

Abstract
A Sn-phony in B!
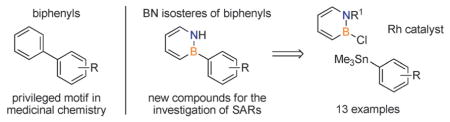
BN isosteres of biphenyl compounds are prepared through Rh-catalyzed cross-coupling between 2-chloro-1,2-azaborines and arylstannanes (see scheme). The synthetic method should enable investigations of structure–activity relationships (SARs) by expanding the chemical space of the pharmaceutically relevant biphenyl structure through BN/CC isosterism.
Keywords: azaborines, biphenyl, boron, heterocycles, isosterism
The biphenyl scaffold can be considered a privileged structural motif in medicinal chemistry.[1] Biaryl compounds have shown a wide range of therapeutic activities, including antifungal, antiinfective, antihypercholesteremic, antirheumatic, analgesic, antiinflammatory, and antiarrhythmic activities.[1] In addition, a statistical analysis of binding data on a variety of protein targets showed that compounds featuring a biphenyl substructure bind to a wide range of proteins with high levels of specificity.[2]
An objective of synthetic chemistry is to create new chemical space that enables novel structure–activity relationship investigations. BN/CC isosterism is emerging as a viable strategy to expand the structural diversity of compounds relevant to biomedical research and materials science.[3,4] In particular, 1,2-dihydro-1,2-azaborines (abbreviated as 1,2-azaborines from hereon) have attracted growing attention as BN isosteres of the biologically important family of aromatic compounds.[5] As an example, we recently demonstrated that BN isosteres of ethylbenzene serve as inhibitors of ethyl-benzene dehydrogenase rather than as substrates.[6,7]
In order to expand the chemical space of biaryl structures through BN/CC isosterism, new synthetic tools for the preparation of BN isosteres of biphenyls (BN biphenyls) need to be developed (Scheme 1). While the synthesis of biphenyls (including the use of various cross-coupling methods)[8] is well established, a general method to access BN biphenyls has not been explored. The boron position in the 1,2-azaborine heterocycle appears to be a straightforward point of attachment for their synthesis. However, the scope with respect to the boron position is currently limited to aryl substituents with functional groups that are compatible with organomagnesium and organolithium reagents.[9]
Scheme 1.
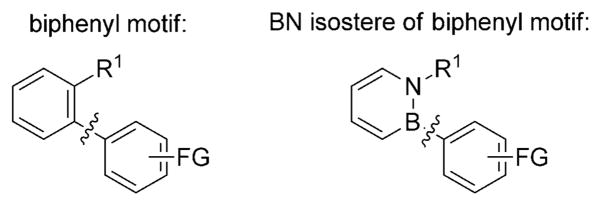
Biphenyl motif and its BN isostere. FG =functional group.
Herein, we present a general functional-group-tolerant synthesis of BN biphenyls through a rhodium-catalyzed cross-coupling reaction of B-Cl-substituted 1,2-azaborines with arylstannanes. To demonstrate the scope of our method, we prepared BN felbinac, the BN isostere of the nonsteroidal anti-inflammatory drug felbinac. Furthermore, we present mechanistic studies that support a consecutive transmetalation pathway for the catalytic coupling reaction.
We chose N-ethyl-2-chloro-1,2-azaborine (1a) and trimethyl(phenyl)tin as model reactants to optimize the coupling conditions. No background reaction occurred between 1a and trimethyl(phenyl)tin at 100°C after 24 h (Table 1, entry 1). Because of the resemblance of our model reaction to the Stille cross-coupling, we surveyed a series of Pd catalysts that are typically used in a Stille reaction,[10] but no desired product was observed (entries 2–4). Group 9 transition metals proved to be more useful as catalysts. Use of chloro(1,5-cyclooctadiene)iridium(I) dimer gave small amounts of product (entry 5). However, an improvement was observed when Wilkinson’s catalyst was employed (entry 6). We then surveyed a series of rhodium–diene complexes (entries 7–10) and found the chloro(norbornadiene)rhodium(I) dimer to be particularly active (entry 9). The observation of rhodium black aggregation and resulting catalyst deactivation from diene-ligated catalysts prompted us to use bidentate phosphine ligands to support homogeneous catalysis. After a survey of different ligands (entries 11–14), we ultimately identified BIPHEP as the optimal ligand in combination with the chlorobis(ethylene)rhodium(I) dimer complex as catalyst (entry 14).[11] Under these optimized conditions, the catalyst loading can be reduced to 2 mol% Rh metal without substantial loss in product yield (entry 15 vs. entry 14).
Table 1.
Survey of catalysts for the coupling of 1a with trimethyl-(phenyl)tin.
| ||
|---|---|---|
| Entry | Catalyst | Yield [%][a] |
| 1 | – | 0 |
| 2 | [Pd(PPh3)4] | 0 |
| 3[b] | [Pd2dba3]/PCy3 | 0 |
| 4 | [Pd2dba3]/rac-BINAP | 0 |
| 5 | [{Ir(cod)Cl}2] | <5 |
| 6 | [Rh(PPh3)3Cl] | 18 |
| 7 | [{Rh(cod)Cl}2] | 43 |
| 8 | [Rh(cod)2]BF4 | 42 |
| 9 | [{Rh(nbd)Cl}2] | 71 |
| 10 | [Rh(nbd)2]BF4 | 67 |
| 11 | [Rh(cod)(dppb)]BF4 | 22 |
| 12 | [{Rh(C2H4)2Cl}2]/rac-BINAP | 83 |
| 13 | [{Rh(C2H4)2Cl}2]/(S)-p-tol-BINAP | 91 |
| 14 | [{Rh(C2H4)2Cl}2]/BIPHEP | 95 |
| 15[c] | [{Rh(C2H4)2Cl}2]/BIPHEP | 89 |
Yields determined by GC analysis using hexadecane as a calibrated internal standard, average of two runs.
10 mol% PCy3.
2 mol% Rh/BIPHEP. BINAP =2,2′-bis(diphenylphosphino)-1,1′-binaphthyl, BIPHEP=2,2′-bis(diphenylphosphino)-1,1′-biphenyl, cod =1,5-cyclo-octadiene, Cy =cyclohexyl, dba =trans, trans-dibenzylideneacetone, dppb =1,1′-bis(diphenylphosphino)butane, nbd =2,5-norbornadiene, p-tol-BINAP =2,2′-bis(di-p-tolylphosphino)-1,1′-binaphthyl.
With optimized conditions determined, we next examined the scope of this coupling reaction. The substituent at the nitrogen atom had little effect on the yield of the isolated product (Scheme 2, 2a vs. 2b). With regard to the range of functionality that is tolerated in the arylstannanes, we found that arylstannanes with para substituents of varying electronic nature (electron-donating and electron-withdrawing) are compatible with the reaction conditions (2 c–2h).[12] However, the electron-deficient pentafluorophenylstannane gave the isolated product in only 40% yield after prolonged reaction time (2i).[13] The successful coupling of arylstannanes bearing aldehyde and ketone functionalities, which are considered relatively sensitive functional groups, is notable (2j–k).[14]
Scheme 2.
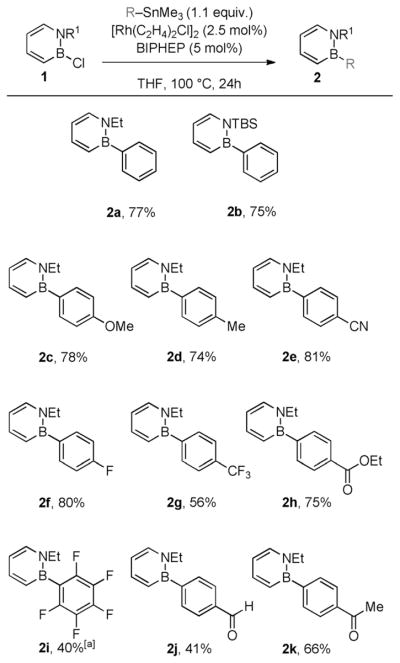
Synthesis of BN biphenyls through Rh-catalyzed addition of arylstannanes. The yields of isolated products are given as the average of two runs. [a] 48 h reaction time.
In view of the wide use of biphenyl compounds in medicinal chemistry, we sought to demonstrate the utility of our coupling method in the synthesis of a drug molecule. We identified felbinac as a suitable target for which the BN isostere can be readily accessed using our method. Felbinac is a topical nonsteroidal anti-inflammatory drug (NSAID) and is used to treat rheumatic pain and inflammation.[15] The two-step synthesis of BN felbinac is illustrated in Scheme 3. Addition of stannane 3, derived from phenylacetic acid, to N-TBS-2-chloro-1,2-azaborine (1b) furnished compound 4 in good yield (77%). We were able to unambiguously establish the connectivity of 4 by single-crystal X-ray diffraction analysis (see ORTEP representation in Scheme 3).[16] Deprotection with tetrabutylammonium fluoride (TBAF) provided the target BN felbinac (5) in 88% yield.[17]
Scheme 3.

Synthesis of BN felbinac (5). Thermal ellipsoids of 4 drawn at the 35% probability level. TBS =tert-butyldimethylsilyl, TMS =trime-thylsilyl.
To further probe the scope of our coupling reaction, we employed the BN isostere of naphthalene 6[18] as the BN-containing substrate. Gratifyingly, the reaction proceeded smoothly to furnish the B-Ph-substituted BN naphthalene 7 in 71% yield [Eq. (1)].[19] Thus, the developed coupling conditions are compatible with protic NH groups in the substrate.[20]
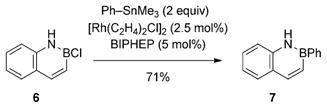 |
(1) |
Our proposed mechanism for the rhodium-catalyzed addition of arylstannanes to B-Cl-substituted 1,2-azaborines is illustrated in Scheme 4. The catalyst resting state is the chlororhodium dimer A. The presumed catalytically active chlororhodium monomer B is generated from A in the presence of a coordinating solvent. Next, transmetalation of a phenyl group from the stannane reagent to rhodium occurs to generate the phenylrhodium species C. Intermediate C subsequently reacts with the 1,2-azaborine 1 to furnish product 2 and regenerate species B.[21] We also propose that the rate-limiting step of the catalytic cycle is the second transmetalation step, which is reflected in the energy diagram shown in Scheme 4.
Scheme 4.
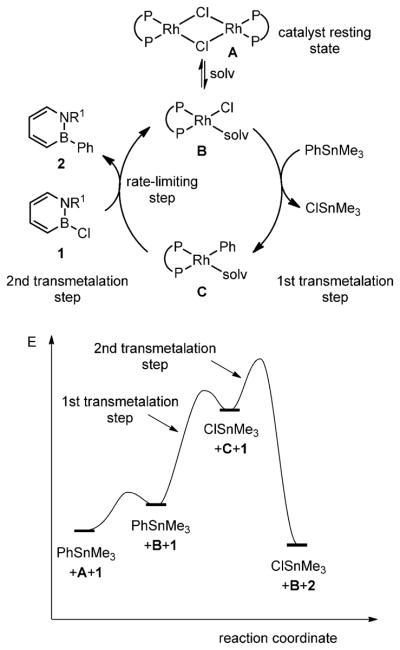
Proposed mechanism of the Rh-catalyzed addition of aryl-stannanes to B-Cl-substituted 1,2-azaborines.
The following mechanistic studies are consistent with the scenario illustrated in Scheme 4. First, we were able to isolate the bis(phosphine)-bound chlororhodium dimer [{Rh-(BIPHEP)Cl}2] and demonstrate that it is an active catalyst [Eq. (2)]. In addition, the [{Rh(BIPHEP)Cl}2] complex was the only observable phosphine species over the course of the reaction. This result is consistent with phosphine-supported Rh being the active species and with [{Rh(BIPHEP)Cl}2] being the resting state of the catalyst.
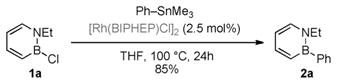 |
(2) |
Secondly, we demonstrated that an analogue (compound C′ in Figure 1)[22] of the proposed monomeric arylrhodium species C (see Scheme 4) can react with the 1,2-azaborine substrate 1a at room temperature to produce the target BN biphenyl 2a and regenerate an analogue (compound B′ in Figure 1) of the proposed monomeric chlororhodium species B. Furthermore, we established that the isolated arylrhodium species C′ is a competent catalyst, producing BN biphenyl 2a in reasonable yield at 5 mol% catalyst loading [Eq. (3)].[23]
Figure 1.

31P NMR spectra of the reaction between C′ and 1 a. P ~P =(S)-BINAP.
We also found that in the absence of 1,2-azaborine substrate 1a, trimethyl(phenyl)tin does not transmetalate the phenyl group to the rhodium catalyst. This observation suggests that C is a high-energy intermediate, and that the overall catalytic reaction is thermodynamically driven by the completion of the second transmetalation step.
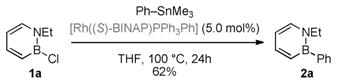 |
(3) |
We performed kinetic studies to establish the order of the reaction with respect to each reactant and the catalyst. The reactions between 1,2-azaborine 1b and trimethyl(phenyl)tin in the presence of [{Rh(BIPHEP)Cl}2] were continuously monitored in a reaction calorimeter, and the kinetic behavior was analyzed using the method of [“excess”] relationship ([“excess”] =difference in the initial concentrations of the two substrates) and graphical rate equation developed by Blackmond.[24,25] Negligible catalyst deactivation or product inhibition is supported by the results of kinetic profiles for two reactions at the same [“excess”] overlaid with one another when plotted as rate versus the concentration of [1b] (see Figure S2). First order behavior for both arylstannane and azaborine 1b is indicated by overlay in the different [“excess”] plots (see Figure S3). The reaction rate divided by the square root of the concentration of rhodium (rate/ [Rh]total0.5) is plotted versus the concentration of 1,2-azaborine 1b, and overlay for plots at two different Rh concentrations shows that the reaction is half-order in [Rh]total throughout the course of the reaction (see Figure S5).[26] Thus, the experimental observations are consistent with the proposed mechanism and energy diagram illustrated in Scheme 4.
In summary, we developed the rhodium-catalyzed addition of arylstannanes to 2-chloro-1,2-azaborines. The scope of the reaction with respect to the arylstannane is broad, including substrates with functional groups that were inaccessible through previously developed syntheses of 1,2-azaborines. We prepared a BN isostere of felbinac to demonstrate the potential of this method for biomedical research. Mechanistic investigations are consistent with an arylrhodium intermediate being responsible for the rate-limiting transfer of the aryl group to the 1,2-azaborine substrate. In view of the importance and prevalence of biphenyl compounds in medicinal chemistry, the synthetic method presented herein should enable further investigation of structure–activity relationships by expanding the chemical space of the privileged biphenyl motif.
Supplementary Material
Footnotes
This research was supported by the National Institutes of Health (National Institute of General Medical Sciences, R01-GM094541 and R41-GM099181). Funding for the University of Oregon Chemistry Research and Instrumentation Services has been provided in part by the NSF (CHE-0923589). We thank Dr. Adam Marwitz for performing the stability assessment of BN felbinac.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201304443.
Contributor Information
Gabriel E. Rudebusch, Department of Chemistry and Biochemistry, University of Oregon, Eugene, OR 97403-1253 (USA)
Dr. Lev N. Zakharov, Center for Advanced Materials Characterization in Oregon, University of Oregon, Eugene, OR 97403-1253 (USA)
Prof. Dr. Shih-Yuan Liu, Email: lsy@uoregon.edu, Department of Chemistry and Biochemistry, University of Oregon, Eugene, OR 97403-1253 (USA)
References
- 1.Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 2.Hajduk PJ, Bures M, Praestgaard J, Fesik SW. J Med Chem. 2000;43:3443–3447. doi: 10.1021/jm000164q. [DOI] [PubMed] [Google Scholar]
- 3.For an overview, see: Campbell PG, Marwitz AJV, Liu SY. Angew Chem. 2012;124:6178–6197.Angew Chem Int Ed. 2012;51:6074–6092. doi: 10.1002/anie.201200063.Bosdet MJD, Piers WE. Can J Chem. 2009;87:8–29.Liu Z, Marder TB. Angew Chem. 2008;120:248–250.Angew Chem Int Ed. 2008;47:242–244. doi: 10.1002/anie.200703535.
- 4.For leading references, see: Hatakeyama T, Hashimoto S, Oba T, Nakamura M. J Am Chem Soc. 2012;134:19600–19603. doi: 10.1021/ja310372f.Hatakeyama T, Hashimoto S, Seki S, Nakamura M. J Am Chem Soc. 2011;133:18614–18617. doi: 10.1021/ja208950c.Wang X-Y, Lin H-R, Lei T, Yang D-C, Zhuang F-D, Wang J-Y, Yuan S-C. J Pei, Angew Chem. 2013;125:3199–3202. doi: 10.1002/anie.201209706.Angew Chem Int Ed. 2013;52:3117–3120. doi: 10.1002/anie.201209706.Taniguchi T, Yamaguchi S. Organometallics. 2010;29:5732–5735.Brough SA, Lamm AN, Liu SY, Bettinger HF. Angew Chem. 2012;124:11038–11041. doi: 10.1002/anie.201203546.Angew Chem Int Ed. 2012;51:10880–10883. doi: 10.1002/anie.201203546.Marwitz AJV, Lamm AN, Zakharov LN, Vasiliu M, Dixon DA, Liu SY. Chem Sci. 2012;3:825–829.Chrostowska A, Xu S, Lamm AN, Mazière A, Weber CD, Dargelos A, Baylère P, Graciaa A, Liu SY. J Am Chem Soc. 2012;134:10279–10285. doi: 10.1021/ja303595z.Marwitz AJV, McClintock SP, Zakharov LN, Liu SY. Chem Commun. 2010;46:779–781. doi: 10.1039/b919632c.Campbell PG, Abbey ER, Neiner D, Grant DJ, Dixon DA, Liu SY. J Am Chem Soc. 2010;132:18048–18050. doi: 10.1021/ja109596m.Lamm AN, Liu SY. Mol BioSyst. 2009;5:1303–1305. doi: 10.1039/b904120f.
- 5.The arene motif is commonly found in biologically active compounds, see: Roughley SD, Jordan AM. J Med Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y.
- 6.Knack DH, Marshall JL, Harlow GP, Dudzik A, Szaleniec M, Liu SY, Heider J. Angew Chem. 2013;125:2660–2662. doi: 10.1002/anie.201208351. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:2599–2601. doi: 10.1002/anie.201208351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For other examples related to biomedical research, see: Liu L, Marwitz AJV, Matthews BW, Liu SY. Angew Chem. 2009;121:6949–6951.Angew Chem Int Ed. 2009;48:6817–6819. doi: 10.1002/anie.200903390.Abbey ER, Zakharov LN, Liu SY. J Am Chem Soc. 2010;132:16340–16342. doi: 10.1021/ja107312u.Abbey ER, Zakharov LN, Liu SY. J Am Chem Soc. 2011;133:11508–11511. doi: 10.1021/ja205779b.
- 8.For leading references, see: Suzuki A. Angew Chem. 2011;123:6854–6869.Angew Chem Int Ed. 2011;50:6722–6737. doi: 10.1002/anie.201101379.Negishi E. Angew Chem. 2011;123:6870–6897.Angew Chem Int Ed. 2011;50:6738–6764. doi: 10.1002/anie.201101380.Littke AF, Fu GC. Angew Chem. 2002;114:4350–4386.Angew Chem Int Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U.
- 9.a) Marwitz AJV, Abbey ER, Jenkins JT, Zakharov LN, Liu SY. Org Lett. 2007;9:4905–4908. doi: 10.1021/ol702383u. [DOI] [PubMed] [Google Scholar]; b) Lamm AN, Garner EB, Dixon DA, Liu SY. Angew Chem. 2011;123:8307–8310. [Google Scholar]; Angew Chem Int Ed. 2011;50:8157–8160. doi: 10.1002/anie.201103192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Littke AF, Schwarz L, Fu GC. J Am Chem Soc. 2002;124:6343–6348. doi: 10.1021/ja020012f. [DOI] [PubMed] [Google Scholar]; b) Milstein D, Stille JK. J Am Chem Soc. 1979;101:4992–4998. [Google Scholar]
- 11.For examples of the use of BIPHEP in Rh-catalyzed reactions, see: Mikami K, Kataoka S, Yusa Y, Aikawa K. Org Lett. 2004;6:3699–3701. doi: 10.1021/ol048604l.Kong JR, Krische MJ. J Am Chem Soc. 2006;128:16040–16041. doi: 10.1021/ja0664786.Faller JW, Wilt JC. J Organomet Chem. 2006;691:2207–2212.Shibata U, Otake Y, Hirano M, Tanaka K. Org Lett. 2009;11:689–692. doi: 10.1021/ol802767s.
- 12.Arylstannanes that bear substituents in ortho position are not suitable coupling partners under our conditions.
- 13.Unreacted stannane and 1,2-azaborine starting materials were observed after 48 h of reaction time.
- 14.The diminished yield of 2j may be due to the rhodium-catalyzed addition of arylstannanes to aldehydes. See: Oi S, Moro M, Inoue Y. Chem Commun. 1997:1621–1622.
- 15.Walsh DA, Shamblee DA, Welstead WJ, Sancilio LF. J Med Chem. 1982;25:446–451. doi: 10.1021/jm00346a022. [DOI] [PubMed] [Google Scholar]
- 16.Crystallographic data for 4: C23H38BNO2Si2; Mr = 427.53; crystal size 0.27× 0.23 × 0.19 mm3, monoclinic, space group P21/n, a = 7.5590(4), b =28.1887(16), c =11.9973(7) Å, β =91.410(1)°, V= 2555.6(2) Å3, Z =4, ρcalc = 1.111 gcm−3, μ =0.156 mm−1, F- (000) =928, T=100(2) K, 2Θmax = 50.00°, 24454 reflections measured [Rint = 0.0368], 4488 reflections observed, 414 refined parameters, R1 =0.0519, wR2 =0.1148, and GOF =1.072 for reflections with I >2σ(I), R1 =0.0529, wR2 =0.1155, and GOF =1.072 for all data, max/min residual electron density +0.380/−0.235 eÅ−3. CCDC 938621 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 17.BN felbinac is a solid that does not show appreciable decomposition when stored as a solid in air under ambient conditions.
- 18.Dewar MJS, Dietz R. J Chem Soc. 1959:2728–2730. [Google Scholar]
- 19.This yield is the average of two runs. See the Supporting Information for details.
- 20.Low yields of 7 were observed with 1.1 equivalents of trimethyl-(phenyl)tin. This could be due to an increased sensitivity toward water impurities.
- 21.This mechanism is in analogy with the arylation of imines and aldehydes. See: Hayashi T, Ishigedani M. J Am Chem Soc. 2000;122:976–977.b) reference [14].
- 22.Hayashi T, Takahashi M, Takaya Y, Ogasawara M. J Am Chem Soc. 2002;124:5052–5058. doi: 10.1021/ja012711i. [DOI] [PubMed] [Google Scholar]
- 23.The lower yield in comparison with the Rh/BIPHEP catalyzed reaction is consistent with the relatively inferior performance of the BINAP ligand (see Table 1, entry 12 vs. entry 14).
- 24.For a review on reaction progress kinetic analysis, see: Blackmond DG. Angew Chem. 2005;117:4374–4393.Angew Chem Int Ed. 2005;44:4302–4320. doi: 10.1002/anie.200462544.
- 25.For examples of the utility of reaction progress kinetic analysis in mechanism elucidation, see: Kina A, Iwamura H, Hayashi T. J Am Chem Soc. 2006;128:3904–3905. doi: 10.1021/ja060225v.Mathew JS, Klussmann M, Iwamura H, Valera F, Futran A, Emanuelsson EA, Blackmond DG. J Org Chem. 2006;71:4711–4722. doi: 10.1021/jo052409i.
- 26.This observation further supports our hypothesis (based on 31P NMR spectroscopy) that [{Rh(BIPHEP)Cl}2] is the resting state of the catalyst.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.