

Abstract
Joubert syndrome (JS) is a congenital cerebellar ataxia with autosomal recessive or X-linked inheritance, which diagnostic hallmark is a unique cerebellar and brainstem malformation recognizable on brain imaging, the “molar tooth sign”. Neurological signs are present from neonatal age and include hypotonia evolving into ataxia, global developmental delay, ocular motor apraxia and breathing dysregulation. These are variably associated with multiorgan involvement, mainly of the retina, kidneys, skeleton and liver. To date, 21 causative genes have been identified, all encoding for proteins of the primary cilium or its apparatus. This is a subcellular organelle that plays key roles in development and in many cellular functions, making JS part of the expanding family of ciliopathies. There is marked clinical and genetic overlap among distinct ciliopathies, which may co-occur even within families. Such variability is likely explained by an oligogenic model of inheritance, in which mutations, rare variants and polymorphisms at distinct loci interplay to modulate the expressivity of the ciliary phenotype.
Introduction
Joubert syndrome (JS) is an inherited congenital cerebellar ataxia characterized by a peculiar mid-hindbrain malformation, the “molar tooth sign” (MTS) (Figure 1), and variable organ involvement. The pathogenetic basis of this clinically and genetically heterogeneous disorder relates to the dysfunction of a subcellular organelle, the primary cilium, making JS part of the expanding group of disorders collectively termed “ciliopathies”.
Figure 1.
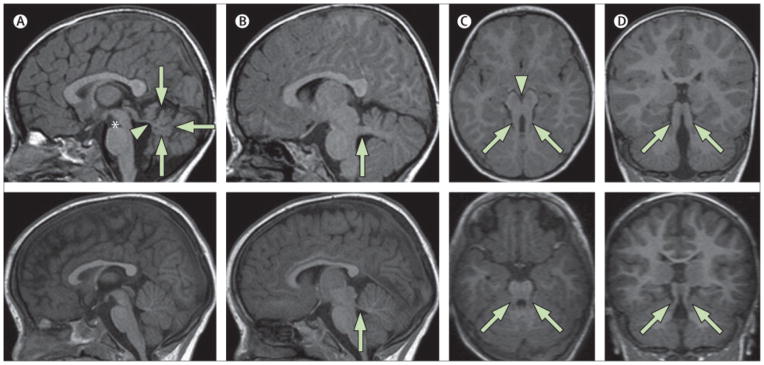
Neuroimaging findings in a 2-year-old child with pure JS (upper panels) compared to a healthy control (lower panels). a) Parasagittal T1-weighted image shows the thickened, elongated and horizontally orientated superior cerebellar peduncles (white arrow). b) Midsagittal T1-weighted image demonstrates a moderate hypoplasia and dysplasia of the cerebellar vermis (white arrows) with secondary distortion and enlargement of the fourth ventricle with rostral shifting of the fastigium (white arrow head). A deepened interpeduncular fossa is also noted. c) Axial T1-weighted image at the level of the pontomesencephalic junction shows the molar tooth sign with a deepened interpeduncular fossa (white arrowhead) and elongated, thickened and horizontally orientated superior cerebellar peduncles (white arrows). Additionally, the cerebellar vermis appears to be hypoplastic and its remnants dysplastic. d) Coronal T1-weighted image reveals the thickened superior cerebellar peduncles (white arrows).
The recent advent of next-generation-sequencing (NGS) strategies has resulted in an impressive progress of our knowledge of JS, with several genes being identified and characterized at the pathophysiological level. In turn, this has led to a dissection of the complexity of the primary cilium, explaining how the malfunction of this ubiquitous organelle could result in such a variety of phenotypes, ranging from developmental malformations to progressive defects.
In the present review, we will discuss the clinical spectrum and genetic basis of JS, the overlay with other ciliopathies, and the evidence supporting oligogenic inheritance. Finally, we will review the multifaceted roles of primary cilia in central nervous system (CNS) development and in the functioning of neuronal cells.
The primary cilium
The primary cilium is an immotile organelle protruding from the surface of nearly all cell types. Long considered a vestigial remnant devoid of any relevant function, this organelle has recently become the focus of intensive research, that has highlighted its many key roles in embryonic development, inherited human diseases and even tumorigenesis.
Primary cilia emerge from the basal body, a modified centriolar structure anchored to the plasma membrane. Their structural core is the axoneme, composed of nine doublets of microtubules, that is surrounded by a membrane contiguous to the cell plasma membrane, but hosting specific signalling molecules. The basal body is joined to the axoneme by the Y-shaped fibres of the “transition zone” (TZ), a recently described region working as a functional ciliary gate, that regulates and restricts the flux of specific proteins to the cilium, in order to maintain it as a compartmentalized organelle (Figure 2).1–3
Figure 2.

Schematic representation of the structure of the primary cilium and its protein complexes. Most proteins mutated in JS and MKS cluster in large complexes residing at the basal body or the transition zone. These complexes participate in the regulation of ciliogenesis, control the trafficking of specific pools of molecules targeted to the cilium and are implicated in signalling pathways mediated by the cilium. Other JS proteins are also found along the ciliary axoneme or interact with Shh or Wnt pathways.
In many adult tissues, primary cilia work as sensors of extracellular signals, and transduce them within cells to regulate tissue maintenance, polarity or proliferation. The disruption of these sensory functions in specialized cells such as the kidney and bile duct epithelium, or the retinal photoreceptors, explains many of the organ defects seen in ciliopathies. Moreover, recent studies have shown that most neuronal types possess a primary cilium, highlighting its many roles in brain development and function (reviewed in 4–6).
Clinical spectrum of Joubert syndrome
Epidemiology
Reliable epidemiological data on JS are lacking. A prevalence between 1/80·000 and 1/100·000 live births is reported by many authors, and likely represents an underestimate which reflects the limited awareness of the MTS, especially in the past.7,8 In the Askhenazi Jews, the predicted prevalence of JS is increased up to 1/34·000–1/40·000 due to the presence of the founder mutation p.R73L in the TMEM216 gene, with a carrier frequency of 1:90–1:100.9,10 Similarly, in the Hutterite population, about 1:17 healthy subjects carry the founder mutation p.R18X in the TMEM237 gene, leading to a predicted disease prevalence of about 1/1·150.11
Neurological features and organ involvement
A clinical suspicion of JS can be posed as early as in the first months of life, upon the observance of hypotonia, abnormal ocular movements (mainly ocular motor apraxia, nystagmus and strabismus), and occasionally alterations of the respiratory pattern, characterized by short alternate episodes of apnea and tachypnea, or episodic tachypnea alone. About 10–15% of affected neonates also show polydactyly, that can be pre-, meso- or post-axial and variably involve hands and feet.12 Later, a delay in the acquisition of developmental steps and intellectual disability of variable severity are observed in nearly all children, with expressive speech usually more affected than comprehension due to concurrent oromotor apraxia;13 yet, rare subjects have been reported with normal cognitive functions.14 Facial dysmorphisms are often present, including prominent forehead, ptosis, prognathia, arched eyebrows, lower lip eversion with trapezoid shaped mouth and tongue protrusion. However, facial features do not aid the diagnosis, as in many patients they can be entirely normal.15 About 50% of children learn to walk independently and develop ataxia with broad-based, unsteady gait and difficulties in running or climbing stairs.12
The clinical picture of JS can be complicated by a large spectrum of associated organ defects, which can manifest at different ages. Among these, the commonest are retinal defects (ranging in severity from Leber congenital amaurosis (LCA) to slowly progressive retinopathies with partially preserved vision), renal defects (nephronophthisis (NPH), or cystic dysplastic kidneys), and congenital liver fibrosis. More rarely observed features include chorioretinal or optic nerve colobomas, congenital heart malformations, situs inversus, severe scoliosis and skeletal dysplasia, Hirschsprung disease, and midline oral and facial defects. These can include cleft lip and/or palate, notched upper lip, lobulated tongue with multiple frenula, lingual or oral soft tumours. In particular, the association of JS with polydactyly and midline oro-facial defects defines the so called “oro-facio-digital syndrome type VI” (OFDVI), that is also part of the JS spectrum.16
This extreme phenotypic variability has generated considerable confusion in classifying JS in the past, with several acronyms that were adopted to describe overlapping conditions sharing the MTS, and which were collectively termed “Joubert syndrome and related disorders” (JSRD).17 However, it has become more and more evident that the variable clinical manifestations associated to the MTS do not configure distinct clinical syndromes, but are simply part of the wide phenotypic spectrum that is characteristic of JS. Nowadays, also in light of the emerging genetic complexity of this condition (see “Genetic basis of JS” below), the distinction between JS and JSRD seems blurred and does not contribute to the diagnostic definition of patients (that is simply based upon recognition of the MTS), at the same time creating confusion especially to the families. For these reasons, we propose to abandon the term “JSRD” in favour of the classical term “Joubert syndrome” to encompass all MTS-related conditions, and to adopt a descriptive classification, that defines JS clinical subgroups based on the extent of organ involvement (Figure 3).
Figure 3.

Spectrum of organ involvement in JS and classification in clinical subgroups (in bold). Chorioretinal colobomas are more frequently found in the subgroup of JS with liver involvement, but can be present also in other subgroups. Similarly, polydactyly (expecially if pre- or meso-axial) is invariably present in the Oro-facio-digital type VI subgroup, but post-axial polydactyly is frequently observed also in association with other JS phenotypes. Other clinical features outside the circles occur more rarely, without a specific association to a clinical subgroup. Legend: CNS: central nervous system; COR: cerebello-oculo-renal; K: kidney involvement; L: liver involvement; MTS: Molar Tooth Sign; OFDVI: Oro-facio-digital type VI syndrome.
Neuroimaging and pathology
After the first description of four siblings by Dr Marie Joubert in 1969,18 several other patients were reported with the core features of JS, but only in 1997 did Maria and co-workers delineate the peculiar cerebellar and brainstem malformation that represents the hallmark of this syndrome, which they termed “the molar tooth sign”.19 The MTS originates by the coexistence of cerebellar vermis hypo-dysplasia, horizontalized, thickened and elongated superior cerebellar peduncles, and an abnormally deep interpeduncular fossa at the level of the isthmus and upper pons, giving the appearance of a “molar tooth” on axial cuts (Figure 1).
At the neuropathological level, this malformation results from the hypoplasia and dysplasia of the cerebellar vermis and of pontine and medullary structures, as well as the lack of decussation of the superior cerebellar peduncles and the pyramidal tracts.7,20,21 This failure of selected tracts to cross the brainstem midline has been further confirmed by diffusion tensor imaging (DTI) studies,22,23 and implies an underlying defect of axon guidance that is unique in JS among all ciliopathies, and still remains unexplained.24
Diagnostic management
In the presence of neurological features suggestive of JS, the diagnosis is easily confirmed upon demonstration of the MTS on brain imaging. This will also reveal possibly associated CNS defects, that can include ventriculomegaly, occipital meningo-encephalocele, polymicrogyria, periventricular nodular heterotopia, hypothalamic hamartoma, absence of the pituitary gland, corpus callosum defects, hippocampal malformations and morphological brainstem abnormalities which affect mostly the midbrain and tectum. An enlarged posterior fossa as seen in the Dandy-Walker malformation (DWM) has been described in nearly half of JS patients, but none of them matched the diagnostic criteria for DWM (cystic dilatation of the fourth ventricle associated to upwards rotation of the hypoplastic cerebellar vermis).25 In rare doubtful cases, DTI can assist the diagnosis, by showing the absence of crossing fibres at the midbrain level.26
Patients with JS need to enter a diagnostic workflow and regular follow-up examinations, to ensure the proper assessment and management of multiorgan complications.12 In neonates and infants, particular care should be taken in managing the episodes of abnormal breathing, since prolonged apneas can be life-threatening and require assisted ventilation. Respiratory defects tend to improve spontaneously and sometimes disappear with age, although sleep-related breathing disorders may persist beyond childhood in a subset of patients.27 Appropriate rehabilitation protocols can help young patients to overcome the delay in acquisition of developmental milestones and cognition.
Ophthalmological investigations should include evaluation of ocular movements, visual acuity and fundus oculi; in selected patients, electroretinogram and slit lamp examination are required to disclose certain retinopathies with normal fundus appearance, and abnormalities of the anterior segment of the eye, respectively. Neuro-ophthalmological rehabilitation is of use to improve fixation and pursuit in children with ocular motor apraxia, and to optimize residual visual function in those with marked visual impairment.
Nephrological assessment is particularly important, especially in young children. In fact, about 20–30% JS patients may develop juvenile NPH, a slowly progressive condition characterized by the formation of small cysts at the corticomedullary junction and interstitial fibrosis.8 NPH may remain asymptomatic for several years, until acute or chronic renal insufficiency manifests in the late first or early second decade, eventually requiring dialysis or kidney transplantation. Infantile NPH is rarer and more severe than the juvenile form, with manifestations in the first years of life. Renal ultrasound may disclose small cysts and loss of cortico-medullary differentiation, or may remain negative. Thus, in young children it is important to monitor the urinary concentration ability (first morning void urine analysis, polydipsia and/or polyuria), which generally precedes impairment of renal function. In addition, children with NPH often have early onset of anemia. Timely diagnosis of NPH is essential to initiate supportive therapy of chronic renal failure and to insure appropriate fluid intake. These measures prevent development of complications such as renal osteodystrophy and poor growth, and are likely to delay progression to end-stage renal disease.
Congenital liver fibrosis may be suspected in presence of hepatomegaly, elevated levels of liver serum enzymes and/or abnormal liver echogenicity, and confirmed by liver magnetic resonance imaging (MRI) and eventually by liver biopsy. Patients may remain asymptomatic or develop severe complications such as portal hypertension and oesophageal varices, which need to be properly addressed. Finally, a thorough examination should disclose rarer associated conditions potentially requiring treatment, such as congenital heart defects, scoliosis or Hirschsprung disease.
Based on the recognition of the MTS, there are no true differential diagnoses of JS. However, clinical differential diagnosis must include other malformative syndromes of the cerebellum and brainstem that can also present with hypotonia, abnormal eye movements and developmental delay, as well as other ciliopathies with similar organ involvement as JS, such as Senior-Löken syndrome (SLS) and Bardet-Biedl syndrome (BBS), and finally cerebro-oculo-renal syndromes.5,28,29
In foetuses with JS, ultrasound examination from the 20–21 week of gestation can detect the hypoplasia of the cerebellar vermis, possibly associated to polydactyly and/or occipital encephalocele. In the suspect of a cerebellar malformation, foetal MRI will confirm the diagnosis, often allowing recognition of the MTS.30–32 In families with a genetic diagnosis of JS made in a previously affected child, prenatal diagnosis becomes possible in the first trimester of pregnancy based on genetic testing of chorionic villous sample. Because of the high carrier frequency among the Ashkenazi Jews and Hutterites, prenatal screening is now commonplace in these populations.
Genetic basis of JS
The genetic bases of JS are extremely complex and only partially understood, despite the tremendous acceleration in gene discovery produced by NGS techniques. To date, 21 causative genes have been identified, with autosomal or X-linked recessive inheritance (Table 1). Whole exome sequencing studies in JS families led to estimate that mutations in known genes account for only about half patients, suggesting further genetic heterogeneity (JG Gleeson, personal communication).33,34
Table 1.
Genes and loci causative of JS
| JBTS | locus | Gene | Protein | Prevalent JS phenotypes | Mutation frequency in JS‡ | Genetic overlap with other ciliopathies | References |
|---|---|---|---|---|---|---|---|
| JBTS1 | 9q34 | INPP5E | Inositol polyphosphate-5-phosphatase | JS±retina; (JS+liver#) | ~3% | MORM | 37,45,47 |
| JBTS2 | 11q12 | TMEM216 | Transmembrane protein 216 | JS±kidney; (JS+liver#, OFDVI) | ~3% | MKS2 | 9,10,37 |
| JBTS3 | 6q23 | AHI1 | Jouberin | JS±retina | ~7–11% | - | 37,44,46,62,63,99,100 |
| JBTS4 | 2q13 | NPHP1 | Nephrocystin 1 | JS+kidney | ~4–7% | NPHP1, SLSN1 | 37,50,57,62,101 |
| JBTS5 | 12q21 | CEP290 | Centrosomal protein 290kDa | JS+retina+kidney | ~7–16% | MKS4, NPHP6*, SLSN6, BBS14, LCA10 | 37,43,56,100,102–106 |
| JBTS6 | 8q22 | TMEM67 | Meckelin | JS+liver# | ~6–10% | MKS3, NPHP11, BBS§* | 37–40,107 |
| JBTS7 | 16q12 | FTM (RPGRIP1L) | RPGRIP1L | JS+kidney; (JS+liver#) | ~1–2% | MKS5, NPHP8 | 37,40,51,53,61,100,108 |
| JBTS8 | 3q11 | ARL13B | ADP-ribosylation factor-like 13B | JS | ~1–2% | - | 37,48 |
| JBTS9 | 4p15 | CC2D2A | coiled-coil and C2 domain containing protein 2A | JS±retina; (JS+liver#) | ~6–10% | MKS6 | 33,37,40,54,109 |
| JBTS10 | Xp22 | CXOrf5 (OFD1) | oral-facial-digital syndrome 1 | variable phenotypes | <1% | OFD1, SGB2 | 110,111 |
| JBTS11* | 2q24 | TTC21B | tetratricopeptide repeat protein 21B | -- | n.a. | MKS§, NPHP12, ATD4 | 37,64 |
| JBTS12 | 15q26 | KIF7 | Kinesin-like protein 7 | JS, (OFDVI) | n.a. | ACS, HLS2, MEDF | 65,68 |
| JBTS13 | 12q24 | TCTN1 | Tectonic-1 | JS±retina | 1 mutated family only | - | 79,100 |
| JBTS14 | 2q33 | TMEM237 | Transmembrane protein 237 | JS+kidney | ~1% | MKS§ | 11,100 |
| JBTS15 | 7q32 | CEP41 | Centrosomal protein 41kDa | JS | <1% | MKS§ | 66 |
| JBTS16 | 11q12 | TMEM138 | Transmembrane protein 138 | variable phenotypes | ~1–2% | MKS§ | 52 |
| JBTS17 | 5p13 | C5Orf42 | C5Orf42 | JS±retina | n.a. | MKS§ | 33,34,100 |
| JBTS18 | 10q24 | TCTN3 | Tectonic-3 | JS±OFD features | n.a. | OFD4+MKS features | 83 |
| JBTS19 | 16q12 | ZNF423 | Zinc finger protein 423 | JS+kidney | 1 mutated family only | NPHP14 | 67 |
| JBTS20 | 16q23 | TMEM231 | Transmembrane protein 231 | JS+retina+kidney | n.a. | MKS§ | 33 |
| JBTS§ | 12q24 | TCTN2 | Tectonic-2 | JS | n.a. | MKS8 | 74 |
only heterozygous mutations reported in JS;
locus number unassigned;
kidney involvement is present in a subset of patients;
only screenings of at least 100 JS probands unselected for specific phenotypes have been included in the calculation of frequencies.
Legend: ACS: Acrocallosal syndrome; ATD: Asphyxiating thoracic dystrophy (Jeune syndrome); BBS: Bardet-Biedl syndrome; HLS: Hydrolethalus syndrome 2; LCA: Leber congenital amaurosis; MEDF: macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance; MORM: mental retardation, truncal obesity, retinal dystrophy, and micropenis; MKS: Meckel syndrome; n.a.: not assessable; NPHP: Nephronophthisis; OFD: Oro-facio-digital syndromes; SGB: Simpson-Golabi-Behmel syndrome; SLSN: Senior-Löken syndrome. Quoted references refer to papers reporting mutations in JS patients.
Several research groups have performed NGS-based targeted resequencing in various ciliopathies,35–37 but comprehensive mutation screenings of all known JS genes in large cohorts of patients are still lacking. Yet, in spite of this, significant correlates have emerged between mutations in certain genes and specific JS subgroups, while other genes seem to associate to a much wider phenotypic spectrum.
Genotype-phenotype correlations
The most relevant correlation has been established between the TMEM67 gene and the subgroup of JS with liver involvement. Mutations in TMEM67 account for about 80% of patients with this rare phenotype, while only a minority bears mutations in other genes such as CC2D2A or RPGRIP1L.38–40 This subgroup includes the condition known with the acronym “COACH” (Cerebellar vermis hypoplasia, Oligophrenia, Ataxia, Coloboma and Hepatic fibrosis),41 as well as the “Gentile syndrome”, presenting with JS and liver fibrosis, in the absence of other clinical features.42 Another relevant correlate involves the CEP290 gene, that is mutated in about 50% of JS patients with the “cerebello-oculo-renal” phenotype, characterized by the co-occurrence of retinopathy (usually LCA) and juvenile NPH.43 Several genes (AHI1, INPP5E, ARL13B and CC2D2A) are nearly exclusively mutated in patients presenting either pure JS or JS with retinal involvement,44–48 and in particular those subjects bearing CC2D2A mutations showed a significantly higher frequency of ventriculomegaly and seizures.49 Finally, mutations in NPHP1, RPGRIP1L and TMEM237 are almost invariably associated to JS with renal involvement.11,50,51
Despite these evidences, genotype-phenotype correlates remain problematic. For instance, the same founder mutation p.R73C in the TMEM216 gene causes a JS phenotype that can be pure or variably associate to polydactyly, renal disease, oro-facial-digital features, and retinopathy.10 While essential to make diagnosis, the MTS is not helpful in classifying JS patients, as there are almost no neuroimaging-genotype correlations, with the possible exception of thinner superior cerebellar peduncles seen in patients with the NPHP1 homozygous deletion.50 Additionally, there is high intrafamilial variability also in terms of neuroimaging findings.25
Shared features among ciliopathies
The variable expressivity of JS goes beyond what is expected in classical autosomal recessive inheritance, and gains a further level of complexity when considering the striking degree of clinical and genetic overlay with other ciliopathies, such as isolated NPH, SLS, Meckel syndrome (MKS), BBS, some oro-facio-digital syndromes and skeletal ciliopathies such as short rib polydactylies. Shared clinical features variably include retinal dystrophy, NPH or cystic dysplastic kidneys, congenital liver fibrosis, polydactyly, situs inversus, occipital encephalocele and midline oral and facial defects (Table 2).
Table 2.
Clinical overlap between JS and other ciliopathies
| JS | MKS | OFD | SkCil | NPH/SLS | BBS | |
|---|---|---|---|---|---|---|
| Central Nervous System | ||||||
| - DD, intellectual disability | + | n.a. | + | + | + | + |
| - hypotonia, ataxia | + | n.a. | + | + | + | |
| - CVH | + | + | + | + | + | |
| - Molar Tooth Sign | + | OFDVI | ||||
| - occipital encephalocele | + | + | + | |||
| - other posterior fossa abn. | + | + | + | + | ||
| - other CNS defects | + | + | + | + | + | |
|
| ||||||
| Eye | ||||||
| - retinal dystrophy | + | + | + | + | + (SLS) | + |
| - colobomas | + | + | ||||
|
| ||||||
| Kidney | ||||||
| - nephronophthisis | + | + | + | + | ||
| - cystic kidneys | + | + | + | + | + | + |
|
| ||||||
| Liver | ||||||
| - congenital fibrosis | + | + | + | + | + | + |
|
| ||||||
| Skeletal | ||||||
| - polydactyly | + | + | + | + | + | + |
| - scoliosis | + | + | ||||
| - bones shortening/bowing | + | + | + | + | ||
| - other skeletal defects | + | + | ||||
|
| ||||||
| Facial | ||||||
| - oral defects | + | + | + | + | ||
| - facial defects | + | + | + | |||
|
| ||||||
| Other defects | ||||||
| - laterality defects | + | + | + | + | + | |
| - congenital heart defects | + | + | + | + | + | |
| - Hirschsprung disease | + | n.a. | + | |||
| - obesity | n.a. | + | ||||
| - genital abnormalities | + | + | + | + | ||
| - diabetes mellitus | n.a. | + | ||||
| - anosmia | n.a. | + | ||||
Legend: BBS: Bardet-Biedl syndrome; DD: developmental delay; MKS: Meckel syndrome; NPHP: Nephronophthisis; OFD: Oro-facio-digital syndromes; SLSN: Senior-Löken syndrome; SkCil: Skeletal ciliopathies, including Jeune syndrome (or asphyxiating thoracic dystrophy), other Short Rib Polydactylies, and Sensenbrenner syndrome (cranioectodermal dysplasia). n.a.: not applicable. The + sign means that the feature may be present, while blank spots indicate features not yet reported in association with that syndrome; grey shaded cells indicate obligate or highly frequent features.
This clinical overlap is mirrored by marked genetic overlap, with allelism at many gene loci (Table 1). The most striking example is MKS, a severe malformative condition that is often lethal in utero, presenting with encephalocele and other posterior fossa anomalies, ductal plate malformation of the liver, polycystic kidneys and polydactyly. MKS and JS are indeed allelic disorders; to date, eleven genes are known to cause both conditions, and it is not uncommon to find affected siblings carrying the same homozygous mutation who present either MKS or JS.10,39,52
Oligogenic inheritance and mutational load
How mutations in one and the same gene can cause such a wide phenotypic spectrum, often encompassing different syndromes of variable severity, it remains an unresolved issue. Correlations between the mutation type and the ciliary phenotype have been observed for some genes such as TMEM67, RPGRIP1L, CC2D2A and TMEM216, with null mutations and hypomorphic missense mutations being significantly enriched in patients with lethal (MKS) and non-lethal (JS, NPH, BBS) disorders, respectively.10,37,39,49,53–55 Yet, things are far more complex than this. For instance, pathogenic mutations in the CEP290 gene have been associated to the whole spectrum of ciliopathies, without any obvious genotype-phenotype correlate.56 Even more strikingly, the same homozygous deletion of a 290kb genomic region on chromosome 2 encompassing the entire NPHP1 gene is known to cause a spectrum of phenotypes ranging from isolated NPH to SLS (NPH plus retinal dystrophy) to JS.50,57
In 2001, Katsanis and co-workers described for the first time a triallelic/digenic mode of inheritance in BBS, a ciliopathy characterized by retinal dystrophy, obesity, polydactyly, intellectual impairment, renal dysfunction and hypogonadism. As other ciliopathies, BBS presents wide genetic heterogeneity, with 17 gene loci identified to date. It was shown that autosomal recessive mutations in one BBS gene were not fully penetrant in some families, and only the co-occurrence of a third heterozygous mutation in another BBS gene resulted in the manifestation of the disease.58,59
This oligogenic inheritance, characterized by the concurrent effect of two or more distinct genes on the resulting phenotype, was subsequently confirmed in other ciliopathies such as NPH,60 and gained further complexity with the finding that not only pathogenic mutations but also rare variants or even common polymorphisms in other ciliary genes could genetically interact with the “main” recessive mutations, to modulate the expressivity of the ciliary phenotype. For instance, the p.A229T polymorphism in the RPGRIP1L gene was found to be associated with photoreceptor loss in various ciliopathies,61 while the p.R830W polymorphism in the AHI1 gene correlated, in two distinct studies, to an increased risk of NPHP1-deleted patients to develop neurological symptoms and retinopathy, respectively.62,63 Moreover, rare variants in TTC21B, a ciliary gene mutated in NPH, were found significantly enriched in cohorts of patients with ciliopathies compared to controls, suggesting they could contribute to increase the penetrance of mutations in other genes.64 In line with these observations, mutation screenings of ciliary genes in large JS cohorts have often identified single heterozygous variants, whose pathogenic significance still remains unclear (see for instance 37,49,54,56,65–67). It can be hypothesised that these could represent genetic modifiers of the phenotype, and indeed mutations in more than one gene have been reported in some JS patients who underwent multiple screenings or whole exome sequencing.56,62,66,68 Intriguingly, a recent paper reported the characterization of Tmem67 knock-out mice, showing that differences in the genetic background of the transgenic animals could significantly influence the severity of the phenotype, resembling either MKS or JS.69 All these findings support the concept of “mutational load”, that refers to the sum of all mutations and genomic variations contributing to define the penetrance and expressivity of ciliopathies.70 Moreover, recent data have unravelled novel pathogenetic mechanisms, related to the effect of variations residing within non-coding regulatory sequences of gene expression. For instance, we have recently shown that the two JS-causative genes TMEM216 and TMEM138 are aligned head-to-tail on chromosome 11, forming a cluster in which the expression of both genes is co-regulated by a conserved regulatory element lying in the intergenic region. This co-regulation is crucial to mediate the coordinated role of both proteins in transporting tethered pools of vesicles to the cilium, and it could be postulated that genetic variations affecting this conserved element may have detrimental effects on both genes’ expression.52
Pathogenetic mechanisms in JS
The finding that all JS causative genes encode for proteins of the primary cilium or its apparatus, and the striking clinical and genetic overlap with other ciliopathies, has put this subcellular organelle at the centre of intense research over the past decade. To date, the so called “ciliome” is predicted to comprise at least 3000 proteins (see for instance the Cildb database at http://cildb.cgm.cnrs-gif.fr/),71 suggesting a complex organization that well reflects the multifaceted roles of primary cilia in different cells and tissues, and the pleiotropic effect resulting from ciliary mutations.
Functional networks of ciliary proteins
Recent data have shown that ciliary proteins interact within functional networks, that largely relate to specific ciliopathy phenotypes. For instance, two motor complexes of proteins (IFT complex B and A) are known to mediate the anterograde and retrograde intraflagellary transport (IFT), that serve to bi-directionally move ciliary proteins along the axoneme using the microtubule motor proteins kinesins and dyneins, respectively.72 Mutations in the IFT proteins are a major cause of skeletal ciliopathies, including Sensenbrenner, Jeune and other short rib polydactyly syndromes (reviewed in 5). The correct assembly of IFT complexes at the ciliary base, and its turnaround from anterograde to retrograde transport at the ciliary tip are mediated by another protein complex, the BBSome, that consists of several proteins mutated in BBS (Figure 2).73
The JS-MKS-NPH protein complexes at the ciliary transition zone
The characterization of a growing number of proteins implicated in JS, MKS and NPH and the adoption of “state of the art” proteomic approaches has demonstrated that most of them localize at the basal body or at the ciliary TZ, where they form a large interactome broken down in complexes with shared functions.74
The most relevant complex for JS pathogenesis is the “B9” or “Tectonic” complex, that includes many proteins (B9D1, B9D2, TCTN1, TCTN2, TCTN3, CC2D2A, CEP290, TMEM216, TMEM138, TMEM67, TMEM237, TMEM231, TMEM17, MKS1, AHI1), all of which, with the exception of TMEM17, are causative of JS and/or MKS. Two other complexes involve mostly NPH proteins, but also JS proteins such as RPGRIP1L (Figure 2).
Several functions have been ascribed to proteins of the complex: they limit the diffusion rate of plasma-membrane proteins into the cilium, maintain the connection between the TZ and the ciliary membrane, and control specific pools of vesicles targeted to the cilium. As a consequence, their mutations or knockdown result in malfunctioning of the whole complex and disruption of the integrity of the ciliary barrier, causing various anomalies such as reduced cilia formation, abnormal ciliary morphology and trafficking, and altered localization of signalling receptors.11,52,74,75
A redundancy in the function of proteins within each complex, and functional interaction between complexes, has been demonstrated by analysing C. Elegans double mutants, that showed worsened phenotypes only when mutations affected two proteins belonging to distinct networks, but not to the same network.2,76
Primary cilia in development: implications for the neurological defects of JS
In the embryonic development, increasing evidence points to a key role for primary cilia in regulating main pathways such as Sonic Hedgehog (Shh), Wnt and planar cell polarity (PCP), that are implicated in left-right axis formation, limb development and neurogenesis.
Shh pathway
In vertebrates, at difference from Drosophila, many Shh core components, such as Patched1 (Ptch1), Smoothened (Smo), Sufu and Gli transcription factors, localize to cilia, and ciliary malfunctioning leads to severe disruption of the Shh pathway (reviewed in 77,78). Indeed, mutations in several JS-related genes have been linked to altered Shh signalling, and mice knock-out for these genes show a spectrum of Shh-related phenotypes that include polydactyly, microphthalmia, cleft palate, laterality defects, and a range of brain developmental abnormalities such as neural tube dorsalization and closure defects, holoprosencephaly, malformed hindbrain and exencephaly.66,74,75,79–83
In the CNS, the Shh pathway also plays a major role in cerebellar development, suggesting an intriguing link between mutations in ciliary genes, altered Shh signalling and cerebellar defects. Shh is expressed by Purkinje cells starting from embryonic day 17.5, and strongly drives the proliferation of granule cell precursors from progenitors in the rhombic lip. In the mouse cerebellum, targeted ablation of ciliary genes was found to induce a failure of expansion of cerebellar progenitors in response to Shh, resulting in cerebellar hypoplasia and foliation abnormalities.84,85 In line with these findings, a reduced proliferation of granule cell progenitors associated with cerebellar hypoplasia has been shown in some mouse models of JS (e.g. Rpgrip1l conditional mutants, ZNF423/Zpf423 knock-out mice).53,86 Interestingly, a recent work on human foetuses showed that cerebellar granule cell progenitors have a primary cilium and proliferate in response to Shh signalling; this proliferation was found to be dramatically reduced after 16 gestational weeks in the cerebellum of patients with JS, MKS or Jeune syndrome caused by distinct genetic mutations.87 Nevertheless, these findings are difficult to reconcile with the normal size of cerebellar hemispheres that is seen in most patients with these ciliopathies, suggesting that other pathological events occurring earlier during development may be responsible for the selective vermal hypoplasia that is typical of JS.
A direct link has also been shown between primary cilia, Shh signalling and human stem cells. Undifferentiated embryonic stem cells were found to harbour primary cilia hosting key components of the Shh signalling, that are essential for early development.88 Moreover, primary cilia were implicated in the Shh-mediated control of adult neural stem cells formation within the hippocampal dentate gyrus. In mouse models, the conditional removal of ciliary genes (such as the JS-causative gene Rpgrip1l), or of genes essential for the Shh pathway (such as Smo), resulted in faulted development of these neuronal precursors and marked hypotrophy of the dentate gyrus.89 Interestingly, hippocampal malformations have been described in a relevant subset of JS subjects,25,90 leading to speculate a potential link with the learning and memory deficits seen in these patients. Yet, this is just one of many possible mechanisms that could explain the occurrence of intellectual disability in JS. For instance, a recent paper making use of Arl13b conditional knock-out mice elegantly demonstrated that primary cilia also play a guiding role in the migration of postmitotic interneurons in the developing brain. These findings suggest that the abnormal cortical neuronal development associated to ciliary dysfunction could partly underlie the cognitive defects seen in JS.91
Canonical Wnt pathway
Recent studies on Ahi1 and Cep290 knock-out mice have shown a cerebellar phenotype characterized by vermis hypoplasia and a midline fusion defect with expansion of the roof plate, that was correlated to the disruption of another key developmental pathway, the canonical Wnt signalling cascade.92 The activation of this complex pathway eventually leads to the stabilization and nuclear localization of β-catenin, with subsequent activation of target genes.93 The role of primary cilia in canonical Wnt signalling has long been debated, with contrasting evidence. Recently, Lancaster and co-workers showed that Jouberin, encoded by the AHI1 gene, is a positive regulator of this signalling by facilitating β-catenin nuclear translocation, and that the primary cilium constrains this pathway by sequestering Jouberin and β-catenin along the cilium, and limiting their nuclear entry.94 In line with these findings, Ahi1 mutant mice displayed specific decrease in Wnt activity and reduced cell proliferation in the developing cerebellum at the midline anlage, that could be partially rescued by Lithium, a Wnt pathway agonist.92 Similarly, Tmem67 knock-out mouse embryos, presenting cerebellar vermis hypoplasia and deep interpeduncular fossa that highly resembled the MTS, showed abnormal cilia and a failure of nuclear β-catenin accumulation following Wnt stimulation, with decrease of Wnt downstream targets.69 These findings provide a direct link between dysregulated canonical Wnt signalling and vermian hypoplasia seen in JS.
Non-canonical Wnt/PCP pathway
The PCP pathway (or non-canonical Wnt signalling) relates to the ability of cells to orient along an axis along the plane of the tissue, and is implicated in several developmental processes, including neural tube closure and left-right patterning.95 There is a close correlation between the PCP and primary cilia, insofar mutations in key PCP effectors result in abnormal ciliogenesis, and vice versa. Mutations in several JS-related genes, such as TMEM237, TMEM216 or TMEM67, were found to significantly disrupt this pathway, leading to a hyper-activation of downstream effectors such as Dishevelled-1 and RhoA signalling, and defective ciliary formation and function.10,11,96
Conclusions and future directions
The field of ciliopathies is expanding at an impressively fast pace, as the complexity of the human ciliome is being unravelled and a growing number of heterogeneous human disorders are being causatively related to mutations in ciliary genes. The concept of mutational load invariably requires to shift the focus of genetic testing from single gene mutation analysis to more extensive and systematic screenings that would capture the genetic variability of each patient, in order to offer personalized counselling and prognostic indications. The advent of NGS technologies is now making this shift possible, as high throughput simultaneous resequencing of several target genes or even the whole exome are being offered as clinical diagnostic tests by many genetic laboratories, some of which provide their services directly to consumers through the Internet market. It is foreseeable that the adoption of such techniques on very large cohorts of well phenotyped patients will help to delineate the scenario of genetic modifiers contributing to specific clinical manifestations of JS. On the other hand, such a vast amount of generated data inevitably raises major interpretation challenges, especially regarding the functional impact of common and rare variants on disease penetrance and expressivity. This is particularly relevant in the clinical setting, since we are expected to translate the bulk of genetic data into meaningful information that would improve the diagnosis, prognosis and counselling for patients and their families. Standards for appropriate design, quality control, analysis and interpretation of sequencing data, as well as ethical regulations, are clearly needed in the upcoming future, before NGS techniques can be safely applied to routine diagnostic protocols.
Most of the current knowledge on JS pathogenesis derives from the study of zebrafish or mouse models. However, although extremely useful, these models often do not recapitulate their human counterpart in terms of organ specificity and severity. In particular, morpholino-based silencing of selected ciliary genes in zebrafish, that is a widely adopted strategy to model human ciliopathies, may produce different results in comparison to germline mutants, and may have toxic side effects that could be difficult to distinguish from gene-specific effects.97 A promising research tool is now offered by induced pluripotent stem cell (iPS) technology. iPS can be generated from somatic cell types through ectopic expression of a set of transcription factors, acquiring the features of embryonic stem cells and thus bearing the potential to give rise to virtually any cell type, including inaccessible tissues such as neurons. Patient-derived iPS are proving an ideal source for studying diseases in vitro, and have laid the basis for many applications, such as patient-specific correction of gene mutations and drug tailoring.98 Moreover, since human iPS maintain the genetic background of the patient, they have the unique advantage to exploit the complex relation between pathogenic mutations, genetic modifiers and phenotype that is seen in “oligogenic” disorders such as ciliopathies.
To date, no therapies are available to cure JS, or at least to improve prognosis by mitigating the disease expressivity. Yet, the rapid pace of genetic discoveries and the use of innovative technologies are expected to greatly improve our understanding of the pathobiology of primary cilia, bridging the gap towards the identification of potential targets for treatment.
Acknowledgments
We are grateful to Drs. Eugen Boltshauser, Joseph Gleeson, Andrea Poretti and Francesco Emma for critical reading of the manuscript and valuable suggestions, and to Dr. Daniel Doherty for his helpful comments on JS nomenclature. Dr Poretti also contributed the brain imaging as in Figure 1. Research in the Valente lab is supported by grants from the European Research Council (ERC Starting Grant #260888), the Italian Ministry of Health (Ricerca Corrente 2013, Ricerca Finalizzata Malattie Rare 2008) and the US National Institute of Health (R01NS048453).
Footnotes
Conflicts of interest
The authors declare they have no conflict of interest.
Search strategy and selection criteria
References for this Review were identified through searches of PubMed with the search terms “Joubert syndrome”, “ciliopathies”, “primary cilium”, and “molar tooth sign” (last searched June 2013). Articles were also identified through searches of the authors’ own files. Only papers published in English were reviewed. The final reference list was generated on the basis of originality and relevance to the broad scope of this Review.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pazour GJ, Bloodgood RA. Targeting proteins to the ciliary membrane. Curr Top Dev Biol. 2008;85:115–49. doi: 10.1016/S0070-2153(08)00805-3. [DOI] [PubMed] [Google Scholar]
- 2.Williams CL, Li C, Kida K, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192:1023–41. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Q, Milenkovic L, Jin H, et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329:436–9. doi: 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–43. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arts HH, Knoers NV. Current insights into renal ciliopathies: what can genetics teach us? Pediatr Nephrol. 2012;28:863–74. doi: 10.1007/s00467-012-2259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JE, Gleeson JG. Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr Opin Neurol. 2011;24:98–105. doi: 10.1097/WCO.0b013e3283444d05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 2012;123:695–709. doi: 10.1007/s00401-012-0951-2. [DOI] [PubMed] [Google Scholar]
- 8.Parisi M, Glass I. Joubert Syndrome and Related Disorders. GeneReviews™. Initial Posting: July 9, 2003; Last Revision: June 14, 2012. http://www.ncbi.nlm.nih.gov/books/NBK1325/
- 9.Edvardson S, Shaag A, Zenvirt S, et al. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet. 2010;86:93–7. doi: 10.1016/j.ajhg.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valente EM, Logan CV, Mougou-Zerelli S, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42:619–25. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang L, Szymanska K, Jensen VL, et al. TMEM237 Is Mutated in Individuals with a Joubert Syndrome Related Disorder and Expands the Role of the TMEM Family at the Ciliary Transition Zone. Am J Hum Genet. 2011;89:713–30. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braddock BA, Farmer JE, Deidrick KM, Iverson JM, Maria BL. Oromotor and communication findings in joubert syndrome: further evidence of multisystem apraxia. J Child Neurol. 2006;21:160–3. doi: 10.1177/08830738060210020501. [DOI] [PubMed] [Google Scholar]
- 14.Poretti A, Dietrich Alber F, Brancati F, Dallapiccola B, Valente EM, Boltshauser E. Normal cognitive functions in joubert syndrome. Neuropediatrics. 2009;40:287–90. doi: 10.1055/s-0030-1249630. [DOI] [PubMed] [Google Scholar]
- 15.Braddock SR, Henley KM, Maria BL. The face of Joubert syndrome: a study of dysmorphology and anthropometry. Am J Med Genet A. 2007;143A:3235–42. doi: 10.1002/ajmg.a.32099. [DOI] [PubMed] [Google Scholar]
- 16.Poretti A, Vitiello G, Hennekam RC, et al. Delineation and diagnostic criteria of Oral-Facial-Digital Syndrome type VI. Orphanet J Rare Dis. 2012;7:4. doi: 10.1186/1750-1172-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gleeson JG, Keeler LC, Parisi MA, et al. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004;125A:125–34. doi: 10.1002/ajmg.a.20437. [DOI] [PubMed] [Google Scholar]
- 18.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–25. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 19.Maria BL, Hoang KB, Tusa RJ, et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–30. doi: 10.1177/088307389701200703. [DOI] [PubMed] [Google Scholar]
- 20.Yachnis AT, Rorke LB. Neuropathology of Joubert syndrome. J Child Neurol. 1999;14:655–9. doi: 10.1177/088307389901401006. [DOI] [PubMed] [Google Scholar]
- 21.Friede RL, Boltshauser E. Uncommon syndromes of cerebellar vermis aplasia. I: Joubert syndrome. Dev Med Child Neurol. 1978;20:758–63. doi: 10.1111/j.1469-8749.1978.tb15307.x. [DOI] [PubMed] [Google Scholar]
- 22.Poretti A, Boltshauser E, Loenneker T, et al. Diffusion tensor imaging in Joubert syndrome. AJNR Am J Neuroradiol. 2007;28:1929–33. doi: 10.3174/ajnr.A0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spampinato MV, Kraas J, Maria BL, Walton ZJ, Rumboldt Z. Absence of decussation of the superior cerebellar peduncles in patients with Joubert syndrome. Am J Med Genet A. 2008;146A:1389–94. doi: 10.1002/ajmg.a.32282. [DOI] [PubMed] [Google Scholar]
- 24.Engle EC. Human genetic disorders of axon guidance. Cold Spring Harb Perspect Biol. 2010;2:a001784. doi: 10.1101/cshperspect.a001784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poretti A, Huisman TA, Scheer I, Boltshauser E. Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. AJNR Am J Neuroradiol. 2011;32:1459–63. doi: 10.3174/ajnr.A2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poretti A, Singhi S, Huisman TA, et al. Tecto-cerebellar dysraphism with occipital encephalocele: not a distinct disorder, but part of the Joubert syndrome spectrum? Neuropediatrics. 2011;42:170–4. doi: 10.1055/s-0031-1287763. [DOI] [PubMed] [Google Scholar]
- 27.Kamdar BB, Nandkumar P, Krishnan V, Gamaldo CE, Collop NA. Self-reported sleep and breathing disturbances in Joubert syndrome. Pediatr Neurol. 2011;45:395–9. doi: 10.1016/j.pediatrneurol.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Schurman SJ, Scheinman SJ. Inherited cerebrorenal syndromes. Nat Rev Nephrol. 2009;5:529–38. doi: 10.1038/nrneph.2009.124. [DOI] [PubMed] [Google Scholar]
- 29.Barkovich AJ, Millen KJ, Dobyns WB. A developmental and genetic classification for midbrain-hindbrain malformations. Brain. 2009;132:3199–230. doi: 10.1093/brain/awp247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fluss J, Blaser S, Chitayat D, et al. Molar tooth sign in fetal brain magnetic resonance imaging leading to the prenatal diagnosis of Joubert syndrome and related disorders. J Child Neurol. 2006;21:320–4. doi: 10.1177/08830738060210041001. [DOI] [PubMed] [Google Scholar]
- 31.Pugash D, Oh T, Godwin K, et al. Sonographic ‘molar tooth’ sign in the diagnosis of Joubert syndrome. Ultrasound Obstet Gynecol. 2011;38:598–602. doi: 10.1002/uog.8979. [DOI] [PubMed] [Google Scholar]
- 32.Saleem SN, Zaki MS. Role of MR imaging in prenatal diagnosis of pregnancies at risk for Joubert syndrome and related cerebellar disorders. AJNR Am J Neuroradiol. 2010;31:424–9. doi: 10.3174/ajnr.A1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srour M, Hamdan FF, Schwartzentruber JA, et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J Med Genet. 2012;49:636–41. doi: 10.1136/jmedgenet-2012-101132. [DOI] [PubMed] [Google Scholar]
- 34.Srour M, Schwartzentruber J, Hamdan FF, et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet. 2012;90:693–700. doi: 10.1016/j.ajhg.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janssen S, Ramaswami G, Davis EE, et al. Mutation analysis in Bardet-Biedl syndrome by DNA pooling and massively parallel resequencing in 105 individuals. Hum Genet. 2011;129:79–90. doi: 10.1007/s00439-010-0902-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Redin C, Le Gras S, Mhamdi O, et al. Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alstrom syndromes. J Med Genet. 2012;49:502–12. doi: 10.1136/jmedgenet-2012-100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otto EA, Ramaswami G, Janssen S, et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2011;48:105–16. doi: 10.1136/jmg.2010.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brancati F, Iannicelli M, Travaglini L, et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat. 2009;30:E432–42. doi: 10.1002/humu.20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iannicelli M, Brancati F, Mougou-Zerelli S, et al. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum Mutat. 2010;31:E1319–31. doi: 10.1002/humu.21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doherty D, Parisi MA, Finn LS, et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis) J Med Genet. 2010;47:8–21. doi: 10.1136/jmg.2009.067249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verloes A, Lambotte C. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. Am J Med Genet. 1989;32:227–32. doi: 10.1002/ajmg.1320320217. [DOI] [PubMed] [Google Scholar]
- 42.Gentile M, Di Carlo A, Susca F, et al. COACH syndrome: report of two brothers with congenital hepatic fibrosis, cerebellar vermis hypoplasia, oligophrenia, ataxia, and mental retardation. Am J Med Genet. 1996;64:514–20. doi: 10.1002/(SICI)1096-8628(19960823)64:3<514::AID-AJMG13>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 43.Brancati F, Barrano G, Silhavy JL, et al. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet. 2007;81:104–13. doi: 10.1086/519026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valente EM, Brancati F, Silhavy JL, et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol. 2006;59:527–34. doi: 10.1002/ana.20749. [DOI] [PubMed] [Google Scholar]
- 45.Travaglini L, Brancati F, Silhavy J, et al. Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders. Eur J Hum Genet. 2013 doi: 10.1038/ejhg.2012.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dixon-Salazar T, Silhavy JL, Marsh SE, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75:979–87. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bielas SL, Silhavy JL, Brancati F, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41:1032–6. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cantagrel V, Silhavy JL, Bielas SL, et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83:170–9. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bachmann-Gagescu R, Ishak GE, Dempsey JC, et al. Genotype-phenotype correlation in CC2D2A-related Joubert syndrome reveals an association with ventriculomegaly and seizures. J Med Genet. 2012;49:126–37. doi: 10.1136/jmedgenet-2011-100552. [DOI] [PubMed] [Google Scholar]
- 50.Castori M, Valente EM, Donati MA, et al. NPHP1 gene deletion is a rare cause of Joubert syndrome related disorders. J Med Genet. 2005;42:e9. doi: 10.1136/jmg.2004.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brancati F, Travaglini L, Zablocka D, et al. RPGRIP1L mutations are mainly associated with the cerebello-renal phenotype of Joubert syndrome-related disorders. Clin Genet. 2008;74:164–70. doi: 10.1111/j.1399-0004.2008.01047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JH, Silhavy JL, Lee JE, et al. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science. 2012;335:966–9. doi: 10.1126/science.1213506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delous M, Baala L, Salomon R, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–81. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 54.Mougou-Zerelli S, Thomas S, Szenker E, et al. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum Mutat. 2009;30:1574–82. doi: 10.1002/humu.21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Otto EA, Tory K, Attanasio M, et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11) J Med Genet. 2009;46:663–70. doi: 10.1136/jmg.2009.066613. [DOI] [PubMed] [Google Scholar]
- 56.Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat. 2010;31:1097–108. doi: 10.1002/humu.21337. [DOI] [PubMed] [Google Scholar]
- 57.Parisi MA, Bennett CL, Eckert ML, et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–9. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 59.Badano JL, Leitch CC, Ansley SJ, et al. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439:326–30. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- 60.Hoefele J, Wolf MT, O’Toole JF, et al. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol. 2007;18:2789–95. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- 61.Khanna H, Davis EE, Murga-Zamalloa CA, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41:739–45. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tory K, Lacoste T, Burglen L, et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18:1566–75. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]
- 63.Louie CM, Caridi G, Lopes VS, et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat Genet. 2010;42:175–80. doi: 10.1038/ng.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davis EE, Zhang Q, Liu Q, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43:189–96. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Putoux A, Thomas S, Coene KL, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet. 2011;43:601–6. doi: 10.1038/ng.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JE, Silhavy JL, Zaki MS, et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet. 2012;44:193–9. doi: 10.1038/ng.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chaki M, Airik R, Ghosh AK, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–48. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dafinger C, Liebau MC, Elsayed SM, et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J Clin Invest. 2011;121:2662–7. doi: 10.1172/JCI43639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abdelhamed ZA, Wheway G, Szymanska K, et al. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum Mol Genet. 2013;22:1358–72. doi: 10.1093/hmg/dds546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zaghloul NA, Katsanis N. Functional modules, mutational load and human genetic disease. Trends Genet. 2010;26:168–76. doi: 10.1016/j.tig.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnaiz O, Malinowska A, Klotz C, et al. Cildb: a knowledgebase for centrosomes and cilia. Database (Oxford) 2009;2009:bap022. doi: 10.1093/database/bap022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 73.Wei Q, Zhang Y, Li Y, Zhang Q, Ling K, Hu J. The BBSome controls IFT assembly and turnaround in cilia. Nat Cell Biol. 2012;14:950–7. doi: 10.1038/ncb2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sang L, Miller JJ, Corbit KC, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–28. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chih B, Liu P, Chinn Y, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- 76.Williams CL, Winkelbauer ME, Schafer JC, Michaud EJ, Yoder BK. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol Biol Cell. 2008;19:2154–68. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–44. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruat M, Roudaut H, Ferent J, Traiffort E. Hedgehog trafficking, cilia and brain functions. Differentiation. 2012;83:S97–104. doi: 10.1016/j.diff.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 79.Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–84. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liem KF, Jr, He M, Ocbina PJ, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci U S A. 2009;106:13377–82. doi: 10.1073/pnas.0906944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jacoby M, Cox JJ, Gayral S, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027–31. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- 82.Vierkotten J, Dildrop R, Peters T, Wang B, Ruther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development. 2007;134:2569–77. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- 83.Thomas S, Legendre M, Saunier S, et al. TCTN3 mutations cause Mohr-Majewski syndrome. Am J Hum Genet. 2012;91:372–8. doi: 10.1016/j.ajhg.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chizhikov VV, Davenport J, Zhang Q, et al. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci. 2007;27:9780–9. doi: 10.1523/JNEUROSCI.5586-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spassky N, Han YG, Aguilar A, et al. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol. 2008;317:246–59. doi: 10.1016/j.ydbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alcaraz WA, Gold DA, Raponi E, Gent PM, Concepcion D, Hamilton BA. Zfp423 controls proliferation and differentiation of neural precursors in cerebellar vermis formation. Proc Natl Acad Sci U S A. 2006;103:19424–9. doi: 10.1073/pnas.0609184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aguilar A, Meunier A, Strehl L, et al. Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. Proc Natl Acad Sci U S A. 2012;109:16951–6. doi: 10.1073/pnas.1201408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kiprilov EN, Awan A, Desprat R, et al. Human embryonic stem cells in culture possess primary cilia with hedgehog signaling machinery. J Cell Biol. 2008;180:897–904. doi: 10.1083/jcb.200706028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Han YG, Spassky N, Romaguera-Ros M, et al. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci. 2008;11:277–84. doi: 10.1038/nn2059. [DOI] [PubMed] [Google Scholar]
- 90.Senocak EU, Oguz KK, Haliloglu G, Topcu M, Cila A. Structural abnormalities of the brain other than molar tooth sign in Joubert syndrome-related disorders. Diagn Interv Radiol. 2010;16:3–6. doi: 10.4261/1305-3825.DIR.2673-09.1. [DOI] [PubMed] [Google Scholar]
- 91.Higginbotham H, Eom TY, Mariani LE, et al. Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex. Dev Cell. 2012;23:925–38. doi: 10.1016/j.devcel.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lancaster MA, Gopal DJ, Kim J, et al. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med. 2011;17:726–31. doi: 10.1038/nm.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–14. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 94.Lancaster MA, Schroth J, Gleeson JG. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol. 2011;13:700–7. doi: 10.1038/ncb2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25:201–13. doi: 10.1101/gad.2008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dawe HR, Adams M, Wheway G, et al. Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J Cell Sci. 2009;122:2716–26. doi: 10.1242/jcs.043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bedell VM, Westcot SE, Ekker SC. Lessons from morpholino-based screening in zebrafish. Brief Funct Genomics. 2011;10:181–8. doi: 10.1093/bfgp/elr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marchetto MC, Brennand KJ, Boyer LF, Gage FH. Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum Mol Genet. 2011;20:R109–15. doi: 10.1093/hmg/ddr336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ferland RJ, Eyaid W, Collura RV, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36:1008–13. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- 100.Alazami AM, Alshammari MJ, Salih MA, et al. Molecular characterization of Joubert syndrome in Saudi Arabia. Hum Mutat. 2012;33:1423–8. doi: 10.1002/humu.22134. [DOI] [PubMed] [Google Scholar]
- 101.Caridi G, Dagnino M, Rossi A, et al. Nephronophthisis type 1 deletion syndrome with neurological symptoms: prevalence and significance of the association. Kidney Int. 2006;70:1342–7. doi: 10.1038/sj.ki.5001768. [DOI] [PubMed] [Google Scholar]
- 102.Valente EM, Silhavy JL, Brancati F, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–5. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 103.Sayer JA, Otto EA, O’Toole JF, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–81. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 104.Helou J, Otto EA, Attanasio M, et al. Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior-Loken syndrome. J Med Genet. 2007;44:657–63. doi: 10.1136/jmg.2007.052027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baala L, Audollent S, Martinovic J, et al. Pleiotropic Effects of CEP290 (NPHP6) Mutations Extend to Meckel Syndrome. Am J Hum Genet. 2007;81:170–9. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Travaglini L, Brancati F, Attie-Bitach T, et al. Expanding CEP290 mutational spectrum in ciliopathies. Am J Med Genet. 2009;149A:2173–80. doi: 10.1002/ajmg.a.33025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Baala L, Romano S, Khaddour R, et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–94. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arts HH, Doherty D, van Beersum SE, et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–8. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- 109.Gorden NT, Arts HH, Parisi MA, et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83:559–71. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Field M, Scheffer IE, Gill D, et al. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet. 2012;20:806–9. doi: 10.1038/ejhg.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coene KL, Roepman R, Doherty D, et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465–81. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]