Immunoblotting of the membranes with their respective IP antibody confirmed the immunoprecipitation of the correct protein (Fig. 5B, D). These data demonstrated that FAK and VEGFR-3 associate in neuroblastoma cells.
Figure 5. Interaction of VEGFR-3 and FAK in neuroblastoma cell lines.
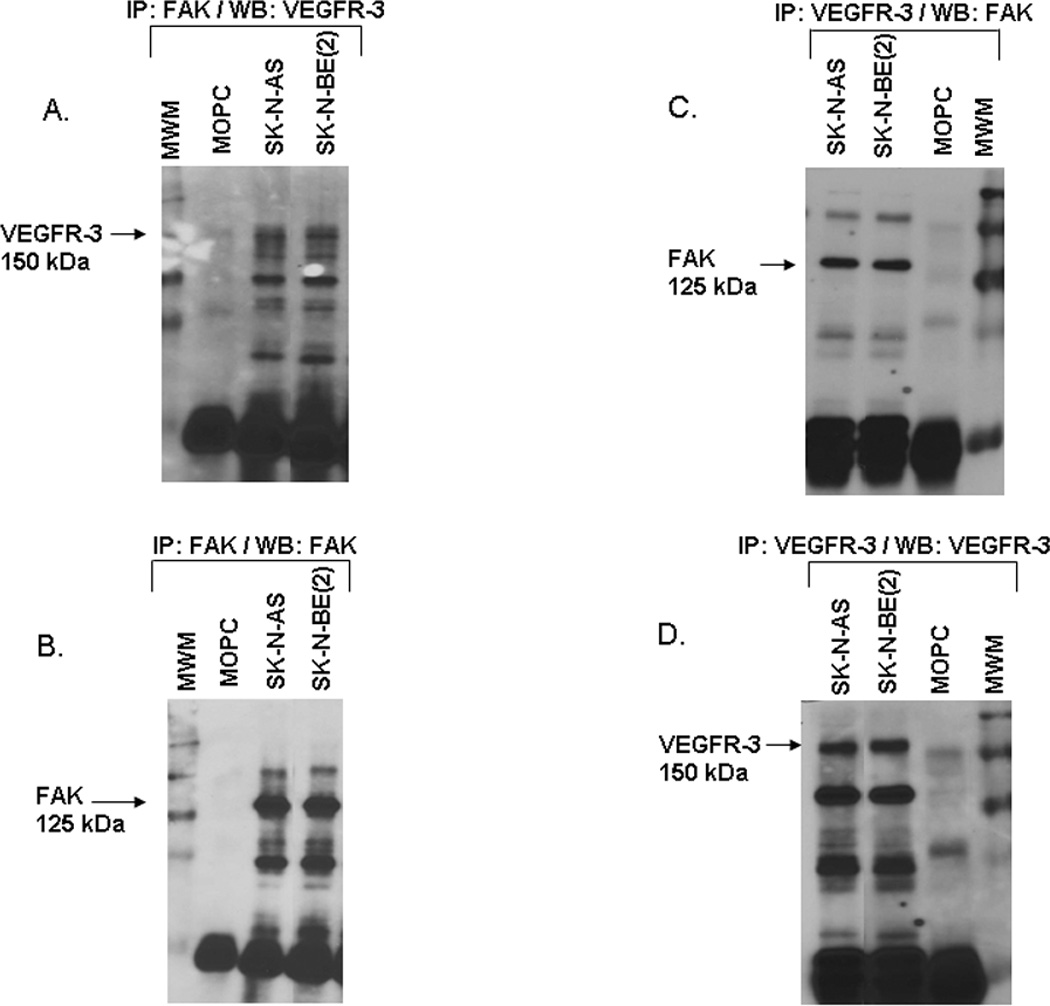
A. Immunoprecipitation followed by Western blotting was used to detect FAK and VEGFR-3 interaction in the neuroblastoma cell lines. Immunoprecipitation with FAK antibody followed by Western blotting for VEGFR-3 showed a band for VEGFR-3 at the expected 150 kDa in both cell lines. B. Western blotting of that same membrane for FAK, revealed a FAK band present at 125 kDa. C. Immunoprecipitation with VEGFR-3 antibody followed by Western blotting for FAK showed a band for FAK at the expected 125 kDa in both cell lines. D. Western blotting of the same membrane for VEGFR-3, detected a band at the expected 150 kDa. These data showed that FAK and VEGFR-3 interact in both neuroblastoma cell lines.
AV3 peptide treatment caused decreased FAK phosphorylation and loss of FAK from the focal adhesions
Previous studies have shown that peptide AV3, a twelve amino acid fragment of VEGFR-3, derived from the binding site with FAK, disrupted the interaction of FAK and VEGFR-3, and resulted in decreased survival in BT474 breast cancer cells [32]. Since we found that FAK and VEGFR-3 also interacted in our neuroblastoma cell lines (Fig. 5C), we sought to characterize the effects of disrupting this interaction. We chose to utilize the AV3 peptide, and again used the neuroblastoma cell lines, SK-N-AS and SK-N-BE(2). We first examined the biochemical effects of AV3 treatment. The neuroblastoma cell lines were treated with AV3 (15.5 µM) or control TAT (15.5 µM) peptides, and immunoblotting was used to detect FAK and VEGFR-3 expression and phosphorylation. AV3 treatment of SK-N-AS cells resulted in minimal change in expression or phosphorylation of FAK or VEGFR-3 (Fig. 6A). However, in the SK-N-BE(2) cell line, AV3 treatment decreased Y397 phosphorylation of FAK (Fig. 6B) and also decreased total VEGFR-3 expression (Fig. 6B). This decrease in total VEGFR-3 expression may have been related to protein degradation correlating with apoptosis from the AV3 treatment. Treatment with control TAT peptide did not affect FAK or VEGFR-3 expression or phosphorylation in either cell line (Fig. 6A, B). Next, dual immunofluorescence staining and confocal microscopy were employed to examine the effects of AV3 upon the FAK and VEGFR-3 association (Fig. 7, Fig. 8). In the SK-N-AS cell line, FAK remained in the focal adhesions (Fig. 7, white arrows bottom fourth panel) after AV3 treatment, resulting in little change in the dual staining of VEGFR-3 and FAK (Fig. 7, bottom third and fourth panels). In contrast, in the SK-N-BE(2) cell line, AV3 treatment resulted in a loss of FAK from the focal adhesions (Fig. 8, bottom third and fourth panels) and loss of colocoalization of FAK and VEGFR-3 staining (Fig. 8, bottom third and fourth panels). Treatment with the control TAT peptide had no effect upon FAK at the focal adhesions in either cell line (Fig. 7, white arrows middle fourth panel and Fig. 8, white arrows middle fourth panel). Manders’ overlap coefficients were calculated for each of the treatment groups (Fig. 9) [45]. The overlap coefficients for the FAK and VEGFR-3 staining were significantly decreased compared to those of control cells after AV3 treatment in the SK-N-BE(2) cell line, but remained unchanged in the SK-N-AS cells (Fig. 9). Again, the overlap in both cell lines was unaffected by TAT treatment alone (Fig. 9). These results demonstrated that AV3 peptide treatment of the SK-N-BE(2) neuroblastoma cell line resulted in a loss of FAK from the focal adhesions and a decrease in FAK and VEGFR-3 association.
Figure 6. AV3 peptide treatment resulted in decreased FAK phosphorylation and decreased VEGFR-3 expression in SK-N-BE(2) cells.
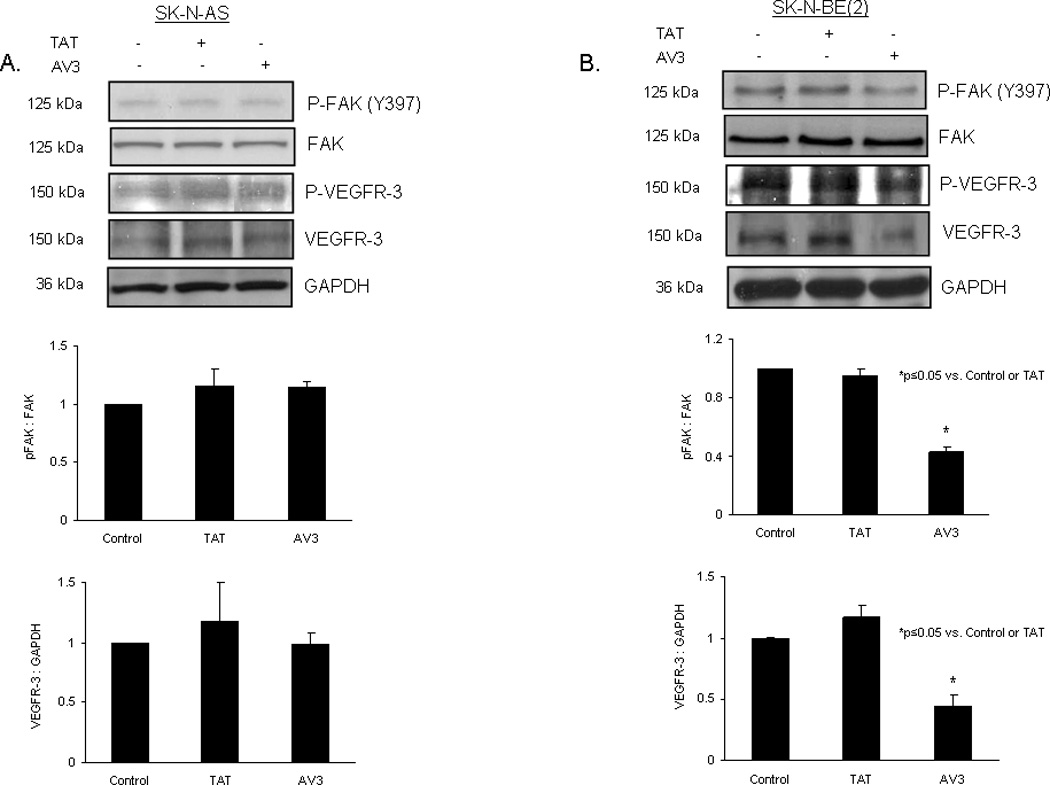
AV3 is a small peptide known to disrupt the binding between FAK and VEGFR-3. Immunoblotting was used to detect FAK and VEGFR-3 phosphorylation and expression following treatment with AV3 or control (TAT) peptide, and immunoblots were analyzed by densitometry. A. In the SK-N-AS cells, AV3 treatment led to little change in phosphorylation of FAK (top blot and middle panel) or total VEGFR-3 (fourth blot and bottom panel) after 24 hours treatment. B. In the SK-N-BE(2) cells, AV3 peptide treatment resulted in a decrease in Y397 FAK phosphorylation (top blot and middle panel), and a decrease in total VEGFR-3 expression after 24 hours (fourth blot and bottom panel). Phosphorylated FAK was presented in relation to total FAK and total VEGFR-3 was presented relative to GAPDH in the densitometry graphs.
Figure 7. AV3 peptide treatment did not result in a loss of FAK from the focal adhesions in the SK-N-AS cell line.
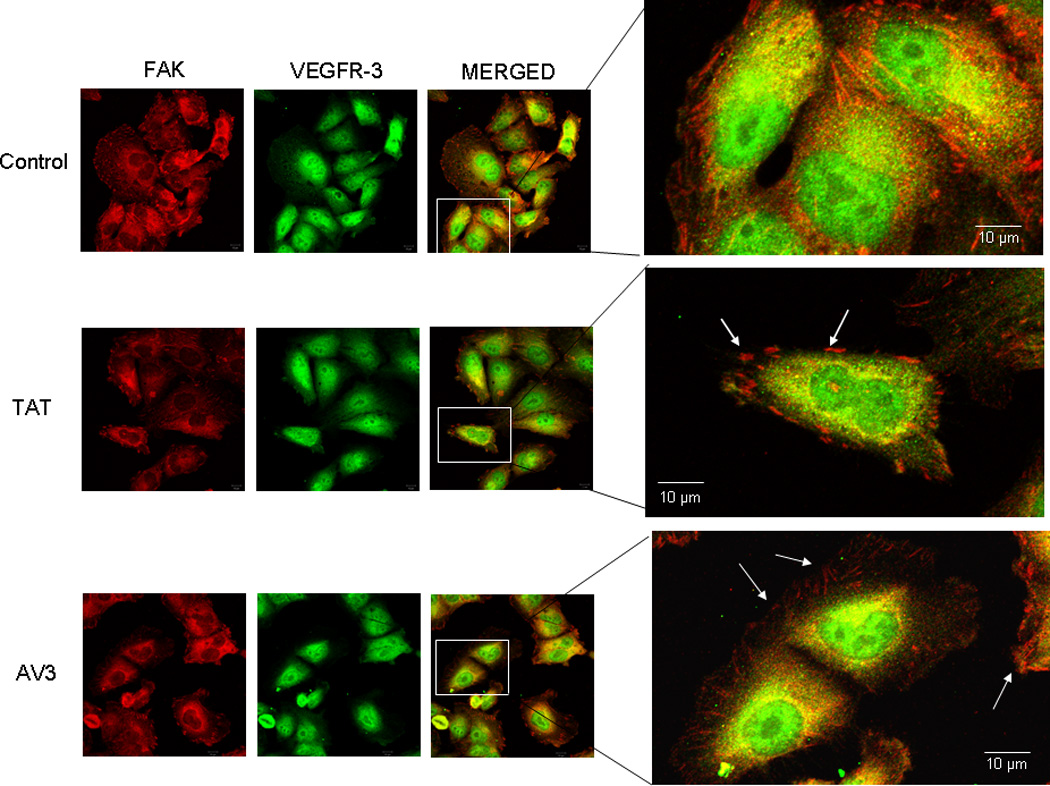
Immunofluorescence staining followed by confocal microscopy was employed to evaluate FAK and VEGFR-3 in the SK-N-AS cell line after AV3 treatment for 24 hours. In these cells, AV3 did not lead to a loss of FAK from the focal adhesions (white arrows bottom fourth panel), and did not affect the dual staining of VEGFR-3 and FAK (bottom third and fourth panels). The control peptide (TAT) had no effect upon FAK at the focal adhesions (white arrows middle fourth panel).
Figure 8. AV3 peptide treatment resulted in a loss of FAK from the focal adhesions in the SK-N-BE(2) cell line.
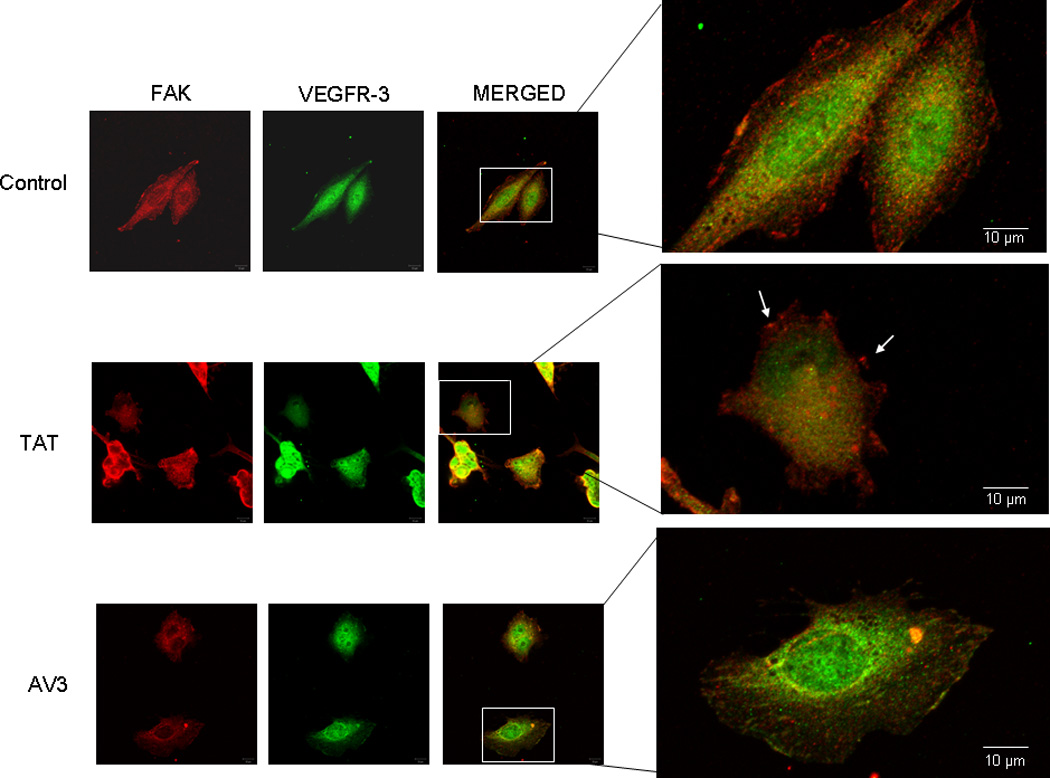
Immunofluorescence staining followed by confocal microscopy was employed to evaluate FAK and VEGFR-3 in the SK-N-BE(2) cell line after AV3 treatment for 24 hours. In these cells, AV3 lead to a loss of FAK from the focal adhesions (bottom third and fourth panels) and a decrease in the dual staining of VEGFR-3 and FAK (bottom third and fourth panels). The control peptide (TAT) had no effect upon FAK at the focal adhesions (white arrows middle fourth panel).
Figure 9. AV3 peptide treatment resulted in a decrease in loss of FAK from the focal adhesions in the SK-N-BE(2) cell line.
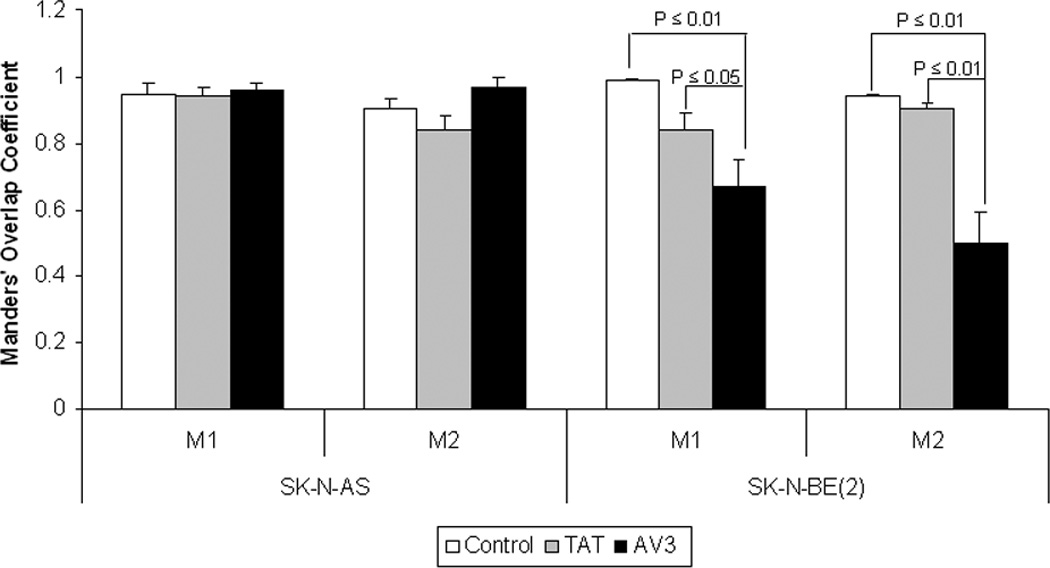
Confocal images were evaluated with Meta Image Series Software to determine the amount of colocalization. Manders’ overlap coefficients were calculated and compared for control cells, cells treated with TAT control peptide, and cells treated with AV3 peptide. There was no difference in the overlap coefficients in the SK-N-AS cells treated with the AV3 peptide compared to controls [M1 (0.95 ± 0.03 vs. 0.96 ± 0.02, control vs. AV3, NS), M2 (0.90 ± 0.03 vs. 0.97 ± 0.03, control vs. AV3, NS)]. In the SK-N-BE(2) cells treated with AV3, there was a significant decrease in the Manders’ coefficients after AV3 treatment compared to controls [M1 (0.99 ± 0.03 vs. 0.67 ± 0.08, control vs. AV3, p < 0.01), M2 (0.94 ± 0.01 vs. 0.5 ± 0.09, control vs. AV3, p < 0.01)]. There were no significant changes in Manders’ overlap coefficients with TAT peptide treatment compared to controls in either cell line.
AV3 treatment of neuroblastoma cells resulted in cellular detachment, decreased viability, and increased apoptosis
Since AV3 caused FAK to be displaced from the focal adhesions and the loss of FAK from the focal adhesions has been associated with cellular detachment [6], we next determined the biological effects of AV3 upon neuroblastoma cells. SK-N-AS and SK-N-BE(2) cells were treated with AV3 or TAT peptide for 48 hours, and the percent detached cells was determined. Cellular detachment was increased in both cell lines, but only reached statistical significance in the SK-N-BE(2) cells following AV3 treatment (Fig. 10A). Cellular attachment was not affected in either of the cell lines with TAT treatment (Fig. 10A).
Figure 10. AV3 peptide treatment resulted in cellular detachment, decreased viability, and increased apoptosis in neuroblastoma cells.
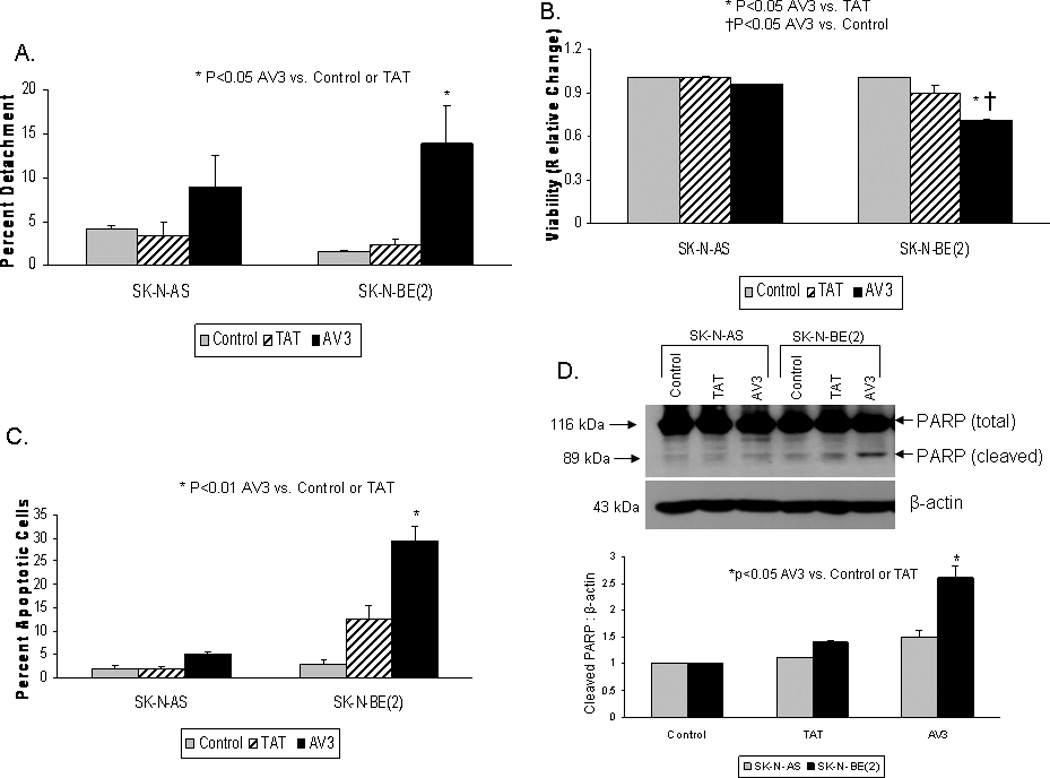
SK-N-AS and SK-N-BE(2) cells were treated with AV3 or control (TAT) peptide for 48 hours. A. The percentage of detached cells was determined by counting with a hemacytometer and dividing the number of detached cells by the total number of cells (attached + detached), and presented as percent detachment. AV3 caused significant cellular detachment in the SK-N-BE(2) cells, and some, although not statistically significant, detachment in the SK-N-AS cells. B. MTT assay was used to detect cell viability following AV3 treatment. AV3 peptide resulted in a decrease in viability in both cell lines, however, the relative change in viability was greater in the SK-N-BE(2) cell line compared to the SK-N-AS. C. Hoechst staining was used to detect apoptosis. There was a significant increase in apoptosis in the SK-N-BE(2) cells after AV3 treatment, but the increase in apoptosis in the SK-N-AS cells did not reach statistical significance. D. Apoptosis was confirmed biochemically by immunoblotting for cleaved PARP following AV3 treatment. AV3 treatment of the SK-N-AS cells resulted in a small increase in cleaved PARP. There was a significant increase in cleaved PARP in the SK-N-BE(2) cells after AV3 treatment. These immunoblots corroborated the Hoechst staining data (Fig. 10C). Densitometry was used to show the ratio between cleaved PARP to β-actin (lower panel). These results showed that AV3 treatment of neuroblastoma cells resulted in apoptosis.
Next, we evaluated the effects of AV3 treatment upon cellular viability. MTT assays were used to determine cellular viability in SK-N-AS and SK-N-BE(2) cell lines after treatment with AV3 or TAT control for 48 hours. There was a decrease in viability in both of the cell lines compared to controls after AV3 treatment, but this only reached significance in the SK-N-BE(2) cell line (1 vs. 0.74 ± 0.02, control vs. AV3, p=0.04) (Fig. 10B). Control TAT peptide did not significantly affect viability in either cell line (Fig. 10B).
Our next question was whether the AV3-induced decrease in cell viability was related to apoptosis. Hoechst staining was used to evaluate apoptosis. After 48 hours of treatment with peptide AV3, there was a significant increase in the percentage of apoptotic SK-N-BE(2) cells (Fig. 10C). As was noted with viability, AV3 treatment resulted in significant apoptosis in the cell line with increased FAK expression {SK-N-BE(2)}, and had less effect upon the cell line with less FAK expression (SK-N-AS) (Fig. 10C). Apoptosis was not significantly increased with TAT treatment in either of the cell lines (Fig. 10C).
Biochemistry was used to confirm apoptosis, by staining for the appearance of cleaved poly (ADP-ribose) polymerase (PARP) with a specific antibody. AV3 treatment of SK-N-AS cells resulted in a small increase in PARP cleavage (Fig. 10D). However, there was a more pronounced increase in the level of cleaved PARP in the SK-N-BE(2) cell line after AV3 treatment (Fig. 10D), indicating apoptosis. These data were consistent with the Hoechst staining results presented in Figure 10C, showing AV3 treatment leading to more apoptosis in the SK-N-BE(2) neuroblastoma cell line. Apoptosis was also measured with flow cytometry (Supplemental Data Fig. 3A) in addition to immunoblotting for the loss of total PARP with a specific antibody (Supplemental Data Fig. 3B, C). Both of these methods also showed a significant increase in apoptosis in the SK-N-BE(2) cells after treatment with AV3. Therefore, AV3 treatment increased cellular detachment, decreased viability, and increased apoptosis in neuroblastoma cells.
Discussion
Investigators have been exploring the importance of VEGFR-3 in the tumorigenesis of lung [22], breast [21,32,44,46], prostate [24,47], melanoma [48] and colorectal cancers [49]. However, the study of VEGFR-3 and its ligand VEGF-C in neuroblastoma has not been as extensive. In the current study, we showed that selected human neuroblastoma cell lines express VEGFR-3 protein, and that the ligand VEGF-C will activate the receptor. We have also seen that mRNA for VEGFR-3 is present in neuroblastoma cell lines and varies amongst cell lines, but corresponds to protein expression (Supplemental Data Fig. 1A, B). Our findings were consistent with the other limited reports in the literature discussing VEGFR-3 and neuroblastoma. Lagodny and colleagues [29] reported the detection of VEGFR-3 mRNA using semi-quantitative RT-PCR in human neuroblastoma specimens. Three other groups have identified VEGF-C, the VEGFR-3 ligand, in primary human neuroblastoma specimens [35,36,40] and found a correlation between VEGF-C and more advanced stage tumors. The findings of these groups and our current study provided the necessary preliminary data to show that VEGFR-3 is relevant to the clinical disease, and should prompt further investigation into the role of VEGFR-3 in neuroblastoma.
Many studies addressing VEGFR-3 in cancer have focused on its effects upon tumor metastasis. The current data addressed the role of VEGFR-3 in neuroblastoma cell survival, and demonstrated that cellular survival was decreased by RNAi-silencing of VEGFR-3 in neuroblastoma cells. These findings were interesting in that despite a significant inhibition of VEGFR-3 by the siRNA, the changes seen in cell survival were statistically significant but not dramatically altered. This finding lends evidence to our hypothesis that the FAK-VEGFR-3 interaction may be more important in these cell lines. These findings may also be due to the method utilized to detect viability. A similar response in both cell lines to RNAi treatment implied that the function of VEGFR-3 was similar in the two cell lines. Other investigators, employing a variety of means to inhibit VEGFR-3, have also shown that VEGFR-3 had an effect upon tumor cell survival and proliferation, and not just metastasis. For example, Kurenova and others found that when VEGFR-3 was downregulated with siRNA in BT474 breast cancer cells, that the proliferation of these cells was decreased by 40% [44]. RNAi silencing of VEGFR-3 in the human colon cancer cell line, LoVo, also resulted in a seven-fold decrease in tumor cell viability [50]. Masood and others demonstrated that the viability of malignant mesothelioma cells was significantly decreased when the cells were treated with antibodies to VEGFR-3 [51].
In the current study, we noted that the effects of VEGFR-3 inhibition with RNAi were similar between the two neuroblastoma cell lines. Other investigators have noted different findings. In a study of oral squamoid carcinoma cells, Matsuura et al showed that inhibition of VEGFR-3 with a small molecule inhibitor resulted in nominal changes in VEGF-C expression in cell lines with low VEGFR-3 expression when compared to those cell lines with high VEGFR-3 expression [52]. In another study, Garces et al showed that BT474 breast cancer cells with significant VEGFR-3 expression had more detachment following treatment with VEGFR-3 blocking peptide than MCF7 breast cancer cells that had lower VEGFR-3 expression [32]. Kurenova et al also demonstrated that downregulation of VEGFR-3 with siRNA in breast cancer cells with VEGFR-3 overexpression had a more profound effect upon motility and viability than in the cells without VEGFR-3 overexpression [44]. These differences may be attributed to the various techniques employed to inhibit VEGFR-3 or may be due to cell line specific differences.
We showed that FAK and VEGFR-3 interact in human neuroblastoma cells. An interaction between these two proteins has been previously demonstrated in breast cancer cells [32]. In addition, the interruption of this FAK - VEGFR-3 interaction with a small molecule inhibitor results in decreased breast cancer cell survival and decreased tumor growth in in vivo studies [31]. The concept that FAK associates with growth factor receptors to provide cellular survival advantage is not unique. Chen and Guan reported in 1996 on the association of FAK with a tyrosine phosphorylated protein. In the face of platelet-derived growth factor stimulation, there was a strong association of FAK with pp200 in NIH 3T3 cells. This association was independent of cell adhesion and was therefore thought to be involved with growth factor-induced cellular effects [53]. Sieg et al have also shown that FAK associates with activated PDGF- and EGF-receptors (PDGFR and EGFR) signaling complexes and established that FAK was an important receptor-proximal link between growth factor receptor and integrin signaling pathways [54]. Recently, an interaction between FAK and insulin-like growth factor-1 receptor (IGF-1R) has been noted. The authors showed through immunoprecipitation and confocal microscopy that FAK and IGF-1R interact in Panc-1 and MiaPaca-2 pancreatic cancer cell lines. Interruption of this interaction resulted in decreased cell viability and increased apoptosis in both cell lines [55].
The AV3 peptide that was used in this study was a twelve amino acid VEGFR-3 sequence (WHWRPWTPCKMF) that was identified previously through phage display experiments as a binding site of FAK and VEGFR-3 [32]. In this study, AV3 displaced FAK from the focal adhesions in neuroblastoma cells. In addition, AV3 had an effect upon the colocalization between FAK and VEGFR-3 in neuroblastoma cells. The important aspect of this study was that AV3 peptide resulted in cellular detachment and apoptosis in neuroblastoma cells when compared to the control TAT peptide. The results from AV3 treatment in our study highlighted the concept that it was not only VEGFR-3 that was important for survival in these neuroblastoma cell lines, but the FAK and VEGFR-3 interaction played a significant role.
In the current study, we demonstrated that AV3 treatment resulted in a decrease in FAK phosphorylation in the SK-N-BE(2) (Fig. 6B), but not in the SK-N-AS cells (Fig. 6A). We have also seen that phosphorylation of FAK was affected by the abrogation of VEGFR-3 via other methodologies. Using MAZ51, a small molecule inhibitor of VEGFR-3 activation [56], we noted a decrease in phosphorylation of FAK in the SK-N-BE(2) but not in the SK-N-AS cells (Supplemental Data Fig. 4). It has not been determined whether the phosphorylation of FAK is critical to the FAK and VEGFR-3 interaction, and these investigations are the focus of future studies.
Confocal microscopy showed that most of the FAK – VEGFR-3 colocalization was located intracellular, in the cytoplasm. AV3 peptide treatment of SK-N-BE(2) cells resulted in a change in the distribution of VEGFR-3 staining to a more peri-nuclear location. The AV3 peptide treatment also resulted in a loss of FAK from the focal adhesions in the SK-N-BE(2) cells, but did not affect the amount of total FAK staining. These results were similar to those seen in BT474 breast cancer cells treated with AV3 [32].
The data in the current study showed that interruption of the FAK and VEGFR-3 interaction in neuroblastoma cell lines had a more profound effect upon cell detachment, viability, and apoptosis in the cell line with more FAK expression than the cell line with less expression of FAK. We also noted these findings in another MYCN amplified cell line, IMR-32, when treated with AV3 in comparison to SK-N-AS (Supplemental Data, Fig. 5A, B). Our previous studies showed that FAK expression was correlated with amplification of the MYCN oncogene. Through dual luciferase assays, site-directed mutagenesis, and ChIP assays, it was demonstrated that MYCN bound to the FAK promoter and activated FAK expression in MYCN amplified neuroblastoma cell lines [15]. Further, we demonstrated with a number of different methods of FAK inhibition that MYCN amplified cells were more dependent upon the FAK pathway and FAK activation [15,17,33]. These findings correlated well with findings from other authors with the inhibition of other kinases. Weinstein and colleagues have discussed the concept of oncogene addiction. It is their thoughts that some cancers depend upon one or two genes to maintain their malignant phenotype [57,58]. Our current study demonstrated that disruption of the FAK-VEGFR-3 interaction with AV3 significantly affected cell lines with more FAK. We can explain this finding based upon the fact that AV3 affects both the VEGFR-3 and FAK pathways (Fig. 6B), going back to the greater dependence upon FAK by the MYCN amplified cells. At the same time, downregulation of VEGFR-3 with siRNA equally affected both cell lines. This effect was not profound, which may indicate prioritization of different signaling pathways for these cells.
In conclusion, our study demonstrated that inhibition of VEGFR-3 and specifically, the interruption of the interaction between FAK and VEGFR-3, resulted in cellular detachment, decreased cellular viability and increased apoptosis in human neuroblastoma cell lines. In addition, disruption of the FAK - VEGFR-3 interaction had the most effect upon cell lines with more FAK, which has important implications in neuroblastoma, since the more aggressive MYCN positive tumors tend to have greater FAK expression. These findings are important in furthering our understanding of the regulation of tumorigenesis in neuroblastoma, and may potentially provide novel therapeutic strategies and targets for neuroblastoma.
Supplementary Material
Acknowledgements
The authors wish to thank the UAB High Resolution Imaging Facility and Shawn Williams for his outstanding technical assistance with confocal microscopy.
Grant Support
The project described was funded in part by grants from the Children’s Neuroblastoma Cancer Foundation (E.A. Beierle) and the Kaul Pediatric Research Institute (E.A. Beierle). EAB and MM are supported in part by grants from the National Cancer Institute, Grant Number K08CA118178 and T32CA091078, respectively. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.…
List of abbreviations
- FAK
focal adhesion kinase
- EDTA
ethylenediaminetetraacetic acid
- NaDOC
sodium deoxycholate
- PMFS
phenylmethanesulfonylfluoride
- VEGFR-3
vascular endothelial growth factor receptor-3
- VEGF-C/D
vascular endothelial growth factor C/D
- MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
- PARP
poly (ADP-ribose) polymerase
References
- 1.Cotterill S, Parker L, More L, Craft A. Neuroblastoma: changing incidence and survival in young people aged 0–24 years A report from the North of England Young Persons’ Malignant Disease Registry. Med Pediatr Oncol. 2001;36(1):231–234. doi: 10.1002/1096-911X(20010101)36:1<231::AID-MPO1056>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 2.Hanks S, Polte T. Signaling through focal adhesion kinase. Bioessays. 1997;19(2):137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- 3.Zachary I. Focal adhesion kinase. Int J Biochem Cell Biol. 1997;29(7):929–934. doi: 10.1016/s1357-2725(97)00008-3. [DOI] [PubMed] [Google Scholar]
- 4.Gabarra-Niecko V, Schaller M, Dunty J. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22(4):359–374. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- 5.Schaller M, Borgman C, Cobb B, Vines R, Reynolds A, Parsons J. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89(11):5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu L, Yang X, Bradham C, et al. The focal adhesion kinase suppresses transformation-associated anchorage-independent apoptosis in human breast cancer cells Involvement of death receptor-related signaling pathways. J Biol Chem. 2000;275(39):30597–30604. doi: 10.1074/jbc.M910027199. [DOI] [PubMed] [Google Scholar]
- 7.Xu L, Yang X, Craven R, Cance W. The COOH-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell Growth Differ. 1998;9(12):999–1005. [PubMed] [Google Scholar]
- 8.Xu L, Owens L, Sturge G, et al. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7(4):413–418. [PubMed] [Google Scholar]
- 9.Golubovskaya V, Beviglia L, Xu L, Earp Hr, Craven R, Cance W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J Biol Chem. 2002;277(41):38978–38987. doi: 10.1074/jbc.M205002200. [DOI] [PubMed] [Google Scholar]
- 10.Han E, Mcgonigal T, Wang J, Giranda V, Luo Y. Functional analysis of focal adhesion kinase (FAK) reduction by small inhibitory RNAs. Anticancer Res. 2004;24(6):3899–3905. [PubMed] [Google Scholar]
- 11.Lipinski C, Tran N, Menashi E, et al. The tyrosine kinase pyk2 promotes migration and invasion of glioma cells. Neoplasia. 2005;7(5):435–445. doi: 10.1593/neo.04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryu S, Cho K, Oh Y, Park S. Role of Src-specific phosphorylation site on focal adhesion kinase for senescence-associated apoptosis resistance. Apoptosis. 2006;11(3):303–313. doi: 10.1007/s10495-006-3978-9. [DOI] [PubMed] [Google Scholar]
- 13.Halder J, Landen CJ, Lutgendorf S, et al. Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin Cancer Res. 2005;11(24 Pt 1):8829–8836. doi: 10.1158/1078-0432.CCR-05-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beierle E, Massoll N, Hartwich J, et al. Focal adhesion kinase expression in human neuroblastoma: immunohistochemical and real-time PCR analyses. Clin Cancer Res. 2008;14(11):3299–3305. doi: 10.1158/1078-0432.CCR-07-1511. [DOI] [PubMed] [Google Scholar]
- 15.Beierle E, Trujillo A, Nagaram A, et al. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J Biol Chem. 2007;282(17):12503–12516. doi: 10.1074/jbc.M701450200. [DOI] [PubMed] [Google Scholar]
- 16.Beierle EA, Trujillo A, Nagaram A, Golubovskaya VM, Cance WG, Kurenova EV. TAE226 inhibits human neuroblastoma cell survival. Cancer Invest. 2008;26(2):145–151. doi: 10.1080/07357900701577475. [DOI] [PubMed] [Google Scholar]
- 17.Beierle E, Ma X, Stewart J, et al. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle. 2010;9(5):1005–1015. doi: 10.4161/cc.9.5.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jussila L, Valtola R, Partanen T, et al. Lymphatic endothelium and Kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res. 1998;58(8):1599–1604. [PubMed] [Google Scholar]
- 19.Dumont D, Jussila L, Taipale J, et al. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998;282(5390):946–949. doi: 10.1126/science.282.5390.946. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Sun X, Wu J. Expression and significance of VEGF-C and FLT-4 in gastric cancer. World J Gastroenterol. 2004;10(3):352–355. doi: 10.3748/wjg.v10.i3.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Iterson V, Leidenius M, von Smitten K, Bono P, Heikkilä P. VEGF-D in association with VEGFR-3 promotes nodal metastasis in human invasive lobular breast cancer. Am J Clin Pathol. 2007;128(5):759–766. doi: 10.1309/7FXVRMXF58PVRJUH. [DOI] [PubMed] [Google Scholar]
- 22.Saintigny P, Kambouchner M, Ly M, et al. Vascular endothelial growth factor-C and its receptor VEGFR-3 in non-small-cell lung cancer: concurrent expression in cancer cells from primary tumour and metastatic lymph node. Lung Cancer. 2007;58(2):205–213. doi: 10.1016/j.lungcan.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 23.Herrmann E, Eltze E, Bierer S, et al. VEGF-C, VEGF-D and Flt-4 in transitional bladder cancer: relationships to clinicopathological parameters and long-term survival. Anticancer Res. 2007;27(5A):3127–3133. [PubMed] [Google Scholar]
- 24.Jennbacken K, Vallbo C, Wang W, Damber J. Expression of vascular endothelial growth factor C (VEGF-C) and VEGF receptor-3 in human prostate cancer is associated with regional lymph node metastasis. Prostate. 2005;65(2):110–116. doi: 10.1002/pros.20276. [DOI] [PubMed] [Google Scholar]
- 25.Tammela T, Zarkada G, Wallgard E, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454(7204):656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 26.Laakkonen P, Waltari M, Holopainen T, et al. Vascular endothelial growth factor receptor 3 is involved in tumor angiogenesis and growth. Cancer Res. 2007;67(2):593–599. doi: 10.1158/0008-5472.CAN-06-3567. [DOI] [PubMed] [Google Scholar]
- 27.Beierle E, Dai W, Langham MJ, Copeland ER, Chen M. VEGF receptors are differentially expressed by neuroblastoma cells in culture. J Pediatr Surg. 2003;38(3):514–521. doi: 10.1053/jpsu.2003.50091. [DOI] [PubMed] [Google Scholar]
- 28.Beierle E, Dai W, Langham MJ, Copeland E, Chen M. Expression of VEGF receptors in cocultured neuroblastoma cells. J Surg Res. 2004;119(1):56–65. doi: 10.1016/j.jss.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Lagodny J, Jüttner E, Kayser G, Niemeyer C, Rössler J. Lymphangiogenesis and its regulation in human neuroblastoma. Biochem Biophys Res Commun. 2007;352(2):571–577. doi: 10.1016/j.bbrc.2006.11.062. [DOI] [PubMed] [Google Scholar]
- 30.Rössler J, Taylor M, Geoerger B, et al. Angiogenesis as a target in neuroblastoma. Eur J Cancer. 2008;44(12):1645–1656. doi: 10.1016/j.ejca.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Kurenova E, Hunt D, He D, Magis A, Ostrov D, Cance W. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J Med Chem. 2009;52(15):4716–4724. doi: 10.1021/jm900159g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garces C, Kurenova E, Golubovskaya V, Cance W. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006;66(3):1446–1454. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- 33.Beierle E, Ma X, Trujillo A, Kurenova E, Cance W, Golubovskaya V. Inhibition of focal adhesion kinase and src increases detachment and apoptosis in human neuroblastoma cell lines. Mol Carcinog. 2010;49(3):224–234. doi: 10.1002/mc.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurenova E, Xu L, Yang X, et al. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. 2004;24(10):4361–4371. doi: 10.1128/MCB.24.10.4361-4371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nowicki M, Konwerska A, Ostalska-Nowicka D, et al. Vascular endothelial growth factor (VEGF)-C - a potent risk factor in children diagnosed with stadium 4 neuroblastoma. Folia Histochem Cytobiol. 2008;46(4):493–499. doi: 10.2478/v10042-008-0067-7. [DOI] [PubMed] [Google Scholar]
- 36.Eggert A, Ikegaki N, Kwiatkowski J, Zhao H, Brodeur G, Himelstein B. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res. 2000;6(5):1900–1908. [PubMed] [Google Scholar]
- 37.Uçar K, Seeger R, Challita P, et al. Sustained cytokine production and immunophenotypic changes in human neuroblastoma cell lines transduced with a human gamma interferon vector. Cancer Gene Ther. 1995;2(3):171–181. [PubMed] [Google Scholar]
- 38.Nguyen T, Hocker JE, Thomas W, et al. Combined RAR alpha- and RXR-specific ligands overcome N-myc-associated retinoid resistance in neuroblastoma cells. Biochem Biophys Res Commun. 2003;302(3):462–468. doi: 10.1016/s0006-291x(03)00177-3. [DOI] [PubMed] [Google Scholar]
- 39.Joukov V, Kumar V, Sorsa T, et al. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J Biol Chem. 1998;273(12):6599–6602. doi: 10.1074/jbc.273.12.6599. [DOI] [PubMed] [Google Scholar]
- 40.Komuro H, Kaneko S, Kaneko M, Nakanishi Y. Expression of angiogenic factors and tumor progression in human neuroblastoma. J Cancer Res Clin Oncol. 2001;127(12):739–743. doi: 10.1007/s004320100293. [DOI] [PubMed] [Google Scholar]
- 41.Sallinen H, Anttila M, Narvainen J, et al. Antiangiogenic gene therapy with soluble VEGFR-1, −2, and −3 reduces the growth of solid human ovarian carcinoma in mice. Mol Ther. 2009;17(2):278–284. doi: 10.1038/mt.2008.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burton J, Priceman S, Sung J, et al. Suppression of prostate cancer nodal and systemic metastasis by blockade of the lymphangiogenic axis. Cancer Res. 2008;68(19):7828–7837. doi: 10.1158/0008-5472.CAN-08-1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heckman C, Holopainen T, Wirzenius M, et al. The tyrosine kinase inhibitor cediranib blocks ligand-induced vascular endothelial growth factor receptor-3 activity and lymphangiogenesis. Cancer Res. 2008;68(12):4754–4762. doi: 10.1158/0008-5472.CAN-07-5809. [DOI] [PubMed] [Google Scholar]
- 44.Kurenova E, Hunt D, He D, et al. Vascular endothelial growth factor receptor-3 promotes breast cancer cell proliferation, motility and survival in vitro and tumor formation in vivo. Cell Cycle. 2009;8(14):2266–2280. doi: 10.4161/cc.8.14.9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manders EMM, Verbeek FJ, Aten JA. Measurement of colocalization of objects in dual-color confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 46.Nakamura Y, Yasuoka H, Tsujimoto M, et al. Flt-4-positive vessel density correlates with vascular endothelial growth factor-d expression, nodal status, and prognosis in breast cancer. Clin Cancer Res. 2003;9(14):5313–5317. [PubMed] [Google Scholar]
- 47.Zeng Y, Opeskin K, Baldwin M, et al. Expression of vascular endothelial growth factor receptor-3 by lymphatic endothelial cells is associated with lymph node metastasis in prostate cancer. Clin Cancer Res. 2004;10(15):5137–5144. doi: 10.1158/1078-0432.CCR-03-0434. [DOI] [PubMed] [Google Scholar]
- 48.Mehnert J, McCarthy M, Jilaveanu L, et al. Quantitative expression of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3 in melanoma tissue microarrays. Hum Pathol. 2010;41(3):375–384. doi: 10.1016/j.humpath.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Onogawa S, Kitadai Y, Tanaka S, Kuwai T, Kimura S, Chayama K. Expression of VEGF-C and VEGF-D at the invasive edge correlates with lymph node metastasis and prognosis of patients with colorectal carcinoma. Cancer Sci. 2004;95(1):32–39. doi: 10.1111/j.1349-7006.2004.tb03167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lui Z, Ma Q, Wang X, Zhang Y. Inhibiting tumor growth of colorectal cancer by blocking the expression of vascular endothelial growth factor receptor 3 using interference vector-based RNA interference. Int J Mol Med. 2010;25(1):59–64. [PubMed] [Google Scholar]
- 51.Masood R, Kundra A, Zhu S, et al. Malignant mesothelioma growth inhibition by agents that target the VEGF and VEGF-C autocrine loops. Int J Cancer. 2003;104(5):603–610. doi: 10.1002/ijc.10996. [DOI] [PubMed] [Google Scholar]
- 52.Matsuura M, Onimaru M, Yonemitsu Y, et al. Autocrine loop between vascular endothelial growth factor (VEGF)-C and VEGF receptor-3 positively regulates tumor-associated lymphangiogenesis in oral squamoid cancer cells. Am J Pathol. 2009;175(4):1709–1721. doi: 10.2353/ajpath.2009.081139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen HC, Guan JL. The association of focal adhesion kinase with a 200-kDa protein that is tyrosine phosphorylated in response to platelet-derived growth factor. Eur J Biochem. 1996;235(3):495–500. doi: 10.1111/j.1432-1033.1996.00495.x. [DOI] [PubMed] [Google Scholar]
- 54.Sieg DJ, Hauck CR, Ilic D, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 55.Liu W, Bloom D, Cance W, Kurenova E, Golubovskaya V, Hochwald S. FAK and IGF-IR interact to provide survival signals in human pancreatic adenocarcinoma cells. Carcinogenesis. 2008;29(6):1096–1107. doi: 10.1093/carcin/bgn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirkin V, Baumann P, Mazitschek R, et al. MAZ51, an indoline that inhibits endothelial cell and tumor cell growth in vitro, suppresses tumor growth in vivo. Int J Cancer. 2004;112(6):986–993. doi: 10.1002/ijc.20509. [DOI] [PubMed] [Google Scholar]
- 57.Weinstein IB. Cancer Addiction to oncogenes- the Achilles heal of cancer. Science. 2002;297(5578):63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 58.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–3080. doi: 10.1158/0008-5472.CAN-07-3293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.