

Abstract
Emerging evidence indicates that complement provides costimulatory signals for murine T cells but whether complement impacts human T cells remains unclear. We observed production of complement activation products C3a and C5a during in vitro cultures of human T cells responding to allogeneic dendritic cells (DC). Both partners expressed the receptors for C3a (C3aR) and C5a (C5aR), and C3aR- and C5aR-antagonists inhibited T cell proliferation. Recombinant C3a/C5a promoted CD4+ T cell expansion, bypassed the inhibitory effects of CTLA4-Ig, and induced AKT phosphorylation, the latter biochemically linking C3aR/C5aR to known T cell signaling pathways. Lowering DC C3a/C5a production by siRNA knockdown of DC C3 reduced T cell alloresponses. Conversely downregulating DC expression of the complement regulatory protein decay accelerating factor increased immune cell C3a/C5a and augmented T cell proliferation, identifying antigen presenting cells as the dominant complement source. Pharmacological C5aR blockade reduced graft versus host disease (GVHD) scores, prolonged survival, and inhibited T cell responses in NOD scid γcnull mouse recipients of human PBMCs, verifying that the mechanisms apply in vivo. Together our findings unequivocally document that immune cell-derived complement impacts human T cell immunity and provide the foundation for future studies targeting C3aR/C5aR as treatments of GVHD and organ transplant rejection in humans.
Keywords: AKT, complement, C3, C5, dendritic cells, GVHD, T cells, transplant
INTRODUCTION
The complement system is traditionally considered an integral component of innate immunity, and is comprised of pathogen recognition receptors (e.g. C1q, MBL, ficollins) that detect pathogen associated molecular patterns, activation products that function as opsonins (e.g. iC3b), anaphylatoxins that induce and amplify inflammation (e.g. C3a and C5a), and assembled protein complexes that can directly lyse pathogens (e.g. C5b-9, membrane attack complex, MAC) [1].
Experimental work, much of it published since the late 1990s in murine models, indicates a broader role for complement as a bridge between innate and adaptive immunity [2–4]. Complement activation is a well-established effector mechanism of antibody-initiated auto- and allo-immunity. C3dg, an activation fragment produced when serum complement activates on pathogens or immune complexes, binds to B cell-expressed CR2, lowers B cell activation thresholds and thereby facilitates antibody production [5]. This latter observation from mice raised the possibility that linking C3dg to an antigen could enhance human vaccine efficacy. Such studies were performed and in some situations confirmed that C3dg could boost antibody formation, but unanticipated complexities were uncovered that limit its effectiveness in humans [6–12]. Consistently, in a systematic comparative study Seok et al. showed that genomic responses to different acute inflammatory stresses are highly similar in humans, but these responses are not reproduced in current mouse models, challenging the notion that current mouse models developed to mimic human diseases translate directly to human conditions [13]. Such observations underscore the need to test whether and how any mechanistic findings related to complement’s impact on adaptive immunity discerned from murine models apply to human systems.
Experimental results derived from several research groups, including ours, since the early 2000’s, revealed that, in mouse models, complement critically regulates the strength and phenotype of T cell immune responses [8–12, 14, 15]. This body of work showed that parenchymal cells, endothelial cells and immune cells (T cells and various professional APCs) produce complement components that activate locally, principally through the alternative pathway, to yield C3a and C5a [9, 16, 17]. These short-lived anaphylatoxins ligate with their respective seven transmembrane-spanning G protein coupled receptors (GPCR), C3aR and C5aR, which are expressed on APCs to promote APC activation (via costimulatory molecule upregulation and cytokine production) and on T cells to directly drive proliferation and survival. The observed mechanisms have unequivocal in vivo consequences, as the absence or blockade of C3aR/C5aR signaling in animal models limits anti-viral T cell immunity [18] blocks T cell autoimmunity linked to diabetes mellitus [19] and experimental autoimmune encephalomyelitis [11], and inhibits T cell alloimmunity that leads to solid organ transplant rejection and graft versus host disease (GVHD) [20].
Functionally active C3aR and C5aR have been described on human monocyte-derived DCs [21, 22], but expression on primary human T cells is controversial [23–25] and whether C3a/C3aR and C5a/C5aR ligations directly or indirectly influence T cell function has not been adequately evaluated. Moreover, while current concepts extrapolated from animal data are that activation of serum complement can impact adaptive immunity, whether immune cell derived complement modulates human T cells remains unclear.
To address these issues, we studied the effects of C3a/C3aR and C5a/C5aR on human T cell alloimmunity in vitro and in an in vivo model of xenogeneic, human anti-mouse, GVHD [26, 27]. Our data demonstrate that C3a/C3aR and C5a/C5aR ligations exert control over human T cell immune responses, and support the need for testing whether targeting signals transmitted by these receptors is an effective immunosuppressive strategy for prevention and treatment of alloimmune injury in human transplant recipients.
METHODS
Human samples
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll gradient centrifugation of de-identified buffy coats from healthy donors purchased from the NY blood bank.
Isolation of cells
Naïve CD4+ T cells were enriched over the autoMACS cell separator by negative selection with human naïve CD4 isolation kit (Miltenyi Biotec Inc., Auburn, CA, USA). After isolation, cells were stained for CD4 and CD45RA to confirm purity that was consistently higher than 95%.
CD14+ monocytes were isolated from PBMC by positive bead selection (EasySep, Stemcell Technologies Inc., Vancouver, BC, Canada) and purity was consistently higher than 85–90% based on CD14 expression.
qPCR
Cellular RNA was prepared using a RNA Mini kit (Life Technologies, Grand Island, NY, USA) and reverse-transcribed to cDNA using a High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). Expression levels for C3, C5, C3aR, C5aR, factor B (fB), and DAF genes were done using TaqMan Gene Expression assays on an Applied Biosystems 7500 Fast system with TaqMan Fast Universal Master Mix. We used RPLPO as housekeeping gene, which provided results comparable to those obtained with 18S gene (data not shown).
Preparation of monocyte-derived DCs
Monocytes were cultured in medium consisting of RPMI with 10% FCS, 2 mM L-glutamine, and 100 U/ml penicillin and streptomycin in the presence of GM-CSF (1000U/ml, Peprotech, Rocky Hill, NJ, USA) and IL-4 (1000U/l, R&D Systems, Minneapolis, MN, USA) at 37°C in 5% CO2 atmosphere for 5 days. On Day 5, cells were stimulated with LPS (5ng/mL, Sigma -Aldrich, St. Louis, MO, USA) for 48 h. Before using DCs in MLR experiments, they were washed 5 times to eliminate any LPS residue.
Some cells were saved at day 1 and 2 after LPS stimulation for FACS analysis of costimulatory molecule expression (CD40, CD80, CD86), HLA-DR, C3aR, C5aR, and DAF.
Antibodies, staining protocols, flow cytometry
8-parameter flow cytometric analysis was performed on a FACSCanto II flow cytometer using DiVA software package (BD, Franklin Lakes, NJ, USA). Data were analyzed using Flow Jo software by Tree Star (Ashland, OR, USA). Cell surface staining was performed using antibodies against CD4, CD45RO, CD45RA, C3aR, C5aR, DAF from eBioscience and CD11c, CD40, CD80, CD86, DAF HLA-DR from (BD Biosciences, San Jose, CA, USA). Appropriate isotype matched monoclonal antibodies were used as controls.
In pAKT studies, CD4+ T cells were stimulated for 24–36 hours with anti-CD3-coated beads and incremental concentrations of recombinant human C3a and/or C5a. Anti-CD28 antibody was used as a positive control. At the end of the culture, cells were permeabilized and stained with anti-pAKT antibody (BD Pharmingen, San Diego, CA, USA) according to manufacture instructions.
In vitro cultures/MLRs
CFSE-labeled CD4+ T cells were cultured in the presence of allogeneic monocyte-derived DCs (moDC:T cell ratio 1:5–1:20) at 37 °C in 5% CO2 atmosphere in serum-free media (X-VIVO 20; Lonza Walkersville, Inc., Walkersville, MD, USA) or in RPMI plus 10% heat-inactivated human AB serum for 5 days and then analyzed for CFSE expression by FACS. CFSE was obtained from Invitrogen. In other sets of MLR, C3a or C5a (R&D Systems) were added on top of CTLA4Ig (100μg/mL, Bristol-Myers Squibb, New York, NY, USA), to test their capability to rescue T cell expansion.
CD4+ T cells were also activated with anti-CD3 coated Dynabeads M-280 Tosylactivated (Invitrogen, Carlsbad, CA, USA), with increasing concentrations of recombinant human C3a, C5a (20–100nM, R&D Systems) or soluble anti-CD28 antibody (5μg/mL, BD Pharmingen) as positive control.
C3aR (SB 290157) and C5aR (W54011) antagonists were purchased from Calbiochem (Gibbstown, NJ, USA).
ELISPOT
Interferon-γ ELISPOT assays were performed as described previously in detail [28]. 300,000 spleen cells from NOD.cg-PrkdcscidIl2rgtm1Wjl (NOD Scid γcnull) mice previously injected with human PBMCs were stimulated with purified (anti-CD2 coated magnetic beads, Miltenyi) splenic APCs from MHC identical NOD/ShiLtJ mice in ELISPOT plates pre-coated with anti-human IFNγ antibody for 24 h. Secondary antibodies and developing reagents were added as previously described [28]. The resulting spots were quantified using the ImmunospotS4 Core Analyzer (CTL, Shaker Heights, OH, USA). We determined mean numbers of IFNγ ELISPOTs per 300,000 responder PBMCs from triplicate wells. PHA-stimulated PBMC were used as positive controls.
siRNA knockdown
Monocyte-derived DCs were transfected by electroporation with C3, DAF or scrambled control small interfering RNA (siRNA) from Thermo Scientific (Waltham, MA, USA). Cells were resuspended in Buffer R from Life Technologies (Grand Island, NY, USA) at 20×106 cells/mL along with siRNA (1μM) and transfected using the NEON electroporation system by Life Technologies providing a single pulse of 1600mV for 20mSec. Cells were used at 24 hours after transfection.
C3a, C5a, and IL-12 ELISA
Supernatants of MLR with naïve CD4+ T cells (10×105) and allogeneic DAF or control siRNA transfected DCs (20×104) were harvested at 24h to evaluate C3a, C5a, and IL-12 concentration. ELISA measurements (optEIA, BD Biosciences) were done according to manufacturer’s instructions.
Humanized mouse model of xeno-graft-versus-host disease (GVHD)
Six to eight weeks old female NOD Scid γcnull were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in the Mount Sinai School of Medicine Center for Comparative Medicine and Surgery in accordance with guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International. Xenogeneic GVHD was induced by retro-orbital injection of 20×106 human PBMCs. PBMC from three different donors were used in the experiments and results were compared between C5aRA or vehicle treated animals receiving PBMC from the same donor. C5aRA (1 mg/kg/d) or vehicle was delivered by osmotic pump (Alzet, DURETIC Corporation, Cupertino, CA, USA) starting from the day before PBMC injection up to day 28 (end of osmotic pump release), and by daily i.p. injections thereafter. Animals were sacrificed when they reached a 20% loss of their initial body weight. The degree of systemic GVHD was also assessed by a scoring system that incorporates five clinical parameters: weight loss (<10%, 10–20%, >20%), posture (hunching), activity, fur texture, and skin integrity [29]. At 40 and 80 days after PBMC infusion, mice were evaluated and graded from 0 to 2 for each criterion. A clinical index was subsequently generated by summation of the five criteria scores (maximum index: 10). The study protocols described herein were reviewed and approved by the Institutional Animal Care and Use Committee at Mount Sinai School of Medicine, New York, New York, USA.
Statistical analysis
To determine whether differences between groups were statistically significant, results were compared using Student’s t test. Log-rank test was employed to compare survival curves. A P value less than 0.05 was considered significant.
RESULTS
Monocyte derived DCs produce C3a and C5a and promote CD4+ T cell expansion
In initial experiments we isolated RNA from in vitro differentiated, monocyte derived, dendritic cells (moDCs) and quantified expression of complement components C3, factor B (fB), factor D (fD) and C5 RNA by qRT-PCR. These assays revealed constitutive expression of C3, fB, fD, and C5 in resting moDCs and marked upregulated of C3 and fB following LPS stimulation, whereas fD and C5 did not change appreciably (Fig 1A). RNA for C3, C5 and fB in resting or anti-CD3/CD28 stimulated, naïve CD45RA+CD45RO-CD4+ T cells was below the detection limit of the qRT-PCR assay (data not shown). Surface staining and flow cytometry revealed that DCs and naïve CD4+ T cells express low levels (above isotype controls) of the C3a receptor (C3aR) and C5a receptor (C5aR, CD88) on their surfaces (Fig 1B–C). Stimulation with anti-CD3 plus anti-CD28 induced upregulated expression of C3aR and C5aR on CD4 cells (Fig 1D). Control experiments showed that stimulation with either anti-CD3 or anti-CD28 had no effect (Fig 1C).
Figure 1. C3aR and C5aR participate in human CD4+ T cell alloreactivity.

(A) qPCR results for C3, C5, factor B (fB) and factor D (fD) mRNA in moDCs at baseline and 1 day after stimulation with LPS [data are the means of 6 (C3, C5, fB) or 3 (fD) separate experiments]. (B) Representative flow plots (L) and quantified MFI (R) of C3aR (top) and C5aR (bottom) expression on human moDCs at baseline (dotted line) and after 24h of LPS stimulation (continuous line) (right panels show means of 3 experiments) (C) Representative flow plots of C3aR (top) and C5aR (bottom) expression on naïve CD4+ T after 1 day of stimulation with aCD3, aCD28, or combined aCD3/CD28 (plots are representative of 5 experiments). (D) Quantified MFI of C3aR (top) and C5aR (bottom) expression on naïve CD4+ T cells before and after 1 or 2 days of stimulation with aCD3/CD28 (means of 3 experiments). (E) Representative CFSE dilution plots of naïve CD4+ T cell proliferation in MLR with allogeneic moDCs +/− antagonists of C3aR and/or C5aRA. (F) Quantified effects of C3aR/C5aR antagonism on allo-induced proliferation of CD4 T cells (n=7; proliferation in vehicle controls ranged from 15 to 55% in individual experiments).*P < 0.05 vs. day 0.
To test whether signaling through C3aR/C5aR impacts primary CD4+ T cell responses, we performed mixed lymphocyte reactions (MLRs) in which CFSE-labeled, naïve (CD45RA+RO-) CD4+ T cells were mixed with allogeneic moDCs ± specific antagonists to C3aR and/or C5aR (C3aRA and C5aRA, respectively). These assays showed that both antagonists (but not vehicle controls) inhibited the proliferative allo-responses (Fig 1E–F).
We tested whether C3aR/C5aR signaling on the naïve CD4 T cells directly impacts their activation/proliferation by adding C3a and/or C5a to cultures of purified naïve CD4 T cells (Fig 2A–B). Whereas anti-CD3 alone did not induce T cell proliferation, addition of recombinant C3a or C5a plus anti-CD3 induced potent T cell proliferation, quantitatively similar to the anti-CD3/CD28 positive control. Stimulation of naïve CD4+ T cells with C3a or C5a without aCD3 had no effect.
Figure 2. C3a and C5a provide costimulatory signals for naïve CD4+ T cells.

(A) Representative flow plots of naïve CD4+ T cells activated with C3a, C5a, aCD3 (negative control, upper row), or aCD3 plus C3a, C5a, or aCD28 (positive control). (B) Quantified proliferation of naïve CD4+ T cell activated with aCD3 and incremental doses of C3a (20–100nM), C5a (20–100nM), or aCD28 [data are the means of 10 (aCD3 +/− C3a, C5a, or aCD28) or 2 (C3a and C5a alone) separate experiments]. (C) Representative flow plots and (D) quantification of AKT phosphorylation at 24 h after stimulation of naïve CD4+ T cells with aCD3 antibody and C3a, C5a, or aCD28 (8 separate experiments). (E) Representative flow plots and (F) quantification results of naïve CD4+ T cell proliferation in MLR in the presence of CTLA4Ig with or without C3a and/or C5a (3 separate experiments).
C3a/C3aR and C5a/C5aR function downstream of CD28 signaling to phosphorylate AKT
The above results raised the possibility that, analogous to findings from murine models [17] C3a and C5a could bypass the requirement for costimulatory CD28 signaling to induce T cell activation/proliferation. Consistent with this hypothesis, we observed that costimulatory blockade with CTLA4-Ig inhibited anti-CD3/CD28 induced proliferation, and that addition of recombinant C3a/C5a overcame the inhibited proliferative responses (Fig 2E–F). Moreover, C3a and C5a synergized with anti-CD3 to augment intracellular AKT phosphorylation (Fig 2C–D). The results link C3a:C3aR- and C5a:C5aR-transmitted signals on T cells to a biochemical pathway known to drive T cell activation/differentiation [30].
Autocrine production of C3a and C5a by DCs drives CD4 T cell expansion
Together, the ability of DCs to produce complement (Fig 1A), the absence of detectable complement RNA by naïve CD4+ T cells, and the functional effects of C3a/C5a on inducing T cell proliferation (Fig 2A–B) suggest that DC-derived complement is the source of C3a/C5a that drives the T cell responses. To test this hypothesis, we knocked down C3 gene expression in DCs with a specific siRNA (or a scrambled siRNA control). We chose this strategy because C3 is required for formation of the C3 convertase (which cleaves C3 to C3a and C3b) and for formation of the C5 convertase (which contains C3b and cleaves C5 to C5a and C5b). Thus C3 knockdown, if effective, was anticipated to reduce immune cell production of both C3a and C5a. Indeed, C3 knockdown lowered C3a and C5a production in culture supernatants (confirming partial knockdown) and reduced CD4+ T cell alloresponses compared to controls (Fig 3A–C). C3 knockdown had no effect on DC expression levels of class II HLA, CD40, CD80 or CD86 supporting the conclusion that the observed decreased proliferative response was due to the relative absence of C3a/C5a produced by the DCs and not to an indirect effect on costimulatory molecule expression (Supplemental Fig 1). Further supporting this conclusion, add back of recombinant C3a/C5a to the cultures containing C3 knockdown DCs rescued the CD4+ cell proliferative responses to the levels of the scrambled siRNA controls without altering DC expression of HLA class II or costimulatory molecules (Fig 3D).
Figure 3. siRNA knockdown of C3 gene in human moDCs reduces C3a and C5a production and limits alloreactive CD4+ T cell proliferation.

(A) ELISAs of C3a (L) and C5a (R) levels in d2 supernatants of MLRs containing naïve CD4+ T cells + allogeneic C3kd or control moDCs. (B) Representative CFSE dilution plots and (C) quantification of naïve CD4+ T cell proliferation in MLR with C3kd or control moDCs at different moDC:Teff ratios (3 separate experiments). T cell proliferation is expressed as percentage of T cell proliferation recorded with control moDCs at each T cell:moDcs dilution. (D) Representative flow plots of naïve CD4+ T cell proliferation in response to control or C3kdmoDCs +/− recombinant C3a and/or C5a (100nM, 3 separate experiments). The experiment was repeated twice with similar results.
As a complementary strategy we used siRNA to knockdown DC expression of decay accelerating factor (DAF, CD55) a cell surface expressed protein that restrains complement activation by accelerating the decay of cell surface-assembled C3 convertases. siRNA knockdown markedly lowered DAF expression on moDC (Fig 4A) and maintained the low levels for up to 5 days in culture (Fig 5A). Supernatants from MLRs performed with DAF-knockdown DCs contained higher quantities of C3a/C5a (Fig 4B–C), and the DAF-knockdown DCs induced stronger CD4 proliferative responses compared to controls. The enhanced proliferation was abolished when we added C3aR- and/or C5aR-antagonists to the cultures (Fig 4D–E). These manipulations had no effect on DC expression of class II HLA or costimulatory molecules (Fig 5A). We did not observe differences in RNA expression levels of IL-1, IL-6, TNFα (qPCR), or IL-12 protein (ELISA) between DAF-knockdown and control DCs (Fig 5B–C). Taken together, the data support the conclusion that CD4+ T cell responses are modulated by DC derived C3a/C5a produced during cognate interactions with T cells.
Figure 4. DAF knockdown on moDCs increases C3a/C5a and promotes naïve CD4+ T cell expansion.

(A) Representative flow plot (L) and quantification (R) of DAF expression on DAF knockdown (DAFkd) and control moDCs (5 separate experiments). (B) C3a and (C) C5a levels in the d2 supernatants of moDCs or CD4+ T cells cultured alone and in MLR with DAFkd and control moDCs. (D) Representative CFSE dilution plots and (E) quantification of naïve CD4+ T cell proliferation in MLR with allogeneic control or DAFkd moDCs with incremental doses of C3aRA and/or C5aRA (n=7 separate experiments).
Figure 5. DAF knockdown on moDC does not affect cell surface phenotype or cytokine production.

(A) Representative flow plots of CD40, CD80, CD86, HLA-DR, and DAF in DAFkd and control moDCs at the end of the MLR (day 5). (B) qPCR for IL-1, IL-6, IL-12b, and TNF-α genes in moDCs before and at 24h after LPS stimulation. (C) IL-12 levels in the supernatants of MLR with DAFkd or control moDCs (6 separate experiments).
C5aR antagonism significantly prolongs survival in a humanized mouse model of GVHD
To test whether and how these in vitro findings apply in vivo, we transferred human PBMCs into NOD scid γcnull recipients, which are deficient in T, B, NK cells [26] and importantly lack murine C5 [31, 32]. In this model the human CD4+ and CD8+ T cells respond to xenogeneic antigens in the mouse and infiltrate the murine tissues (Supplemental Fig 2). The animals develop clinical features of GVHD including weight loss, skin and hair abnormalities and diarrhea over 3–6 weeks [26, 27]. We treated one group of animals with a C5aR-A beginning on day 0 and compared outcomes to animals treated with vehicle control. Expanding upon previous findings [33], using 3 individual PBMC donors we observed that administration of the C5aR-A diminished clinical expression of GVHD and prolonged survival (Fig 6A–B). Flow cytometric analysis of splenic T cells showed that essentially all of the cells in both groups expressed an activated CD45RO+ phenotype with rare Foxp3+ CD4+ cells (Supplemental Fig 2). To assess effects of C5aR-A on induction of anti-host T cell immunity, we performed human IFNγ ELISPOT assays in which we stimulated spleen cells obtained 40 days after PBMC transfer from mice in each group with NOD spleen stimulator cells. These assays showed fewer anti-mouse IFNγ producers in the spleens of the C5aR-A treated mice (Fig 6D) supporting the conclusion that the therapy inhibited alloreactive T cell expansion/differentiation in vivo.
Figure 6. C5aR antagonist prolongs survival of NOD Scid γcnull mice with GVHD induced by human PBMC injection.
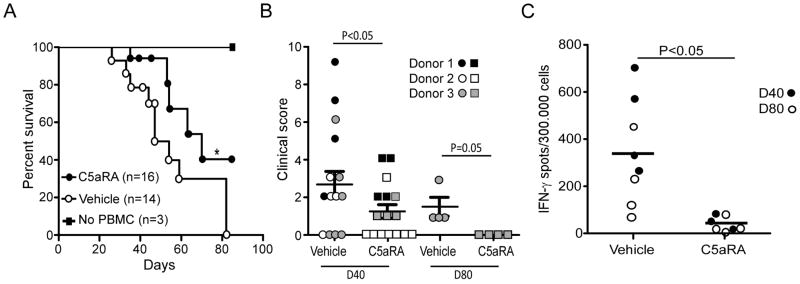
(A) Survival curves for naïve NOD Scid γcnull mice injected with human PBMC (or no injection) +/− C5aRA (1mg/kg/d) or vehicle control. *P < 0.05 vs. vehicle control. PBMC were obtained from three different donors (B) Clinical score of mice treated with C5aRA or vehicle control at 40 and 80 days after PBMC injection. Symbols differentiate the animals adoptively transferred with PBMC from each of the 3 donors. (C) Frequencies of human IFNγ ELISPOTs using spleen cells from each animal stimulated with splenic APCs from NOD/ShiLtJ mice [4 animals in each group were sacrificed at D40 (closed circles) and 4 at D80 (open circles)].
DISCUSSION
Our findings build upon previously published work in murine systems to unambiguously document that immune cell derived C3a and C5a ligate their specific, T cell expressed receptors and thereby function as costimulatory intermediaries for activation of human alloreactive T cells. In support of this conclusion, resting human T cells express cell surface C3aR and C5aR and upregulate them during anti-CD3/CD28-induced T cell activation (Fig 1C–D). Recombinant C3a and/or C5a a) substitute for anti-CD28 to induce T cell proliferation (Fig 2A–B), b) induce intracellular AKT phosphorylation (Fig 2C–D) and c) bypass the inhibitory effects of costimulatory blockade with CTLA4-Ig (Fig 2E–F). Our data indicate that the C3a/C5a derive predominantly from DC-produced complement that locally activates during cognate T cell/APC interactions (Fig 4B–C). In line with previously published data [22], we showed that human moDCs synthesize complement proteins (Fig 1A). Moreover, knockdown of C3 in DCs lowered C3a/C5a in supernatants of MLRs and partially abrogated alloreactive T cell proliferation (Fig 3A–C). Conversely, DC DAF knockdown augmented local C3a/C5a production and enhanced T cell proliferation, the latter of which was blocked by pharmacological antagonists to C3aR/C5aR (Fig 4B–E).
Our observations that recombinant C3a/C5a costimulates anti-CD3 induced proliferation (Fig 2A–B) coupled with an absence of discernible effects of C3a/C5a on DC phenotype or cytokine production during T cell/APC interactions (Fig 5), supports the concept that the DC-derived anaphylatoxins function as paracrine mediators by ligating their receptors on the responding T cells.
While we could not detect human T cell complement production by qRT-PCR (using relatively limited numbers of starting human T cells), it is possible that low levels of T cell derived complement [17, 34] could contribute to the T cell activation process. Regardless, our data and the previously published murine data [17] strongly suggest that the predominant source of complement that results in local accumulation of C3a/C5a is the stimulating APC, though it is possible that also other lymphocyte populations are capable of producing complement components.
Our work extends the discovery of fundamental mechanisms linking complement to human T cell immune responses by translating the observations into a clinically relevant therapy (Fig 6). Using an established model of human anti-mouse GVHD we showed that treatment with a C5aR-A slowed weight loss, improved clinical scores, and significantly reduced the frequencies of xenoreactive IFNγ-producing T cells in vivo. Of note, because NOD Scid γcnull mice lack of C5 [31, 32], the beneficial effect of human C5aR antagonism in the model must be attributed to inhibiting C5aR-mediated effects induced by ligation of immune cell derived, human C5a. This finding unequivocally highlights the importance of locally produced C5a in alloimmune responses in vivo, since the only source of C5a in our model was the human PBMCs. Based on the observations that blocking C3aR and C5aR individually inhibits T cell responses (Fig 2A–B) our data validate in human cells previous work from murine models which indicate overlapping downstream signaling mechanisms of C3aR and C5aR [17].
Collectively, our data add to the literature by demonstrating that immune cell-derived C3a/C5a promotes T cell activation during cognate interaction with APCs in vitro and in vivo. Because C3a and C5a also inhibit human Treg induction, function and stability [33, 34], our combined results support the need for testing anti-C5 mAb [35] and C5aR-antagonist (ClinicalTrials.gov Identifier:NCT01363388) as inhibitors of human T cell immunity in transplant recipients.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Marvin Lin for his technical assistance (Mount Sinai School of Medicine). The work was supported by NIH 3R01 AI43578 awarded to PSH. PC is a recipient of a fellowship award from the American Heart Association (12POST12050294).
ABBREVIATIONS
- C3aR
C3a receptor
- C5aR
C5a receptor
- C3aRA
C3a receptor antagonist
- C5aRA
C5a receptor antagonist
- DC
Dendritic cells
- GPCR
G protein coupled receptors
- GVHD
Graft versus host disease
- PBMC
Peripheral blood mononuclear cells
- siRNA
Small interfering RNA
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8:582–587. doi: 10.1038/nm0602-582. [DOI] [PubMed] [Google Scholar]
- 3.Heeger PS, Kemper C. Novel roles of complement in T effector cell regulation. Immunobiology. 2012;217:216–224. doi: 10.1016/j.imbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Qian Z, Wasowska BA, Behrens E, et al. C6 produced by macrophages contributes to cardiac allograft rejection. Am J Pathol. 1999;155:1293–1302. doi: 10.1016/S0002-9440(10)65231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 6.Bower JF, Yang X, Sodroski J, Ross TM. Elicitation of neutralizing antibodies with DNA vaccines expressing soluble stabilized human immunodeficiency virus type 1 envelope glycoprotein trimers conjugated to C3d. J Virol. 2004;78:4710–4719. doi: 10.1128/JVI.78.9.4710-4719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gor DO, Ding X, Li Q, Greenspan NS. Genetic fusion of three tandem copies of murine C3d sequences to diphtheria toxin fragment B elicits a decreased fragment B-specific antibody response. Immunol Lett. 2006;102:38–49. doi: 10.1016/j.imlet.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 8.Liu J, Miwa T, Hilliard B, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. 2005;201:567–577. doi: 10.1084/jem.20040863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112:1759–1766. doi: 10.1182/blood-2008-04-151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pavlov V, Raedler H, Yuan S, et al. Donor deficiency of decay-accelerating factor accelerates murine T cell-mediated cardiac allograft rejection. J Immunol. 2008;181:4580–4589. doi: 10.4049/jimmunol.181.7.4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Lin F, Strainic MG, et al. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol. 2008;180:5882–5889. doi: 10.4049/jimmunol.180.9.5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang C, Miwa T, Shen H, Song WC. Complement-dependent enhancement of CD8+ T cell immunity to lymphocytic choriomeningitis virus infection in decay-accelerating factor-deficient mice. J Immunol. 2007;179:3178–3186. doi: 10.4049/jimmunol.179.5.3178. [DOI] [PubMed] [Google Scholar]
- 13.Seok J, Warren HS, Cuenca AG, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng Q, Li K, Anderson K, et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood. 2008;111:2452–2461. doi: 10.1182/blood-2007-06-095018. [DOI] [PubMed] [Google Scholar]
- 15.Peng Q, Li K, Wang N, et al. Dendritic cell function in allostimulation is modulated by C5aR signaling. J Immunol. 2009;183:6058–6068. doi: 10.4049/jimmunol.0804186. [DOI] [PubMed] [Google Scholar]
- 16.Raedler H, Yang M, Lalli PN, Medof ME, Heeger PS. Primed CD8(+) T-cell responses to allogeneic endothelial cells are controlled by local complement activation. Am J Transplant. 2009;9:1784–1795. doi: 10.1111/j.1600-6143.2009.02723.x. [DOI] [PubMed] [Google Scholar]
- 17.Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim AH, Dimitriou ID, Holland MC, et al. Complement C5a receptor is essential for the optimal generation of antiviral CD8+ T cell responses. J Immunol. 2004;173:2524–2529. doi: 10.4049/jimmunol.173.4.2524. [DOI] [PubMed] [Google Scholar]
- 19.Lin M, Yin N, Murphy B, et al. Immune cell-derived c3 is required for autoimmune diabetes induced by multiple low doses of streptozotocin. Diabetes. 2010;59:2247–2252. doi: 10.2337/db10-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwan WH, Hashimoto D, Paz-Artal E, et al. Antigen-presenting cell-derived complement modulates graft-versus-host disease. J Clin Invest. 2012;122:2234–2238. doi: 10.1172/JCI61019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li K, Fazekasova H, Wang N, et al. Functional modulation of human monocytes derived DCs by anaphylatoxins C3a and C5a. Immunobiology. 2012;217:65–73. doi: 10.1016/j.imbio.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li K, Fazekasova H, Wang N, et al. Expression of complement components, receptors and regulators by human dendritic cells. Mol Immunol. 2011;48:1121–1127. doi: 10.1016/j.molimm.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin U, Bock D, Arseniev L, et al. The human C3a receptor is expressed on neutrophils and monocytes, but not on B or T lymphocytes. J Exp Med. 1997;186:199–207. doi: 10.1084/jem.186.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Werfel T, Kirchhoff K, Wittmann M, et al. Activated human T lymphocytes express a functional C3a receptor. J Immunol. 2000;165:6599–6605. doi: 10.4049/jimmunol.165.11.6599. [DOI] [PubMed] [Google Scholar]
- 25.Nataf S, Davoust N, Ames RS, Barnum SR. Human T cells express the C5a receptor and are chemoattracted to C5a. Journal of Immunology. 1999;162:4018–4023. [PubMed] [Google Scholar]
- 26.Ito R, Katano I, Kawai K, et al. Highly sensitive model for xenogenic GVHD using severe immunodeficient NOG mice. Transplantation. 2009;87:1654–1658. doi: 10.1097/TP.0b013e3181a5cb07. [DOI] [PubMed] [Google Scholar]
- 27.Hippen KL, Bucher C, Schirm DK, et al. Blocking IL-21 signaling ameliorates xenogeneic GVHD induced by human lymphocytes. Blood. 2012;119:619–628. doi: 10.1182/blood-2011-07-368027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heeger PS, Greenspan NS, Kuhlenschmidt S, et al. Pretransplant frequency of donor-specific, IFN-gamma-producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of posttransplant rejection episodes. J Immunol. 1999;163:2267–2275. [PubMed] [Google Scholar]
- 29.Cooke KR, Kobzik L, Martin TR, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–3239. [PubMed] [Google Scholar]
- 30.Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. 2003;192:7–20. doi: 10.1034/j.1600-065x.2003.00008.x. [DOI] [PubMed] [Google Scholar]
- 31.Lynch DM, Kay PH. Studies on the polymorphism of the fifth component of complement in laboratory mice. Exp Clin Immunogenet. 1995;12:253–260. [PubMed] [Google Scholar]
- 32.Baxter AG, Cooke A. Complement lytic activity has no role in the pathogenesis of autoimmune diabetes in NOD mice. Diabetes. 1993;42:1574–1578. doi: 10.2337/diab.42.11.1574. [DOI] [PubMed] [Google Scholar]
- 33.van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, Heeger PS. Cutting Edge: Receptors for C3a and C5a Modulate Stability of Alloantigen-Reactive Induced Regulatory T Cells. J Immunol. 2013 doi: 10.4049/jimmunol.1300847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14:162–171. doi: 10.1038/ni.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355:1233–1243. doi: 10.1056/NEJMoa061648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.