

Abstract
Fibroblast growth factor receptors (FGFRs) play diverse roles in control of cell proliferation, cell differentiation, angiogenesis, and development. Activating mutations of FGFRs in the germline have long been known to cause a variety of skeletal developmental disorders, but it is only recently that a similar spectrum of somatic FGFR mutations has been associated with human cancers. Many of these somatic mutations are gain-of-function and oncogenic and create dependencies in tumor cell lines harboring such mutations. A combination of knock-down studies and pharmaceutical inhibition in preclinical models has further substantiated genomically-altered FGFR as a therapeutic target in cancer, and the oncology community is responding with clinical trials evaluating multi-kinase inhibitors with anti-FGFR activity and a new generation of specific pan-FGFR inhibitors.
Keywords: FGFR, tyrosine kinase, somatic mutation, targeted therapy
Physiological FGFR activation
Members of the fibroblast growth factor receptor (FGFR) receptor tyrosine kinase (RTK) family, FGFR1-4, are differentially activated by binding to a subset of 18 fibroblast growth factors (FGFs) in conjunction with heparan sulfate proteoglycan, which stabilizes and sequesters FGFs [1]. Ligand specificity of FGFR1-3 is, in part, controlled by an alternative splicing event that affectsthe third immunoglobulin (Ig) loop (IgIII) in the ligand-binding domain, resulting in a “IIIb” isoform preferentially expressed in epithelial cells and a “IIIc” isoform preferentially expressed in mesenchymal cells. FGF3, FGF7, FGF10, and FGF22 exclusively bind the IIIb isoform; FGF1 binds both the IIIb and IIIc isoforms; and the remaining 13 FGF family ligands for which FGFR-stimulatory activity has been demonstrated preferentially bind the IIIc isoform [2]. Importantly, ligand expression is controlled in a cell-specific manner such that physiological receptor stimulation tends to occur in a paracrine rather than autocrine manner; indeed, a switch to autocrine regulation can promote tumorigenesis [3].
Binding of cognate ligands induces FGFR dimerization, trans-autophosphorylation, and kinase activation [4]. Downstream signaling events are summarized in Box 1. Most of the original work dissecting the biochemistry of FGFR signal transduction involved primarily FGFR1; surprisingly little is known of the similarities and differences between the four family members. However, activating somatic mutations in all four FGFR genes have now been found in diverse human cancers, indicating the potential of FGFR inhibition as a powerful new approach to targeted cancer therapy.
Text Box. Physiological FGFR signaling.
Ligand-stimulated FGFRs phosphorylate the FGFR-associated cytosolic docking protein, FGFR substrate 2 (FRS2), which mediates activation of the RAS/MAPK (mitogen-activated protein kinase) pathway by binding the growth factor receptor-bound 2 (GRB2) - son of sevenless (SOS1) complex and Src homology region 2 domain phosphatase (SHP2). GRB2 also forms a complex with GRB2-associated binding protein 1 (GAB1), facilitating activation of the PI3K (phosphoinositide 3-kinase) pathway. GRB2 can additionally recruit the ubiquitin ligase CBL (casitas B-lineage lymphoma proto-oncogene) to FRS2, resulting in negative regulation of FGFR signaling. Phospholipase C-γ (PLCγ) directly binds the C-terminal tails of FGFRs when phosphorylated, to Tyr 766 in the case of FGFR1, but the significance of PLCγ binding remains unclear.
FGFR3 mutations in cancer
FGFR3 was the first FGFR family member reported to be somatically mutated in cancer, specifically in multiple myeloma [5] (Table 1). Recurring translocations between the immunoglobulin heavy chain (IGH) locus and FGFR3 were identified in 25% of patient samples and cell lines tested, frequently resulting in elevated expression levels of FGFR3 [5, 6]. Translocation only roughly correlates with increased FGFR3 protein expression [5, 7], and there is currently insufficient functional data to conclude that translocation-driven increases in wild-type protein expression are sufficient for tumorigenesis, perhaps implicating the consistently-overexpressed reciprocal translocation partner, multiple myeloma SET domain protein (MMSET) in these cases instead [8]. However, sequence analysis showed that about 10% of FGFR3 translocations harbor recurring somatic mutations of FGFR3, including alleles encoding Y373C, K650E, and K650M, correlating more accurately with increased FGFR3 expression[5]. Interestingly, these alleles and the subsequently-identified allele encoding R248C [9] have been reported in three sporadic skeletal dysplasias that result from germline mutation of FGFR3: R248C and Y373C in thanatophoric dysplasia type I, K650E in thanatophoric dysplasia type II, and K650M in severe achondroplasia with developmental delay and acanthosis (SADDAN) syndrome [10-12], thus establishing a paradigm that would repeatedly apply to FGFR family mutation in cancer.
Table 1.
Somatic mutations of FGFR family genes in cancer with known or likely oncogenic effects.
| Gene | Encoded Substitutiona | Cancer | Germline Disease | Oncogenicq | References |
|---|---|---|---|---|---|
| FGFR1 | P252T | Lung | FGFR1 P252R, PSe | [30, 84] | |
| FGFR1 | P252S | Melanoma | FGFR1 P252R, PS | [55, 84] | |
| FGFR1 | N546K | Glioblastoma | FGFR3 N540K, Hf | yes | [47, 48, 50] |
| FGFR1 | K656E | Glioblastoma | FGFR3 K650E, TDIIg | yes | [12, 46, 49] |
| FGFR2 | D101Y | Endometrial | yes | [25] | |
| FGFR2 | S252W | Endometrial | ASh | yes | [25, 26, 85] |
| FGFR2 | P253R | Endometrial | AS | yes | [25, 85] |
| FGFR2 | S267P | Gastric | CSi | [31, 33] | |
| FGFR2 | W290C | Lung | PS | [30, 34] | |
| FGFR2 | A(-2)G spl acceptor exIIIc | Gastric | PS | [31, 32] | |
| FGFR2 | A314D | Endometrial | FGFR2 A314S, Uj | [25, 86] | |
| FGFR2 | A315T | Endometrial | FGFR2 A315S, UNSCk | [26, 87] | |
| FGFR2 | S373C | Endometrial | BSCGSl | [26, 88] | |
| FGFR2 | Y376C | Endometrial | BSCGS | [26, 88] | |
| FGFR2 | C383R | Endometrial | yes | [26, 28] | |
| FGFR2 | N550K | Endometrial | FGFR2 N550H, CS | yes | [25, 26, 89] |
| FGFR2 | K660E | Endometrial | FGFR3 K650E, TDII | [12, 26] | |
| FGFR2 | K660M | Cervical | FGFR3 K650M, SSm | [11, 25] | |
| FGFR2 | K660N | Endometrial | U | [25, 89] | |
| FGFR2 | L764fs*4 | Endometrial | yes | [26, 90] | |
| FGFR3 | R248C | Bladder, MMb, SKc | TDIn | yes | [9, 12, 13, 17, 18] |
| FGFR3 | S249C | Bladder, Cervical, SK | TDI | yes | [17-19, 67] |
| FGFR3 | G370C | Bladder, SK | TDI | [10, 17, 18] | |
| FGFR3 | S371C | Bladder, SK | TDI | [12, 18, 91] | |
| FGFR3 | Y373C | Bladder, MM, SK | TDI | yes | [5, 10, 16, 18, 20] |
| FGFR3 | G380R | Bladder | Ao | [92, 93] | |
| FGFR3 | A391E | Bladder, SK | CS/ANp | [23, 94, 95] | |
| FGFR3 | N540S | Bladder | FGFR3 N540K, H | [48, 96] | |
| FGFR3 | K650E | Bladder, MM, SK, SSd | TDII | yes | [5, 12, 16-18, 24] |
| FGFR3 | K650M | Bladder, MM, SK | SS | [5, 11, 18, 93] | |
| FGFR3 | K650N | Bladder | H | [97, 98] | |
| FGFR3 | K650Q | Bladder | H | [99, 100] | |
| FGFR3 | K650T | Bladder | H/AN | [93, 101] | |
| FGFR3 | 795fs*139 | MM | TDI | yes | [16, 102] |
| FGFR4 | Y367C | Breast | FGFR3 Y373C, TDI | [10, 55] | |
| FGFR4 | N535D | Rhabdomyosarcoma | FGFR3 N540K, H | [48, 54] | |
| FGFR4 | N535K | Rhabdomyosarcoma | FGFR3 N540K, H | yes | [48, 54] |
| FGFR4 | V550E | Rhabdomyosarcoma | yes | [54] | |
| FGFR4 | V550L | Rhabdomyosarcoma | [54] |
Amino acid numbers are derived from the following reference transcripts: FGFR1, NM_000604; FGFR2, NM_022970; FGFR3, NM_000142; FGFR4, NM_002011.
MM, multiple myeloma
SK seborrheic keratosis
SS, spermatocytic seminoma
Pfeiffer syndrome
hypochondroplasia
thanatophoric dysplasia, type II
Apert syndrome
Crouzon syndrome
unclassified or unspecified craniosynostosis
unicoronal non-syndromic craniosynostosis
Beare-Stevensen Cutis Gyrata Syndrome
SADDAN syndrome
thanatophoric dysplasia, type I
achondroplasia
acanthosis nigricans
as defined by a positive result in a cell-based transformation assay or animal model of cancer
Studied in the context of thanatophoric dysplasia and SADDAN syndrome, ectopically-expressed Y373C, K650E, and K650M FGFR3 displayed constitutively elevated kinase activity[11, 13, 14]. The K650E substitution is located in the activation loop of the FGFR3 kinase domain and presumably directly affects the active site conformation of the mutant protein [15]. In contrast, the Y373C and R248C substitutions located in the extracellular region of FGFR3 introduce an unpaired cysteine which results in the formation of intermolecular disulfide bonds, leading to constitutive receptor dimerization and therefore constitutive kinase activation [13]. The activated alleles were also tested in cell-based transformation assays. Whereas expression of the wild-type FGFR3 had no effect on colony formation in soft agar, both the Y373C and K650E-encoding alleles supported anchorage-independent growth of NIH-3T3 cells, indicating that these alleles are in fact oncogenic [16]. Ectopic expression of FGFR3 K650E in Ba/F3 cells additionally conferred interleukin-3 (IL-3)-independent proliferation, a phenotype often associated with expression of oncogenic RTKs.
Recurring activating mutations of FGFR3 were subsequently detected in additional tumor types (Table 1), including 35% of urothelial cell (bladder) carcinomas and 25% of cervical carcinomas, as well as 39% of benign seborrheic keratoses [17, 18]. In addition to the mutations described above, the S249C and G370C substitutions that introduce an unpaired cysteine, resulting in constitutive dimer formation, were found in bladder carcinomas as well as thanatophoric dysplasia type I patients [10, 13, 17, 19, 20]. In contrast to transformed phenotypes observed in NIH-3T3 cell assays, neither FGFR3 S249C nor Y373C supported anchorage-independent growth of immortalized normal human urothelial cells, which are assumed to be more physiologically relevant to bladder cancer than NIH-3T3 cells, with the caveat that no positive control data was presented for this assay [21]. These two mutants did, however, confer increased cell saturation density [21].
FGFR3 mutations in urothelial cell carcinomas correlate with lower tumor grade. However, within the stratum of low-grade non-muscle invasive tumors, FGFR3 mutation correlates with higher risk of recurrence compared to tumors without FGFR3 mutation [22, 23]. Therefore, although FGFR3 mutations are found primarily in low-grade tumors, treatment targeted to FGFR3 mutations might still benefit a subset of bladder cancer patients.
Seborrheic keratosis is a common benign skin tumor originating from keratinocytes of the epidermis, the prevalence of which increases with age [18]. A spectrum of somatic alleles similar to that found in multiple myeloma and urothelial cell carcinoma are also found in seborrheic keratosis (Table 1), with the addition of S371C, another substitution observed in patients with thanatophoric dysplasia type I that results in receptor dimerization [12, 13, 18].
More recently, the FGFR3 K650E substitution was identified in spermatocytic seminomas, rare testicular malignancies that occur in aging patients. An observed clonal expansion of activated K650 mutants in sperm (but not blood) that correlated with increased donor age indicates that positive selection of sperm harboring activated FGFR3 K650E mutants underlies both the sporadic incidence of spermatocytic seminoma and the germline transmission of thanatophoric dysplasia type II [24].
Recurring oncogenic mutations of FGFR3 have thus been identified in several tumor types. It is reasonable to expect that additional somatic alleles of FGFR3 previously associated with similar germline skeletal dysplasia syndromes will be oncogenic as well. However, for novel FGFR3 mutations, detailed functional studies will be required to distinguish “driver” mutations that contribute to tumorigenesis from “passenger” mutations that provide no fitness benefit for the tumor.
FGFR2 mutations in cancer
The next most significant discovery of FGFR family mutation in cancer was the identification of FGFR2 mutations in endometrial carcinoma [25, 26] (Table 1). Sequence analysis of primary tumors and cell lines uncovered somatic extracellular and kinase domain mutations in about 12% of samples tested, with particularly high recurrence of mutations encoding S252W, P253R, and N550K, as well as several different substitutions of K660 [25, 26]. Analogous to the situation with FGFR3, the S252W and P253R –encoding alleles of FGFR2 are also found as autosomal dominant mutations associated with the congenital developmental disorder Apert Syndrome [27]. Mutations that alter N550 and K660 are associated with similar craniosynostosis disorders [27], and are paralogous to FGFR3 N540K and K650E (Figure 2). Substitution of these two residues activates kinase activity by disengaging a “molecular brake” that maintains FGFR2 in an inactive conformation through a network of hydrogen bonds that inhibits movement of the N lobe toward the C lobe and is required for alignment of catalytic residues [15].
Figure 2.
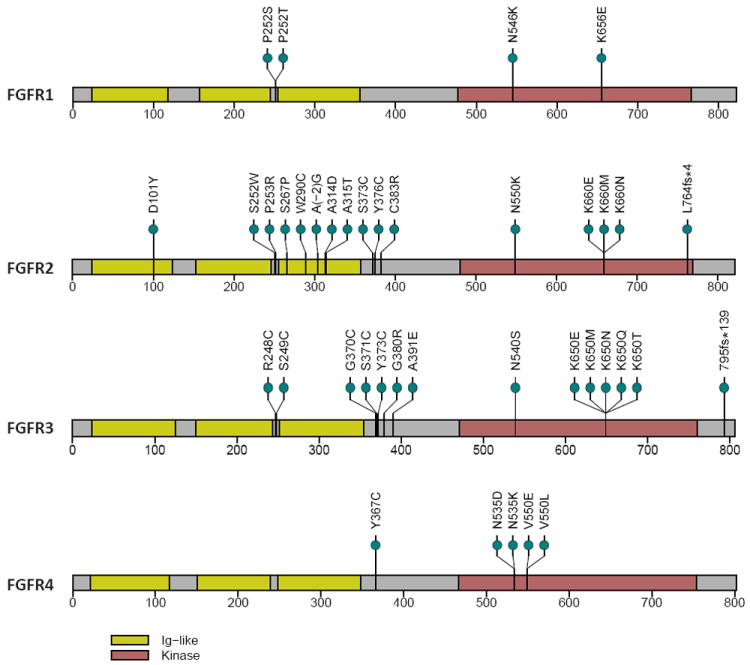
Alignment of altered amino acids encoded by somatic mutations found in FGFRs that are known or likely to be oncogenic (Table 1).
Unique amino acid substitutions are represented by green circles. Amino acid numbers are derived from the following reference transcripts: FGFR1, NM_000604; FGFR2, NM_022970; FGFR3, NM_000142; FGFR4, NM_002011. Yellow, Ig-like domains; red, kinase domains.
A subset of FGFR2 mutations found in endometrial carcinoma have been tested for oncogenic potential in cell-based transformation assays, and most have scored positive (Table 1), further suggesting a causative role in cancer for many of these alleles [25, 28]. The S252W and P253R substitutions, in particular, activate FGFR2 in a manner that is unique in oncology to the FGFR family. These altered residues lie in the ligand binding region and confer a gain in ligand binding promiscuity, such that ligands expressed by the epithelial cells that normally only bind the mesenchymally expressed “c” isoform of FGFR2 can now bind the S252W “b” isoform and establish an autocrine loop [29].
FGFR2 somatic mutations have also been identified in other cancers, but it is difficult to impute causation in the absence of functional data. Nevertheless, based on our experience to date, it is reasonable to expect that somatic alleles found in cancer would contribute to tumor formation if they are also observed in the germline of craniosynostosis patients. For example, the W290C-encoding allele found in lung squamous cell carcinoma [30] and the S267P-encoding allele found in gastric cancer [31] are also found in craniosynostosis syndromes [32-34], and thus might play a role in tumor development. In fact, a W290G mutant studied in the context of Crouzon syndrome indirectly caused intermolecular disulfide bond formation, constitutive dimerization, elevated kinase activity, and oncogenic transformation of NIH-3T3 cells, indicating a possible mechanism of action for other oncogenic substitutions at this residue [35].
There are to our knowledge only two reports of patients harboring germline mutations of an FGFR gene who develop cancer. Specifically, two Apert Syndrome patients systemically expressing FGFR2 P253R, 4 and 13 years of age, were reported to develop bladder cancer and ovarian dysgerminoma, respectively [36, 37]. In the absence of a systematic analysis, made difficult by the low number of cases, it remains unclear if craniosynostosis patients harboring germline FGFR mutations have an increased risk of cancer. Another set of germline events, single nucleotide polymorphisms (SNPs) with high minor allele frequencies (38-39%) present in the second intron of FGFR2 were found to be associated with increased risk of breast cancer in two genome-wide association studies [38, 39]. SNPs in this region were also associated with increased FGFR2 expression [40], suggesting that, under certain circumstances, overexpression of wild-type FGFR2 can also affect cancer incidence.
Although many FGFR mutations found in cancer have been demonstrated or are reasonably hypothesized to confer gain-of-function, FGFR2 mutations identified in 10% of melanoma cell lines and patient samples, including nonsense and frameshift mutations, appear to be loss-of-function mutations [41]. The biological consequences of these mutations for melanoma development remain unclear, but raise the possibility of context-specific functions of FGFRs in tumorigenesis.
FGFR1 mutations in cancer
Reminiscent of FGFR3 in multiple myeloma, FGFR1 was shown to be recurrently translocated in the 8p11 myeloproliferative syndrome (EMS), also known as stem cell leukemia/lymphoma (SCLL) [42]. However, whereas translocation of FGFR3 to the IGH locus results primarily in overexpression and occasional subsequent missense mutation of FGFR3, FGFR1 translocations typically result in the production of a fusion transcript, replacing 5’ coding sequences of FGFR1 with coding sequences from one of several possible fusion partners. Many observed translocation products, including zinc finger protein 198 (ZNF198)-FGFR1, 110 kDa centrosomal protein (CEP110)-FGFR1, and breakpoint cluster region protein (BCR)-FGFR1, constitutively dimerize when ectopically expressed or synthesized in vitro, with dimerization mediated by the fusion partner at least in some cases [43-45]. Although these fusion proteins exhibit constitutive kinase activity and transform Ba/F3 cells to IL-3 independence, poorly-controlled or conflicting data in the literature make it difficult to determine whether these data truly indicate a gain of function over the wild-type FGFR1 protein; it therefore remains unclear what fitness benefit tumors derive from these hybrid proteins.
In contrast to FGFR3, no point mutations in the translocated FGFR1 sequences found in EMS/SCLL have been reported. In fact, FGFR1 has not been shown to be a frequent target of somatic missense mutation in cancer sequencing studies, and the mutations that have been reported in primary tumor samples generally have not been recurrent. However, two FGFR1 mutations found in glioblastoma patients, encoding N546K and K656E [46, 47], are paralogous to FGFR3 germline alleles found in skeletal dysplasias and support morphological transformation of Rat-1 cells and increased focus formation in NIH-3T3 cells, respectively, suggesting that they may be driving tumorigenesis [12, 48-50].
Although few functionally validated FGFR1 mutations have been reported, it is possible that FGFR1 contributes to tumorigenesis primarily through gene amplification and overexpression. In fact, FGFR1 is a statistically significant target of focal amplification across cancers and in lung cancer in particular [51, 52]. Others have suggested that FGFR1 is a target of amplification in breast cancer [53]. Evidence for FGFR1 as a therapeutic target in tumor cells that harbor translocated, mutated, or amplified FGFR1 will be considered below.
FGFR4 mutations in cancer
FGFR4 is perhaps the least well-studied of the FGFR family members, and it is only recently that its contributions to cancer have begun to be uncovered. Recurring somatic kinase domain mutations of FGFR4 have recently been described in 8% of rhabdomyosarcoma patients [54]. An FGFR4 N535K-encoding allele, paralogous to the FGFR2 N550K-encoding allele found in endometrial carcinoma, supported oncogenic transformation of NIH-3T3 cells, as did a V550E-encoding allele. These FGFR4 mutants were also associated with metastatic phenotypes, as ectopic expression of these mutants in the rhabdomyosarcoma cell line RMS772 enhanced the ability of the cells to colonize the lung after intravenous injection [54].
Among other isolated reports of FGFR4 mutation in cancer, likely gain of function can be logically predicted only for the Y367C mutant found in a breast cancer cell line [55]. This residue is paralogous with other mutants found in the germline of patients with skeletal dysplasia or craniosynostosis syndromes and somatically in patients with diverse cancers, including FGFR2 Y375C and FGFR3 Y373C. Ectopic expression of the FGFR4 Y367C-encoding allele in HEK293 cells resulted in elevated levels of endogenous extracellular signal-regulated kinase (ERK) phosphorylation, but no transformed phenotypes were observed in NIH-3T3 cells expressing the Y367C mutant [56]. Definitive characterization of the effects of this substitution on protein function requires further study.
Although not found as a somatic mutation, a SNP in FGFR4 is associated with increased disease aggressiveness. The Gly388 –encoding allele (G388) of FGFR4 has been associated with a shorter time to progression after initial surgery and adjuvant therapy in breast cancer patients, and also correlated with lymph node metastasis and disease recurrence in prostate cancer patients [57, 58]. Although no phenotype was observed in unperturbed FGFR4 Arg385knock-in mice (murine equivalent to human FGFR4 Arg388), crosses with mice conditionally overexpressing transforming growth factor α in breast epithelia resulted in mice with increased mass of mammary tumors and increased number of metastases [59]. Biochemically, FGFR4 Arg388 increases protein stability and prolongs protein phosphorylation in response to ligand stimulation [60]. FGFR4 Arg388 appears to affect only tumor progression and not initiation; this more subtle effect is consistent with the substantial prevalence of this allele in the germline of the Caucasian population, with over 50% of individuals carrying one or two copies of this allele [57].
These data suggest that FGFR4 also plays a role in tumorigenesis for several tumor types. It should be noted that FGFR4 was found to be significantly mutated in lung adenocarcinoma [61]. However, some of these mutations are frameshifting deletions which likely lead to loss of protein function, reminiscent of the situation with loss-of-function FGFR2 mutations in melanoma. Perhaps in these cases the gene expression state of the tissue of origin influences whether a particular gene acts as an oncogene or tumor suppressor. Functional experiments are required to determine the biochemical nature of these FGFR4 mutations and whether they play a role in lung tumorigenesis or are merely passenger mutations, providing no fitness benefit for the tumor.
Validation of FGFRs as therapeutic targets
If FGFR family members mutated or amplified in human cancers are to serve as therapeutic targets, tumor cells harboring such genomic lesions must depend on FGFR activity for survival. Such dependencies have been investigated using either RNA interference techniques or treatment with a commercially-available kinase inhibitor, PD173074, which demonstrates high specificity toward FGFRs [62] but is not suitable for clinical use. Additional experiments have been performed using multi-target kinase inhibitors that have progressed to Phase II/III trials but exhibit a range of activities against FGFRs and other kinases.
The potential clinical utility of inhibiting FGFRs was first shown in multiple myeloma cell lines harboring oncogenic FGFR3 mutations. Treatment of KMS11 cells, harboring an allele encoding FGFR3 Y373C, with the pan-FGFR inhibitor PD173074 induced apoptotic cell death as evidenced by annexin V staining [63]. Treatment with the clinically-relevant, albeit less specific, kinase inhibitor CHR258/TKI258 caused a reduction in tumor growth and increased survival in KMS11 subcutaneous xenograft models [64], and similarly decreased tumor growth and increased survival in a KMS11 orthotopic xenograft model [65].
In addition to kinase inhibitors, neutralizing antibodies directed against FGFR3 have also shown preclinical efficacy in vivo in multiple myeloma. A neutralizing antibody directed towards FGFR3 that blocks ligand binding in vitro and prevents receptor dimerization associated with constitutively activating extracellular mutations, R3Mab, exerted a potent anti-tumor effect in KMS11 subcutaneous xenografts primarily due the induction of antibody-dependent cell-mediated cytotoxicity (ADCC) [66].
Following the identification of FGFR3 activating mutations in bladder cancer, preclinical in vitro studies showed that knockdown of FGFR3 or inhibition with the SU5402 pan-FGFR kinase inhibitor resulted in a reduction in proliferation and soft agar colony formation of MGH-U3 and 97-7 bladder cancer cells expressing the activated Y373C and S249C mutants, respectively, without inducing apoptosis [67, 68]. Tumor growth inhibition was furthermore observed with UM-UC-14, expressing FGFR3 S249C, and MGH-U3 bladder cancer cell lines when engrafted subcutaneously into SCID mice and treated with PD173074 [69].
More progress has been made in demonstrating that endometrial cancer cells are dependent on mutant FGFR2 for cell survival. Because the most common FGFR2 mutant observed in endometrial tumors, S252W, is located in the extracellular domain and confers only ligand-dependent, albeit ligand-promiscuous activation, it was not known whether cells expressing this variant would depend on mutant FGFR2 for survival, given that the majority of mutated kinases for which oncogene dependence has been reported are constitutively activated in a ligand-independent manner. Nevertheless, knockdown of FGFR2 in cell lines with both kinase domain mutations and ligand binding domain mutations inhibited transformation and induced cell cycle arrest and cell death [25, 70]. Endometrial cancer cell lines harboring activating mutations in FGFR2 were selectively sensitive to the pan-FGFR inhibitor, PD173074, in both monolayer growth and soft agar colony formation assays [25, 70]. Notably, MAP kinase/ERK kinase 1 and 2 (MEK1 and MEK2) inhibition ameliorates the craniosynostosis phenotype of knock-in mice harboring an FGFR2 S252W –encoding germline allele [71], raising the possibility that inhibition of downstream signaling proteins activated by mutant FGFR2 could also provide additional therapeutic options in cancer.
Data supporting FGFR1 fusion proteins as therapeutic targets in EMS/SCLL are more limited. Because EMS/SCLL frequently transforms into acute myeloid leukemia (AML) [42], the AML cell line KG-1, harboring a translocation encoding an FGFR1OP2-FGFR1 fusion product, was used to study dependence of FGFR1-driven hematopoietic malignancies on FGFR1 function. Treatment of KG-1 cells with FGFR1-specific small interfering RNA (siRNA) caused inhibition of cell growth and induction of apoptosis [72]. The multi-kinase inhibitor TKI-258 similarly inhibited survival of KG-1 cells, as well as primary cells from EMS/SCLL patients harboring FGFR1 translocations, but not from EMS/SCLL patients without FGFR1 translocations [73].
FGFR inhibitors have also shown efficacy in vitro and in vivo in tumor types with amplification of FGFR1 and FGFR2, including gastric, breast, and lung cancers. Treatment of the KATO-III and OCUM-2M gastric cancer cell lines carrying FGFR2 amplifications with the anti-VEGFR/FGFR kinase inhibitor AZD2171 showed an inhibition of proliferation in vitro as well as in subcutaneous xenografts implanted in nude mice [74]. Furthermore, antibodies specific for the IIIb isoform of FGFR2 decrease FGFR2 phosphorylation in gastric cells which overexpress FGFR2 and display potent anti-tumor activity in xenograft models of gastric cancer [75]. The response of breast cancer cell lines harboring FGFR1 amplifications to FGFR inhibition is less convincing. siRNA knockdown of FGFR1 or treatment with PD173074 had no effect on cell proliferation under two-dimensional culture conditions. However, PD173074 treatment blocked soft agar colony formation by the CAL120 cell line, a breast cancer cell line capable of anchorage-independent growth [53]. In contrast, in the breast cancer cell line MFM223, in which FGFR2 is amplified, treatment with PD173074 resulted in induction of cell death [76]. Lung cancer cell lines harboring focal FGFR1 amplifications similarly exhibited a cytotoxic response to PD173074 treatment [52].
Overall, the data described above support dependence of tumor cell lines harboring FGFR alterations on FGFR activity with the exception of oncogenic FGFR4 mutations, which suffer from a lack of appropriate rhabdomyosarcoma cell lines in which to study such lesions. However, data regarding whether FGFR inhibition results in cell cycle arrest or induction of cell death in cell line models with amplification and/or mutational activation of FGFRs are inconclusive. Whether the apparent disparities reflect true differences in tissue or tumor type dependencies of FGFR signaling or the vagaries of a limited number of cell line models is currently unknown. It should be noted that little data has been published on the molecular mechanism of cell death induced by FGFR inhibition in these different cellular contexts, a necessary piece of the puzzle in understanding the exact mechanism of oncogene addiction that exists in these different cell types and exploiting this oncogene dependency for therapeutic gain.
Agents with anti-FGFR activity in clinical trials
Several small molecules with activity against FGFRs are currently in preclinical or clinical development. The majority of these are multi-kinase inhibitors often designed primarily as anti-angiogenic inhibitors with activity against vascular endothelial growth factor receptors (VEGFRs) and platelet-derived growth factor receptors (PDGFRs) and, as such, often don’t demonstrate sufficiently potent anti-FGFR activity. However, at least two second-generation pan-FGFR inhibitors that have increased specificity for the FGFR family have entered Phase I trials and several more are in preclinical development, further supporting the emerging role of FGFRs as therapeutic targets in oncology (Table 2).
Table 2.
Multi-kinase and pan-FGFR inhibitors in clinical development
| Pharmaceutical company | Chemical name Drug name | Multi-kinase activity | Clinical Trials | Ref. |
|---|---|---|---|---|
| Novartis | TKI258 Dovotinib | FLT3, FGFRs, VEGFRs, PDGFRs, KIT, CSFR | Phase II (Breast, Bladder, Myeloma) | [77] |
|
| ||||
| Bristol Myers Squib | BMS582664 Brivanib | VEGFRs, FGFRs, | Phase II (Endometrial, Gastric, Bladder) | [103] |
|
| ||||
| Astra Zeneca | AZ2171 Cediranib | VEGFRs, FGFRs, KIT | Phase I (Gastric, Breast) | [74] |
| Phase II (Endometrial) | ||||
|
| ||||
| Eisai | E7080 | VEGFR, FGFRR, PDGFRs | Phase II (Endometrial ca.) | [79] |
| Phase II – solid malignancies | ||||
|
| ||||
| Astra Zeneca | AZD4547 | Pan-FGFR | Phase I – solid malignacies | |
| Phase I/II – Breast in combination | ||||
|
| ||||
| Novartis | BGJ398 | Pan-FGFR | Phase I – solid malignancies | |
|
| ||||
| Five Prime Therapeutics | FP-1039 | FGFR1c:Fc ligand trap | Phase I – solid malignancies | |
| Phase II-endometrial cancer | ||||
TKI-258 (Novartis) is the multi-target kinase inhibitor with the best evidence for physiologically-relevant efficacy against activated FGFRs. TKI-258 is an orally administered, multi-targeted growth factor receptor inhibitor that has activity against FGFRs, VEGFRs, PDGFR, KIT, and FLT3 [77]. In a move that reflects current clinical trial design with new targeted agents, TKI258 is being evaluated in bladder cancer patients, with the inclusion criterion of availability of archival tumor tissue for FGFR3 mutation testing to enable correlative studies (NCT00790426). Additionally, it is being evaluated in advanced breast cancer patients with and without FGFR1 amplification (NCT00958971), and relapsed multiple myeloma cases with and without the t(4;14) translocation often associated with FGFR3 amplification (NCT01058434). Of note, a more specific pan-FGFR inhibitor, BGJ398 (Novartis), is currently being evaluated in a phase I study in advanced solid malignancies restricted to patients with advanced solid tumors demonstrating either FGFR1 or FGFR2 amplification or FGFR3 mutation for whom no further effective treatment exists (NCT01004224).
Brivanib (Bristol Myers Squibb) is a dual tyrosine kinase inhibitor of VEGFR and FGFR signaling which has shown activity against metastatic solid tumors refractory to standard therapy in Phase I clinical trials and is currently being evaluated in a large number of trials primarily as an anti-angiogenic agent [78]. Based on its additional anti-FGFR activity, it is also being tested in endometrial cancer (NCT00888173); however, in contrast to TKI258, no data has been published regarding whether brivanib shows in vitro or in vivo tumor growth inhibition in cell line models driven by mutated or amplified FGFRs.
E7080 (Eisai) has activity against VEGFRs, FGFRs, PDGFRs and KIT [79] and is currently in phase II clinical trials for a wide range of solid malignancies. Based on a partial response in the Phase I trial, a Phase II trial was opened to examine the efficacy of E7080 in patients with metastatic endometrial cancer (NCT01111461). Several additional molecules being developed as anti-angiogenic compounds are being trialed in endometrial cancer (AZ2171, NCT01132820; BIBF1120, NCT01225887). Should there be any partial or complete responses in these trials it will be interesting to correlate these with the mutation status of FGFR2.
AZD4547 (AstraZeneca) is a more specific pan-FGFR inhibitor that is currently in Phase I trials for patients with solid malignancies (NCT00979134) and in a Phase I/II trial in breast cancer in combination with an aromatase inhibitor where the Phase II arm includes examination of FGFR1 amplification as a biomarker of response (NCT01202591). No data have yet been published regarding the efficacy of this compound in preclinical models driven by activated FGFRs.
FP-1039 (FivePrime), an FGFR1c:Fc decoy receptor which acts as a broad FGF ligand trap is in Phase I trials in solid malignancies (NCT00687505). A small Phase II trial testing FP1039 in endometrial cancer patients preselected for somatic alleles encoding the S252W or P253R mutants, and thus presumably dependent on ligands sequestered by this decoy receptor, has opened (NCT01244438). This is the first trial to specifically select patients based on FGFR mutation status alone and the results are eagerly anticipated. Several companies have presented pre-clinical data at meetings reporting additional pan-FGFR inhibitors with increased specificity; we expect to hear more about these in the future.
Concluding remarks
The field of anti-FGFR therapy is in its infancy; therefore it is too soon to predict whether treatments targeting FGFRs will be as successful as those targeting tumors demonstrating oncogene dependence on other activated kinases, such as the BCR-ABL fusion protein in chronic myelogenous leukemia, mutant KIT and PDGFRA in gastrointestinal stromal tumors, and mutant epidermal growth factor receptor (EGFR) in lung adenocarcinoma [80-83]. Although many trials are underway in FGFR-dependent tumor types with multi-target inhibitors, results from Phase I trials of more specific second-generation FGFR inhibitors are eagerly awaited. Should these multi-kinase and/or pan-FGFR inhibitors result in clinically-significant response rates, an emerging area of research will be to identify the main mechanisms of resistance to FGFR inhibition so that combination therapies with chemotherapies or targeted agents that inhibit parallel or downstream signaling pathways can be identified that delay or minimize the likelihood of acquired resistance to anti-FGFR therapy.
It is possible that other tumor types, in addition to those considered here, might respond to FGFR inhibitor therapy. Ongoing massively-parallel next-generation cancer sequencing experiments will exhaustively characterize the cancer genomes of many tumor types, and will facilitate identification of additional diseases in which somatic FGFR mutation or amplification plays a major role. Furthermore, large scale studies that correlate cancer cell genotype with inhibitor sensitivity, such as the Center for Molecular Therapeutics 1000 (Sanger Institute and Massachusetts General Hospital) and the Cancer Cell Line Encyclopedia project (Broad Institute and Novartis) could reveal novel tumor types sensitive to FGFR inhibition.
Studies of the functional effects of somatic FGFR mutations in cancer have been greatly assisted by the prior characterization of germline mutations in skeletal malformation syndromes. However, FGFR signaling in cancer might also exhibit context dependence, exemplified by the selection for FGFR2 gain-of-function mutations in endometrial carcinoma and loss-of-function mutations in melanoma. In addition, many somatic mutations of FGFR family members, especially those that are not found to be recurrent, or more precisely, those that are not mutated at rates statistically significantly higher than the background mutation rate for the gene in question, could be passenger mutations that do not provide any fitness benefit for the tumor. As in all somatic mutation studies, experimental determination of which mutations play a causative role in tumorigenesis and how mutations affect protein function is currently the rate-limiting step to fully understanding the clinical implications of our genomic data.
Acknowledgments
We thank Lauren Solomon for assistance in preparing Figure 1, and Alex H. Ramos for assistance in preparing Figure 2.
Figure 1.
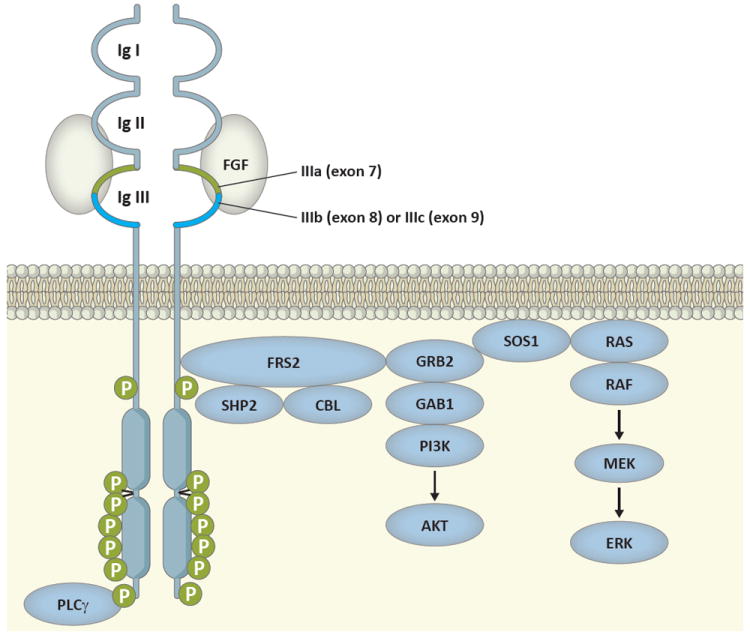
Schematic diagram of signal transduction pathway activated by ligand-stimulated FGFRs.
Ligand specificity is determined in part by an alternative splicing event in the C-terminal half of the third immunoglobulin domain (bright blue) in which either exon 8 or exon 9 can be used, producing the IIIb and IIIc isoforms, respectively. Ligand binding results in receptor autophosphorylation in the kinase domain region and phosphorylation of FRS2, which initiates a signaling cascade resulting in activation of the AKT and ERK downstream signaling pathways.
Glossary Box
- Apert syndrome
Autosomal dominant disorder characterized by craniosynostosis (premature fusion of skull bones) and syndactyly (digit fusion) of the hands and feet
- Crouzon syndrome
Autosomal dominant disorder characterized by craniosynostosis
- SADDAN syndrome
Severe achondroplasia (short-limbed dwarfism) with developmental delay and acanthosis nigricans (skin disorder characterized by thick, dark skin); bone growth disorder presumed to be autosomal dominant
- Sebhorreic keratosis
Common benign skin growth occurring primarily in older adults
- Spermatocytic seminoma
Rare germ cell tumor of the testis
- Thanatophoric dysplasia
Severe skeletal disorder presumed to be autosomal dominant, characterized by short limbs
References
- 1.Ornitz DM, Itoh N. Fibroblast growth factors. Genome biology. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang X, et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan G, et al. Exon switching and activation of stromal and embryonic fibroblast growth factor (FGF)-FGF receptor genes in prostate epithelial cells accompany stromal independence and malignancy. Mol Cell Biol. 1993;13:4513–4522. doi: 10.1128/mcb.13.8.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 5.Chesi M, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richelda R, et al. A novel chromosomal translocation t(4; 14)(p16.3; q32) in multiple myeloma involves the fibroblast growth-factor receptor 3 gene. Blood. 1997;90:4062–4070. [PubMed] [Google Scholar]
- 7.Keats JJ, et al. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003;101:1520–1529. doi: 10.1182/blood-2002-06-1675. [DOI] [PubMed] [Google Scholar]
- 8.Lauring J, et al. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood. 2008;111:856–864. doi: 10.1182/blood-2007-05-088674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Intini D, et al. Analysis of FGFR3 gene mutations in multiple myeloma patients with t(4;14) British journal of haematology. 2001;114:362–364. doi: 10.1046/j.1365-2141.2001.02957.x. [DOI] [PubMed] [Google Scholar]
- 10.Rousseau F, et al. Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1) Hum Mol Genet. 1996;5:509–512. doi: 10.1093/hmg/5.4.509. [DOI] [PubMed] [Google Scholar]
- 11.Tavormina PL, et al. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. American journal of human genetics. 1999;64:722–731. doi: 10.1086/302275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tavormina PL, et al. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet. 1995;9:321–328. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- 13.d’Avis PY, et al. Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ. 1998;9:71–78. [PubMed] [Google Scholar]
- 14.Webster MK, et al. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol Cell Biol. 1996;16:4081–4087. doi: 10.1128/mcb.16.8.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27:717–730. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chesi M, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- 17.Cappellen D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 18.Logie A, et al. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum Mol Genet. 2005;14:1153–1160. doi: 10.1093/hmg/ddi127. [DOI] [PubMed] [Google Scholar]
- 19.Tavormina PL, et al. Another mutation that results in the substitution of an unpaired cysteine residue in the extracellular domain of FGFR3 in thanatophoric dysplasia type I. Hum Mol Genet. 1995;4:2175–2177. doi: 10.1093/hmg/4.11.2175. [DOI] [PubMed] [Google Scholar]
- 20.van Rhijn BW, et al. FGFR3 and P53 characterize alternative genetic pathways in the pathogenesis of urothelial cell carcinoma. Cancer Res. 2004;64:1911–1914. doi: 10.1158/0008-5472.can-03-2421. [DOI] [PubMed] [Google Scholar]
- 21.di Martino E, et al. Mutant fibroblast growth factor receptor 3 induces intracellular signaling and cellular transformation in a cell type- and mutation-specific manner. Oncogene. 2009;28:4306–4316. doi: 10.1038/onc.2009.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hernandez S, et al. Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. J Clin Oncol. 2006;24:3664–3671. doi: 10.1200/JCO.2005.05.1771. [DOI] [PubMed] [Google Scholar]
- 23.van Rhijn BW, et al. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. 2001;61:1265–1268. [PubMed] [Google Scholar]
- 24.Goriely A, et al. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet. 2009;41:1247–1252. doi: 10.1038/ng.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dutt A, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008;105:8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollock PM, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26:7158–7162. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, et al. Activation of FGF receptors by mutations in the transmembrane domain. Oncogene. 1997;14:1397–1406. doi: 10.1038/sj.onc.1200983. [DOI] [PubMed] [Google Scholar]
- 29.Yu K, et al. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc Natl Acad Sci U S A. 2000;97:14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies H, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:7591–7595. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- 31.Jang JH, et al. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res. 2001;61:3541–3543. [PubMed] [Google Scholar]
- 32.Cornejo-Roldan LR, et al. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Human genetics. 1999;104:425–431. doi: 10.1007/s004390050979. [DOI] [PubMed] [Google Scholar]
- 33.Oldridge M, et al. Mutations in the third immunoglobulin domain of the fibroblast growth factor receptor-2 gene in Crouzon syndrome. Hum Mol Genet. 1995;4:1077–1082. doi: 10.1093/hmg/4.6.1077. [DOI] [PubMed] [Google Scholar]
- 34.Tartaglia M, et al. Trp290Cys mutation in exon IIIa of the fibroblast growth factor receptor 2 (FGFR2) gene is associated with Pfeiffer syndrome. Human genetics. 1997;99:602–606. doi: 10.1007/s004390050413. [DOI] [PubMed] [Google Scholar]
- 35.Robertson SC, et al. Activating mutations in the extracellular domain of the fibroblast growth factor receptor 2 function by disruption of the disulfide bond in the third immunoglobulin-like domain. Proc Natl Acad Sci U S A. 1998;95:4567–4572. doi: 10.1073/pnas.95.8.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreou A, et al. Early-onset low-grade papillary carcinoma of the bladder associated with Apert syndrome and a germline FGFR2 mutation (Pro253Arg) Am J Med Genet A. 2006;140:2245–2247. doi: 10.1002/ajmg.a.31430. [DOI] [PubMed] [Google Scholar]
- 37.Rouzier C, et al. Ovarian dysgerminoma and Apert syndrome. Pediatr Blood Cancer. 2007 doi: 10.1002/pbc.21156. [DOI] [PubMed] [Google Scholar]
- 38.Easton DF, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunter DJ, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer KB, et al. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gartside MG, et al. Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Mol Cancer Res. 2009;7:41–54. doi: 10.1158/1541-7786.MCR-08-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goradia A, et al. The 8p11 myeloproliferative syndrome: review of literature and an illustrative case report. International journal of clinical and experimental pathology. 2008;1:448–456. [PMC free article] [PubMed] [Google Scholar]
- 43.Ollendorff V, et al. Characterization of FIM-FGFR1, the fusion product of the myeloproliferative disorder-associated t(8;13) translocation. J Biol Chem. 1999;274:26922–26930. doi: 10.1074/jbc.274.38.26922. [DOI] [PubMed] [Google Scholar]
- 44.Smedley D, et al. ZNF198-FGFR1 transforms Ba/F3 cells to growth factor independence and results in high level tyrosine phosphorylation of STATS 1 and 5. Neoplasia (New York, N Y) 1999;1:349–355. doi: 10.1038/sj.neo.7900035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao S, et al. ZNF198-FGFR1 transforming activity depends on a novel proline-rich ZNF198 oligomerization domain. Blood. 2000;96:699–704. [PubMed] [Google Scholar]
- 46.Network TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rand V, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci U S A. 2005;102:14344–14349. doi: 10.1073/pnas.0507200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bellus GA, et al. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet. 1995;10:357–359. doi: 10.1038/ng0795-357. [DOI] [PubMed] [Google Scholar]
- 49.Hart KC, et al. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene. 2000;19:3309–3320. doi: 10.1038/sj.onc.1203650. [DOI] [PubMed] [Google Scholar]
- 50.Lew ED, et al. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Science signaling. 2009;2:ra6. doi: 10.1126/scisignal.2000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss J, et al. Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer. Science translational medicine. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turner N, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor JGt, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. The Journal of clinical investigation. 2009;119:3395–3407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruhe JE, et al. Genetic alterations in the tyrosine kinase transcriptome of human cancer cell lines. Cancer Res. 2007;67:11368–11376. doi: 10.1158/0008-5472.CAN-07-2703. [DOI] [PubMed] [Google Scholar]
- 56.Roidl A, et al. The FGFR4 Y367C mutant is a dominant oncogene in MDA-MB453 breast cancer cells. Oncogene. 2010;29:1543–1552. doi: 10.1038/onc.2009.432. [DOI] [PubMed] [Google Scholar]
- 57.Bange J, et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002;62:840–847. [PubMed] [Google Scholar]
- 58.Wang J, et al. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clin Cancer Res. 2004;10:6169–6178. doi: 10.1158/1078-0432.CCR-04-0408. [DOI] [PubMed] [Google Scholar]
- 59.Seitzer N, et al. A single nucleotide change in the mouse genome accelerates breast cancer progression. Cancer Res. 2010;70:802–812. doi: 10.1158/0008-5472.CAN-09-3239. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, et al. Altered fibroblast growth factor receptor 4 stability promotes prostate cancer progression. Neoplasia (New York, N Y) 2008;10:847–856. doi: 10.1593/neo.08450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kunii K, et al. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008;68:2340–2348. doi: 10.1158/0008-5472.CAN-07-5229. [DOI] [PubMed] [Google Scholar]
- 63.Trudel S, et al. Inhibition of fibroblast growth factor receptor 3 induces differentiation and apoptosis in t(4;14) myeloma. Blood. 2004;103:3521–3528. doi: 10.1182/blood-2003-10-3650. [DOI] [PubMed] [Google Scholar]
- 64.Trudel S, et al. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105:2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- 65.Xin X, et al. CHIR-258 is efficacious in a newly developed fibroblast growth factor receptor 3-expressing orthotopic multiple myeloma model in mice. Clin Cancer Res. 2006;12:4908–4915. doi: 10.1158/1078-0432.CCR-06-0957. [DOI] [PubMed] [Google Scholar]
- 66.Qing J, et al. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. The Journal of clinical investigation. 2009;119:1216–1229. doi: 10.1172/JCI38017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernard-Pierrot I, et al. Oncogenic properties of the mutated forms of fibroblast growth factor receptor 3b. Carcinogenesis. 2006;27:740–747. doi: 10.1093/carcin/bgi290. [DOI] [PubMed] [Google Scholar]
- 68.Tomlinson DC, et al. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene. 2007;26:5889–5899. doi: 10.1038/sj.onc.1210399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miyake M, et al. 1-tert-butyl-3-[6-(3,5-dimethoxy-phenyl)-2-(4-diethylamino-butylamino)-pyr ido[2,3-d]pyrimidin-7-yl]-urea (PD173074), a selective tyrosine kinase inhibitor of fibroblast growth factor receptor-3 (FGFR3), inhibits cell proliferation of bladder cancer carrying the FGFR3 gene mutation along with up-regulation of p27/Kip1 and G1/G0 arrest. J Pharmacol Exp Ther. 2010;332:795–802. doi: 10.1124/jpet.109.162768. [DOI] [PubMed] [Google Scholar]
- 70.Byron SA, et al. Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer Res. 2008;68:6902–6907. doi: 10.1158/0008-5472.CAN-08-0770. [DOI] [PubMed] [Google Scholar]
- 71.Shukla V, et al. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet. 2007;39:1145–1150. doi: 10.1038/ng2096. [DOI] [PubMed] [Google Scholar]
- 72.Gu TL, et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood. 2006;108:4202–4204. doi: 10.1182/blood-2006-06-026666. [DOI] [PubMed] [Google Scholar]
- 73.Chase A, et al. Activity of TKI258 against primary cells and cell lines with FGFR1 fusion genes associated with the 8p11 myeloproliferative syndrome. Blood. 2007;110:3729–3734. doi: 10.1182/blood-2007-02-074286. [DOI] [PubMed] [Google Scholar]
- 74.Takeda M, et al. AZD2171 Shows Potent Antitumor Activity Against Gastric Cancer Over-Expressing Fibroblast Growth Factor Receptor 2/Keratinocyte Growth Factor Receptor. Clin Cancer Res. 2007;13:3051–3057. doi: 10.1158/1078-0432.CCR-06-2743. [DOI] [PubMed] [Google Scholar]
- 75.Bai A, et al. GP369, an FGFR2-IIIb specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer Res. 2010 doi: 10.1158/0008-5472.CAN-10-1489. [DOI] [PubMed] [Google Scholar]
- 76.Turner N, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29:2013–2023. doi: 10.1038/onc.2009.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee SH, et al. In vivo target modulation and biological activity of CHIR-258, a multitargeted growth factor receptor kinase inhibitor, in colon cancer models. Clin Cancer Res. 2005;11:3633–3641. doi: 10.1158/1078-0432.CCR-04-2129. [DOI] [PubMed] [Google Scholar]
- 78.Rosen LS, et al. Phase I dose escalation study to determine the safety, pharmacokinetics and pharmacodynamics of BMS-582664, a VEGFR/FGFR inhibitor in patients with advanced/metastatic solid tumors. Journal of Clinical Oncology; 2006 ASCO Annual Meeting Proceedings Part I; 2006. p. 3051. [Google Scholar]
- 79.Matsui J, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122:664–671. doi: 10.1002/ijc.23131. [DOI] [PubMed] [Google Scholar]
- 80.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 81.Joensuu H, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344:1052–1056. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- 82.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 83.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 84.Muenke M, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet. 1994;8:269–274. doi: 10.1038/ng1194-269. [DOI] [PubMed] [Google Scholar]
- 85.Wilkie AO, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 86.Steinberger D, et al. The mutations in FGFR2-associated craniosynostoses are clustered in five structural elements of immunoglobulin-like domain III of the receptor. Human genetics. 1998;102:145–150. doi: 10.1007/s004390050668. [DOI] [PubMed] [Google Scholar]
- 87.Johnson D, et al. A novel mutation, Ala315Ser, in FGFR2: a gene-environment interaction leading to craniosynostosis? Eur J Hum Genet. 2000;8:571–577. doi: 10.1038/sj.ejhg.5200499. [DOI] [PubMed] [Google Scholar]
- 88.Przylepa KA, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nat Genet. 1996;13:492–494. doi: 10.1038/ng0896-492. [DOI] [PubMed] [Google Scholar]
- 89.Kan SH, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. American journal of human genetics. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lorenzi MV, et al. Ligand-independent activation of fibroblast growth factor receptor-2 by carboxyl terminal alterations. Oncogene. 1997;15:817–826. doi: 10.1038/sj.onc.1201242. [DOI] [PubMed] [Google Scholar]
- 91.Bakkar AA, et al. Sensitive allele-specific PCR assay able to detect FGFR3 mutations in tumors and urine from patients with urothelial cell carcinoma of the bladder. Clinical chemistry. 2005;51:1555–1557. doi: 10.1373/clinchem.2005.049619. [DOI] [PubMed] [Google Scholar]
- 92.Rousseau F, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- 93.van Rhijn BW, et al. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur J Hum Genet. 2002;10:819–824. doi: 10.1038/sj.ejhg.5200883. [DOI] [PubMed] [Google Scholar]
- 94.Hafner C, et al. High frequency of FGFR3 mutations in adenoid seborrheic keratoses. The Journal of investigative dermatology. 2006;126:2404–2407. doi: 10.1038/sj.jid.5700422. [DOI] [PubMed] [Google Scholar]
- 95.Meyers GA, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet. 1995;11:462–464. doi: 10.1038/ng1295-462. [DOI] [PubMed] [Google Scholar]
- 96.Lindgren D, et al. Molecular characterization of early-stage bladder carcinomas by expression profiles, FGFR3 mutation status, and loss of 9q. Oncogene. 2006;25:2685–2696. doi: 10.1038/sj.onc.1209249. [DOI] [PubMed] [Google Scholar]
- 97.Bellus GA, et al. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. American journal of human genetics. 2000;67:1411–1421. doi: 10.1086/316892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Platt FM, et al. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res. 2009;15:6008–6017. doi: 10.1158/1078-0432.CCR-09-0898. [DOI] [PubMed] [Google Scholar]
- 99.Heuertz S, et al. Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. Eur J Hum Genet. 2006;14:1240–1247. doi: 10.1038/sj.ejhg.5201700. [DOI] [PubMed] [Google Scholar]
- 100.Sibley K, et al. Loss of heterozygosity at 4p16.3 and mutation of FGFR3 in transitional cell carcinoma. Oncogene. 2001;20:686–691. doi: 10.1038/sj.onc.1204110. [DOI] [PubMed] [Google Scholar]
- 101.Castro-Feijoo L, et al. Hypochondroplasia and Acanthosis nigricans: a new syndrome due to the p.Lys650Thr mutation in the fibroblast growth factor receptor 3 gene? European journal of endocrinology / European Federation of Endocrine Societies. 2008;159:243–249. doi: 10.1530/EJE-08-0393. [DOI] [PubMed] [Google Scholar]
- 102.Rousseau F, et al. Stop codon FGFR3 mutations in thanatophoric dwarfism type 1. Nat Genet. 1995;10:11–12. doi: 10.1038/ng0595-11. [DOI] [PubMed] [Google Scholar]
- 103.Huynh H, et al. Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma. Clin Cancer Res. 2008;14:6146–6153. doi: 10.1158/1078-0432.CCR-08-0509. [DOI] [PubMed] [Google Scholar]