

Abstract
The extract of marine sponge Hyrtios communis (Carter, 1885) (Order Dictyoceratida, Family Thorectidae) was found to inhibit activation of the transcription factor hypoxia-inducible factor-1 (HIF-1) in T47D human breast tumor cells. Bioassay-guided isolation led to the identification of six new (1–6) and five previously reported (7–11) sesterterpene analogues and two unrelated sesterterpenes. Two new sesterterpenes, thorectidaeolide A (1) and 4-acetoxythorectidaeolide A (2), and luffariellolide (11) were among the most potent inhibitors of hypoxia (1% O2)-induced HIF-1 activation (IC50 values 3.2, 3.5, and 3.6 μM, respectively). Luffariellolide (11) exhibited a significant level of cytotoxicity that mirrored its HIF-1 inhibitory activity. Neither 1, nor 2, or any of the other less active sesterterpenes suppressed breast tumor T47D or MDA-MB-231 cell viability.
Sestertepenes represent an important group of active secondary metabolites from marine sponges.1 Marine sesterterpenes that contain both a linear component and an α,β-unsaturated γ-lactone ring moiety are found primarily in sponges of the dictyoceratid families Thorectidae (Luffariella Thiele, Hyrtios Duchassaing & Michelotti, Thorectandra Lendenfeld, Cacospongia Schmidt, Fasciospongia Burton, Aplysinopsis Lendenfeld, Fascaplysinopsis Bergquist, Thorecta Lendenfeld) and Irciniidae (Sarcotragus Schmidt), the unrelated dendroceratid family Dictyodendrillidae (Acanthodendrilla Bergquist), and astrophorid family Pachastrellidae (Brachiaster Wilson).1–3 These relatively lipophilic sponge metabolites possess a wide variety of biological activities including antibacterial, anti-inflammatory, Cdc25 phosphatase inhibitory, cytotoxic, molluscicidal, nicotinic receptor antagonistic, and fish deterrent activities (reviewed by Proksch and co-workers1). As part of our molecular-targeted antitumor natural product discovery program, an extract of the marine sponge Hyrtios communis (Carter, 1885) (Order Dictyoceratida, Family Thorectidae) (NCI Open Repository collection no. C030113) was found to inhibit HIF-1 activation in T47D cells. Bioassay-guided isolation led to the identification of six new (1–6) and five previously reported (7–11) sesterterpene analogues, and two unrelated sesterterpenes. The structures of the previously reported sesterterpenes were confirmed by comparison of the NMR spectra and specific rotation values with published data. Herein, the chemical and biological characterization of these compounds is described.
Results and Discussion
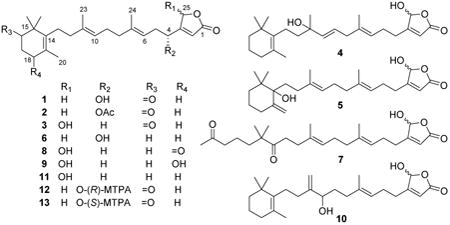
Compound 1 was obtained as a colorless oil. The molecular formula C25H36O4 was deduced from HRESIMS data (index of hydrogen deficiency = 8). The IR spectrum indicated the presence of an α,β-unsaturated γ-butenolide group (1778,1745 cm−1), a ketone (1710 cm−1), and a hydroxy moiety (3422 cm−1). The 1H NMR spectrum displayed resonances corresponding to five methyl groups [δH 1.72 (3H, s, CH3-20), 1.66 (3H, s, CH3-24), 1.65 (3H, s, CH3-23), and 1.17 (6H, s, CH3-21, 22)], three olefinic protons [δH 5.97 (1H, br s, H-2), 5.14 (1H, t, J=7.2 Hz, H-6), and 5.11 (1H, t, J=6.8 Hz, H-10)], one oxymethylene [δH 4.87 (2H, br s, H-25)], one oxymethine [δH 4.62 (1H, t, J=6.4 Hz, H-4)], and seven methylene units (δH 2.01–2.54). The 13C and DEPT NMR spectra revealed a total 25 carbon resonances for five methyl, eight methylene, four methine, and eight quaternary carbons. Six of the eight degrees of hydrogen deficiency could be accounted for by the NMR data (a ketone group, an ester carbonyl moiety, and four carbon-carbon double bonds). Thus, it was apparent that two rings were present in the structure. The presence of a trimethylcyclohex-3-enone moiety was indicated by 13C resonances [δC 136.4 (C-14), 48.1 (C-15), 215.9 (C-16), 36.1 (C-17), 31.7 (C-18), 127.4 (C-19), 19.6 (CH3-20), and 24.6 (CH3-21/22)] that were comparable to those reported for 2,2,4-trimethyl-3-(3-hydroxy-3-methylpent-4-enyl)cyclohex-3-enone.4 Subsequent analysis of the HMBC spectrum identified the following long-range correlations: gem-dimethyl system [δH 1.17 (H3-21 and H3-22) coupled to δC 215.9 (C-16), 136.4 (C-14), and 48.1 (C-15)]; methyl singlet [δH 1.72 (H3-20) coupled to δC 136.4 (C-14), 127.4 (C-19), and 31.7 (C-18)]; methylene triplet [δH 2.52 (H2-17) coupled to δC 215.9 (C-16), 127.4 (C-19), and 31.7 (C-18); and a methylene triplet [δH 2.33 (H2-18) coupled to δC 215.9 (C-16), 136.4 (C-14), 127.4 (C-19), and 36.1 (C-17). These results, together with the observed COSY correlations between H2-17 and H2-18, further confirmed the C14–C22 partial structure shown in Figure 1. Analysis of the 1H and 13C NMR spectra of 1 supported the presence of a β-substituted α,β-unsaturated γ-butenolide moiety [δH 5.97 (1H, s, H-2) and 4.87 (2H, br s, H-25); δC 173.7 (C-1), 114.9 (C-2), 171.9 (C-3), and 71.2 (C-25)], as observed in the chemical structures of luffariolide B,5 deoxymanoalide,6 and the luffarins P–W.7 Resonances for two trisubstituted double bonds [δH 5.14 (H-6), 5.11 (H-10); δC 141.7 (C-7), 136.1 (C-11), 123.6 (C-10), 117.3 (C-6)], and their corresponding associated olefinic methyl [δH 1.66 (H3-24), 1.65 (H3-23); δC 16.5 (C-24), 16.1 (C-23) ] and methylene substituents [δC 40.1 (C-12), 39.7 (C-8), 35.4 (C-5), 26.3 (C-9)] in the NMR spectra of 1 revealed the occurrence of a geranyl moiety stretching from C5–C12, that was further confirmed by HMBC correlations as shown in Figure 1. Correlations in COSY and HMBC spectra succeeded in positioning a hydroxy group at C-4, α to the γ-butenolide functionality and β to a trisubstituted double bond (partial structure C1–C7) (Figure 1). Further comparison of 13C NMR chemical shifts with those reported for a similarly C-4 hydroxy-substituted analogue, luffarin R,7 supported assembly of the C1–C13 subunit. The connectivity of the trimethylcyclohex-3-enone moiety with that of C1–C13 subunit was achieved by observation of HMBC correlations between H2-13 and C-14, C-15, and C-19, as well as between H2-12 and C-14. The configurations of both tri substituted double bonds were determined to be E due to upfield 13C NMR chemical shifts for the olefinic methyl substituents [δC 16.1 (C-23) and 16.5 (C-24)].8 Thus, 1 could be envisioned to be an oxidized analogue of the relatively simplified sesterterpene luffariellolide γ-lactone, a compound Faulkner and coworkers originally prepared from 11 by NaBH4 reduction of its γ-hydroxybutenolide.9 The absolute configuration at C-4 was determined by the modified Mosher's method. The (R)- and (S)-α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) esters (12 and 13) of 1 were prepared in NMR tubes with deuterated pyridine solutions of (S)- and (R)-MTPA chloride, respectively. The 1H NMR spectra of 12 and 13 were obtained by recording data in the reaction NMR tubes. The Δδ (δS–δr) values observed in the 1H NMR spectra were calculated. The resulting negative Δδ values for H-2 and H2-25, and positive Δδ values for H2-5, H-6, H2-8, H-10 and H3-24 (Figure 2), were consistent with the 4R-configuration. Based on the family-level classification of the sesquiterpene-rich source sponge, H. communis, 1 was assigned the name thorectidaeolide A.
Figure 1. Selected COSY and HMBC correlations of 1 (arrows pointing protons to carbons).

Figure 2. Δδ (δS – δr) values (in ppm, data obtained in pyridine-d5) for the MTPA esters of 1.

The molecular formula C27H38O5 was deduced for 2 from its HRESIMS data. As in 1, the IR spectrum indicated the presence of an α,β-unsaturated γ-butenolide moiety (1780 and 1745 cm−1) and a ketone moiety (1710 cm−1), respectively. The 1H and 13C NMR spectra of 2 (Tables 1 and 2) indicated that it was essentially identical to 1, except for the presence of resonances for an additional acetate ester substituent (δH 2.10; δC 169.8, 20.8). The downfield shift of C-4 (δC 69.6) and H-4 (δH 5.62, t, J=6.4 Hz) relative to the resonances observed in 1, indicated that the C-4 hydroxy group was acetylated in 2. This was further confirmed by HMBC correlations between H-4 (δH 5.62) and the carbonyl carbon (δC 169.8) of acetate ester. Therefore, 2 was given the trivial name 4-acetoxythorectidaeolide A.
Table 1. 1H NMR Spectroscopic Data for 1–6 (multiplicities, J in Hz)a,b.
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 2 | 5.97, br s | 5.97, br s | 5.84, br s | 5.85, br s | 5.84, br s | 5.98, br s |
| 4 | 4.62, t (6.4) | 5.62, t (6.4) | 2.51, 2.41 m | 2.49, m | 2.45, t (6.0) | 4.61, t (6.8) |
| 5 | 2.45, t (6.8) | 2.52, t (6.8) | 2.31, t (6.8) | 2.34, m | 2.32, t (6.0) | 2.45, t (6.8) |
| 6 | 5.14, t (7.2) | 5.07, t (7.2) | 5.12, t (6.8) | 5.18, t (6.8) | 5.09, m | 5.14, t (7.2) |
| 8 | 2.10, m | 2.02, m | 1.99-2.05, m | 2.72, d (6.0) | 2.01, t (6.4) | 1.99-2.06, m |
| 9 | 2.10, m | 2.04, m | 2.06-2.14, m | 5.62, dt (15.6, 6.0) | 2.07, t (6.4) | 2.08-2.16, m |
| 10 | 5.11, t (6.8) | 5.11, t (6.4) | 5.12, t (6.8) | 5.57, d (15.6) | 5.08, m | 5.09, t (6.4) |
| 12 | 2.03, m | 2.00, m | 2.06-2.14, m | 1.61, m | 1.90, 1.70, m | 2.08-2.16, m |
| 13 | 2.10, m | 2.06, m | 1.99-2.05, m | 2.00, m | 1.87, 1.57, m | 1.99-2.06, m |
| 16 | 1.41, m | 1.65, 1.38, m | 1.42, m | |||
| 17 | 2.52, t (6.8) | 2.52, t (6.8) | 2.53, t (6.8) | 1.56, m | 1.54 m | 1.57, m |
| 18 | 2.33, t (6.8) | 2.32, t (6.8) | 2.34, t (6.8) | 1.90, t (6.0) | 2.30, 1.96, m | 1.91, t (6.0) |
| 20 | 1.72, s | 1.71, s | 1.72, s | 1.58, s | 4.89, 4.83, s | 1.60, s |
| 21 | 1.17, s | 1.16, s | 1.17, s | 0.98, s | 0.96, s | 1.00, s |
| 22 | 1.17, s | 1.16, s | 1.17, s | 0.98, s | 0.88, s | 1.00, s |
| 23 | 1.65, s | 1.59, s | 1.63, s | 1.33, s | 1.60, s | 1.65, s |
| 24 | 1.66, s | 1.62, s | 1.65, s | 1.64, s | 1.60, s | 1.67, s |
| 25 | 4.87, br s | 4.83, dd (17.6, 1.6) | 5.99, br s | 5.99, br s | 5.98, br s | 4.87, br s |
| 4.75, dd (17.6, 1.6) | ||||||
| OAc | 2.10 s |
Assignments based on HSQC and HMBC spectra.
All spectra were recorded in CDCl3.
Table 2. 13C NMR Spectroscopic Data for 1–6a,b.
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 173.7, C | 172.9, C | 171.2, C | 171.0, C | 171.5, C | 173.6, C |
| 2 | 114.9, CH | 116.4, CH | 117.6, CH | 118.0, CH | 117.7, CH | 115.0, CH |
| 3 | 171.9, C | 167.4, C | 169.3, C | 169.1, C | 169.6, C | 171.7, C |
| 4 | 68.3, CH | 69.6, CH | 27.7, CH2 | 27.6, CH2 | 27.6, CH2 | 68.3, CH |
| 5 | 35.4, CH2 | 32.4, CH2 | 25.1, CH2 | 25.5, CH2 | 25.5, CH2 | 35.4, CH2 |
| 6 | 117.3, CH | 116.4, CH | 122.2, CH | 123.3, CH | 122.3, CH | 117.2, CH |
| 7 | 141.7, C | 140.7, C | 137.2, C | 136.2, C | 137.0, C | 142.0, C |
| 8 | 39.7, CH2 | 39.7, CH2 | 39.6, CH2 | 42.2, CH2 | 39.4, CH2 | 39.8, CH2 |
| 9 | 26.3, CH2 | 26.5, CH2 | 26.4, CH2 | 126.0, CH | 26.0, CH2 | 26.3, CH2 |
| 10 | 123.6, CH | 123.6, CH | 123.9, CH | 138.0, CH | 123.9, CH | 122.9, CH |
| 11 | 136.1, C | 135.8, C | 135.5, C | 73.4, C | 136.0, C | 136.9, C |
| 12 | 40.1, CH2 | 40.1, CH2 | 40.1, CH2 | 42.8, CH2 | 33.6, CH2 | 40.2, CH2 |
| 13 | 28.2, CH2 | 28.3, CH2 | 28.3, CH2 | 22.9, CH2 | 30.9, CH2 | 27.9, CH2 |
| 14 | 136.4, C | 136.5, C | 136.5, C | 136.6, C | 80.6, C | 137.0, C |
| 15 | 48.1, C | 48.1, C | 48.2, C | 35.1, C | 39.7, C | 35.0, C |
| 16 | 215.9, C | 215.8, C | 216.2, C | 39.8, CH2 | 38.0, CH2 | 39.8, CH2 |
| 17 | 36.1, CH2 | 36.1, CH2 | 36.1, CH2 | 19.5, CH2 | 22.7, CH2 | 19.5, CH2 |
| 18 | 31.7, CH2 | 31.6, CH2 | 31.6, CH2 | 32.8, CH2 | 34.0, CH2 | 32.7, CH2 |
| 19 | 127.4, C | 127.3, C | 127.3, C | 127.1, C | 150.4, C | 127.0, C |
| 20 | 19.6, CH3 | 19.6, CH3 | 19.6, CH3 | 19.8, CH3 | 108.3, CH2 | 19.8, CH3 |
| 21 | 24.6, CH3 | 24.6, CH3 | 24.6, CH3 | 28.6, CH3 | 24.1, CH3 | 28.6, CH3 |
| 22 | 24.6, CH3 | 24.6, CH3 | 24.6, CH3 | 28.6, CH3 | 22.1, CH3 | 28.6, CH3 |
| 23 | 16.1, CH3 | 16.0, CH3 | 16.0, CH3 | 27.5, CH3 | 16.0, CH3 | 16.1, CH3 |
| 24 | 16.5, CH3 | 16.4, CH3 | 16.2, CH3 | 16.5, CH3 | 16.3, CH3 | 16.4, CH3 |
| 25 | 71.2, CH2 | 71.1, CH2 | 99.0, CH | 99.0, CH | 99.3, CH | 71.2, CH2 |
| 4-OAc | 169.8, C | |||||
| 20.8, CH3 |
Assignments based on HSQC and HMBC spectra.
Spectra were recorded in CDCl3.
Compound 3 was obtained as a colorless oil. The molecular formula C25H36O4 was deduced by HRESIMS (index of hydrogen deficiency = 8). As in 1, the presence of an ester carbonyl and ketone moiety was verified by the IR (1759, 1710 cm−1) and 13C NMR spectra (δC 171.2, 216.2). A broad IR absorption centered at 3299 cm−1 suggested the presence of a hydroxy group. The 1H and 13C NMR spectra (Tables 1 and 2) were comparable to those of 1, with similar resonances for the region C8–C24. The 13C resonances at δC 171.2 (C-1), 117.6 (C-2), 169.3 (C-3), and 99.0 (C-25), and their corresponding 1H resonances [(δH 5.84 (1H, br s, H-2), and 5.99 (H, br s, H-25)], indicated that 3 possessed a β-substituted α,β-unsaturated γ-hydroxybutenolide moiety, rather than the β-substituted α,β-unsaturated γ-butenolide moiety in 1. Comparison of the NMR spectra of the C1–C11 region with those reported for the structural analogues acantholide B (8),10 acantholide C (7),10 16-hydroxyluffariellolide (9)11 and luffariellolide (11)12 facilitated assembly of its C1–C11 component. The Δ6,7 and Δ10,11-double bonds were both assigned E-configurations based on the observation of the upfield 13C NMR chemical shifts of the C-24 (δC 16.2) and C-23 (δC 16.0) methyl group resonances, respectively. Compound 3 was assigned the trivial name thorectidaeolide B.
The molecular formula of 4 (C25H38O4) was deduced by HRESIMS (index of hydrogen deficiency = 7). The 13C NMR spectrum (Table 2) displayed resonances for a disubstituted double bond (δC 126.0, 138.0), two trisubstitued double bonds (δC 118.0, 169.1, 123.3, 136.2), a fully substituted double bond (δC 136.6, 127.1), and a carbonyl carbon (δC 171.0). According to the index of hydrogen deficiency, the structure of 4 was deduced to be bicyclic due to the absence of any other observable sp or sp2 carbon resonances. These data suggested that 4 is a luffariellolide-type sesterterpene. The 1H and 13C NMR spectra (Tables 1 and 2) were comparable to those of 11,8 indicating that both compounds possessed identical cyclohexene and γ-hydroxybutenolide terminal ring systems. The major differences between 4 and 11 were found only in the C9–C11 region of the structure. The C9–C11 segment of the structure was deduced from analysis of the DEPT and HMBC spectra (Figure 3). The DEPT spectrum indicated the presence of an oxygenated quaternary carbon (δC 73.4), which was assigned to C-11. The HMBC correlations between the methyl singlet at δH 1.33 (H3-23) and carbons at δC 73.4 (C-11) and 138.0 (C-10) were observed. The HMBC spectrum also showed long-range correlations between the H-9 (δH 5.62) and H-10 (δH 5.57) olefinic protons and the oxygenated C-11 quaternary carbon (δC 73.4). The structure was further supported by comparison of the relevant NMR data of 4 with those reported for two C-11 hydroxy-substituted analogues luffariolide G13 and tasnemoxide A.14 The C9–C11 segment and both the C1–C8 and C12–C22 partial structures were connected based on observed COSY and HMBC correlations between these three structural components (Figure 3). The E-configuration of the Δ9,10-double bond was deduced from the large coupling constant (J =15.6 Hz) between H-9 and H-10. Compound 4 was assigned the trivial name thorectidaeolide C. Insufficient material was available for further experiments to determine the C-11 absolute configuration.
Figure 3. Selected COSY (solid lines) and HMBC correlations of 4 (arrows pointing from protons to carbons).

The C25H38O4 molecular formula of 5 was deduced from its HRESIMS spectrum (index of hydrogen deficiency = 7). The IR spectrum exhibited absorption bands corresponding to a hydroxy group (3285 cm−1), an ester carbonyl (1752 cm−1), and an exomethylene substituent (895 cm−1). Similarities in the NMR spectra between 5 (Tables 1 and 2) and 11 suggested that 5 is also a luffariellolide-type sesterterpene. The main differences in the 1H NMR spectrum of 5 and that of 11 were the absence of one olefinic methyl group resonance in 5, and the appearance of resonances attributable to an exomethylene moiety [δH 4.89, 4.83 (H2-20)]. Observation of a methylene resonance (δC 108.3) and a corresponding quaternary carbon (δC 150.4) further supported the presence of an exomethylene functionality in the structure of 5. An oxygenated quaternary carbon [δC 80.6, (C-14)] was observed by 13C NMR and DEPT experiments. The assignment and placement of the C-14 hydroxy substituent and the exomethylene at C-19 were deduced by observation of long-range 1H-13C HMBC correlations (H2-13 to C-14; H3-21/22 to C-14; H2-18 to C-14; H2-13 to C-19; H2-18 to C-19; H2-17 to C-19; H2-20 to C-19; H2-20 to C-14; and from H2-20 to C-18). The hydroxy and exomethylene-substituted cyclohexane system in the structure was further supported by comparison of the relevant NMR spectra of 5 with those reported for the similarly substituted marine sesterterpene metabolites tasnemoxide C14 and epimuqubilin B.15 The 13C chemical shifts of the C-23 and C-24 methyl groups (δC 16.0 and 16.3, respectively) were indicative of E-configurations for the Δ6,7 and Δ10,11-double bonds. Compound 5 was assigned the name thorectidaeolide D.
Compound 6 was isolated as a colorless oil. The molecular formula C25H38O3 was deduced from its HRESIMS data, 13C NMR, and DEPT spectra. The IR spectrum indicated that the structure contained an α,β-unsaturated γ-butenolide moiety (1779,1742 cm−1) and hydroxy group (3423 cm−1). When the NMR spectra of 6 and 1 were compared, it was evident that they were structurally similar except that 6 lacked a ketone functionality. Examination of 13C NMR spectrum of 6 suggested that the resonances associated with the trimethylcyclohex-3-enone moiety in 1 were replaced in this new compound by a set of resonances [δC 137.0 (C-14), 35.0 (C-15), 39.8 (C-16), 19.5 (C-17), 32.7 (C-18), 127.0 (C-19), 19.8 (CH3-20), and 28.6 (CH3-21/22)] that was attributable to a trimethylcyclohexene moiety. The structure was further supported, by observation that the chemical shifts for the C13–C22 partial structure in 6 matched those of the corresponding unit in luffariellolide (11)12 and the luffariolides A–E.5 The proton and carbon NMR resonances in the spectra of 6 were readily assigned from the COSY, HSQC, and HMBC spectra. As in 1, the upfield 13C NMR chemical shifts for the olefinic methyl resonances (δC 16.1 and 16.4) indicated that both double bonds had E-configurations. As observed with similarly substituted sesterterpenes of the series,2,6 compound 6 was deduced to have a 4R-configuration, based on the observation that it exhibited a similar specific rotation with that of 1 (+6.0 and +4.0, respectively). Thus, 6 was given the name thorectidaeolide E.
The transcription factor hypoxia-inducible factor-1 (HIF-1) regulates cellular oxygen homeostasis, the ability of tumor cells to adapt and grow under hypoxic conditions, and represents an important molecular target for anticancer drug discovery.16 Concentration-response studies were performed to determine the effects of 1–10, and 11 on HIF-1 activation in a T47D cell-based reporter assay. The three new sesterterpenes (1–3) that possess a simple α,β-unsaturated γ-butenolide moiety inhibited hypoxia (1% O2, 16 h)-induced HIF-1 activation with IC50 values of 3.2 μM [95% Confidence Interval (CI) 2.8–3.5 μM], 3.5 μM (CI 3.3–3.8 μM), and 6.2 μM (CI 5.8–6.7 μM), respectively. Among the other sesterterpenes that possess a γ-hydroxybutenolide moiety, only acantholide A (10) [IC50 7.1 μM (CI 6.5–7.8 μM)] and 11 [IC50 3.6 μM (CI 3.4–3.9 μM)] significantly (i.e., IC50 <10 μM) inhibited HIF-1 activation. A standard 48 h treatment study was performed in T47D and MDA-MB-231 cells to determine the effects of test compounds on cell proliferation/viability. None of the compounds except 11 significantly inhibited cell proliferation (IC50 >10 uM). Consistent with the previous reports of cytotoxicity,17 11 exhibited antiproliferative activity in both T47D and MDA-MB-231 cells [IC50 values: 5.1 μM (CI 4.6–5.7) and 4.5 μM (CI 4.1–4.8), respectively] that mirrored its HIF-1 inhibitory activity. The cytotoxic activity of 11 was previously attributed to its ability to act as an agonist of retinoic acid receptors (RARs).16 The Schiff base forming reactivity of the γ-hydroxybutenolide ring system is believed to be responsible for the anti-inflammatory and enzyme inhibitory (e.g., phosphatase cdc25A) properties of 11 and other related marine natural products.9 Alternatively, compounds that do not possess a hydroxylated γ-butenolide ring system (i.e., 1, 2, and 6) exhibit pronounced HIF-1 inhibitory activity. Further mechanistic studies are required to resolve the precise means by which compounds such as these new sestertepenes disrupt HIF-mediated hypoxic signaling in tumor cells.
Experimental Section
General Experimental Procedures
Specific rotation values were obtained on an AP IV/589-546 digital polarimeter. A Bruker Tensor 27 Genesis Series FTIR was used to obtain the IR spectra. The NMR spectra were recorded in CDCl3 on AMX-NMR spectrometers (Bruker) operating at 400 MHz for 1H and 100 MHz for 13C, respectively. Residual solvent resonances (δ 7.26 for 1H and δ 77.16 for 13C) were used as internal references for the NMR spectra recorded running gradients. The HRESIMS spectra were determined on a Bruker Daltonic micro TOF fitted with an Agilent 1100 series HPLC and an electrospray ionization source. HPLC was performed on a Waters system, equipped with a 600 controller and a 2998 photodiode array detector. A semi-preparative HPLC column (Phenomenex Luna, RP-18, 5 μm, 250 × 10.00 mm) was employed for isolation. TLC was performed using Merck Si60F254 plates, sprayed with a 10% H2SO4 solution in EtOH, heated, and/or visualized under UV at 254 nm. The purities of the compounds were judged on the percentage of the integrated signal at UV 220 nm. All purified compounds submitted for bioassay were at least 95% pure as judged by HPLC and supported by 1H NMR analysis.
Sponge Material
Sponge material was obtained from the U.S. National Cancer Institute's Open Repository Program and collected from a depth of 18–21 m from the Northern Reefs region off the coast of Palau on 22 June 2009 (collection no. 0CDN9952; NPID no. C030113). Upon collection, material was frozen at −20 °C and later ground in a meat grinder. The sponge was massive to lobate, the surface conulose and rough, 5 mm diameter oscules are scattered on the tops of lobes. The texture is cheese-like, compressible, and rubbery, the color in life is grey to black externally and beige to tan internally. The skeleton consists of broad fascicles of sand grains that extend vertically in the sponge towards the surface conules. There is no obvious spongin surrounding the fascicles, and the cellular material between the fascicules is dense, uniform and very fleshy. The sponge is most closely comparable to Hyrtios communis (Carter, 1885) (Order Dictyoceratida: Family Thorectidae), and differs from other common Indo-Pacific species H. erecta (Keller, 1899) and H. reticulatus (Thiele, 1899) in gross morphology and coloration which in the latter species is digitate, greenish brown and brick red, respectively, and in the nature of the skeleton, which in the latter species forms a distinct regular rounded reticulation of primary and secondary fibers packed with sand. Voucher specimens of 0CDN9952 are held in the collections of the Coral Reef Research Foundation, Palau, Michelle Kelly, NIWA, Auckland, and in the Department of Invertebrate Zoology, National Museum of Natural History, Smithsonian Institution, Washington, DC.
Extraction and Isolation
Ground material of H. communis was extracted with H2O. The residual sample was then lyophilized and extracted with 50% MeOH in CH2Cl2,18 residual solvents were removed under vacuum, and the extract (no. C030113) was stored at −20 °C in the NCI repository at the Frederick Cancer Research and Development Center. In order to reduce the loss of potential HIF-1/antitumor activity,19 Si gel chromatography was avoided in the bioassay-guided separation of the active constituents. The H. communis extract (2.8 g) inhibited HIF-1 activation in T47D cells by 97% (5 μg mL−1) was passed over Sephadex LH-20 CC with 50% MeOH in CH2Cl2 to yield four fractions. The active fourth fraction (1.81 g, 98% HIF-1 inhibition at 5 μg mL−1) was further separated into eight fractions by VLC (C18) eluted with step gradients of MeOH in H2O (30:70, 50:50, 60:40, 70:30, 80:20, 85:15, 90:10, 100:0). The active fourth fraction (71 mg, 95% HIF-1 inhibition at 5 μg mL−1) that eluted with 70% MeOH was purified by semi-preparative HPLC (Luna 5 μm, C18(2) 100 Å, 250 × 10.0 mm, isocratic 82% MeOH in H2O, 4.0 mL min−1), to produce 7 (acantholide C,10 1.1 mg, 0.04% yield, tR 8 min), 1 (2.1 mg, 0.08% yield, tR 12 min), 8 (acantholide B,10 3.1 mg, 0.11% yield, tR 15 min), 2 (4.0 mg, 0.14% yield, tR 17 min), a mixture of 3 and 9 (5.1 mg, tR 18 min), 4 (1.1 mg, 0.04% yield, tR 20 min), 10 (acantholide A, 2.1 mg, 0.08% yield, tR 21 min) and 5 (3.0 mg, 0.11% yield, tR 23 min). The mixture of 3 and 9 was further purified by a semi-preparative HPLC (Luna 5 μm, C18(2) 100 Å, 250 × 10.0 mm, isocratic 75% MeCN in H2O, 4.0 mL min−1), to produce 9 (16-hydroxyluffariellolide,11 1.4 mg, 0.05% yield, tR 12 min), and 3 (2.4 mg, 0.09% yield, tR 16 min). The active fifth fraction (1557 mg, inhibited HIF-1 activation in T47D cells by 99% at 5 μg mL−1) that eluted with 80% MeOH from the C18 VLC was purified by a semi-preparative HPLC (Luna 5 μm, C18(2) 100 Å, 250 × 10.0 mm, isocratic 90% MeOH in H2O, 4.0 mL min−1), to produce 6 (1.8 mg, 0.06% yield, tR 18 min), 11 (ircinolide B, petrosaspongiolide B,20 2.5 mg, 0.09% yield, tR 23 min) and 11 (luffariellolide,8 1000 mg, 35.71% yield, tR 24 min). The active sixth fraction (50 mg, inhibited HIF-1 activation in T47D cells by 86% at 5 μg mL−1) that eluted with 85% MeOH from the C18 VLC was purified by recrystallization to produce ircinolide A (petrosaspongiolide A,20 20 mg, 0.7% yield).
Thorectidaeolide A (1)
colorless oil; +4.0 (c 0.20, MeOH); IR (film) νmax 3422, 2928, 1778, 1745, 1710, 1443, 1131, 1025, 885, 859 cm−1; 1H NMR and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 423.2516 [M+Na]+ (calcd for C25H36O4Na, 423.2511).
4-Acetoxythorectidaeolide A (2)
colorless oil; +3.8 (c 0.26, MeOH); IR (film) νmax 2929, 1780, 1745, 1710, 1443, 1374, 1228, 1135, 1026, 885, 857 cm−1; 1H NMR and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 465.2596 [M+Na]+ (calcd for C27H38O5Na, 465.2617).
Thorectidaeolide B (3)
colorless oil; +3.2 (c 0.13, MeOH); IR (film) νmax 3299, 2929, 1759, 1710, 1443, 1357, 1277, 1126, 1024, 945 cm−1; 1H NMR and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 399.2523 [M−H]− (calcd for C25H35O4, 399.2535).
Thorectidaeolide C (4)
colorless oil; +6.0 (c 0.07, MeOH); IR (film) νmax 3340, 2925, 1741, 1439, 1360, 1128, 945 cm−1; 1H NMR and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 401.2691 [M−H]− (calcd for C25H37O4, 401.2692).
Thorectidaeolide D (5)
colorless oil; −6.4 (c 0.19, MeOH); IR (film) νmax 3285, 2930, 1752, 1444, 1383, 1137, 944, 895 cm−1; 1H NMR and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 401.2680 [M−H]− (calcd for C25H37O4, 401.2692).
Thorectidaeolide E (6)
colorless oil; +6.0 (c 0.10, MeOH); IR (film) νmax 3423, 2926, 1779, 1742, 1440, 1381, 1131, 1065, 1026, 886, 859 cm−1; 1H NMR and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 409.2704 [M+Na]+ (calcd for C25H38O3Na, 409.2719).
Preparation of the (R)- and (S)-MTPA Ester Derivatives of 1
The (R)- and (S)-MTPA ester derivatives of 1 were prepared as previously described.21 Briefly, two portions of 1 (each 0.8 mg) were transferred into clean NMR tubes and were dried under vacuum. Pyridine-d5 (200 μL, 99.5% deuterated, 0.05% TMS) was added under an argon gas stream into each of two NMR tubes, and (R)-(-)- and (S)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (3 μL each) were immediately added into the NMR tubes, respectively. The NMR tubes were shaken to mix the samples and MTPA chlorides evenly. The reaction NMR tubes were allowed to stand in a desiccator at room temperature and monitored hourly by 1H NMR. The reactions were found to be complete after 5 h. The 1H NMR spectra were obtained directly from the NMR reaction tubes and were assigned on the basis of their respective COSY spectra.
Thorectidaeolide A, (R)-MTPA Ester (12)
1H NMR (pyridine-d5) δ 6.374 (1H, br s, H-2), 6.324 (1H, t, J=6.4 Hz, H-4), 5.295 (1H, t, J=6.4 Hz, H-10), 5.227 (1H, t, J=6.8 Hz, H-6), 5.172 (1H, dd, J=18.0, 1.2 Hz, H-25a), 5.088 (1H, dd, J=18.0, 1.2 Hz, H-25b), 2.756 (2H, t, J=6.4 Hz, H-5), 2.550 (2H, t, J=6.8 Hz, H-17), 2.060 (2H, t, J=6.8 Hz, H-8), 1.719 (3H, s, H-20), 1.700 (3H, s, H-23), 1.615 (3H, s, H-24), 1.273 (6H, s, H-21, 22).
Thorectidaeolide A, (S)-MTPA Ester (13)
1H NMR (pyridine-d5) δ 6.349 (1H, t, J=6.4 Hz, H-4), 6.168 (1H, br s, H-2), 5.365 (1H, t, J=6.8 Hz, H-6), 5.312 (1H, t, J=6.8 Hz, H-10), 5.092 (1H, dd, J=18.0, 1.2 Hz, H-25a), 4.952 (1H, dd, J=18.0, 1.2 Hz, H-25b), 2.820 (2H, t, J=6.4 Hz, H-5), 2.551 (2H, t, J=6.8 Hz, H-17), 2.125 (2H, t, J=6.4 Hz, H-8), 1.722 (3H, s, H-20), 1.698 (3H, s, H-23), 1.684 (3H, s, H-24), 1.273 (6H, s, H-21, 22).
Cell-Based Reporter and Proliferation/Viability Assays
Human breast tumor T47D and MDA-MB-231 cells were obtained from ATCC. Cell maintenance, experimental procedures, and data presentation for the cell-based reporter and proliferation/viability assays (48 h) were as previously described.22 Cycloheximide (protein synthesis inhibitor) and rotenone (mitochondrial respiration inhibitor) were used as positive controls for both the reporter and cell viability assays (results shown in S36, Supporting Information). All extract, fraction, and compound samples were prepared as stock solutions in DMSO (final solvent concentration less than 0.5% in all assays).
Statistical Analysis
Data analyses were performed with GraphPad Prism 4.
Supplementary Material
Acknowledgments
The authors thank the Natural Products Branch Repository Program at the National Cancer Institute for providing marine extracts from the NCI Open Repository used in these studies, Dr. D. J. Newman and E. C. Brown (NCI, Frederick, MD) for assistance with sample logistics and collection information, and Dr. S. L. McKnight (University of Texas Southwestern Medical Center at Dallas) for providing the pTK-HRE3-luc construct. This work was supported in part by the National Institutes of Health National Cancer Institute (grant CA98787), and the National Oceanic and Atmospheric Administration National Institute for Undersea Science and Technology (grant NA16RU1496). This investigation was conducted in a facility constructed with Research Facilities Improvement Grant C06 RR-14503-01 from the National Institutes of Health.
Footnotes
Notes: The authors declare no competing financial interest.
Associated Content: Supporting Information. NMR spectra for (1–6), a photo of the H. communis (collection no. 0CDN9952; NPID no. C030113), and additional positive control results from the reporter and cell viability assays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ebada SS, Lin W, Proksch P. Mar Drugs. 2010;8:313–346. doi: 10.3390/md8020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ueoka R, Nakao Y, Fujii S, van Soest RWM, Matsunaga SJ. Nat Prod. 2008;71:1089–1091. doi: 10.1021/np8001207. [DOI] [PubMed] [Google Scholar]
- 3.Wonganuchitmeta S, Yuenyongsawar S, Keawpradub N, Plubrukarn A. J Nat Prod. 2004;67:1767–1770. doi: 10.1021/np0498354. [DOI] [PubMed] [Google Scholar]
- 4.Marco JA, Sanz JF, Yuste A, Carda M, Jakupovic J. Phytochemistry. 1991;30:3661–3668. [Google Scholar]
- 5.Tsuda M, Shigemori H, Ishibashi M, Sasaki T, Kobayashi J. J Org Chem. 1992;57:3503–3507. [Google Scholar]
- 6.Uddin MH, Otsuka M, Muroi T, Ono A, Hanif N, Matsuda S, Higa T, Tanaka J. Chem Pharm Bull. 2009;57:885–887. doi: 10.1248/cpb.57.885. [DOI] [PubMed] [Google Scholar]
- 7.Butler MS, Capon RJ. Aust J Chem. 1992;45:1705–1743. [Google Scholar]
- 8.Couperus PA, Clague ADH, van Dongen JPCM. Org Magn Reson. 1976;8:426–431. [Google Scholar]
- 9.Potts BCM, Faulkner DJ, De Carvalho MS, Jacobs RS. J Am Chem Soc. 1992;114:5093–5100. [Google Scholar]
- 10.Elkhayat E, Edrada R, Ebel R, Wray V, van Soest R, Wiryowidagdo S, Mohamed MH, Muller WEG, Proksch P. J Nat Prod. 2004;67:1809–1817. doi: 10.1021/np040118j. [DOI] [PubMed] [Google Scholar]
- 11.Cao S, Foster C, Lazo JS, Kingston DGI. Bioorg Med Chem. 2005;13:5094–5098. doi: 10.1016/j.bmc.2005.04.070. [DOI] [PubMed] [Google Scholar]
- 12.Albizati KF, Holman T, Faulkner DJ, Glaser KB, Jacobs RS. Experientia. 1987;43:949–950. [Google Scholar]
- 13.Kobayashi J, Zeng C, Ishibashi M, Sasaki T. J Nat Prod. 1993;56:436–439. doi: 10.1021/np50093a020. [DOI] [PubMed] [Google Scholar]
- 14.Youssef DT. J Nat Prod. 2004;67:112–114. doi: 10.1021/np0340192. [DOI] [PubMed] [Google Scholar]
- 15.Sperry S, Valeriote FA, Corbett TH, Crews P. J Nat Prod. 1998;61:241–247. doi: 10.1021/np970467w. [DOI] [PubMed] [Google Scholar]
- 16.Semenza GL. Oncogene. 2010;29:625. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S, Wang Z, Lin S, Zheng W, Wang R, Jin S, Chen J, Jin L, Li Y. Biochem J. 2012;446:79–87. doi: 10.1042/BJ20120726. [DOI] [PubMed] [Google Scholar]
- 18.McCloud TC. Molecules. 2010;15:4526–4563. doi: 10.3390/molecules15074526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Datta S, Zhou YD, Nagle DG. J Nat Prod. 2013;76:642–647. doi: 10.1021/np300858c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lal AR, Cambie RC, Rickard CEF, Bergquist PR. Tetrahedron Lett. 1994;35:2603–2606. [Google Scholar]
- 21.Su BN, Park EJ, Mbwambo ZH, Santarsiero BD, Mesecar AD, Fong HH, Pezzuto JM, Kinghorn AD. J Nat Prod. 2002;65:1278–1282. doi: 10.1021/np0202475. [DOI] [PubMed] [Google Scholar]
- 22.Du L, Mahdi F, Jekabsons MB, Nagle DG, Zhou YD. J Nat Prod. 2010;73:1868–1872. doi: 10.1021/np100501n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.