

Abstract
The present study is the first to show in pancreatic cancer (PC) the growth inhibition and apoptosis by novel MDM2 inhibitors (MI-319 & 219) through reactivation of p53 pathway. Our results highlight two new secondary targets of MDM2 inhibitor ‘SIRT1’ and Ku70. SIRT1 has a role in ageing and cancer and is known to regulate p53 signaling through acetylation. Ku70 is a key component of non-homologous end joining machinery in the DNA damage pathway and is known to regulate apoptosis by blocking Bax entry into mitochondria. Given the growth inhibition and apoptosis by MI-219, MI-319 was accompanied by increase in levels of p53 along with p21WAF1 and the proapoptotic Puma. SiRNA against p21WAF1 abrogated the growth inhibition of PC cells confirming p21WAF1 as a key player downstream of activated p53. Immunoprecipitation-western blot analysis revealed reduced association of MDM2-p53 interaction in drug exposed PC cells. In combination studies, the inhibitors synergistically augmented anti-tumor effects of therapeutic drug gemcitabine both in terms of cell growth inhibition as well as apoptosis. Surface plasmon resonance studies confirmed strong binding between MI-319 and Ku70 (KD 170 nM). Western blot revealed suppression of SIRT1 and Ku70 with simultaneous upregulation of acetyl-p53 (Lys379) and Bax. Co-Immunoprecipitation studies confirmed that MI-319 could disrupt Ku70-Bax and SIRT1-Bax interaction. Further, using wt-p53 xenograft of Capan-2, we found that oral administration of MI-319 at 300 mg/kg for 14 days resulted in significant tumor growth inhibition without any observed toxicity to the animals. No tumor inhibition was found in mut-p53 BxPC-3 xenografts. In light of our results, the inhibitors of MDM2 warrant clinical investigation as new agents for PC treatment.
Keywords: MDM2 and p53, Small molecule inhibitors, cell cycle arrest, apoptosis, pancreatic cancer
INTRODUCTION
Pancreatic cancer is a dreadful disease and is the fourth leading cause of cancer related deaths in the United States [1]. Among the various important genes altered in pancreatic malignancy, the tumor suppressor p53 is considered crucial. p53 gene is a master transcriptional regulator controlling several key cellular pathways [2,3] and is found to be mutationally inactivated in about 50% of infiltrating pancreatic adenocarcinoma [PC] [4]. Cells with p53 functional deficiencies loose their abilities to trigger cell cycle arrest, as well as apoptosis [5]. This acts synergistically to favor the transformation of normal cells into malignant cells. Moreover, cancer treatments are based on genotoxic stresses inducing cell death through different pathways [DNA damage, cell cycle inhibition and apoptosis], so the loss of p53-mediated apoptosis drastically reduces the response to various clinically-employed treatments [6–8]. In light of the numerous genes regulated by p53 as well as its role in the development of resistance to therapy, it makes p53 an ideal candidate target for reactivation using novel agents for PC therapy.
The activity of wt-p53 is mainly regulated at the post-translational level through its proteolytic turn over [9]. This is achieved through the interaction with MDM2, a RING domain protein, which induces wt-p53 degradation by ubiquitin-mediated proteolysis [10]. In normal non-stressed cells, MDM2 induces p53 degradation, making it a short-lived protein [10]. However, in response to DNA damage, MDM2 is auto-poly-ubiquitinated, resulting in its self-degradation accompanied by an associated increase in p53 level and activity [9]. This regulatory mechanism is subjected to a feedback loop since p53 in turn regulates the level of MDM2 transcription giving rise to subtle balances between the amounts of p53 and MDM2 [11,12]. Although studies over the years have concentrated on p53 as the primary target of MDM2, p53 independent functions of MDM2 are also important. In this regard Zhang and co-workers have elegantly reviewed MDM2 to have p53-independent activities in carcinogenesis and human cancer progression and therapy [13] and therefore, MDM2 can be a rational drug target for cancer chemotherapy.
SIRT1 is a multifunctional, NAD+-dependent protein deacetylase that is involved in a wide variety of cellular processes from cancer to ageing [14,15]. SIRT1 has been shown to have oncogenic properties by down regulating p53 activity [14]. Primarily, SIRT1 blocks p53 function by inhibiting its acetylation [16]. It has been recently suggested that use of small-molecule inhibitors of SIRT1 in tumor cells that over express the protein may sensitize the cells to a combination of therapeutics, inducing a stronger p53 response and promoting apoptosis of tumor cells [17]. Another protein, Ku70 that is under the regulation of SIRT1 through acetylation is known to play an important role in apoptosis by blocking Bax entry into the mitochondria [18]. Studies have shown that Ku70 ubiquitinylating activity of Hdm2 may contribute to its p53-independent oncogenic activity [19]. The elevated levels of Hdm2 may cause a decrease in Ku70 supply from the cytosol, which may negatively influence Ku-dependent DNA repair activity in the nucleus. In certain types of cancer cells, a temporary Ku70 decrease caused by an elevated Hdm2 level may facilitate acquisition of further mutations, and cancer cells may become resistant to apoptosis by an elevation of Ku70 [19].
Based on functional significance of MDM2 in the biology of p53, one attractive pharmacological approach to wt-p53 reactivation is to use a small-molecule to block the MDM2–p53 interaction [20–23]. The discovery of the Nutlins provided the important proof of the concept for this approach [21]. Nutlins were shown to bind to MDM2, block the MDM2–p53 interaction, and activate wild-type p53 both in vitro as well as in xenograft models [21,24,25]. However Nutlin 3 remained the only inhibitor in its class specific for MDM2 and thus newer inhibitors with higher specificity and minimum toxicity are required. In this paper we show that specific and orally active MDM2 inhibitors MI-219 and MI-319 transiently reactivates p53, resulting in growth inhibition and apoptosis in vitro in wild wt-p53 PC cells as well as tumor growth retardation in vivo in a Capan-2 [wt-p-53] and not in BxPC-3 [mut-p53] PC xenograft model. Further, we also identify two new targets of MDM2 (SIRT1 and Ku70) that play crucial role in the biology of p53.
MATERIALS AND METHODS
Cell Culture and Experimental Reagents
Five human PC cell lines Capan-1, Capan-2, Colo-357, HPAC and BxPC-3 were used in this study. All cells except Colo-357 were purchased from [American Type Culture Collection [ATCC]]. Colo-357 was a gift from Dr. Paul Chiao [M.D. Anderson Cancer Center, Houston, TX]. Capan-1, HPAC, BxPC-3 and Colo-357 were cultured in DMEM [In-vitrogen] supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. Capan-2 was grown in McCoy 5 A media [Invitrogen]. All cells were cultured in a 5% CO2-humidified atmosphere at 37°C. Primary antibodies for p53, acetyl-p53, p21WAF1 p21], Puma, MDM2, Bax and SIRT1 were purchased from Cell Signaling [Danvers, MA]. Ku70 antibody was purchased from Abcam [Cambridge, MA]. Anti-β-actin and all secondary antibodies were obtained from Sigma [Saint Louis]. LipofectAMINE 2000 was purchased from Cell Signaling [Danvers, MA]. Vectastain ABC Kit coupled with avidin-biotin linked system was purchased from Vectastain [Burlingame, CA]. All chips buffers and reagents used for Surface plasmon resonance Biacore experiments were from GE Healthcare (Biacore Life Sciences), unless noted otherwise. The instrument used was Biacore 3000 (instrument ID 3442), which is maintained by Dr. Stanley R. Terlecky, in the Department of Pharmacology at Wayne State University School of Medicine and Calibrated by Jose A. Gutierrez from GE Healthcare every six months. Instrument configuration is IFC6 carrying Biacore 3000 Control software version 3.2. All analysis was done in BI-Aevaluation version 4.1 and all experiments were performed at 25°C.
Chemical Synthesis of MI-219 and MI-319
MI-219 and MI-319 were synthesized by using our previously published methods [26]. Nutlin-3 ((±)-4-[4,5-Bis(4-chlorophenyl)-2-(2-isopropoxy-4-methoxy-phenyl)-4,5-dihydro-imidazole-1-carbonyl]-piperazin-2-one) was purchased from Sigma [Sigma St Louis USA]. The binding of small molecules to human MDM2 was predicted by using the GOLD program [27].
SiRNA and Transfections
The p21WAF1 siRNA and siRNA control were obtained from Cell Signaling. Human PC cells were transfected with p2WAF1 siNA and control siNA respectively, using LipofectAMINE 2000 as described in the manufacturers protocol [Cell Signaling].
Cell Growth Inhibition Studies by MTT Assay
The cells [3 × 103] were seeded in a 96-well culture plate and treated with MDM2 inhibitors or gemcitabine or combination of both for indicated time and subsequently incubated with MTT reagent [0.5 mg/mL] at 37°C for 2 h, and MTT assay was done as described earlier [28]. In another set of experiments seeded cells were transfected with p21 siRNA or control siRNA for 5 hours followed by MDM2 inhibitor treatment for 72 hrs and MTT assay was performed. The results were plotted as means ± SD of three separate experiments having six determinations per experiment for each experimental condition.
Flow Cytometry and Cell Cycle Analysis
Cell cycle analysis on MI-219 and MI-319 treated cells were performed using PI staining. The percent of cells in different phases of the cell cycle was analyzed with CELLQUest software [BDIS] using a Power Macintosh 7500/100 computer [Apple Computer].
Quantification of Apoptosis by ELISA
The cell apoptosis ELISA detection kit [Roche, Palo Alto, CA] was used to detect apoptosis in PC cells treated with MDM2 inhibitors and/or gemcitabine alone or in combination for 72 h according to manufacturer’s protocol. Cell death was also detected using Trypan Blue exclusion assay under similar conditions using our previously described methods [29].
Immunoprecipitation
Cell lysate [200 μg] were subjected to immunoprecipitation by adding 2.5–5 μg of anti-p53, anti-MDM2, anti-SIRT1, anti Ku70 or anti-Bax antibody and incubation overnight at 4 °C. After adding 50 μl of Protein G-agarose [Santa Cruz Biotechnology, Santa Cruz CA] and incubation for 1 h, the samples were centrifuged. The agarose pellet was then washed three times, resuspended in Laemmli buffer, and boiled for 5 min. Boiled samples were centrifuged, and supernatant was used for Western blot analysis.
Western Blot Analysis
Procedure for cells lyses, protein concentration determination and SDS-PAGE analysis has been described in our previous publication [30]. Appropriate secondary antibodies coupled with avidin-biotin linked system [Vectastain ABC kit] were used according to manufactures protocol [30].
Reverse-Transcription PCR
Total RNAs were isolated from cells using the RNeasy® Isolation Kit [Qiagen, Valencia, CA]. The amount of total RNA was estimated by UV absorption at 260nm. The extracted RNAs [2 μg of each sample] were reverse-transcribed with the ImProm-II[TM] Reverse Transcription System, following the manufacturer’s instructions [Promega, Madison, WI]. The reactions were primed with random hexamer. The resulting cDNA preparations [usually 1/50th of the total volume] were subjected to PCR amplifications with HotMaster® Mix 2.5x [Eppendorf, Westbury, NY]. The sequence of the primers used is as follows: Ku70-F [5′-ctttgaggaatccag caagc-3′]/Ku70-R [5′-attaaaggtccgggtcttgg-3′] and actin-F [5′-tcatgaggtagtcagtcagg-3′]/actin-R [5′-tgacccagatcatgtttgag-3′]. The PCR products were analyzed by electrophoresis on 1% agarose gel containing ethidium bromide, and photographed over UV light.
SIRT1 FRET Based Screening Assay
SIRT1 FRET Based Assay kit (Cayman Chemicals Ann Arbor) was used to screen for SIRT1 inhibitory activity of MDM2 inhibitors in a 96 well plate according to the manufacturers protocol.
Surface Plasmon Resonance (SPR) Experiments
Ku70 recombinant protein was gently immobilized to SPR chips (CM5, research grade Biacore, a division of GE Healthcare, Piscataway, NY) in EHS-EP, and covalently attached using NHS/EDC chemistry. First, the chip was reconditioned with two cycles of pulses of 100 mM HCl and 50 mM NaOH for 10 seconds at a flow rate of 100 μL/min which opens up the carboxymethylcellulose matrix for facile flow of proteins. With the “Aim for immobilized level” feature of the Biacore 3000, chips were prepared using a standard NHS/EDC coupling chemistry. Typical ranges of peptide binding varies between 100–400 RU while for protein binding the range considered was be 600–7000 RU, depending on the protein being immobilized. Initially the pH scouting was done on a CM5 chip in 10 mM acetate buffer at a pH ranging between 4.0 and 4.5, depending on the pKa of the protein or peptide being immobilized. After this the chip was preconditioned with two cycles of pulses of 100 mM HCl and 50 mM NaOH at a flow rate of 100 uL/min. Based on the results of pH scouting the “Aim for immobilized level” feature was used in the amine coupling immobilization method. This method uses a standard NHS/EDC coupling chemistry. Typical ranges of peptide binding are from 300–400 RU while for protein binding the range is 800–7500 RU, depending on the protein being immobilized. The chips with peptide immobilized have Piftide (24 amino acids) as ligands, with flow cell always being blank. The piftide chips have flow cell 3 with low density Piftide, flow cell 4 with high density Piftide. Chips with protein immobilized also had a flow cell 1 blank with flow cells 2, 3 and 4 immobilized with proteins.
Binding Analysis
Concentration series of analytes was prepared by serial diluting in HBS-EP. The buffer concentrations of proteins (Piftide as ligand) ranged from 1.0 μg/mL to 0.03125 μg/mL, with each serial dilution being a factor of 2. The concentration range of the drug MI-319 or Nutlin-3 (+ve control) (proteins as ligands) was 3000 nM to 12.35 nM, with each serial dilution being by a factor of 3. The analyte was injected for 5 minutes, depending on the dissociation rate. Regeneration conditions varied depending on the ligand being immobilized. Surfaces of Piftide was regenerated using 50 mM NaOH at a flow rate of 30 uL/min for 30 seconds. Surfaces of proteins was regenerated using combinations of 1 % octyl-b-D-glucopyranoside (Biochemika), 0.5 % Triton X (Sigma), and 50 % ethylene glycol, which were all prepared in HBS-EP buffer, with two injections at a flow rate of 30 μL/min for 60–120 seconds. Stabilization times from 2–5 minutes was used as well. The first step in analysis was cutting out the regeneration steps in the sensogram and averaging the baseline of all curves to zero before using a y-transform option. After this for each cycle the blank surface (flow cell 1) was subtracted from the other surfaces (flow cells 2–4). The resulting sensograms were analyzed using Langmuir binding. All analysis was done using separate fit kinetics. The dissociation phase was analyzed first which calculates KD (Dissociation) values for each curve. These KD values are used in the analysis of association phase for each curve. Once again using separate fit kinetics.
Competition Experiments
A concentration series of MI-319 or Nutlin-3 ranging from 9000 nM to 12.35 nM with dilutions of a factor of 3 was made in HBS-EP buffer with 0.5 μg/mL MDM2, Ku-70, Bax or SIRT1. These solutions were allowed to pre-incubate for approximately one hour before injecting over CM5 chip with Piftide on the surface. The injection conditions were as mentioned above in Binding analysis.
Development of PC Xenografts
All in vivo studies were conducted in accordance with Wayne Sate University approved animal care and ethics committee guidelines and procedures. For the Capan-2 and BxPC-3 subcutaneous model, 10X10 cells were subcutaneously [sc] injected into the flanks of 4–5 week old female severe combined immune deficient mice [ICR-SCID] [Taconic Farms, Germantown, NY] using 26 G ½ Precision Glide needles [Becton Dickinson]. Palpable tumors started to appear in 3–5 weeks. Tumors were measured twice weekly using a caliper and expressed in [mg]. To prevent any pain or discomfort, mice were euthanized and their tumors removed once they reach 1500–2000 mg burden. Tumors were then dissected into 20–30 mg pieces and re-transplanted into naïve ICR-SCID for serial propagation.
Preclinical Efficacy Trail Design of MI-319 Given Orally
A total of 14 animals were trocared bilaterally with Capan-2 or BxPC-3 tumor fragments. One week later, the tumor xenograft animals were separated into two groups of seven with equal tumor burden. The animals were randomly assigned to two different cohorts: orally, group one received diluent only and group two was treated with MI-319 at 300 mg/kg for fourteen consecutive days. All animals were checked every other day for any signs of toxicity.
Statistical Analysis
The cell growth inhibition, apoptosis pre and post treatment as well as pre and post transfection was statistically evaluated using GraphPad StatMate software [GraphPad Software, Inc.]. Comparisons were made between control and transfection. P < 0.01 was used to indicate statistical significance.
RESULTS
MDM2 Inhibitors MI-219 and MI-319 Induce Cell Growth Inhibition and Apoptosis in wt-p53 PC Cells
The structures of MI-219, MI-319, MI-10 and Nutlin-3 are given in Fig. (1A). MI-219 has been extensively described in our previous publication [31]. MI-319 is an analogue of MI-219 with similar binding affinities towards MDM2 while MI-10 is the inactive analog which does not bind to MDM2. To test the effect of MDM2 inhibitors on cell growth inhibition, five pancreatic cancer cell lines were used. Capan-2 [wt-p53] [32–35], Capan-1, Colo-357, HPAC and BxPC-3 [mut-p53] were incubated with increasing concentrations of MI-219, MI-319, MI-10 or Nutlin-3 [0–50 μM] for 72 hrs. As can be seen in Fig. (1B), all the three inhibitors induced cell growth inhibition more significantly in wt-p53 cells. MI-219 and MI-319 showed slightly better growth inhibition than Nutlin-3 [IC50 of MI-219, MI-319 and Nutlin-3 in Capan-2 was found to be ~10.5 μM, 14 μM and 18 μM respectively]. The inactive analog MI-10 did not induce cell growth inhibition at the same concentrations. The IC50 in the mut-p53 cells was much higher clearly indicating that the inhibitors have more pronounced effect on cells that have wt-p53 and not mut-p53. Because p53 is also a central regulator of apoptosis, we examined whether activation of p53 by MI-219, MI-319 or MI-10 could lead to apoptosis as assessed by Histone DNA ELISA. Both MI-219 and MI-319 effectively induced apoptosis in Capan-2 but not in mut-p53 cancer cells Colo-357, Capan-1, HPAC or BxPC-3 (Fig. 1C). We also assessed cell death using trypan blue assay and similar to MTT and apoptosis MI-219, MI-319 as well as Nutlin-3 induced significant cell death while MI-10 was inactive. Nutlin-3 was used as a positive control and as expected Nutlin-3 effectively induced apoptosis in Capan-2 cells. Again the inactive analog MI-10 was not an effective inducer of apoptosis at the same concentrations. We then tested whether MDM2 inhibitors could induce cell cycle arrest in the cells under study.
Fig. (1). MDM2 inhibitors MI-219 and MI-319 induce cell growth inhibition and apoptosis in wt-p53 cells.
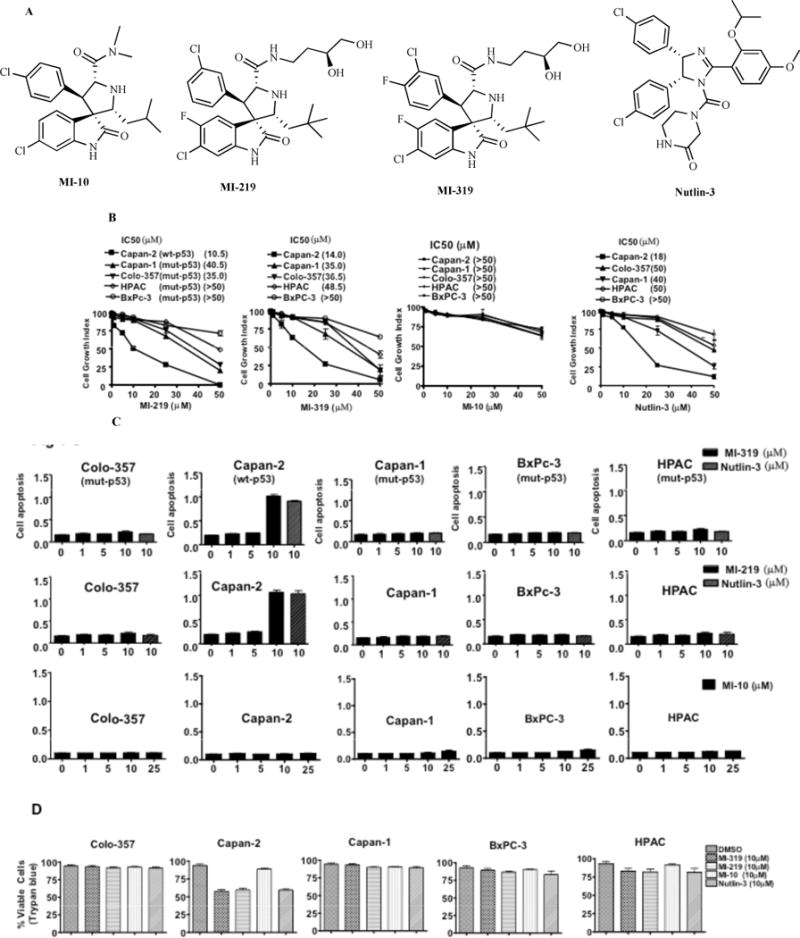
(A) Structures of MI-219, MI-319, MI-10 and Nutlin-3. (B) PC cells were treated with DMSO, MI-219, MI-319, MI-10 or Nutlin-3 (0–50 μM) for 72 hrs and cell viability was assessed using MTT assay. The inhibition of cell growth was dose-dependent. Error bars denote SEM of 3 independent experiments. (C) Capan-2, Capan-1 and Colo-357, HPAC and BxPC-3 cells were incubated with MI-219, MI-319 {0–10 μM) and MI-10 (0–25 μM) for 72 hours and apoptosis was analyzed by Histone/DNA ELISA using Roche Cell Death Detection Kit (Roche). Nutlin-3 (10 μM) was used as a positive control in the apoptosis experiments. (D) Cell death as assessed by trypan blue exclusion assay. PC cells were treated with MDM2 inhibitors at indicated concentrations for 72 hrs. ** Denotes p<0.01 when compared to untreated controls.
MDM2 Inhibitors Induce Cell Cycle Arrest in wt-p53 PC Cells
Cell cycle analysis revealed that indeed MI-219 and MI-319 arrested cells in G2-M phase. Both the inhibitors were effective in depleting S-phase in wt-p53 cells. The same treatment had negligible effect on cells lacking wt-p53 (Fig. 2A and B). The tumor suppressor p21 acts as an inhibitor of cell cycle and cell growth. Interestingly, knockdown of p21 using siRNA reduced the growth inhibitory efficiency of both inhibitors only in cell line with wt-p53 confirming the involvement of p21 in the regulation of p53 (Fig. 2C Upper and Lower Panels and 2D). These results are consistent with the work published by other laboratories which show that Nutlins, as well as MI-219 arrest cells in G2-M phase in lymphoma, prostate and colon cancer [21,36]. We then tested whether the inhibitors are able to reactivate the p53 pathway using Western blot analysis.
Fig. (2). MDM2 inhibitors induce cell cycle arrest in PC cells.
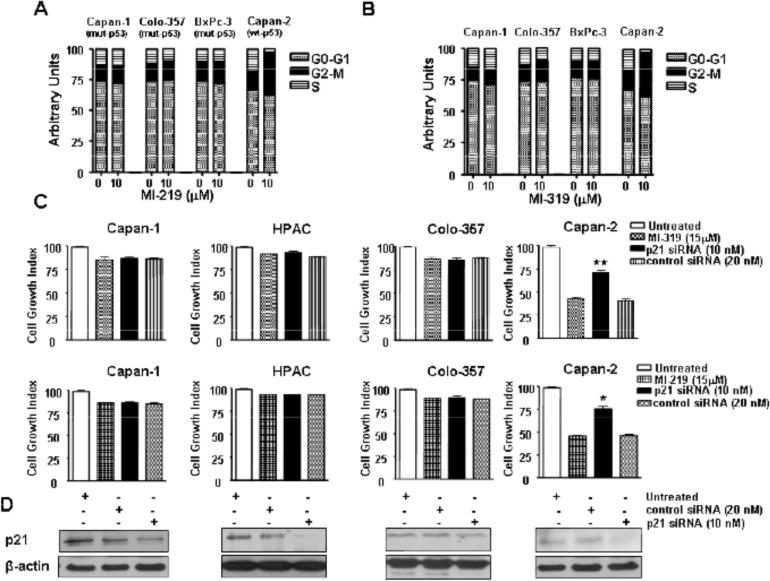
(A) PC cells were exposed to MI-219 or MI-319 (0 and 10 μM) for 72 hrs and cell cycle was analyzed by PI staining. The results are representative of three independent experiments. (B) siRNA against p21 blocks the cell growth inhibition (GI) by MDM2 inhibitors. PC cells transfected with either p21 siRNA or control siRNA were treated with MI-319 or MI-219 for 72 hours and cell growth inhibition was analyzed by MTT assay. ** Denotes p<0.01 when compared to inhibitor treated control. (C) Western blot analysis of lysates of Colo-357, Capan-1, HPAC and Capan-2 cells treated indicated concentrations of either untreated; control siRNA or p21 siRNA for 5 hrs. Protein was isolated according to procedures described in methods section.
MI-219 and MI-319 Block MDM2-p53 Complex and Reactivate p53 Pathway in wt-p53 PC Cells
In order to test the effect of our MDM2 inhibitors on protein levels of p53 and other related molecules, Western blot analyses were performed. As seen in Fig. (3A and B), exposure of wt-p53 cells to MI-219 as well as MI-319 [0–25 μM] for 24 hrs lead to a progressive increase in the levels of p53. We further tested the effect of MI-219 and MI-319 exposure on critical cell cycle regulator p21 whose expression is known to be directly regulated by p53 [37]. P21-dependence of cell cycle arrest has also been previously been shown in colon cancer for MI-43, a MI-219 family MDM2 inhibitor [38]. Our results show that MDM2 inhibitors increased p21 progressively with increasing concentrations in Capan-2 cells. Moreover, we observed that the induction of p53 was accompanied by increased expression of pro-apoptotic protein Puma. Protein levels of MDM2 were not changed, suggesting that the inhibitors only block MDM2 function and not its expression. In all the experiments Nutlin-3 was used as a positive control which, as expected, effectively induced p53, p21 and puma in Capan-2 cells only. Consistent levels of β-actin protein [used as a loading control] confirmed that the observed effects were p53 specific and not due to any non-specific effects of dying cells. Mut-p53 cells lines Capan-1 and Colo-357 were also analyzed for activation of p53 related genes. Results of Fig. (3C) reconfirm our cell growth and apoptosis data with no observed upregulation of p53, p21 or puma. Co-immunoprecipitation studies were done to verify blocking of MDM2-p53 complex formation by the inhibitors. First, we immunoprecipitated the MDM2-p53 complex using MDM2 antibodies followed by Western analysis with p53 antibodies, and vice versa using protein extracts from inhibitor-treated cell lysates. As can be seen from our results presented in Fig. (3D), both MI-219 and MI-319, blocked MDM2-p53 interaction only in wt-p53 cells as documented by negligible levels of p53 or MDM2 coimmunoprecipitated in treated cells compared to controls. These results confirm that both inhibitors are capable of disrupting the MDM2-p53 interaction in wt-p53 which ultimately leads to the reactivation of wt-p53 function. Subsequently, we studied the consequence of the re-activation of wt-p53 in gemcitabine-induced cell growth inhibition and cell death primarily because gemcitabine is the only effective chemotherapeutic agent that is currently used for the treatment of human PC [39].
Fig. (3). MDM2 inhibitors MI-219 and MI-319 block intracellular MDM2-p53 interaction and activate p53 pathway in wt-p53 cells.
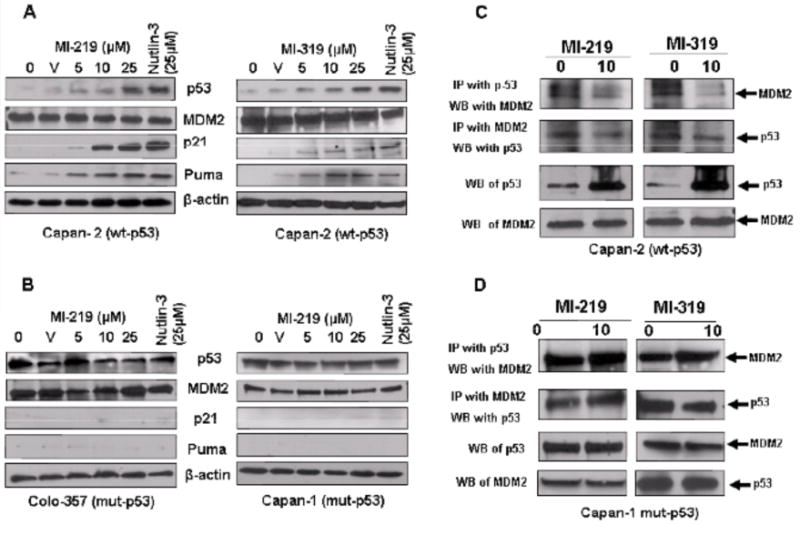
(A) Western blot analysis of whole cell lysate isolated from Capan-2 cells exposed to either DMSO (V) or increasing concentrations of MI-219 (Left Panel) and MI-319 (Right Panel) (0–25 μM for 24 hrs). Note up-regulation of p53, p21, Puma in Capan-2 cells (wt-p53). Nutlin-3 (25 μM) was used as a positive control in all experiments. (B) Western blot analysis of whole cell lysate isolated from Capan-1 and Colo-357 cells exposed to increasing concentrations of MI-219 (0–25 μM for 24 hrs). Note no up-regulation of p53, p21, Puma in Capan-1 or Colo-357 cells (mut-p53). Nutlin-3 (25 μM) was used as a positive control in all experiments. (C) Co-immunoprecipitation of MI-219 or MI-319 treated Capan-2 or Colo-357 cell lysate was performed with MDM2 followed by western blot analysis with p53. Note reduced association of MDM2-p53 interaction in drug treated Capan-2 (wt-p53) but not in Colo-357 (mut-p53) cells. Blots are representative of three independent experiments.
MDM2 Inhibitors Enhance the Cytotoxic Effect of Standard Chemotherapeutic Agent Gemcitabine
We assessed the effect of MDM2 inhibitors either as single agent or in combination with gemcitabine on cell viability. For these studies, Capan-2, Capan-1 and Colo-357 cells were treated with sub IC50 doses of MI-219 or MI-319 [5 μM] either alone or in combination with 100 nM of gemcitabine, and viable cells were evaluated at 72 hours using MTT assay. The dose used here was chosen based upon a preliminary dose escalation study done by us prior to this experiment. As can be seen in Fig. (4A and B) [upper panels], the treatment of Capan-2 cells with 5 μM of MI-219 or MI-319 resulted in 24 % and 26 % loss of cell viability, respectively. While treatment with gemcitabine [100 nM] for 72 hours resulted in only 9 % loss of cell viability. However, the combination treatment using MI-219 or MI-319 with gemcitabine resulted in 74% and 77% cell growth inhibition, respectively. Isobologram analysis using Calcu Sync software revealed a synergistic combination between the inhibitors and gemcitabine with combination index values less than 1 [CI<1] (Fig. 4A and B lower panels). These results suggest that MDM2 inhibitors sensitize the cells for better cell growth inhibition when combined with conventional chemotherapeutic drug such as gemcitabine. Histone/DNA ELISA apoptosis analysis revealed that relative to single agent, combination treatment with gemcitabine induced much more apoptosis in Capan-2 cells (Fig. 4C and D upper panel]. The CI values of the gemcitabine–inhibitor combinations were less than 1 which is synergistic (Fig. 4C and D lower panels), and thus the cell death data is consistent with the results of cell growth inhibition as observed by MTT assay. No synergistic cell growth or apoptosis was observed with the combination treatments in mut-p53 Colo-357 and Capan-1 cells [results no shown]. Collectively, the above results clearly suggest that MDM2 inhibitors could sensitize wt-p53 pancreatic cancer cells to gemcitabine induced killing. We then looked at the mechanism of action of MDM2 inhibitors by identifying targets upstream and downstream of p53.
Fig. (4). MDM2 inhibitors augment antitumor effect of gemcitabine.
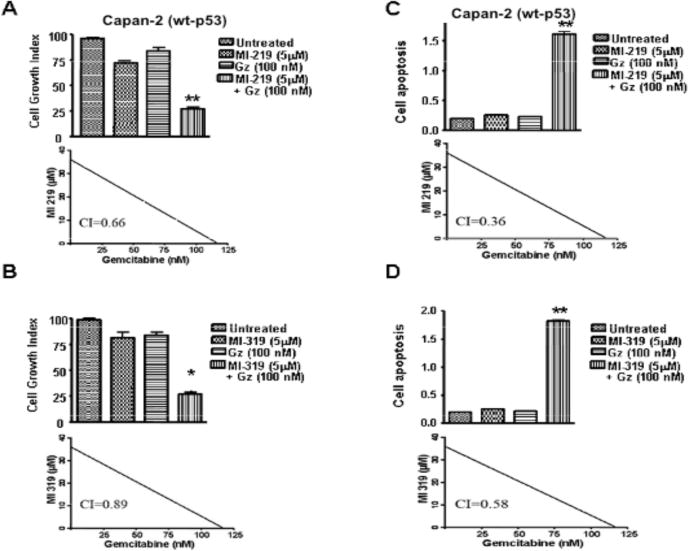
Panels A – D (Upper Panels); Capan-2 cells were exposed to either vehicle (DMSO), MI-219 (5 μM), MI-319 (5 μM), Gemcitabine (100 nM) alone, MI-219 (5 μM) + Gemcitabine (100 nM) and MI-319 (5 μM) + Gemcitabine (100 nM). Cell growth inhibition was detected using MTT assay while apoptosis was detected using Histone DNA/ELISA assay as described under Material and Methods. Error Bars denote SEM of four independent experiments. ** p value < 0.01 when compared to untreated controls. Panels A – D (Lower Panels); Isobologram analysis of the combination of gemcitabine and MI-219 or gemcitabine and MI-319 in Capan-2 cells. Combination index (CI) values were calculated using Calcusyn software. Points below the line indicate synergy.
MI-219 Interferes with the Sirtuin1 Pool of Proteins in PC
The p53 protein is activated through acetylation in response to various types of stresses [39]. SIRT1 can deacetylate p53, and studies have suggested that deacetylation by SIRT1 reduces p53-dependent apoptosis in response to stress [40,41]. SIRT1 is also known to deacetylate another protein Ku70 which regulates apoptosis by blocking Bax entry into mitochondria. In order to understand the mechanism of p53 re-activation by MDM2 inhibitors, we determined the expression patterns of SIRT1 partner proteins such as KU70, Bax and acetyl-p53 pre- and post- MI-219 treatment. Our results clearly show that MI-219 [0–25 μM] suppressed SIRT1, Ku70 and simultaneously increased p53 acetylation as well as induced the pro-apoptotic Bax (Fig. 5A). MI-219 did not suppress SIRT1 or Ku70 in mut-p53 Colo-357 cells (Fig. 5B). RT-PCR analysis revealed that MI-319 treatment could suppress Ku70 transcription at the mRNA level only in capan-2 and not in colo-357 mut-p53 PC cells (Fig. 5C Left Panel). Nutlin-3 was used as a positive control which also suppressed capan-2 Ku70 mRNA. SIRT1 mRNA levels did not change which is consistent with other reported observations where it was suggested that calorie restriction or genotoxic stress modulates SIRT1 only at the post-translational level without interfering with its transcription [42,43]. We also performed a SIRT1 FRET based screening assay to verify SIRT1 activity inhibition by our SMIs. As can be seen from results of Fig. (5D), MI-219, MI-319 and Nutlin-3 significantly suppressed SIRT1 activity while the inactive analog MI-10 did not. Co-immunoprecipitation experiments showed that MI-219 could disrupt SIRT1-Bax and Ku70-Bax interaction as lesser protein was visible in drug treated samples (Fig. 5E). Further, we also used surface plasmon resonance [SPR] experiments to show competitive binding between MI-319 and Ku70. As can be seen from results of Fig. (5F) our SPR results confirm high binding affinity between MI-319 and Ku70 with a KD of 170 nM. For comparative studies we also analyzed binding affinity of Nutlin-3 to Ku70 using SPR under similar conditions. Interestingly Nutlin-3 was also found to have nearly similar affinity towards Ku70 (KD of 169 μM). These results together with the results of Western blot analysis, RT-PCR and immunoprecipitation show crucial off target effects of MDM2 inhibitors that were previously unexplored. Therefore, we have demonstrated, for the first time that MI-219 not only blocks MDM2 but also interferes in the pathways influenced by SIRT1. The promising results of the mechanistic in vitro studies led us to test the efficacy of MDM2 inhibitors in animal model in vivo.
Fig. (5). MI-219 suppresses SIRT1 and disrupts SIRT1-Bax as well as Ku70-Bax interaction in Capan-2 cells.
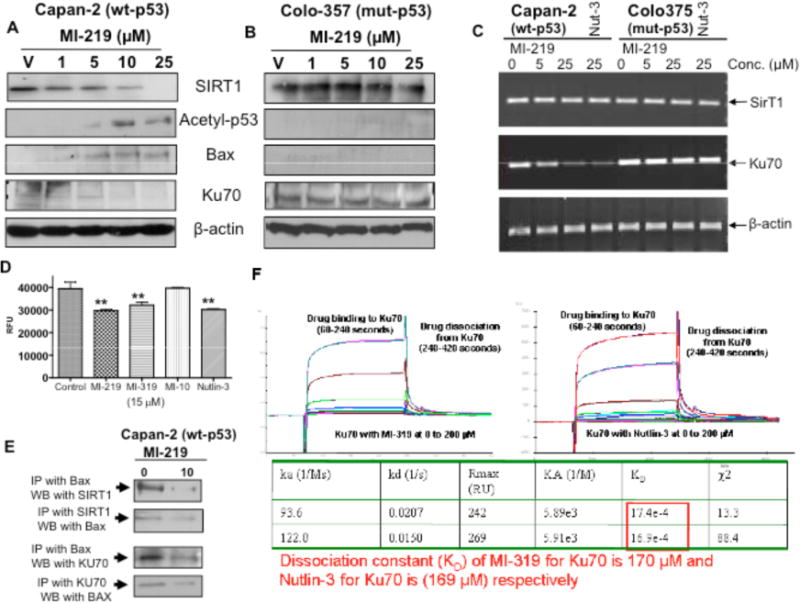
(A & B) Western blot analysis of lysates extracted from Capan-2 and Colo-357 cells exposed to increasing doses of MI-219 (0–25 μM for 24 hrs). Equal amount of protein was loaded and Western blot analysis was performed using anti-SIRT1, anti Ku70, anti-Bax and anti-acetyl-p53 antibodies. Anti-β-actin antibody was used as protein loading control as shown for each blot. (D) SIRT1 FRET based screening assay for activity of MDM2 inhibitors against SIRT1. Assay was performed in 96 well plate according to manufacturers protocol. Note suppression of SIRT1 post 15 μM MI-219; MI-319 or Nutlin-3 treatment. No suppression in SIRT1 by MI-10. (E) (Left Panel) Co-immunoprecipitation of MI-219 treated Capan-2 cell lysate was performed with SIRT1 followed by Western blot analysis with Bax and vice versa. Note reduced association of SIRT1-Bax interaction in drug treated cells. (Right Panel) Co-immunoprecipitation of MI-219 treated Capan-2 cell lysate was performed with Ku70 followed by Western blot analysis with Bax and vice versa. Note reduced association of Ku70-Bax interaction in drug treated cells. Blots are representative of three independent experiments. (F) Label-Free analysis of binding using surface plasmon resonance (SPR) reveals that both MI-319 and Nutlin-3 bind to Ku70 protein with low to mid micro-molar dissociation constants (KD). 8000 refractive index units (RU) of recombinant KU70 were covalently immobilized on the surface of Fc4 on a gold CM5 SPR chip, and refractive index changes were monitored in real time after injection of concentration series of MI-319 at 25 °C. Binding was permitted over 300 seconds followed by wash with HBS-EP for 480 seconds to reveal dissociation. Sensogram were normalized to a common baseline, and fitted using BIAevaluation software to estimate drug ON-rates (Ka) and Drug OFF-rates (Kd) followed by calculation of KD (ka/Kd).
Efficacy of MDM2 Inhibitor MI-319 on Pancreatic Tumor growth and Animals Weights
For any targeted small molecule inhibitor to be brought to clinical trial, its efficacy need to be tested in animal models. Therefore, to determine whether MDM2 inhibitors could suppress tumor growth in animals, we sought to establish Capan-2 and BxPC-3 xenograft model in SCID mice using previously published protocols [44]. We were successful in establishing both Capan-2 and BxPC-3 xenograft models with 100 % take rate in SCID mice. As shown in Fig. (6A), the MDM2 inhibitor, MI-319 significantly [p=0.015] inhibited the growth only in Capan-2 tumors and not in BxPC-3 tumor model. Moreover, MI-319 did not cause any loss in the body weight of our experimental animals and there was no obvious sign of toxicity during the course of the treatment (Fig. 6B).
Fig. (6). MI-319 orally inhibits tumor growth in Capan-2 (wt-p53) and not in BxPC-3 (mut-p53) xenograft animal model.
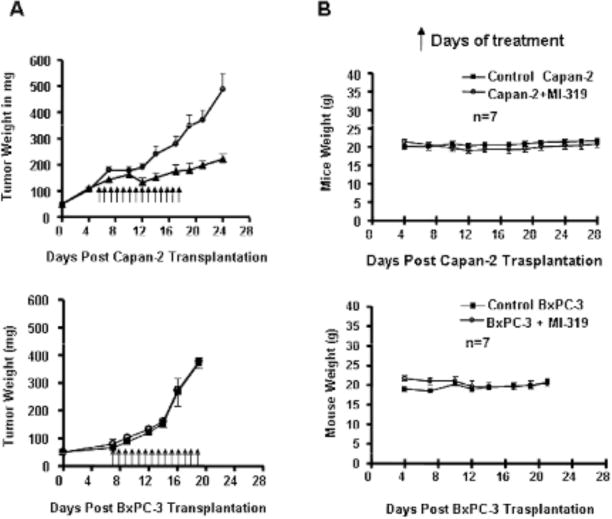
(A) Capan-2 xenografts were inoculated subcutaneously in SCID mice. Once transplanted, fragments developed into palpable tumors (about 60 mg), groups of 7 animals were removed randomly and assigned to two treatment groups. Mice were administered MI-319 orally at 300 mg/kg for fourteen consecutive days. (○) Vehicle Control and (▲) MI-319 treated group. (B) MI-319 did not cause any loss in mice body weight during the course of treatment. (○) Vehicle Control and (■) MI-319 treated group. (C) BxPC-3 xenografts were established and treated with MI-319 similar to Capan-2 in SCID mice. (○) Vehicle Control and (■) MI-319 treated group. (D) MI-319 did not cause any loss in mice body weight during the course of treatment. (○) Vehicle Control and (■) MI-319 treated group.
DISCUSSION
The present study is the first to show in PC the growth inhibition and apoptosis by novel MDM2 inhibitors through the direct reactivation of wt-p53 along with up-regulation of proteins under its influence such as p21 and the pro-apoptotic Puma. Our results also highlight two novel secondary new targets of MDM2 inhibitor ‘SIRT1’ and Ku70, that regulate p53 through acetylation and blocking Bax mediated apoptosis, respectively.
PC is a deadly disease and prognosis is generally poor, with <5% survival rate [1]. Among the most critical gene deregulated in pancreatic carcinogenesis, the alterations in the p53 tumor suppressor gene [45,46]. The activity of wt-p53 is mainly regulated at the post-translational level by MDM2, which causes wt-p53 degradation by ubiquitin-mediated proteolysis [9]. Based on functional significance of MDM2–p53 interaction, one attractive pharmacological approach to wt-p53 reactivation is to use a small-molecule to block the MDM2 [20–22]. In this regard Nutlin-3 a cis-imidazole has been well studied in different cancers. Our MI series of MDM2 inhibitors belong to different class (spiro-oxindole) and have a slightly higher affinity towards MDM2 when compared to Nutlins. Using a structure-based de novo design strategy it was shown that the interaction between p53 and MDM2 is primarily mediated by four key hydrophobic residues (Phe 19, Leu 22, Trp 23 and Leu 26) of p53 and a small but deep hydrophobic cleft in MDM2. Nutlin-3 mimics the interactions of the p53 peptide to a high degree, with one bromophenyl moiety sitting deeply in the Trp pocket, the other bromophenyl group occupying the Leu pocket, and the ethyl ether side chain directed toward the Phe pocket. In essence, the imidazoline scaffold replaces the helical backbone of the peptide and is able to direct, in a fairly rigid fashion, the projection of three groups into the pockets normally occupied by Phe19, Trp23, and Leu26 of p53. However, unlike Nutlin-3 in case of our inhibitors (MI series), computational modeling predicted that MI-219 mimics the four (instead of three in case of Nutlin-3) key binding residues in p53 (Phe-19, Leu-22, Trp-23 and Leu-26) resulting in optimal hydrogen bonding and hydrophobic interactions with MDM2. Recently our laboratory has successfully shown the applicability/activity of MI-319 against lymphoma (cell lines, patients samples and animal xenografts) [47]. Therefore, we tested the biological activity of MDM2 inhibitors MI-319 and MI-219, on five PC cell lines [one containing wild type 53 and four containing mutant-p53] and our in vitro results confirmed that both inhibitors could induce cell growth inhibition and apoptosis in PC in a wt-p53 dependant manner as mut-p53 cells did not show robust growth inhibition or apoptosis towards these inhibitors. This confirmed the “proof of principle” of MDM2 inhibitor action in wt-p53 PC cells and clearly provided direct evidence for the functional activation of p53, by both MI-219 and MI-319 through destabilization of the wt-p53-MDM2 complex leading to the stabilization of wt-p53 in PC cells. One can argue that the doses of MDM2 inhibitors required for growth inhibition in PC were much higher compared to other tumor types such as prostate, colon and breast. In this regard it should be noted that PC is an inherently resistant cancer that is not easily amenable to currently available chemotherapeutic drugs. This is further supported by studies which suggest that the wild type p53 pathway is compromised in PC due to SNP309 polymorphism of MDM2 that ultimately facilitates accelerated pancreatic adenocarcinoma formation [48,49]. Nevertheless, our combination studies show that MI-319 as well as MI-219 at lower concentrations can synergize with gemcitabine leading to enhanced cell growth inhibition and apoptosis in PC cell suggesting that these inhibitors certainly have applicability in this tumor type.
In-depth mechanistic studies on the mode of action of inhibitors on MDM2 and the consequent p53 reactivation are lacking and it was of interest to us to explore the roles of crucial proteins that are involved in the regulation of p53. Activated p53 is known to be influenced by multiple post-translational control processes such as phosphorylation and acetylation that positively regulate p53 function [50]. Acetylation is an important epigenetic phenomenon in the biology of p53 [51,52]. Upon stress, p53 is acetylated at Lys382 which enhances its DNA binding activity [53]. Moreover, deacetylation of p53 by SIRT1 has been shown to repress p53 mediated cell cycle regulation and apoptosis. SIRT1 is also known to deacetylate another protein Ku70 which, in turn, interacts with Bax and is responsible for blocking Bax entry into mitochondria. Therefore, we sought to determine whether acetylation of p53 could be influenced by our inhibitors in PC cells. Indeed our results showed that MI-219 treatment suppresses SIRT1 protein and simultaneously enhances acetylation of p53 (Fig. 5A). Using state of the art Surface plasmon resonance techniques we studied the binding between MI-319 or Nutlin-3 and Ku70 and our results confirm high affinity association between the two (Fig. 5F). Interestingly MI-219 treatment resulted in the suppression of Ku70 expression along with disruption of Ku70-Bax interaction (Fig. 5E). This observation is of great importance because it proves that MDM2 inhibitor not only blocks MDM2 which is its primary target but also suppresses two secondary targets the negative regulator ‘SIRT1’, which is a molecule that regulates p53 function and Ku70. Although it is too preliminary to confirm the true binding/interaction site of Ku70 or SIRT1 to MI-319, yet is can be speculated that MI-319 or Nutlin-3 may interact with peptide sequence (LSQETFSDLWKLL) similar to p53 transactivation domain towards which both Nutlin-3 or MI series of inhibitors were built.
As MI-219 does not alter MDM2 expression yet Ku70 and SIRT1 are suppressed suggesting that these drugs may have a MDM2 independent role in the biology of cells. However, compelling evidence in literature supports to a MDM2 dependent mechanism of action of these drugs on Ku70 and SIRT1. Our cell free FRET based SIRT1 activity assay showed inhibition of SIRT1 activity by MDM2 inhibitors. Yet in a cellular system the dynamics of SIRT1 is complex. Studies so far suggest that only wt-p53 can inhibit SIRT1 while cells that have lost or have mutations in p53 have over expressed SIRT1 and cannot repress it [17]. This certainly points out that the suppression of SIRT1 in our system is p53 dependent however elucidation of the exact mechanism of action requires further work. As far as Ku70 is concerned, very recently Nutlin, a drug with similar mode of action as MI-219 was shown to disrupt hdm2-Ku70 interaction [19]. Based on our results and those of others we propose multiple mode of action MI-219 on SIRT1 and Ku70. MDM2 inhibitors down regulate SIRT1 (western blot results Fig. 5A–C) that in principle may prevent Ku70 and p53 deacetylation. Surface plasmon resonance and Co-IP results confirm that MI drugs directly bind to Ku70 as well as disrupt Ku70-Bax interaction (Fig. 5). Although yet to be proved, it is suggested that such a direct binding may induce conformational changes in Ku70 rendering it ineffective in binding to Bax and therefore allowing the latter to induce apoptosis. MI drugs also directly suppress Ku70 mRNA and protein expression (Fig. 6A and B) which in turn allows p53 induced free Bax to mediate apoptotic events.
In phase I/II pilot study showed that adenoviral p53 gene delivery in conjunction with gemcitabine increased survival in patients with advanced metastatic PC [54]. The basic idea behind such a study certainly holds some ground, yet it does not acknowledge the role of MDM2 that would block nascent p53 produced by the virus thus effectively diminishing the outcome of the adenoviral therapy. Therefore, we believe that our inhibitors might play pivotal role in a combination therapy setting by blocking MDM2 binding to p53 and therefore enhancing the cytotoxic effect of gemcitabine. Based on this assumption we analyzed combination treatments on both mutants as well as wt-p53 PC cells. Although the combination treatment was ineffective on the mutant cell lines, both MI-219 and MI-319 treatment sensitized PC cells to induce synergistically significant apoptosis by gemcitabine [CI<1] in wt-p53 cells. The observed results have significant therapeutic implications because gemcitabine alone cannot be considered as an ideal agent for PC therapy. In view of our in vitro combination results, we hypothesize that using a targeted agent such as MDM2 inhibitor in conjunction with conventional drugs would be beneficial for the treatment of patients diagnosed with PC.
Finally to determine whether animal models can add to the weight of preclinical evidence warranting further development of MDM2 inhibitors towards clinical application, we evaluated the anti-tumor activity of MI-319 as an oral agent employing Capan-2 [wt-p53] and BxPC-3 [mut-p53] as xenograft mouse model. The MI-319 was highly effective [p=0.015] in inhibiting tumor growth of Capan-2 and not BxPC-3 xenografts (Fig. 6). Interestingly, MI-319 was given orally for 14 consecutive days with no apparent toxicity observed in all the treated animals. Based on the in vitro combination results with gemcitabine, we hypothesized that our MDM2 inhibitors would also have enhanced antitumor activity of gemcitabine in vivo. Those studies are beyond the scope of this manuscript. In vivo studies on the role of SIRT1 and Ku70 in tumor growth inhibition either with MI-319 alone or in combination are currently under investigation. Further, very recently we have shown that MI-319 when combined with cisplatin can be effective inducer of apoptosis in PC irrespective of the mutational or functional status of p53. In the same study we have confirmed the involvement of a distinct pathway involving p73 that is also under the influence of MDM2 and unlike p53, is rarely mutated in PC [55]. Nevertheless our results presented in this study clearly suggest that re-activation of p53 by our potent and specific MDM2 inhibitor could be a promising therapeutic strategy that warrants clinical investigation for the treatment of human PC for which there is no curative therapy.
Acknowledgments
The authors acknowledge the following grant support the National Cancer Institute, NIH Grant R01CA109389 [Mohammad RM] and NIH Grant U19CA113317 [S. Wang]. University of Michigan has filed a patent on TW-37, which has been licensed by Ascenta Therapeutics Inc. University of Michigan and Shaomeng Wang own equity in Ascenta. Shaomeng Wang also serves as a consultant for Ascenta and is the principal investigator on a research contract from Ascenta to University of Michigan. GUIDO foundation support is acknowledged.
ABBREVIATIONS
- PC
Pancreatic cancer
- MDM2
Murine double minute 2
- SIRT1
Sirtuin 1
- CI
Combination Index
- Wt-p53
wild type p53
- Mut-p53
mutant p53
- SPR
Surface Plasmon Resonance
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 netwo. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Barton CM, Staddon SL, Hughes CM, Hall PA, O’Sullivan C, Kloppel G, Theis B, Russell RC, Neoptolemos J, Williamson RC. Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br J Cancer. 1991;64:1076–1082. doi: 10.1038/bjc.1991.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 6.Lowe SW. Cancer therapy and p53. Curr Opin Oncol. 1995;7:547–553. doi: 10.1097/00001622-199511000-00013. [DOI] [PubMed] [Google Scholar]
- 7.Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science. 1994;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- 8.Lowe SW, Bodis S, Bardeesy N, McClatchey A, Remington L, Ruley HE, Fisher DE, Jacks T, Pelletier J, Housman DE. Apoptosis and the prognostic significance of p53 mutation. Cold Spring Harb Symp Quant Biol. 1994;59:419–426. doi: 10.1101/sqb.1994.059.01.047. [DOI] [PubMed] [Google Scholar]
- 9.Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haupt Y, Barak Y, Oren M. Cell type-specific inhibition of p53-mediated apoptosis by mdm2. EMBO J. 1996;15:1596–1606. [PMC free article] [PubMed] [Google Scholar]
- 11.Lahav G. Oscillations by the p53-Mdm2 feedback loop. Adv Exp Med Biol. 2008;641:28–38. doi: 10.1007/978-0-387-09794-7_2. [DOI] [PubMed] [Google Scholar]
- 12.Bose I, Ghosh B. The p53-MDM2 network: from oscillations to apoptosis. J Biosci. 2007;32:991–997. doi: 10.1007/s12038-007-0103-3. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Wang H, Li M, Rayburn E, Agrawal S, Zhang R. Novel MDM2 p53-independent functions identified through RNA silencing technologies. Ann N Y Acad Sci. 2005;1058:205–214. doi: 10.1196/annals.1359.030. [DOI] [PubMed] [Google Scholar]
- 14.Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- 15.Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005;309:886–887. doi: 10.1126/science.1116801. [DOI] [PubMed] [Google Scholar]
- 16.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009;9:123–128. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Yokota T, Gama V, Yoshida T, Gomez JA, Ishikawa K, Sasaguri H, Cohen HY, Sinclair DA, Mizusawa H, Matsuyama S. Bax-inhibiting peptide protects cells from polyglutamine toxicity caused by Ku70 acetylation. Cell Death Differ. 2007;14:2058–2067. doi: 10.1038/sj.cdd.4402219. [DOI] [PubMed] [Google Scholar]
- 19.Gama V, Gomez JA, Mayo LD, Jackson MW, Danielpour D, Song K, Haas AL, Laughlin MJ, Matsuyama S. Hdm2 is a ubiquitin ligase of Ku70-Akt promotes cell survival by inhibiting Hdm2-dependent Ku70 destabilization. Cell Death Differ. 2009;16:758–769. doi: 10.1038/cdd.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein C, Vassilev LT. Targeting the p53-MDM2 interaction to treat cancer. Br J Cancer. 2004;91:1415–1419. doi: 10.1038/sj.bjc.6602164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Secchiero P, di Iasio MG, Gonelli A, Zauli G. The MDM2 inhibitor Nutlins as an innovative therapeutic tool for the treatment of haematological malignancies. Curr Pharm Des. 2008;14:2100–2110. doi: 10.2174/138161208785294663. [DOI] [PubMed] [Google Scholar]
- 22.Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3:419–421. [PubMed] [Google Scholar]
- 23.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 24.Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci USA. 2006;103:11003–11008. doi: 10.1073/pnas.0604507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Secchiero P, Corallini F, Gonelli A, Dell’Eva R, Vitale M, Capitani S, Albini A, Zauli G. Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ Res. 2007;100:61–69. doi: 10.1161/01.RES.0000253975.76198.ff. [DOI] [PubMed] [Google Scholar]
- 26.Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, Stuckey J, Krajewski K, Roller PP, Tomita Y, Parrish DA, Deschamps JR, Wang S. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 27.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 28.Azmi AS, Wang Z, Burikhanov R, Rangnekar VM, Wang G, Chen J, Wang S, Sarkar FH, Mohammad RM. Critical role of prostate apoptosis response-4 in determining the sensitivity of pancreatic cancer cells to small-molecule inhibitor-induced apoptosis. Mol Cancer Ther. 2008;7:2884–2893. doi: 10.1158/1535-7163.MCT-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azmi AS, Ahmad A, Banerjee S, Rangnekar VM, Mohammad RM, Sarkar FH. Chemoprevention of pancreatic cancer: characterization of Par-4 and its modulation by 3,3′ diindolylmethane (DIM) Pharm Res. 2008;25:2117–2124. doi: 10.1007/s11095-008-9581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu SM. Clinical applications of immunoperoxidase technique. R I Med J. 1979;62(11):447–452. [PubMed] [Google Scholar]
- 31.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, Bernard D, Zhang J, Lu Y, Gu Q, Shah RB, Pienta KJ, Ling X, Kang S, Guo M, Sun Y, Yang D, Wang S. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisold S, Linnebacher M, Ryschich E, Antolovic D, Hinz U, Klar E, Schmidt J. The effect of adenovirus expressing wild-type p53 on 5-fluorouracil chemosensitivity is related to p53 status in pancreatic cancer cell lines. World J Gastroenterol. 2004;10:3583–3589. doi: 10.3748/wjg.v10.i24.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujioka S, Schmidt C, Sclabas GM, Li Z, Pelicano H, Peng B, Yao A, Niu J, Zhang W, Evans DB, Abbruzzese JL, Huang P, Chiao PJ. Stabilization of p53 is a novel mechanism for proapoptotic function of NF-kappaB. J Biol Chem. 2004;279:27549–27559. doi: 10.1074/jbc.M313435200. [DOI] [PubMed] [Google Scholar]
- 34.Bouvet M, Bold RJ, Lee J, Evans DB, Abbruzzese JL, Chiao PJ, McConkey DJ, Chandra J, Chada S, Fang B, Roth JA. Adenovirus-mediated wild-type p53 tumor suppressor gene therapy induces apoptosis and suppresses growth of human pancreatic cancer [seecomments] Ann Surg Oncol. 1998;5:681–688. doi: 10.1007/BF02303477. [DOI] [PubMed] [Google Scholar]
- 35.Sui X, Shin S, Zhang R, Firozi PF, Yang L, Abbruzzese JL, Reddy SA. Hdm2 is regulated by K-Ras and mediates p53-independent functions in pancreatic cancer cells. Oncogene. 2009;28:709–720. doi: 10.1038/onc.2008.423. [DOI] [PubMed] [Google Scholar]
- 36.Zauli G, Rimondi E, Corallini F, Fadda R, Capitani S, Secchiero P. MDM2 antagonist Nutlin-3 suppresses the proliferation and differentiation of human pre-osteoclasts through a p53-dependent pathway. J Bone Miner Res. 2007;22:1621–1630. doi: 10.1359/jbmr.070618. [DOI] [PubMed] [Google Scholar]
- 37.Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 38.Shangary S, Ding K, Qiu S, Nikolovska-Coleska Z, Bauer JA, Liu M, Wang G, Lu Y, McEachern D, Bernard D, Bradford CR, Carey TE, Wang S. Reactivation of p53 by a specific MDM2 antagonist (MI-43) leads to p21-mediated cell cycle arrest and selective cell death in colon cancer. Mol Cancer Ther. 2008;7:1533–1542. doi: 10.1158/1535-7163.MCT-08-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burris HA, III, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 40.Vaziri H, Dessain SK, Ng EE, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 41.Smith J. Human Sir2 and the ‘silencing’ of p53 activity. Trends Cell Biol. 2002;12:404–406. doi: 10.1016/s0962-8924(02)02342-5. [DOI] [PubMed] [Google Scholar]
- 42.Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle. 2009;8:712–715. doi: 10.4161/cc.8.5.7753. [DOI] [PubMed] [Google Scholar]
- 43.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Sengupta R, Banerjee S, Li Y, Zhang Y, Rahman KM, Aboukameel A, Mohammad R, Majumdar AP, Abbruzzese JL, Sarkar FH. Epidermal growth factor receptorrelated protein inhibits cell growth and invasion in pancreatic cancer. Cancer Res. 2006;66:7653–7660. doi: 10.1158/0008-5472.CAN-06-1019. [DOI] [PubMed] [Google Scholar]
- 45.Hollstein M, Moeckel G, Hergenhahn M, Spiegelhalder B, Keil M, Werle-Schneider G, Bartsch H, Brickmann J. On the origins of tumor mutations in cancer genes: insights from the p53 gene. Mutat Res. 1998;405:145–154. doi: 10.1016/s0027-5107(98)00131-6. [DOI] [PubMed] [Google Scholar]
- 46.Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, Hovig E, Smith-Sorensen B, Montesano R, Harris CC. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551–3555. [PMC free article] [PubMed] [Google Scholar]
- 47.Mohammad RM, Wu J, Azmi AS, Aboukameel A, Sosin A, Wu S, Yang D, Wang S, Al-Katib AM. An MDM2 antagonist (MI-319) restores p53 functions and increases the life span of orally treated follicular lymphoma bearing animals. Mol Cancer. 2009;8:115. doi: 10.1186/1476-4598-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grochola LF, Muller TH, Bond GL, Taubert H, Udelnow A, Wurl P. MDM2 SNP309 Associates With Accelerated Pancreatic Adenocarcinoma Formation. Pancreas. 2010;29:76–80. doi: 10.1097/MPA.0b013e3181b9f105. [DOI] [PubMed] [Google Scholar]
- 49.Asomaning K, Reid AE, Zhou W, Heist RS, Zhai R, Su L, Kwak EL, Blaszkowsky L, Zhu AX, Ryan DP, Christiani DC, Liu G. MDM2 promoter polymorphism and pancreatic cancer risk and prognosis. Clin Cancer Res. 2008;14:4010–4015. doi: 10.1158/1078-0432.CCR-07-4187. [DOI] [PubMed] [Google Scholar]
- 50.Stommel JM, Wahl GM. A new twist in the feedback loop: stress-activated MDM2 destabilization is required for p53 activation. Cell Cycle. 2005;4:411–417. doi: 10.4161/cc.4.3.1522. [DOI] [PubMed] [Google Scholar]
- 51.Gu W, Luo J, Brooks CL, Nikolaev AY, Li M. Dynamics of the p53 acetylation pathway. Novartis Found Symp. 2004;259:197–205. [PubMed] [Google Scholar]
- 52.Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J Neurosci. 2008;28:8772–8784. doi: 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolkersdorfer GW, Thiede C, Fischer R, Ehninger G, Haag C. Adenoviral p53 gene transfer and gemcitabine in three patients with liver metastases due to advanced pancreatic carcinoma. HPB (Oxford) 2007;9:16–25. doi: 10.1080/13651820600839555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Azmi AS, Aboukameel A, Banerjee S, Wang Z, Mohammad M, Wu J, Wang S, Yang D, Philip PA, Sarkar FH, Mohammad RM. MDM2 inhibitor MI-319 in combination with cisplatin is an effective treatment for pancreatic cancer independent of p53 function. Eur J Cancer. 2010;46:1122–1131. doi: 10.1016/j.ejca.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]