

Thousands of copies of mitochondrial DNA are present in each cell, and they have a very high mutation rate. Heteroplasmic mutations can accumulate and play important roles in aging, cancer, and metabolic diseases.
Abstract
The unorthodox genetics of the mtDNA is providing new perspectives on the etiology of the common “complex” diseases. The maternally inherited mtDNA codes for essential energy genes, is present in thousands of copies per cell, and has a very high mutation rate. New mtDNA mutations arise among thousands of other mtDNAs. The mechanisms by which these “heteroplasmic” mtDNA mutations come to predominate in the female germline and somatic tissues is poorly understood, but essential for understanding the clinical variability of a range of diseases. Maternal inheritance and heteroplasmy also pose major challengers for the diagnosis and prevention of mtDNA disease.
THE GENETIC CHALLENGES OF mtDNA DISEASES
It is has become increasingly clear that mitochondrial dysfunction lies at the nexus of a wide range of metabolic and degenerative diseases, cancer, and aging. Two major reasons for why mitochondrial dysfunction has been overlooked in “complex” diseases is that subtle bioenergetic alterations can have major clinical consequences and mitochondrial defects can be generated by the unique quantitative genetics of the maternally inherited mitochondrial DNA (mtDNA).
The mitochondrial genome encompasses between 1000 to 2000 nuclear DNA (nDNA) genes plus thousands of copies of the maternally inherited mtDNA. The mtDNA codes for the most important bioenergetic genes. So mtDNA defects impinge on a wide spectrum of cellular functions.
A large number of pathogenic mtDNA mutations have been identified and the more severe mutations are frequently mixed with normal mtDNAs within the cell, a state known as heteroplasmy. Heteroplasmic alleles can shift in percentage during both mitotic and meiotic cell division, leading to a potentially continuous array of bioenergetic defects, a process known as replicative segregation. As the percentage of mutant mtDNAs increases, the resulting bioenergetic defect becomes increasingly severe. Because different tissues have different bioenergetic thresholds, as a patient's bioenergetic capacity declines it eventually falls below the minimum threshold for that tissue and symptoms ensue. Because the tissues and organs with the highest bioenergetic requirements are also those that are primarily affected in the common metabolic and degenerative diseases, it follows that mitochondrial dysfunction may be a major contributor to complex diseases.
Women that harbor deleterious heteroplasmic mutations have a high probability of having affected children, the nature and severity of the phenotype depending on the mtDNA mutation and the percentage of heteroplasmy. Cells and individuals can accumulate an array of different mtDNA mutations over time, the aggregate of which degrade the energetic capacity of the cell. Such mutations are important in aging and cancer. Given the enormous potential explanatory power of heteroplasmic mtDNA mutations, it is striking that very little is known about the origin, genetics, and phenotypic effects of heteroplasmic mtDNA mutations.
HUMAN mtDNA GENETICS
That mtDNA mutations could cause disease was first reported at the molecular level in 1988 with the demonstration that isolated patients with mitochondrial myopathy could harbor heteroplasmic mtDNA deletions (Holt et al. 1988); that the maternally inherited sudden onset blindness disease, Leber hereditary optic neuropathy (LHON), was caused by a homoplasmic missense mutation in the ND4 gene at nt 11778G>A (arginine codon 340 to histidine, R340H) (Wallace et al. 1988a); and that myoclonic epilepsy and ragged red fiber disease (MERRF) was caused by a heteroplasmic mutation in the tRNALys gene at nt 8344A>G (Wallace et al. 1988b; Shoffner et al. 1990). These discoveries set the stage for investigating and understanding a broad range of enigmatic familial and age-related diseases.
Incidence of mtDNA Mutations and Disease
Mutations in mtDNA are surprisingly common. Genetic epidemiological studies quantifying only the most common pathogenic mtDNA mutations have estimated that the incidence of clinical mitochondrial diseases is about one in 5000 (Schaefer et al. 2004, 2008). More surprising, a survey of newborn cord bloods revealed that one in 200 infants harbored one of 10 common pathogenic mtDNA mutations (Elliott et al. 2008; Chinnery et al. 2012). Hence, pathogenic mtDNA mutations are very common and constantly arising.
Human OXPHOS and the Range of Phenotypes: Conception to Old Age
To understand the clinical implications of mtDNA mutations, it is essential to understand the central role that mitochondrial oxidative phosphorylation (OXPHOS) plays in cellular biology. The mitochondria oxidize the calories in our diet with the oxygen that we breathe to generate ∼ 90% of cellular energy. In OXPHOS, electrons (reducing equivalents) derived from our food flow down the mitochondrial inner membrane electron transport chain (ETC) from reduced to oxidized states, ultimately terminating with reduction of oxygen to water. The ETC is initiated with oxidation of NADH by complex I (NADH:CoQ oxidoreductase or NADH dehydrogenase) or succinate by complex II (succinate:CoQ oxidoreductase or succinate dehydrogenase). The electrons are then transferred to coenzyme Q (CoQ), complex III, cytochrome c, complex IV (cytochrome c oxidase or COX), and finally to oxygen. As the electrons traverse complexes I, III, and IV, the energy released is used to pump protons from the mitochondrial matrix across the mitochondrial inner membrane to the intermembrane space (Wallace 2005, 2007, 2011). This creates a transmembrane electrochemical gradient of ∼0.2 volts. This potential energy can then be used to drive OXPHOS complex V (H+-translocating ATP synthase) to condense ADP and phosphate (Pi) to generate ATP (Mitchell 1961), thus coupling oxidation by the ETC with phosphorylation by the ATP synthase. The mitochondrial ATP is then exported to the cytosol via the adenine nucleotide translocators (ANTs), where the ATP energizes cellular reactions and drives work.
In addition to generating ATP energy, the mitochondria regulate cytosolic Ca2+ levels, which in turn modulate cellular and mitochondrial metabolic pathways, control the cellular REDOX state that regulates a wide array of cellular enzymatic reactions and transcription factors via thiol-disulfide interconversion, regulate mitochondrial ROS production that is both a signal transduction agent that impinges on molecules such as HIF and RAS, and is the major source of oxidative stress that can activate the innate immunity response through NF-κB signaling. When mitochondria experience extreme stress (elevated Ca2+ and ROS, depleted adenine nucleotides, and reduced membrane potential), this can activate the mitochondrial permeability transition pore (mtPTP) thus initiating apoptosis and necrosis (Wallace 2005, 2011, 2012, 2013a,b; Wallace et al. 2013).
Mitochondrial Genetics
The mtDNA
The mtDNA (Fig. 1) codes for the 13 most important OXPHOS polypeptides. These include seven of the ∼45 polypeptides of OXPHOS complex I (ND1-3, ND4L, ND4-6): one of the 11 polypeptides of complex III (cytochrome b, cytb), three of the 13 polypeptides of complex IV (COI-III), and two of the ∼15 polypeptides of complex V (ATP6 and 8). In addition, the mtDNA encodes the mitochondrial 16S and 18S rRNAs and 22 tRNAs for mitochondrial protein synthesis. The mtDNA also encompasses an ∼1000 nt control region that contains an origin for replication of the G-rich heavy (H) stand and the promoters for transcription of both the H stand and the C-rich light (L) stand. Both mtDNA stands are transcribed into large polycistronic transcripts in which the larger rRNA and mRNA transcripts are punctuated by tRNAs. The tRNAs are processed out and the larger RNA products are polyadenylated. The mtDNA mRNAs are translated on mitochondrial-specific 55S ribosomes, which are sensitive to bacterial ribosomal initiators chloramphenicol (CAP) and aminoglycosides and are initiated with an N-formylmethionine just like bacterial protein synthesis (Wallace 2007).
Figure 1.

Human mitochondrial DNA map showing representative pathogenic and adaptive base substitution mutations. CR (control region) = D-loop. The letters around the outside perimeter or on the inside circle indicate cognate amino acids of the tRNA genes. Other gene symbols are defined in the text. Arrows followed by continental names and associated letters on the inside of the circle indicate the position of defining polymorphisms of selected region-specific mtDNA lineages. Arrows associated with abbreviations followed by numbers around the outside of the circle indicate representative pathogenic mutations, the number being the nucleotide position of the mutation. The full array of pathogenic mtDNA mutations and polymorphisms are available through Mitomap.org (MITOMAP 2012). DEAF, deafness; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; LHON, Leber hereditary optic neuropathy; ADPD, Alzheimer's disease and Parkinson's disease; MERRF, myoclonic epilepsy and ragged red fiber disease; NARP, neurogenic muscle weakness, ataxia, retinitis pigmentosum; LDYS, LHON + dystonia; PC, prostate cancer. (From Wallace 2007; reproduced, with permission, from the author.)
mtDNA Mutations
The mtDNA genes have a very high sequence evolution rate, on the order of 10–20 times that of comparable nDNA genes (Brown et al. 1982; Neckelmann et al. 1987; Wallace et al. 1987). This is the product of both an exceptionally high mutation rate, perhaps 100- to 1000-fold higher than nDNA genes, times an mtDNA mutant fixation rate: E = µF where E is the sequence evolution rate, µ the mutation rate, and F the fixation rate. When a new mutation arises in the mtDNA, it creates an intracellular heteroplasmic mixture of mutant and normal mtDNAs, but the mutant mtDNA is but one among thousands of nonmutant mtDNAs. In some manner, the initial mutant mtDNA becomes enriched within certain cells, ultimately coming to predominate and influence the cellular and patient phenotype. The mechanism by which this enrichment occurs in either germline or somatic cells remains a mystery.
Once an mtDNA mutation reaches an appreciable level within cells, the percentage of mutant mtDNAs can drift by replicative segregation. For an embryo generated by the fertilization of a heteroplasmic oocyte, the percentage of mutant and normal mtDNAs in different descendant tissues and organs can have quite different values. This genetic mosaicism results in bioenergetic mosaicism and phenotypic complexity. Added to the stochastic segregation of heteroplasmic mtDNA is the differential sensitivity of different organs to different mitochondrial physiological alterations. The brain is the most sensitive to partial bioenergetic defects followed by heart, muscle, kidney, and endocrine systems (Wallace 2005). Hence, subtle systemic mitochondrial deficiencies can result in organ-specific symptoms.
There are three classes of clinically relevant mtDNA variants: recent deleterious mutations, ancient adaptive mtDNA mutations, and somatic mtDNA mutations.
Maternally Inherited Diseases
The most clinically overt class of mtDNA variants is newly arising maternally inherited disease mutations. Because of the high mtDNA mutation rate, new pathogenic mtDNA mutations are continuously being introduced into the human population. Hundreds of pathogenic mtDNA mutations have now been documented (MITOMAP 2012; Wallace et al. 2013) and these can affect virtually every tissue in the body, depending on the mutation's severity, nature, and heteroplasmy level. Hence, pathogenic mtDNA mutations and mitochondrial dysfunction result in a wide range of multisystem degenerative diseases.
Ancient Adaptive Polymorphisms
There is also substantial mtDNA sequence diversity between individuals and human populations. These ancient mtDNA polymorphisms accumulated along radiating maternal lineages as women migrated out of Africa to colonize the globe. As new mtDNA mutations arose, new branches of the mtDNA tree were generated. If a founder mutation changed mitochondrial physiology in a manner beneficial to individuals within that regional environment, then that mtDNA lineage became enriched in that geographic locality. Subsequent additional base substitutions in descendant mtDNAs generated a group of related regional haplotypes designated a haplogroup. Hence, each continent and geographical region is associated with a distinctive array of mtDNA sequence types.
All African mtDNAs are related and encompassed within one large continent-specific lineage designated “macrohaplogroup L.” Macrohaplogroup L arose between 130,000 and 200,000 years before present (YBP) (Fig. 2), founded by mtDNAs similar to haplogroup L0, which is common among the Khoi-San Bushmen of South Africa. In Ethiopia, ∼65,000 YBP African haplogroup L3 gave rise to two mtDNAs designated M and N. Only the mtDNA descendants from M and N mtDNAs left Africa to colonize the rest of the world, generating macrohaplogroups M and N. From Africa, macrohaplogroup M moved along tropical Southeast Asia, ultimately reaching Australia. Later, M descendants moved north out of Southeast Asia to form a plethora of central and eastern Asian mtDNA haplogroups including C, D, G, and M1–M20. Out of Africa, macrohaplogroup N went in two directions. In one, N moved through Southeast Asia to Australia and from southern Asia, north into central Asia to generate haplogroups A and Z. In the second, macrohaplogroup N moved north out of Africa to form the European haplogroups I, X, and W. In western Eurasia, N also gave rise to submacrohaplogroup R. R then gave rise to the remaining European haplogroups H, J, Uk, T, U, and V. R also moved east to produce the Asian mtDNA haplogroups B and F (Fig. 3).
Figure 2.

Diagram of the migratory history of the human mtDNA haplogroups. Homo sapiens mtDNAs arose in Africa ∼130,000–200,000 years before present (YBP), with the first African-specific haplogroup branch being L0, followed by the appearance in Africa of lineages L1, L2, and L3. In northeastern Africa, L3 gave rise to two new lineages, M and N. Only M and N mtDNAs successfully left Africa ∼65,000 YBP and colonized all of Eurasia and the Americas. In Eurasia and the Americas, M and N gave rise to a diverse array of mtDNA lineages designated macrohaplogroups M and N. The founders of macrohaplogroup M moved out of Africa through India and along the Southeast Asian coast down along the Malaysian peninsula and into Australia, generating haplogroups Q and M42 ∼48,000 YBP. Subsequently, M moved north out of Southeast Asia to produce a diverse array of Central Asian mtDNA lineages including haplogroups C, D, G, and many other M haplogroup lineages. In northeast Asia, haplogroup C gave rise to haplogroup Z. The founders of macrohaplogroup N also moved through Southeast Asia and into Australia, generating haplogroup S. In Asia, macrohaplogroup N mtDNAs also moved north to generate central Asian haplogroup A and Siberian haplogroup Y. In western Eurasia, macrohaplogroup N founders also moved north to spawn European haplogroups I, W, and X, and in western Eurasia, gave rise to submacrohaplogroup R. R moved west to produce the European haplogroups H, J, Uk, T, U, and V and also moved east to generate Australian haplogroup P and eastern Asian haplogroups F and B. By 20,000 YBP, mtDNA haplogroups C and D from M, and A from N, were enriched in northeastern Siberia and thus were positioned to migrate across the Bering land bridge (Beringia) to give rise to the first Native American populations, the Paleo-Indians. Haplogroups A, C, and D migrated throughout North America and on through Central America to radiate into South America. Haplogroup X, which is most prevalent in Europe but is also found in Mongolia though not in Siberia, arrived in North America ∼15,000 YBP but remained in northern North America. Haplogroup B, which is not found in Siberia but is prevalent along the coast of Asia, arrived in North America ∼12,000–15,000 YBP and moved through North and Central America and into South America, combining with A, C, D, and X to generate the five dominant Paleo-Indian haplogroups (A, B, C, D, X). A subsequent migration of haplogroup A out of the Chukotka peninsula ∼7000–9000 YBP gave rise to the Na-Déné (Athabaskins, Navajo, Apache, etc.). Subsequent movement across the Bering Strait, primarily carrying haplogroups A and D after 6000 YBP, produced the Eskimo and Aleut populations. Most recently, eastern Asian haplogroup B migrated south along the Asian coast through Micronesia and out into the Pacific to colonize all of the Pacific islands. Ages of migrations are approximated using mtDNA sequence evolution rates determined by comparing regional archeological or physical anthropological data with corresponding mtDNA sequence diversity. Because selection may have limited the accumulation of diversity in certain contexts, ages for regional migrations were estimated from the diversity encompassed within an individual regional or continental haplogroup lineages. This is because selection would have acted on the haplogroup mtDNA but most subsequent mutations would accumulate by random genetic drift and thus be “clock-like.” (From Wallace 2013a,b; reproduced, with permission, from the author.)
Figure 3.

(A) Phylogeny of the haplogroups J and T demonstrating that each new branch of the mtDNA phylogeny is founded by a functionally significant polypeptide variant that is subsequently transmitted to all downstream descendants. Key internal replacement mutations are designated by the gene name and the nucleotide substitution. (B) The table provides function information and interspecific sequence conservation (conservation index = CI) for selected polymorphic amino acid sites. (From Ruiz-Pesini et al. 2004; reproduced, with permission, from the author and the American Association for the Advancement of Science © 2004.)
Of all the Asian mtDNA variants, only A, C, and D became enriched in northeastern Siberia and were in a position to cross the Bering Land Bridge ∼20,000 YBP to establish the Paleo-Indian populations. Later additional migrations brought haplogroups B and X to join haplogroups A, C, and D (Fig. 2) (Wallace et al. 1999, 2013).
The regionality of the mtDNA haplogroups is extraordinary in several respects. First, of all of the African diversity, only two mtDNA lineages (M and N) colonized the rest of the world. Second, of all of the Asian mtDNAs, only three mtDNA lineages (A, C, and D) moved to extreme northeast Siberia to found the Paleo-Indians. Third, and most surprising, the mtDNA sequence evolution rate is such that it produced important mtDNA evolutionary changes that coincide with the major human geographic migrations. Such associations could not have occurred by chance. Rather, it is most likely that mtDNA variation permitted adaptation of our human ancestors to different regional environments, thus being the adaptive system that permitted human colonization of the diverse environments that they encountered around the globe (Ruiz-Pesini et al. 2004; Mishmar et al. 2006; Ruiz-Pesini and Wallace 2006; Wallace 2013a).
The founding mtDNA for macrohaplogroup N, which moved directly from subtropical Africa north into the European temperate zone, harbored two polypeptide variants, ND3 nt 10398G>A (A114T) and ATP6 nt 8701G>A (A59T) (Wallace et al. 1999, 2013). These variants have been associated with alterations in the mitochondrial membrane potential and Ca2+ metabolism (Kazuno et al. 2006). Presumably, these variants reduced the coupling efficiency of mitochondrial OXPHOS (loose coupling), resulting in an increase in the number of calories burned by the mitochondria to generate the ATP required to perform work. Because a calorie is a unit of heat, burning more calories would increase core body-heat production, rendering these individuals more resistant to the cold stress encountered in more northern environments. By contrast, macrohaplogroup M mtDNAs, which initially remained in the tropics, did not acquire comparable functional mtDNA mutations. Presumably, this mtDNA lineage retained the tight coupling of OXPHOS found in Africa in which ATP production is maximized and heat production is minimized (Ruiz-Pesini et al. 2004; Mishmar et al. 2006; Ruiz-Pesini and Wallace 2006; Wallace 2013a). Consistent with the concept that mtDNA variation has permitted climatic adaptation, mtDNA variation but not nDNA variation has been found to correlate with climatic differences (Balloux et al. 2009). Also, the basal metabolic rate of Siberian populations is higher than that of more southern populations (Leonard et al. 2002; Snodgrass et al. 2005, 2008).
The portion of the European mtDNA tree encompassing haplogroups J and T provides one example of the importance of functional variants in founding and defining the branches of the mtDNA haplogroup trees (Fig. 3). The J-T lineages were founded by two polypeptide gene amino acid substitution variants, ND1 nt 4216T>C (Y304H) and cytb nt 15452C>A (L236I). The lineages then subdivided into the T and J haplogroups, the T haplogroup being founded by an ND2 amino acid substitution at nt 4917A>G (N150D) and haplogroup J founded by the reversion of the out-of-Africa ND3 10398A>G (T114A) variant and the acquisition of a new ND5 variant at nt 13708G>A (A458T). Haplogroup J then subdivided into two lineages: J1 founded by a 16S rRNA 3010G>A variant followed by cytb variant at nt 14798T>C (F18 L) and J2 founded by a cytb variant at nt 15257G>A (D171N). Each of the founding polypeptide substitutions changes an evolutionarily highly conserved amino acid. The haplogroup T nt 4917 variant changes an amino acid that has been conserved from the first metazoans to man, having an interspecific conservation index (CI) = 90%. The subhaplogroup J1 cytb 14798 variant changes an amino acid conserved to Caenorhabditis elegans (CI = 79%), and the J2 cytb 15257 variant alters an amino acid that is conserved down to bacteria (CI = 95%). Thus, mtDNA polymorphic variants have accumulated within our species that change amino acids highly conserved throughout evolution. This phenomenon, in which conserved amino acids among species are polymorphic within a species, is inconsistent with classical neo-Darwinian theory. However, it can be explained through mitochondrial physiology and the high mtDNA sequence evolution rate. Once a species arises and begins to expand its range, it encounters environmental variation that favors bioenergetic alterations in central OXPHOS functions. These demands are met by the high mutation rate of the energetically important genes of the mtDNA. However, when a new species arises, it likely may require a more efficient mitochondrial bioenergetic system. The high mtDNA mutation rate accommodates this by reverting the regional variants back to the more efficient and universal energy production system (Wallace 2013a).
That mtDNA variants alter mitochondrial function has been confirmed by OXPHOS analysis of European haplogroups H and Uk (Rollins et al. 2009; Gomez-Duran et al. 2010) and Asian haplogroups B and F (Ji et al. 2012).
The strict maternal inheritance of the mtDNA means that mtDNAs can never mix and thus do not recombine. Hence, mtDNA single-nucleotide variants that have accumulated throughout human history remain in total linkage disequilibrium. The significance of an mtDNA variant is strongly influenced by the preexisting mtDNA variants on which it arose. This is particularly clearly demonstrated for the mtDNA variant in ND1 at nt 3394C, which causes amino acid substitution Y30H (Ji et al. 2012). When this variant arises on macrohaplogroup N mtDNAs, it reduces mitochondrial complex I activity by 15%–28% and markedly increases the penetrance of the milder LHON mutations (Brown et al. 1995; Liang et al. 2009). However, if the mutation arises on a macrohaplogroup M mtDNA and this mtDNA resides in high altitude, then this same variant is associated with maximum complex I activity and adaptation to high altitude (Ji et al. 2012).
Consistent with their functional importance, mtDNA haplogroups have been correlated with predisposition to a wide range of metabolic and degenerative diseases, various cancers, and longevity (Khusnutdinova et al. 2008; Wallace 2008; Gomez-Duran et al. 2010; Wallace et al. 2013). For example, Asian haplogroup F, which has been correlated with predilection to diabetes and obesity (Fuku et al. 2007), is associated with a 30% lower complex I activity relative to other macrohaplogroup N mtDNAs (Ji et al. 2012). In addition to metabolic and degenerative diseases, mtDNA haplogroups have been associated with the severity of sepsis (Baudouin et al. 2005) and the outcome of ischemia, strokes (Chinnery et al. 2010), and trauma (Gomez et al. 2009; Zhang et al. 2010a; Krysko et al. 2011).
Somatic mtDNA Mutations
Finally, additional clinically relevant mtDNA mutations accumulate over time in tissues. These somatic mtDNA mutations arise in tissues as well as in stem cell lineages with age and progressively erode mitochondrial function, generating the aging clock (Wallace 2005). De novo mtDNA mutations can accumulate at anytime throughout life from the oocyte to the cells of the elderly. The earlier in development the mutation occurs, the more broadly it will be distributed. Hence, mtDNA mutations that arise in the embryonic period can be dispersed throughout the body, while those that arise in an adult organ will be tissue specific (Holt et al. 1988; Coskun et al. 2010).
nDNA
Mitochondrial diseases can also result from mutations in any one of the hundreds of nDNA-coded mitochondrial genes. Most of the >200 pathogenic nDNA mitochondrial mutations that have been reported to date are highly deleterious and result in severe childhood disease (Koopman et al. 2012; Wallace et al. 2013).
nDNA–mtDNA Interaction
Mild nDNA mitochondrial gene variants can also become clinically relevant when combined with an incompatible mtDNA. Severe encephalomyopathy associated with a complete complex I deficiency was observed in the boys of one family. Genetic analysis revealed that their disease was the result of inheriting from their mother an X-linked NDUFA1 gene mutation (G32R) that caused a 30% reduction in complex I activity. This occurred in the context of inheriting their mother's mtDNA, which harbored two additional complex I gene mutations, ND1 (M21T) and ND5 (M88T), and which also caused a 25% deficiency in complex I activity. In the mother, the NDUFA1 G32R mutation was masked by her normal X-chromosome gene. However, in her son, the hemizygous NDUFA1 G32R was unmasked and interacted directly with the mother's mtDNA ND1 (M21T) and ND5 (M88T) mtDNA variants to cause severe complex I deficiency and disease (Potluri et al. 2009). Similarly, the severity of the cardiomyopathy of members of a 13-generation Mennonite pedigree whose members are homozygous for a frameshift mutation in the heart muscle ANT1 isoform gene was found to be determined by their mtDNA haplogroup inherited from their mothers. Homozygous ANT1 mutant individuals that harbored haplogroup H mtDNAs had mild cardiomyopathy, while those who harbored haplogroup U mtDNAs presented with severe cardiomyopathy leading to heart failure (Strauss et al. 2013).
Mitochondrial Pathophysiology of Complex Diseases
The complexity of the genetics and pathophysiology of common diseases can now be reinterpreted in the context of mitochondrial bioenergetic and genetic principles (Fig. 4) (Wallace 2011, 2013b). Assuming common diseases result from partial mitochondrial deficiencies, this could then perturb an array of physiological processes including energy production, REDOX state, Ca2+ homeostasis, ROS production, anabolic and catabolic metabolic pathways, etc. (Wallace et al. 2010). Alteration in mitochondrial bioenergetics would increase mtDNA damage and mutation rate, perturb mtDNA replication and mitophagy, and lead to the accumulation of somatic mtDNA mutations. This would result in the progressive decline in mitochondrial function with age. Cells that are sufficiently energetically impaired would malfunction and ultimately undergo apoptosis and necrosis. Thus, the accumulation of somatic mtDNA mutations becomes the aging clock in individuals born with a normal mitochondrial function. For individuals born with partial mitochondrial dysfunction, the accumulation of mtDNA mutations and mitochondrial damage could account for the delayed onset and progressive course of their diseases. The stochastic nature of this process could also explain variable expressivity and/or penetrance of disease.
Figure 4.

Integrated mitochondrial paradigm to explain the genetic and phenotypic complexities of metabolic and degenerative disease, aging, and cancer. The top three arrows show the three types of variation that impact on individual mitochondrial OXPHOS robustness and hence risk for developing disease symptoms. These include nuclear DNA (nDNA) variation encompassing DNA sequence changes and epigenomic modification of gene regulation and signal transduction pathways, mitochondrial DNA (mtDNA) variation including recent deleterious mutations and ancient adaptive polymorphisms, and environmental influences encompassing the availability and demand for calories and inhibition of mitochondrial function by environmental insults. The central oval encompasses the pathophysiological basis of disease processes and the basis of disease progression. The primary defect is reduction in the energy-transformation capacity of OXPHOS. This can result in reduced energy output, increased reactive oxygen species (ROS) production, altered REDOX status, and altered calcium homeostasis. The decline in OXPHOS efficiency can, in turn, perturb mitochondrial biogenesis, increase ROS production, impair mitophagy, etc., resulting in progressive increase in mtDNA damage and somatic mutations and further decline in mitochondrial function. Once mitochondrial function falls below the bioenergetic threshold of a tissue, symptoms ensue. Continued energetic failure can initiate cell destruction by apoptosis or necrosis. The lower five arrows summarize the disease categories and the phenotypic outcomes of perturbed mitochondrial energy transformation. The bottom arrow shows the effect of the stochastic accumulation of somatic mtDNA mutations resulting in delayed onset and a progressive course of diseases and aging. The right arrow indicates clinical problems that can result from reduced energy production in the most energetic tissues: the brain, heart, muscle, and kidney. The number and severity of symptoms in these organs reflect the degree and specific nature of the mitochondrial defect. The left arrow indicates the metabolic effects of mitochondrial dysfunction, which result in the perturbation of the body's energy balance. This results in the symptoms of the metabolic syndrome. The lower right arrow indicates that mitochondrial alterations are critical for cancer initiation, promotion, and metastasis. The lower left arrow outlines the hypothesized inflammatory and autoimmune responses that may result from the chronic introduction of the mitochondria's bacteria-like DNA and N-formylmethionine proteins into the bloodstream (Wallace 2011).
Perturbation of mitochondrial bioenergetics can predispose to a wide range of “complex” diseases. Bioenergetic perturbations can result from genetic, epigenetic, and environmental factors. Alterations in nDNA-coded mitochondrial genes could impair energy metabolism by inactivating an OXPHOS polypeptide, perturbing antioxidant defenses, altering mtDNA replication and repair, or affecting mitochondrial quality control through alterations in mitochondrial fission and fusion or in mitophagy (Chen et al. 2010; Youle and van der Bliek 2012; Jokinen and Battersby 2013). Mitochondrial OXPHOS could also be perturbed by modulation of the expression of the nDNA-coded mitochondrial genes through variation in the epigenome (Wallace and Fan 2010).
Mitochondrial function could also be perturbed by mtDNA variation, either by recent deleterious mtDNA mutations or ancient adaptive mtDNA polymorphisms. Finally, mitochondrial energy production could be perturbed by the nature and availability of calories; the demands made on cellular energy for growth, maintenance, and reproduction; and the acute sensitivity of mitochondrial OXPHOS to a broad range of environmental toxins. As mitochondrial energetics declines, the organs with the highest energy requirements would be the first to show functional alterations. Even very subtle bioenergetic defects can adversely affect the central nervous system with its high mitochondrial energetic demand. Other sensitive organs include the heart, muscle, and kidney.
Alterations in the nature of available calories (carbohydrates, fats, proteins) would be differentially metabolized by individuals with different mtDNA haplogroups. Hence, mitochondrial alterations that perturb the flux of calories through the system could result in metabolic diseases such as diabetes, obesity, hypertension, and cardiovascular disease.
The mitochondria are also the most common bacterium in the human body, our bodies harboring on the order of 1017 mitochondria. Hence, damage to cells can release into the extracellular space and bloodstream mitochondrial N-formylmethionine-bearing polypeptides, mtDNA fragments, cardiolipin, and various other mitochondrial breakdown products, known as damage-associated molecular patterns (DAMPs), which can elicit an inflammatory response (Zhang et al. 2010a,b; Krysko et al. 2011; Oka et al. 2012). Because apoptosis, which destroys the mitochondria before they are released into the bloodstream, is an energy-dependent process, the chronic energy deficiency of mitochondrial disease would foster the release of mitochondrial antigens. This, in turn, would result in local inflammation in degenerative diseases and systemic inflammation in autoimmune diseases.
Finally, the growth of cancer cells is directly limited by energetics. Hence, cancer cells must acquire modifications in their mitochondrial physiology to optimize energy production to their changing environments (Wallace 2012).
SEGREGATION OF HETEROPLASMIC GERMLINE mtDNA MUTATIONS
While the causes of mitochondrial dysfunction can be genetically complex because of the interaction of the large number of nDNA and mtDNA genes and variants, the most unpredictable aspect of mitochondrial genetics is the segregation of mtDNA heteroplasmy. Heteroplasmic mutants segregate along both the female germline and in somatic tissues. Therefore, understanding the quantitative genetics of mtDNA segregation is essential if we are to understand the mitochondrial etiology of complex diseases.
Germline Segregation of mtDNA Heteroplasmy
Human mtDNA Disease Segregation
Familial Transmission of Heteroplasmic mtDNA Mutations
Maternal transmission of heteroplasmic mtDNA mutations is now well documented for both tRNA and polypeptide mutations (Wallace et al. 2013). The first example of the interaction between mtDNA heteroplasmy variation and phenotype was the report that the tRNALys nt 8344A>G (Fig. 5A) mutation that causes myoclonic epilepsy and ragged red fiber (MERRF) disease was heteroplasmic (Wallace et al. 1988b; Shoffner et al. 1990). In this initial family, the phenotypic variability ranged from severe MERRF (III-1) to mild mitochondrial myopathy and electrophysiological aberrations (II-4, III-2, 3, and 4) (Fig. 5B). The severity of the clinical phenotypes varied with the percentage of mutant mtDNAs after the individuals were stratified by age (Fig. 5C).
Figure 5.

MERRF tRNALys A8344G pedigree showing variable clinical expression in association with variable mtDNA mutant heteroplasmy modified by age. (A) Structure of tRNALys showing position of A8344G transition in TΨC loop. (B) Pedigree of proband (III-1) showing that all maternal relatives had some clinical manifestations (filled symbols), though highly variable, while the three paternal sons were symptom free. VER, visual evoked response; EEG, electroencephalograph; Mito. myop., mitochondrial myopathy with ragged red fibers and abnormal mitochondria; deafness, sensory neural hearing loss; ME, myoclonic epilepsy; dementia, progressive cognitive decline; hypovent, hypoventilation. (C) Pedigree showing variable proportions of mutant-type (mt) and wild-type (wt) mtDNA along the maternal pedigree. A 183-nt PCR fragment was digested with CviJI. The wild-type (8344A) gave an 88-nt uncut fragment, whereas the mutant (8344G) created a new site cutting the 88-nt fragment into 48 and 40 nt fragments. “CL” is a cloned mutant fragment. Cases (A), (B), and (C) are independent pedigrees. Individual (C) is the maternal aunt of proband III-1 in (A), which manifested MERRF. All of the maternal relatives of the pedigree are heteroplasmic for the mutant mtDNA and the severity of the phenotype correlated with the percentage of heteroplasmy when corrected for age. (From Wallace et al. 1988b; reproduced, with permission, from the author and from Shoffner et al. 1990; reproduced, with permission, from the author and the National Academy of Sciences © 1990.)
The effect of mtDNA mutant heteroplasmy on phenotype is even more striking with the tRNALeu(UUR) nt 3243A>G mutation associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) (Goto et al. 1990). When the heteroplasmy of this mutation is high, it can present as lethal childhood Leigh syndrome, MELAS, chronic progressive external ophthalmoplegia (CPEO), cardiomyopathy, migraines, diabetes mellitus, and deafness. In pedigrees with high heteroplasmy members, meiotic segregation of the mutant mtDNAs can result in the full range of phenotypes from asymptomatic to lethal disease (see example in Fig. 1 in Brown et al. 2001). Yet in other pedigrees, when the 3243A >G mutation is present in 10%–30% heteroplasmy, this same mutation results in only maternally inherited diabetes and deafness (see example in Fig. 1 in van den Ouweland et al. 1992).
Variability in clinical presentation as a result of heteroplasmic variation can also be observed in mtDNA polypeptide missense mutation pedigrees. The mtDNA ATP6 nt 8993T>G mutation, which causes the amino acid substitution L156R, is the most common example (Holt et al. 1990). This mutation was originally associated with the clinical designation of neurogenic muscle weakness, ataxia, and petinitus pigmentosum (NARP), but also can cause lethal childhood Leigh syndrome, olivopontocerebellar atrophy, cerebellar ataxia, and/or retinitis pigmentosa when present in ∼70% to 100% of the mtDNAs (Tatuch et al. 1992; Ortiz et al. 1993).
However, heteroplasmic variation is not the only cause of clinical variation in mtDNA disease. In Leber hereditary optic neuropathy, pedigrees that are essentially homoplasmic for one of the common causal LHON mtDNA mutations, males are three to four times more likely to manifest mid-life subacute blindness than females. Other factors also affect the onset of blindness including mtDNA haplogroup background and environmental stressors such as smoking and alcohol abuse (Brown et al. 1997, 2002; Torroni et al. 1997; Sadun et al. 2003, 2011).
Variability of mtDNA Heteroplasmy in Maternal Oocytes
One reason for the high variability observed in the mtDNA heteroplasmy levels of maternal relatives is variation in the percentage of mutant mtDNAs in the oocytes of heteroplasmic women. Variability in oocyte heteroplasmy has been most intensively investigated in women harboring the tRNALeu(UUR) 3243A>G and ATP6 8993T>G mutations. A 41-year-old mother who harbored the 3243A>G mutation in 18.1% of her quadriceps muscle and 7.2% of her leukocyte mtDNAs had two sons, one of whom was found at 15 years of age to harbor 11.7% 3243A>G mutant in his blood cells. The woman underwent a hysterectomy for endometriosis and 82 oocytes were recovered and analyzed for their 3243A>G mutation levels, which ranged from 0% to 45% with a mean heteroplasmy of 12.6% and a median of 8.2%. Eight of the oocytes (9.8%) lacked detectable mutant mtDNA and ∼35 of the oocytes had heteroplasmy levels ranging from 1% to 45%. To model the distribution of oocyte genotypes, the researchers assumed that the distribution of oocyte heteroplasmy levels would approximate a binomial distribution, with the initial maternal mtDNA mutant allele frequency (p0) being represented by the mean of the allele frequencies of the oocytes. Furthermore, it was hypothesized that the extent of the variation in mtDNA heteroplasmy levels was determined by a “bottleneck.” This bottleneck was envisioned to reduce the number of mtDNA segregating units “N” within the maternal germline over “g” germline cell divisions, to be followed by expansion of the mtDNA copy number back to an infinite size. The variance (V) was calculated by the formula
From the mean oocyte heteroplasmy level of 12.6%, p0 = 0.126, and with a variance of V = 0.0143, the authors estimated that the number of segregating units of the bottleneck (N) would be 173 if maintained over 24 germline cell divisions or eight if the bottleneck lasted for one cell generation (Brown et al. 2001).
Studies on the germline segregation of heteroplasmic 3243A>G mutant mtDNAs was extended to 38 preimplantation embryos from two women. The oral mucosa heteroplasmy of one woman was 30% and the mean heteroplasmy level of her embryos was 30% ± 15%. The mucosal heteroplasmy of the second woman was 27% and the mean of her embryos was 32% ± 23%. Six of the embryos had no detectable mutant <2%. Among the 35 embryos from the 27% heteroplasmy woman, 83% of the embryos harbored the 3243A>G mtDNA, with a heteroplasmy range of 5% to 77%, but none of her embryos were pure mutant.
Germline transmission of the heteroplasmy mtDNA ATP6 8993T>G gene missense mutation has also been found to result in oocytes with widely different hetroplasmy levels. In one case, an asymptomatic mother with 50% blood mutant mtDNAs had three boys; one died of Leigh syndrome with 98% mutant mtDNAs in muscle and fibroblasts, one died of sudden infant death syndrome (SIDS) with 92% mutant in blood, and one was affected with Leigh syndrome and harbored 87% mutant in blood. The mother was superovulated and seven oocytes could be recovered and genotyped. One of the oocytes had no detectable mutant mtDNA, whereas the remaining six oocytes had >95% mutant (Blok et al. 1997).
Low-Level Maternal Germline Heteroplasmy
While the maternal transmission of biallelic heteroplasmy of mtDNA disease has been extensively studied, the advent of next-generation sequencing (NGS) is now providing the capacity to determine whether the maternal germline might also harbor a spectrum of mtDNA variants each at a very low percentage of heteroplasmy. This is possible because NGS permits sequencing a region of the human mtDNA from a human sample more than a thousand times revealing rare variants. When the mtDNA sequence of the control region hypervariable region 2 (MT-HV2 from nt 162–455) and the COIII region (MT-CO3 from nt 9307–9591) were analyzed using the Roche 454 sequencing platform from blood and skeletal muscle samples of seven subjects, every subject was reported to harbor heteroplasmic variants in one or more bases at >0.2% heteroplasmy in both tissues. Overall, the number of variants per base position was greater in skeletal muscle than in blood and also was greater in MT-HV2 than in MT-CO3. Heteroplasmy levels >2% were also observed, but only in the muscle of three subjects within the MT-HV2; all other heteroplasmy levels were low (Payne et al. 2013).
Patients with mtDNA polymerase γ mutations, which decreased the fidelity of mtDNA replication, had elevated MT-HV2 mutations and these increased in frequency with age. When first-degree relatives of patients with mutations in the nDNA genes required for mtDNA maintenance were analyzed, 40% of the variants in a given individual were shared with their maternal relatives, as compared to only 12% of the variants shared with unrelated individuals. Seventy-one percent of the shared variants among relatives were primarily found in skeletal muscle as opposed to 13% shared muscle variants in unrelated individuals. Therefore, according to this study, low-level heteroplasmies (0.2%–2%) are present in all individuals and a significant proportion of these can be transmitted through the maternal lineage (Payne et al. 2013).
Using the Illumina platform, quality control criteria for mtDNA sequence validation and a heteroplasmy cutoff of 2%, a much lower frequency of heteroplasmic mutations was reported for blood and mucosal mtDNAs of nine individuals from three families. Four heteroplasmic mtDNA variants were reported, with one apparent germline mutation. Of the remaining variants one was prominent and two were low-heteroplasmy variants (Goto et al. 2011).
While these studies suggest that low-heteroplasmy variants are ubiquitous and can be maternally inherited, the rise of NGS to detect very low-level heteroplasmic mutant mtDNAs may be subject to artifacts. For example, most current protocols for making NGS libraries use PCR and PCR polymerases are error prone and thus could generate spurious mutations. One effort to overcome the potential of PCR artifacts is “duplex sequencing.” This method requires that all sequence variants be confirmed by identification of the complementary nucleotide change on both DNA strands. The estimated error rate of the duplex sequencing method was estimated to be 1/109. When this approach was applied to a human brain sample, the mtDNA mutation rate was found to be 3.5 × 10−5, lower than that expected for the above studies. Still this is much higher than reported nDNA mutation rates (Schmitt et al. 2012). Given a mutant density of 3.5 × 10−5, this tissue had about one mtDNA mutation per every two mtDNA molecules. Therefore, there is considerable genetic heterogeneity within the thousands of mtDNAs within a somatic cell. The frequency and heteroplasmy levels of potentially maternally transmitted low-heteroplasmy mutations merit further examination.
Bovine
Proof that low-heteroplasmy variants can be transmitted through the maternal germline would be if mtDNAs harboring one of two variant alleles were to alternately appear in successive generations. Such mtDNA allelic switching across maternal generations has been reported for bovine lineages. A synonymous sequence variant in the URF-5 (now ND5) gene at nt 12792C>T, detected as an HaeIII restriction fragment length polymorphism, was observed to switch from one allele to the other within two maternal generations in a 1982 study (Hauswirth and Laipis 1982). This variant was then linked to four mtDNA control region variants at nts 16074, 16079, 16231, and 16250, generating four different haplotypes. These also switched among generations over an eight-generation bovine pedigree (Olivo et al. 1983). While the observed haplotypes appeared to be homoplasmic in the animals studied, three offspring from one cow were found to be heteroplasmic, suggesting that genotypic switching was occurring by the germline transmission of low-heteroplasmic genotypes (Ashley et al. 1989).
A more extensive bovine survey of mtDNA variation revealed that a control region variant that changed a G to a C at the end of a homopolymeric run of Gs switched between the G and the C allele in 13 different mother–daughter pairs (Koehler et al. 1991). These early bovine observations were the first to lead to the hypothesis that there was an mtDNA copy-number bottleneck in the mammalian female germline.
Mouse
The Shoubridge laboratory has studied maternal germline segregation of mtDNAs in heteroplasmic mice. In their system, the mtDNAs of two different mouse lineages, NZB/BinJ (NZB) and BALB/cByJ (BALB), were combined by removing a bleb of cytoplasm from a one-cell embryo and fusing it to the one-cell embryo of the other mtDNA strain. The heteroplasmic embryos were then implanted into foster mothers at the two-cell stage. In the initial study, five founder females carrying 3.1% to 7.1% of the donor mtDNA were studied. These were crossed with BALB males and the progeny analyzed. The mean mtDNA heteroplasmy levels of the offspring were found to be similar to that of the founder mother, but the heteroplasmy levels of the individual offspring varied with the highest heteroplasmy levels being 29.6% in one pup (Jenuth et al. 1996).
In the mouse, the primary oocytes are thought to be derived from 50 primordial germ cells (PGCs) located at the base of the allantois of a 7.5-d postcoitum (dpc) mouse embryo. These cells are alkaline phosphatase (ALP) positive, permitting them to be identified and isolated. The PGCs migrate to the germinal ridge where they grow and differentiate into oogonia. The oogonia then proliferate by mitosis during embryonic development. In the mouse, the oogonia undergo 15 divisions to generate ∼25,000 primary oocytes. In humans, about six to seven million primary oocytes are produced through roughly 24 cell divisions. The oogonia will either degenerate or differentiate into primary oocytes through asymmetric division, generating a primary oocyte and a daughter oogonium. By birth, most oogonia have either differentiated into primary oocytes or degenerated. The primary oocytes undergo oogenesis in which they enter meiosis and become arrested in prophase I where they remain until puberty begins and individual proto-oocytes complete differentiation, form follicles, and can be ovulated. Within the follicle, the oocyte completes the first meiotic division generating the first polar body and enters the second meiotic division where it becomes arrested at metaphase II. At fertilization, meiosis II is completed, the second polar body is extruded and the female and male pronuclei approach each other and fuse.
Based on ultrastructural analysis of mouse oogonia, the Shoubridge laboratory estimated that there were ∼40 mitochondria per oogonium and assuming five mtDNAs per mitochondrion they concluded that an oogonium contained ∼200 mtDNAs. Based on the assumption that the average number of mtDNAs per PGC and oogonium remained relatively constant throughout the PGC replication phase and on the observed distribution of oocyte and offspring NZB/BALB mtDNA heteroplasmy levels, the Shoubridge laboratory concluded that there must be ∼185 (range 76–867) mtDNAs in an oogonium. The Shoubridge laboratory concluded that the rapid segregation of the mtDNA heteroplasmy in mammals could be explained by a drastic reduction in the number of mtDNA segregation units, a bottleneck, occurring in the PGCs of the female germline. Such a reduction in mtDNA segregating units would greatly increase the rate of genetic drift, leading to rapid segregation of different mtDNA types in different female germline cells. They estimated that the number of segregating mtDNA units in PGCs was ∼200 and that the multiple cell divisions of the oogonia required to generate that large a number of primary oocytes were sufficient to account for the observed variance in heteroplasmy frequency of oocytes and offspring. They summarized: “Our study suggests that the probability of inheriting one of two mtDNA genotypes can be modeled as a binomial sampling process …” Thus, “It seems unlikely that strong positive or negative selection for pathogenic mtDNAs occurs in the oocyte in early embryogenesis …” (Jenuth et al. 1996). In short, germline heteroplasmy segregation is the product of the stochastic sampling process known as genetic drift.
Eleven years after the initial Shoubridge study reported an mtDNA PGC mtDNA bottleneck, Cao and associates published a paper reporting that the number of mtDNAs in PGCs was not as low as surmised by Shoubridge. Using ALP staining to identify PGCs in embryos between 7.5 and 13.5 dpc, Cao and associates determined by quantitative real-time polymerase chain reaction (PCR)(qRT-PCR) that the average mtDNA copy number was 1561 ± 161 (range 1350–1732), and that the minimum mtDNA copy number of the smallest PGCs was 953. They also estimated that there were ∼100 mitochondria in a single PGC. Additional germ cell mtDNA copy-number estimates included PGCs at 13.5 dpc at 3.66 × 103, primary oocytes at 1.16 × 103, and mature oocytes at 1.57 × 105 mtDNAs per cell. By contrast, quantification of the mtDNAs in the somatic cells of 7.5-dpc embryos was reported as low, ranging from 57 to 3345 mtDNAs per cell. One complexity of these assessments discovered by this research group was that staining embryo cells with ALP partially inhibited mtDNA quantification. As an alternative approach to identifying germline cells, the authors used mice that expressed the enhanced green fluorescent protein (EGFP) driven by the 18-kb Oct-4 promoter (GOF-18/GFP). Oct-4 is transcribed in germ cells at d 9.5 to 13.5 dpc. Cao and colleagues then reported that the average mtDNA copy number for PCGs isolated using GOT-18/GFF was 1408 and 1294 in two experiments versus 673 and 736 for ALP-stained cells. Hence, they corrected their ALP-stained PGC mtDNA estimates by multiplying by 1.92. Based on these results, this group concluded that there was no constriction of mtDNA content in PGCs, and thus that the rapid mtDNA germline heteroplasmy segregation was not the product of a physical bottleneck in the number of mtDNA within the PGCs (Cao et al. 2007).
This report was followed a year later by a report from the Chinnery laboratory reaffirming that the mtDNA copy number in PGCs was on the order of 203 at 7.5 dpc, but that the mtDNA copy number in older PGCs increased to 1529 mtDNAs/cell by 14.5 dpc (Cree et al. 2008). To isolate PCGs without ALP staining, this group identified PGCs by the fluorescence of GFP transcribed from the Stella (Dppa3) promoter, which is specific for PGCs. Their studies revealed that the mouse oocyte contains 2.28 × 105 mtDNAs, that the mtDNAs do not replicate until the PGCs are formed, that the median mtDNA copy number in 5.5-dpc PGCs is 203, and the mean is 451. However, by 13.5 dpc when the number of primary oocytes is ∼25,791, the median mtDNA copy number is 1529. Hence, the mtDNA copy number per cell increases from d 5.5 to 13.5. They also observed that by 14.5 dpc, female germline cells had a lower mtDNA content then male germline cells: 1376 ± 601 versus 2152 ± 951. From these observations, these authors built a mathematical model that encompassed both a severe mtDNA copy-number bottleneck in early mammalian PGCs as well as a subsequent mtDNA amplification phase, the combination of the two being able to account for the rapid germline segregation of mtDNA heteroplasmy (Cree et al. 2008).
At the end of 2008, the Shoubridge laboratory published another paper in which they quantified the mtDNA copy number in the germ cells of NZB/BALB heteroplasmic mice. In addition, they determined the proportion of the NZB and BALB mtDNAs in the germ cells at different stages. Based on the concern that ALP staining might result in spuriously low mtDNA copy numbers, this team used EGFP transcribed from the Oct4 (Pou5fl) promoter and then manually isolated the germ cells. They then quantified the mtDNA copy number in 8.5-dpc cells, observing a mean mtDNA copy number of ∼280 with a median of 145. By 10.5 dpc, they found that the mtDNA copy number had increased to a mean of ∼2800 and median of 2200. At 14.5 dpc when the PGCs have colonized the gonad, the mtDNA copy number had risen to ∼6000 per cell. Thus, these data indicate that the mtDNA copy number decreases 700-fold from oocyte to PGC, but then increases 10- to 20-fold during expansion of the PCG population and the colonization of the gonad (Wai et al. 2008). While this was consistent with the observations of Cree et al. (2008), the Shoubridge laboratory then analyzed the proportion of NZB and BALB mtDNAs during embryonic development. This led to the surprising conclusion that the proportion of NZB and BALB mtDNAs did not change markedly during the mtDNA constriction and proliferation cycle of the PGCs and the oogonia. Hence, the Shoubridge laboratory concluded that “despite the severe reduction in mtDNA copy number, the mitochondrial genetic bottleneck does not occur during embryonic oogenesis.” However, when they examined the mtDNA heteroplasmy variance in PGCs, oogonia, and primary oocytes in primordial follicles with <10,000 mtDNA per cell to that of postnatal mature ovulated oocytes and primary oocytes in secondary follicles that harbor >10,000 mtDNAs per cell, they found that the mtDNA heteroplasmy variance had increased significantly. They then hypothesized that the “… genetic bottleneck must be the result of the selective replication of a random subset of mtDNA templates during the growth and maturation of the ovarian follicles,” which must start in the primordial follicles. To identify this differential replication phase, the researchers pulse-labeled female pups with bromo-deoxyuridine (BrdU) injected at d 1 (P1) and (P4) postpartum. They then analyzed the mtDNA labeling of oocyte mtDNAs within mtDNA nucleoids, the nucleoids identified by their association with mtDNA-binding protein (Tfam), polymerase γ (PLOG), and single-strand binding protein (mt-SSB). This revealed that only a limited number of mtDNAs were replicating, replicating mtDNAs did not always correlate with Tfam reactivity, and the replicating mtDNAs were not consistently associated with the Balbiani body. The Balbiani body is an aggregate of Golgi elements surrounded by mitochondrial and endoplasmic reticulum. The Shoubridge group then concluded that the purpose of the reduced mtDNA copy number in early PGCs to ∼200 mtDNAs is to permit selection against cells with high percentages of deleterious mtDNA mutations. However, “The genetic bottleneck for neutral (and less deleterious mtDNA sequence variants that are associated with most human disease) occurs during folliculogensis in early postnatal life …” (Wai et al. 2008).
Although this Wai-Shoubridge study did not correlate the proposed differential mtDNA replication bottleneck with the Balbiani-body-associated mitochondrial cloud, comparative interspecific studies have been used to argue that the Balbiani body is important for regional mtDNA proliferation (Zhou et al. 2010).
The conclusion of the Wai-Shoubridge study that segregation did not occur during embryonic oogenesis contradicted their previous genetic-based conclusions (Jenuth et al. 1996) as well as the conclusions of the Chinnery group that a low mtDNA copy number in the early PGCs was one of two major factors in the rapid segregation of the mtDNA heteroplasmy along the female germline. In a follow-up report, the Chinnery laboratory argued that the discrepancy in the conclusions of the two Shoubridge laboratory studies was the result of the frequently extreme bias in the relative levels of the NZB versus the BALB mtDNAs, one mtDNA type always being present at a low percentage heteroplasmy. This is the product of the heteroplasmy being derived from a small bleb of cytoplasm from one oocyte fused to a much larger cytoplasm of the recipient oocyte. Because a greater variance and thus range of heteroplasmy levels can be generated from a starting mtDNA heteroplasmic ratio of 50:50 than can be generated from a starting ratio of 5:95, Chinnery argued that the variance levels observed by Shoubridge did not reflect the true extent of the heteroplasmy segregation (Samuels et al. 2010).
The question of whether or not there was a severe reduction in mtDNA copy number in PGCs was again raised by Cao and collaborators. They questioned the effectiveness of previous studies to identify true PGCs at 7.4 dpc. To increase the reliability of studying only PGCs, Cao and associates identified the PGCs using three different protein markers and also distinguished among PGCs isolated from early-bud (EB) and late-bud (LB) 7.5-d embryos. The first marker used was Blimp1, which is expressed in nascent PGCs and is PGCs specific. By introducing into mice a bacterial artificial chromosome harboring a monomeric red fluorescent protein gene (mRFP) expressed from the Blimp1 promoter (Blimp1-mRFP), they were able to isolate positive cells manually. The appropriate cells were further validated by immunohistochenical staining for Stella (PGC7) and by ALP staining. Quantification of the mtDNA copy number of the Blimp1-mRFP-positive EB cells at 7.5 dpc gave a mean value of 1396 mtDNAs/cell and LB cells at 7.5 dpc of 1479. At 13.5 dpc, female germ cells gave a mean copy number of 1747 and male germ cells of 2039. Thus, Cao again concluded that mtDNA segregation does not occur because of a low mtDNA copy number in the early PGCs. Rather, segregation must occur later in female germ cell development without reduction in mtDNA copy number, presumably because of differential replication of mtDNAs during oocyte maturation. Consistent with this conclusion, they point out that mouse models heteroplasmic for highly deleterious mtDNA mutations including an mtDNA 4696-nt deletion (Sato et al. 2007) and an ND6 frameshift mutation (Fan et al. 2008) produced pups with lower percentages of mutant mtDNAs in successive litters. If the mtDNA variance were entirely determined by segregation and/or selection early in female germline development, then all litters should be generated from oocytes with the same distribution of heteroplasmy levels. Thus, the distribution of mtDNA heteroplasmy across female litters with advancing maternal age should be the same. If the mutant mtDNAs were segregating later in oocyte maturation, then the sicker oocytes would be preferentially lost from the ovary during the female reproductive lifespan, resulting in the successive decline in mutant mtDNAs over sequential litters. Why then might the PGCs retain high mtDNA copy numbers while adjacent somatic cells have lower mtDNA levels? These authors speculate that the high PGC mtDNA copy number permits the retention of heteroplasmic mutations that can be transmitted through the female germline and permit subsequent adaptation to changing environments (Cao et al. 2009).
Clearly, there is no consensus as to the cellular and molecular mechanism of rapid germline mtDNA heteroplasmic segregation. It could result from the rapid segregation of heteroplasmic mtDNAs because of genetic drift resulting from a physical bottleneck in the number of mtDNA segregating units in the PGCs (Jenuth et al. 1996; Cree et al. 2008), a combination of an initial severe reduction in the mtDNA population size followed by further segregation during subsequent replication (Cree et al. 2008; Khrapko 2008), an aggregation of a larger number of mtDNAs into homogeneous segregating units such as multiple mtDNA containing nucleoids (Carling et al. 2011), the replication of only a small proportion of the mtDNAs in the primordial follicle cells leading to biased transmission of a few mtDNAs (Wai et al. 2008; Carling et al. 2011; Jokinen and Battersby 2013), or to some as-yet unidentified factors.
Primate
Although the progeny of a heteroplasmic female mice can have an array of heteroplasmic ratios, the segregation rate does not seem as extreme as has been observed in human pedigrees harboring the tRNALeu(UUR) nt 3243 A>G mutation or the ATP6 nt 8993T>G L126R mutations. To determine whether there are significant differences in mtDNA heteroplasmic segregation between rodents and primates, mtDNA segregation was studied in cytoplasmic mixing experiments of rhesus macaque oocytes.
The mtDNAs of macaque oocytes were mixed by karyoplast-cytoplast fusion. A karyoplast is a portion of the cytoplasm of a cell that contains the nucleus surrounded by the cellular plasma membrane. A cytoplast is a fragment of the cell that lacks the nucleus but contains most of the cytoplasm, mitochondria, and mtDNAs. In this macaque experiment, a micropipette was inserted under the zona pellucida of a metaphase stage-II embryo and 50% of the cytoplasm plus the nucleus was extracted. This karyoplast was then inserted under the zona pellucida of another oocyte from which the nucleus and half of the cytoplasm had been removed. The two cell fragments were then fused to generate a “reconstituted cell,” which was fertilized by intracytoplasmic sperm injection (ICSI).
The karyoplast and cytoplast donors were derived from Indian and Chinese origin macaques, which differed in their mtDNA control region (D-loop) sequence, permitting the fate of the two mtDNA haplotypes to be monitored through development. The mean heteroplasmy in 15 analyzed reconstituted oocytes was 54.9%± 10%. In two cell embryos, half of the embryos had ∼50% of each haplotype in each blastomere, but the other half of the embryos exhibited significant differences among the blastomeres, the most divergent case being 36% and 70%. The divergence in percentage heteroplasmy increased in four- and eight-cell embryos, the coefficient of variance increasing from 17.7% to 25.0% to 30.9% and the range increasing from 13.3% to 25.3% to 43.2% in two-, four-, and eight-cell embryos, respectively. By the eight-cell stage, some embryo blastomeres differed by as much as 10% and 80% of the Indian and Chinese mtDNAs (Lee et al. 2012).
Three embryonic stem cell (ESC) lines were derived from the inner cell mass cells of heteroplasmic blastocytes and found to harbor heteroplasmy levels of the cytoplast mtDNA of 97.9%, 93%, and 5%. From nine clones of the 93% cell line, six were homoplasmic for the cytoplast mtDNA and three were heteroplasmic in the range of 90.7% to 92.9%, with the direction of segregation being independent of the nuclear origin (Lee et al. 2012).
Reconstituted oocyte-derived embryos were implanted in females and two fetuses were analyzed for their heteroplasmy levels. The male fetus harbored 26.3% of the cytoplast mtDNA, while the female fetus harbored 93.8% of the cytoplast mtDNA, with her tissue levels ranging from 91.1% in blood to 98.4% in kidneys. Recovery of the ovaries from the female fetus and analysis of 51 primordial oocytes revealed a continuous range of cytoplast donor mtDNAs from 3.7% to 99.2%. Hence, the somatic cell heteroplasmy level of the female fetus was essentially independent of the level of heteroplasmy in her germline cells (Lee et al. 2012).
A difference in heteroplasmy segregation in somatic versus germline cells was already apparent in the epiblast cell lineages, resulting in marked asymmetric segregation of the mtDNAs into the somatic tissues, even though the female germline remained capable of generating the full range of possible heteroplasmic levels. Hence, there appears to be two mtDNA segregation systems, one for somatic tissues that tends to move rapidly toward homoplasmy and is already functioning in the early embryo and the other that is acting in the female germline and generates a diverse array of oocyte heteroplasmy levels (Lee et al. 2012).
This difference between primate germline and somatic cell lineage heteroplasmy levels is reminiscent of the observation of Cao et al. (2007, 2009) who reported that the mtDNA copy number in the somatic cells of mouse embryos was markedly lower than that of the female germline cells. Such a somatic lineage mtDNA bottleneck would be conducive to the rapid segregation of mtDNA heteroplasmy in somatic cells.
Selection in mtDNA Germline Segregation
All of the above studies demonstrate that an intrinsic feature of heteroplasmic mtDNA mutations is their “imperfect transmission” (Chapman et al. 1982), which can be greatly accentuated along the maternal lineage by bottlenecks in the mtDNA copy number that foster intracellular heteroplasmic mtDNA genetic drift (Hauswirth and Laipis 1982; Charlesworth and Charlesworth 2010). Genetic drift is driven by fluctuations in gene pool size and, as the constriction of the gene pool size increases, the segregation rate of a heterogeneous pool of genetic variants toward homogeneity also increases (Takahata and Slatkin 1983). The rapid segregation of heteroplasmic mtDNA genotypes along the female germline by an extreme “bottleneck” counters the accumulation of large numbers of mtDNA mutations over many generations, which would progressively erode energetic function and ultimately compromise the viability of the species, a process known as “Muller's ratchet.” In a sense, rapid mtDNA allelic drift acts like nuclear gene recombination in that it increases diversity while limiting the accumulation of deleterious mutations. By rapidly sorting out heteroplasmic alleles along the maternal lineage, mtDNA genetic drift permits the introduction of new mtDNA mutations into the population in the near homoplasmic state. These genotypes can then be acted on by selection with deleterious alleles being removed by purifying selection thus maintaining the integrity of the maternal germline. Nonpathogenic variants would accumulate along radiating maternal lineages at random, and rare advantageous alleles could become enriched within regional bioenergetic environments to found regional haplogroups. While severe mtDNA bottlenecks will increase the probability that a deleterious allele will be presented, resulting in genetic disease and in the rapid demise of that lineage (Bergstrom and Pritchard 1998), a severe mtDNA germline bottleneck would also increase the presentation of rare beneficial alleles, thus increasing species diversity and adaptability to changing environments. Thus, in contrast to nuclear genetics, in mtDNA genetics, genetic drift and selection are not two mutually exclusive or competing forces. Rather, they work together to inhibit the accumulation of deleterious mutations and facilitate the fixation of advantageous ones.
If we are to have a complete understanding of origin and biological importance of mtDNA genetic diversity, we need to understand the role that selection plays in shaping mtDNA genetic diversity. To date, only a few mouse models have been generated and studied that provide insight into the importance of selection in sorting out mtDNA heteroplasmic mtDNA mutations.
Mouse Germline Segregation of an mtDNA Deletion
In one mouse model system, an mtDNA deletion was introduced into the mouse female germline via somatic cell cytoplasmic hybrid (cybrid) fusions (Bunn et al. 1974; Wallace et al. 1975). A rearranged mtDNA was recovered from mouse brain by fusion of synaptosomes, neuronal synaptic bouton fragments containing mitochondria and mtDNAs, to cultured cells lacking mtDNA (ρ0) (King and Attardi 1989; Chomyn 1996). Because ρ0 cells cannot grow without uridine and pyruvate, ρ0 cells that acquire partially functional mitochondria can be selected by growth in media lacking either pyruvate or uridine. The resulting synaptosome cybrids were screened for mtDNA mutations and one clone was identified that harbored a 4696-nt mtDNA deletion that removed six tRNAs and seven structural genes.
Next, enucleated cell cytoplasts from this mtDNA 4696-nt deletion cell line were fused to single-cell mouse embryos, the embryos implanted into foster mothers, and the resulting mice found to be heteroplasmic for the deleted mtDNA. Unlike human mtDNA deletion mutations, which are not generally maternally inherited, this deletion was transmitted through the female germline (Inoue et al. 2000). The heteroplasmic mice have COX-negative fibers in their heart and muscle, renal dysfunction, and male mice with ≥70% rearranged mtDNAs are infertile (Inoue et al. 2000; Nakada et al. 2006).
In humans, it has been observed that mtDNA duplications can be maternally inherited and that these duplicated molecules generate deletions by intramolecular recombination within postmitotic tissues (Wallace and Fan 2009). Hence, it is possible that the maternal transmission of the 4696-nt mtDNA deletion is the product of maternal transmission of a duplication, although this has not been determined.
Regardless of the molecular nature of the maternally transmitted rearranged mtDNA, analysis of the level of the deleted mtDNA in pups derived from heteroplasmic female mice revealed a striking directional loss of the rearranged mtDNA in successive litters. Among five females with tail deletion levels between 20% and 60%, the mean deletion heteroplasmy of all of the pups declined precipitously. By the third litter, all of the pups had less than 10% deleted mtDNA, with many having no detectable deleted mtDNA in their tail tissue. Interestingly, in two of the females, the mean mtDNA heteroplasmy of the pups of the first litter was higher than that of the tail of the mother. The same striking decline in the percentage of deleted mtDNA was seen in the oocytes of two females that were superovulated at d 44 and 117. In both cases, the oocyte heteroplasmy level in the first superovulation oocytes was less than that of the females and the reduction of heteroplasmy in the oocytes in the subsequent superovulation was even more marked. The same reduction in mtDNA deletion levels was observed in the litters of mice in which the ovaries of heteroplasmic deletion mice were grafted into ovarectimized C57BL/6 mice harboring Mus spretus mtDNAs (Sato et al. 2007). Apparently, then, oocytes with high mtDNA deletion levels can be ovulated while the mouse is young, but, as the female mouse ages, the high deletion oocytes are progressively lost. Hence, there must be selection against the formation or ovulation of the oocytes with highest deletion levels throughout the female mouse's lifespan.
Mouse Germline Segregation of a Frameshift mtDNA Mutation
An analogous finding has been reported for a mouse line that harbored a heteroplasmic mtDNA ND6 frameshift mtDNA mutation. This mutation was the result of an insertion of a C at nt 13885 within the mtDNA (ND6 13885insC). The original mtDNA frameshift mutation was isolated in mouse L cells linked to an mtDNA homoplasmic COI T6589C missense mutation. When this cell line was enucleated and fused to a female mouse embryonic stem cell (mfESC), one of the cybrid clones was found to harbor mtDNAs that had reversed the ND6 13885insC mutation by deletion of an adjacent T, thus restoring the reading frame. Hence, this mfESC cybrid was homoplasmic for the COI T6589C missense mutation but heteroplasmic for the ND6 13885insC frameshift mutation.
Injection of this mfESC cybrid into mouse blastocysts resulted in the generation of one female mouse that was heteroplasmic for the ND6 13885insC frameshift mutation at 44% ± 3% in all of her tissues, with the percentage frameshift mutation being 47% in her tail. When this female was mated, all of the pups of her first and second litter had tail mtDNAs that were 14% frameshift, in the third litter all of the pups had 6% frameshift mtDNA, and in the fourth, fifth, and sixth litters the frameshift mtDNA was lost. When females that harbored 14% frameshift mtDNAs were mated, they produced pups with either 6% or 0% frameshift mtDNAs. When a female with 14% frameshift mtDNA was superovulated and 11 oocytes were genotyped, two oocytes were found to harbor 16% frameshift mtDNAs, one harbored 14%, one 12%, two 6%, and five 0% frameshift mtDNAs (Fan et al. 2008). Hence, there was a directional and concerted loss of the frameshift mtDNA over successive generations. Furthermore, the distribution of oocyte heteroplasmy levels was not Gaussian but was truncated such that there were oocytes with substantially less mutant mtDNA than the mother's tail genotype, but none had significantly more mutant mtDNAs than the mother. Therefore, germ cells or oocytes with a higher percentage of frameshift mtDNAs must have been selectively removed prior to ovulation. Thus, mammalian females have a prefertilization mtDNA selection that eliminates those oocytes harboring the most deleterious mtDNA mutations.
Independent evidence that a selection process exists in the female germline was obtained by analyzing the fate of mutant mtDNAs produced by the mouse polymerase γ (D257A PolgA exo−) mutator gene. This revealed that while deleterious tRNA mutations were introduced into the germline, there was a dearth of deleterious polypeptide gene mutations (Stewart et al. 2008).
Germline Segregation of Single-Base tRNA Deletion Mutations
Using the mtDNA polymerase γ (D257A PolgA exo−) mutator gene to generate germline mtDNA mutations, a mouse was isolated that was heteroplasmic for a tRNAMet nt 3875 C deletion (tRNAMet 3875 delC) mutation. The inheritance of the mtDNA harboring this mutation was monitored and found to be biased toward loss of the tRNA mutation. The tRNAMet 3875delC mutation inhibits aminoacylation, but the biochemical defect was partially ameliorated by the up-regulation of the other mtDNA tRNAs and all of the mtDNA mRNAs except for that of ND6 (Freyer et al. 2012).
Mice harboring the tRNAMet 3875delC mutation had relatively similar heteroplasmy levels across tissues and these did not change with age. Yet, even with selective breeding, the levels of tRNAMet 3875delC mutation heteroplasmy could not be increased above 86%. Hence, selection is clearly acting against this tRNA mutation at high levels of heteroplasmy. The mtDNA genotypes of 44 mothers and their 533 offspring were analyzed. When the heteroplasmy of the mothers was binned into groups of 40%–60%, 61%–70%, and >70%, the heteroplasmy levels of the pups of the 40%–60% and >70% mothers were found to have undergone nonrandom segregation. This conclusion was based on comparison of the offspring distribution of genotypes observed versus prediction made from the Kimura distribution that would be expected for a random genetic drift model. In contrast to the ND6 13885insC frameshift mutation and the nt 4696 deletion mtDNA mice in which the heteroplasmy levels of the pups of successive litters declined with age, the heteroplasmy distribution of the tRNAMet nt 3875delC mutation mtDNAs did not significantly differ among generations (Freyer et al. 2012).
Analysis of female germ cell mtDNA tRNAMet 3875delC mutation levels in 819 Stella-GFP-isolated PGCs from 18 embryos at 13.5 d revealed a wide variation in heteroplasmy levels, with some PGCs cells being 100% mutant. Thus, germline cells could be homoplasmic for the mutant even though no homoplasmic animals were generated. This indicated that early in female germ cell development there was a stochastic distribution of heteroplasmic levels, but that the highest levels of mutant mtDNAs were lost as the oogonia or oocytes matured. The authors conclude that “… the variance in heteroplasmy among offspring is determined embryonically, consistent with a prenatal germline bottleneck governing the segregation of mutated mtDNA.” Analysis of 340 oocytes from the ovaries of five 3.5-d-old neonate female mice revealed a relatively random distribution of mtDNA genotypes, consistent with a Kimura distribution prediction of random genetic drift. From this, the authors concluded that the distribution of mtDNA tRNAMet 3875delC mutation genotypes of the oocytes was not truncated by selection. However, when the mean mutation level of the pups of a heteroplasmic female were plotted against the mother's tRNAMet 3875delC heteroplasmy levels, the offspring of females with 45%–60% heteroplasmy had pups with an up to ∼14% increased mutation load, while females with 65%–83% heteroplasmy levels exhibited a progressive decline in the average tRNAMet nt 3875delC mutation levels of ∼10%. The authors concluded “That selection is likely to have occurred after the oocyte stage …,” (Freyer et al. 2012), but it is noteworthy that none of the oocytes genotyped contained the tRNAMet 3875delC mutation heteroplasmy at levels greater than ∼85%, even though such genotypes were present in the PGCs. Therefore, the descendants of the PGCs with 100% mutant mtDNA must have been eliminated prior to the formation of the mature oocytes.
For the tRNAMet 3875 delC mutation, two factors appear to be acting on mutant heteroplasmy levels. At 45%–60% mutant in the mother, there is a tendency for the mutant mtDNAs to increase in the offspring, while at heteroplasmy levels of 65%–83%, there was a tendency for their loss. Hence, the tRNAMet 3875delC mutation levels seem to be maintained in the female germline by two opposing selective forces, one enriching for the mutant mtDNA at intermediate heteroplasmy levels and the other strongly selecting against the mutant at high levels of heteroplasmy.
Haplogroup Incompatibility in Heteroplasmic Mice
To determine whether mtDNA haplogroups might also show nonrandom segregation along the mouse female germline, NZB and 129S6 mtDNAs were combined within the female mouse germline by fusing cytoplasts from a culture mouse cell line harboring NZB mtDNAs with a mfESCs derived from a 129S6 mouse. The NZB-129 heteroplasmic mfESC cybrids were injected into blastocysts to generate a heteroplasmic mouse line. NZB and 129 mtDNAs differ in 91 nucleotide positions including 15 nonsynonymous amino acid substitutions, five tRNA variants, seven rRNA variants, and 11 control region variants. The heteroplasmic female mice were backcrossed for ∼20 generations with C57BL6/J2 mice originally obtained from the Jackson Laboratory, and all subsequent females were mated with males from the same C57BL/6 J line maintained by brother/sister mating.
The C57BL6/J2 NZB-129 heteroplasmic mice were then allowed to segregate the two mtDNAs. This revealed a strong bias in segregation toward the loss of the NZB mtDNAs. Over 24 generations, 171 mice segregated to <3% NZB mtDNAs within one to two generations. Three out of seven mice that lost the NZB mtDNAs bred true as pure 129 mtDNAs. By contrast, more than 10 generations of selective breeding were required to generate 12 females with >97% NZB mtDNAs, and of three of these only one bred true for the NZB mtDNA.
In unrestricted segregation experiments, of 864 pups born to heteroplasmic mothers, 67% had a mean increase in 129 heteroplasmy, 28% had an increase in NZB heteroplasmy, and only 5% had a mean heteroplasmy similar to their mother. Moreover, the tendency to segregate NZB mtDNAs was highest when the mother's heteroplasmy level was in the 60%–80% range, with the tendency to lose the NZB mtDNAs declining when the percentage of heteroplasmy of the mother was strongly biased toward either 129 or NZB mtDNAs. “Therefore, for mice with approximately equal proportions of NZB and 129 mtDNAs, there was a strong bias to decrease NZB mtDNA among generations. The enrichment of the 129 mtDNAs was seen in the ovaries of the heteroplasmic females and to a lesser extent in the oocytes of the heteroplasmic females. The tendency to segregate the NZB mtDNAs was not simply the result of incompatibility between the C57BL/6 J nucleus and the NZB mtDNAs because the fertility and fecundity of the homoplasmic NZB mice was equal to or better than that of the 129 mice with the same nucleus” (Sharpley et al. 2012). Thus, the segregation pattern of the NZB-129 heteroplasmic mice was similar to that of the tRNAMet nt 3875delC mutation mice, although less directional or concerted than was seen for the mtDNA 4696-nt deletion and ND6 13885insC frameshift mutations.
This series of experiments unequivocally demonstrates that selection acts on mtDNA genotypes within the female germline, acting to weed out the most severe mtDNA mutations. Furthermore, the bias toward the elimination of a deleterious mtDNA is correlated with the severity of the mitochondrial defect associated with the mtDNA mutation.
Mathematical Models to Detect Selection in mtDNA Segregation
The striking inconsistencies observed among different estimates of the mtDNA copy numbers in mouse female germline cells, the variation in estimates of heteroplasmy level variances in progeny cells and offspring of different studies and model systems, and the role of selection in defining the transmission of heteroplasmic mtDNA genotypes mean that it is currently impossible to define the factors that determine the origin and inheritance of pathogenic mtDNA mutations. In an effort to more accurately compare different experiments and ultimately to better understand mtDNA mutant transmission, significant efforts have been made to model the transmission of heteroplasmic mtDNAs along the female germline.
Unfortunately, three factors have limited the investigator's ability to generalize the results from different studies: (1) development of methods for comparing differences in heteroplasmy variance among samples and experiments and to calculate the statistical significance of these differences, (2) development of appropriate mathematical models to describe the origin and transmission of mtDNA heteroplasmy, and (3) development of theory and methods to determine the role of selection in mtDNA heteroplasmy transmission.
Statistical Analysis of mtDNA Heteroplasmy Variance
A major limitation for defining the principles covering heteroplasmy transmission has been difficulty in comparing the heteroplasmy variance levels observed across different experimental systems. Difficulties arise at two levels, correcting for the effects of parental allele frequencies on the potential range of heteroplasmy variances of their descendants and difficulties in calculating the standard error of heteroplasmy variance estimates for testing the statistical significance of observed differences.
The heteroplasmy variance of offspring or derived cells is a function of the heteroplasmy level of the mothers for offspring or of the parental cells for derived cells, the parental heteroplasmy designated as p0. This fact becomes particularly important when the heteroplasmic level of one of the mtDNA types in very low, <10%. Under these conditions, the range of heteroplasmy values of the offspring or derived cells will be constrained by the frequency of the low parental allele, making the range of heteroplasmic variance levels aberrantly low. Heteroplasmic variance will then be maximum when p0 = 0.5 and minimal when p0< 0.1 (Samuels et al. 2010). To correct for this systematic distortion in variance capacity, Samuels and collaborators have used Mendelian population genetics theory (Wright 1969) to normalize the heteroplasmy variances in proportion to p0. This was accomplished by dividing the observed heteroplasmy variance by the term p0(1-p0). This term is at maximum (0.25) when p0 is 0.5, and becomes progressively smaller as p0 gets smaller. Hence, dividing the observed variance by p0(1-p0) proportionately increases population variance estimates for low values of p0. With this correction, Samuels and associates discovered that a number of conclusions about changes in heteroplasmy variance drawn from various studies involving animals with low maternal p0 could not be substantiated (Samuels et al. 2010).
Once the effects of extreme values of p0 on variance estimates were corrected, it became necessary to calculate the standard error (SE) of the variance so that the statistical significance of differences in variance among different sample determinations could be calculated. At low levels of p0, the required sample size to obtain statistical significance rapidly increases. Wonnapinij and colleagues (2010) pointed out that many experimental estimates of mtDNA heteroplasmy variance have been based on sample sizes ≤20. Their simulations indicate that these variance estimates have very large SEs. When Wonnapinij and colleagues estimated the SE of the variance for the original 1996 NZB-BALB mtDNA segregation of Jenuth and collaborators (1996), they concluded that once variance normalization was applied and variance SEs were calculated, the reported differences in heteroplasmy levels between primary and mature oocytes were not statistically significant. However, the heteroplasmy variance of the PGCs was significantly lower than that of primary and mature oocytes. This analysis supports the hypothesis that an mtDNA bottleneck occurs during the transition of PGCs to primary oocytes.
Similarly, Wonnapinij and colleagues reanalyzed the mouse mtDNA heteroplasmy variation of the Wai and associates (2008) study. Once the heteroplasmy variances were normalized and SE was estimated, Wonnapinij and colleagues concluded that the heteroplasmy variance values determined on postnatal d 11 and after relative to those of d 8 and earlier were not significantly different. Therefore, the conclusion of Wai and associates (2008) that an mtDNA heteroplasmy bottleneck occurs during postnatal folliculogenesis could not be substantiated (Wonnapinij et al. 2010). Wonnapinij and colleagues also pointed out that the mtDNA heteroplasmy variance levels in the early postnatal period differed between the Jenuth study (Jenuth et al. 1996) and Wai study (Wai et al. 2008). From these analyses, Wonnapinij and colleagues concluded that the major bottleneck responsible for mtDNA heteroplasmy variance occurs in the maternal germline between the PGCs and the oocytes.
Application of these analytical approaches to mouse versus human mtDNA heteroplasmic variance data revealed that human heteroplasmy variance was greater than mouse. Comparison of the 3243A>G mutation heteroplasmy levels of the 82 oocytes derived from the hysterectomy of the carrier mother (quadriceps = 18.11%; leukocytes = 7.25%) with the heteroplasmy variance of oocytes from the NZB-BALB mouse mtDNA studies revealed that the human normalized oocyte A3243G, mutation level variance was 0.13, while the mouse normalized oocyte mutation level variance was 0.02–0.04. Similarly, using an aggregate of human pedigrees harboring the NARP 8993T>G, LHON 11778G>A and 3460G>A, and MERRF 8344 A>G mutations encompassing 72 mother–child pairs with heteroplasmy levels in the range of 40%–60%, Wonnapinij and colleagues concluded that the normalized heteroplasmy mutation level variance of human offspring was ∼0.37, while that for mouse offspring was 0.04–0.12 (Wonnapinij et al. 2010). This comparison assumes that the nature of the genetic differences between human mtDNA 3243A>G mutant and between NZB and BALB mtDNAs does not influence the heteroplasmy segregation rate.
A Neutralist Mathematical Model to Describe mtDNA Heteroplasmy Segregations
To determine whether mtDNA heteroplasmy variance has been affected by nonrandom factors such as selection, it was necessary to define the expectation for heteroplasmy variance if only stochastic factors were contributing to mtDNA segregation. The calculation of the expectations of random heteroplasmy transmission required a theoretical model. Generally, researchers have used the Sewall Wright variance formula, V = p0(1-p0)(1-e−t/Ne) for determining the effect of random genetic drift on mtDNA heteroplasmy inheritance. Despite its simplicity, this approach ignores much of the information that is present in the total heteroplasmy distribution, especially when this is not symmetric. Additionally, a common assumption when comparing heteroplasmy distributions between cells or individuals is that the population follows a normal distribution. However, Wonnapinij and colleagues have argued that the normal distribution is not the optimal choice for representing the potential distribution of mtDNA heteroplasmic genotypes for two main reasons. First, the normal (or Gaussian) distribution is defined over a range of infinite minus and plus tails, yet genetic variation is constrained to allele frequencies (p0) between zero and one. Second, the normal distribution is always symmetric, whereas mtDNA heteroplasmy distribution is often skewed. Therefore, Wonnapinij and colleagues (2008, 2010) argued that while the normal distribution can provide a reasonable model for the stochastic dispersion of heteroplasmy allele frequencies with ρ0 values close to 0.5, it is less accurate at extreme allele frequencies.
To predict the entire heteroplasmy probability distribution including the probabilities of a single allele becoming fixed or lost, Wonnapinij and colleagues developed a tool based on Kimura's distribution, henceforth designated the Wonnapinij/Chinnery/Samuels-Kimura (WCS-K) tool. In the 1950s, Kimura (1955) developed a model of the possible allele frequency distribution for a finite population resulting from the random sampling of gametes. His continuous model includes a set of probability distribution functions that describe gene frequency distributions of populations under pure random genetic drift. These functions are represented by three equations: a probability f(0, t) for losing an allele, a probability f(1, t) for fixing that allele, and a probability distribution function φ(x,t) that the allele is present at frequency x in the population. This model has three parameters: the initial gene frequency, p0; the effective population size, Ne; and the number of generations, t. In the case of mtDNA heteroplasmy, p0 is interpreted as the founder's heteroplasmy proportion or as a surrogate of the parental allele frequency calculated as the mean heteroplasmy in the derived cells or offspring of the progenitor. Ne is interpreted as a parameter related to the number of segregating mtDNA units. t is interpreted as the number of generations. The WCS-K tool avoids complications related to the definition of Ne by combining it with t into a single parameter called b, and treats b as one parameter (b = e−t/Ne). Parameter b is determined from the heteroplasmy variance either of a number of offspring from a single mother or by pooling the heteroplasmy values of offspring from mothers with similar heteroplasmy levels (b = 1-[V/p0 (1-p0)] or b = 1-Vnormalized). Wonnapinij and colleagues refer to parameter b as the bottleneck parameter, which then determines the width of the heteroplasmy distribution in the offspring (Wonnapinij et al. 2008; Samuels et al. 2013). To determine the statistical significance of the observed differences in the fit of experimental data with the Kimura distribution, the WCS-K tool uses the Kolmogorov-Smirmov (KS) test (Wonnapinij et al. 2008).
Using the Kimura model, Wonnapinij and colleagues reassessed the variance estimates and conclusions of a number of previous experimental studies on heteroplasmic segregation. When the authors applied the WCS-K tool to the heteroplasmy distribution of 82 oocytes recovered from the hysterectomy of the 3243A>G mutation woman (Brown et al. 2001), they concluded that the oocyte mtDNA distribution fit the expectations of the Kimura distribution and thus was by inference stochastic. Application of the tool to the Jenuth and collaborators data on the transmission to oocytes of the mouse NZB versus BALB mtDNAs from heteroplasmic mothers (Jenuth et al. 1996), the authors concluded that six of the eight mouse line datasets were consistent with the expectations of the Kimura distribution for random genetic drift, but that two datasets at the extreme ends of the heteroplasmic distribution failed to fit the Kimura distribution because of unexpectedly low numbers of oocytes that lacked one or the other mtDNA. Of course, the determination of whether an mtDNA is present or absent depends on the sensitivity of the method used to detect the minor allele.
Analysis of two datasets on heteroplasmic Drosophila eggs (Solignac et al. 1984; de Stordeur et al. 1989) resulted in five of six datasets fitting the Kimura distribution, the inference being that their distribution arose by random segregation. In one D. simulans dataset in which the mtDNA heteroplasmy was generated by cytoplasmic injection of siIII and siII mtDNA genomes (two naturally occurring Drosophila mtDNA sequences), the heteroplasmy distribution was found to fit the Kimura distribution and thus was random. However, in this case the founder female harbored 38.5% heteroplasmy and by the third generation the mean heteroplasmy of the offspring was p0 =12.7% (V = 0.0096). Therefore, although this striking directional shift could not have occurred by chance, the WCS-K tool was unable to detect deviation from randomness. Finally, in a Drosophila dataset derived from eggs 30 generations away from the founder, the results did not fit the expectations of the Kimura distribution, suggesting that factor(s) in addition to genetic drift had acted on this mtDNA segregation pattern (Wonnapinij et al. 2008).
Thus, by applying the WCS-K tool to heteroplasmy distributions, Wonnapinij and colleagues concluded that many were consistent with stochastic processes. Still, even this tool showed that some cases deviated significantly from the expectations of randomness.
In contrast to the seemingly modest role played by selection in determining transmission of heteroplasmy when analyzed using the WCS-K tool, the mouse experiments that analyzed the transmission of deleterious mutations including the mtDNA 4696 deletion (Sato et al. 2007), the ND6 13885insC mutation (Fan et al. 2008), the tRNAMet 3875delC mutation (Freyer et al. 2012), and the NZB mtDNA in 129-NZB haplogroup heteroplasmy (Sharpley et al. 2012) all unequivocally show evidence of mtDNA germline purifying selection. One set of potential limitations of using the Kimura distribution to model a stochastic distribution of mtDNA genotypes resides in the three parameters that relate to the neutralist basis of the distribution. These include: (1) a large population of N diploid parents, (2) no mutation, selection, or migration, and (3) no overlapping generations (Kimura 1955). One of the mathematical assumptions of the continuous Kimura model is that the effective size N must be sufficiently large so that terms of order 1/N2 and higher can be omitted without serious error. Studies of mammalian embryonic development suggest that the number of mitochondria or mtDNA molecules may be drastically reduced (Jansen and de Boer 1998; Krakauer and Mira 1999). If the number of segregating units remains ≥200 (Jenuth et al. 1996; Cree et al. 2008; Wai et al. 2008), then this assumption is still valid. If, however, the number of mtDNA segregating units were to approach N = 8, as suggested in one scenario for the 3243A>G genotype distribution of the 82 oocytes removed from a hysterectomy (Brown et al. 2001), then this term could introduce error into the conclusions of the Kimura model.
Another concern is whether the KS test for significance is appropriate and sufficiently sensitive to evaluate differences between the experimentally observed heteroplasmy distribution and the predictions of the Kimura model, especially when the heteroplasmy levels are ∼0.5. By implementing the KS nonparametric test, the WCS-K tool avoids assuming that the data were sampled from Gaussian distributions, but there are drawbacks to using a nonparametric test. If the populations are approximately Gaussian (as in the case of ∼0.5 heteroplasmy level), the nonparametric tests have less power (are less likely to give you a small p value), especially with small sample sizes (Stephens 1974). Additionally, the KS test is most sensitive when the distribution functions differ in a global fashion near the center of the distribution. But if there are repeated deviations between the distribution functions or the distribution functions have (or are adjusted to have) the same mean values, then the distribution functions cross each other multiple times and the maximum deviation between the distributions is reduced (Babu and Feigelson 2006).
Because the predictive ability of the WCS-K tool is based on the assumption of random genetic drift in a finite large population, the strength of the predictions of the WCS-K tool is strongly influenced by sample size. This makes it possible to change the statistical significance of a comparison simply by increasing the range of mtDNA heteroplasmy genotypes binned together. When the Kimura distribution was applied to analysis of heteroplasmy transmission of the tRNAMet 3875delC mutation, even for more central p0 values where the effects on “b” are minimized, quite large sample sizes were required to show the directionality associated with selection (Freyer et al. 2012). While statistical evidence of selection was obtained when binning the data from mothers in the range of 40%–60% heteroplasmy (Freyer et al. 2012), it was not when binning females with heteroplasmy levels of 50%–60% (data not shown). This limitation is compounded because the currently available program can only analyze transmissions through one generation, thus limiting detection of cumulative effects of selection. Moreover, the effects of population size fluctuations on selection are also not considered and therefore not modeled (Wonnapinij et al. 2008, 2010).
While these technical considerations are pertinent, perhaps a more important reason why the WCS-K tool is relatively insensitive in identifying the effects of selection on mtDNA mutations is that the Kimura model was developed for changes in the allele frequency of Mendelian genes within finite populations. The Mendelian mode of inheritance was the only genetic model known in the 1950s when Kimura developed his model. However, the Mendelian rules of inheritance derive from the behavior of chromosomal genes and the biological unit upon which selection acts on Mendelian genes is the diploid organism. This unit of selection is fundamentally different from the situation with the mtDNA in which selection acts on cells and organisms with several thousand-fold ploidy.
This does not adversely affect the mathematics of allelic transmission if all alleles are neutral. However, it has a profound effect on whether the Kimura model can detect the effects of purifying selection during the transmission of a deleterious heteroplasmy mtDNA mutation. In diploid animals, selection acts on units of two gene copies for which there are only three allelic combinations: a/a, a/b, and b/b and two to three quantized phenoptypic states. For mtDNA genetics, selection acts on the cellular phenotype, which is a composite of the several thousand mtDNAs with different percentages of two or more allele systems.
For an nDNA allele at a population frequency of 1%, virtually every minor allele will be located in a heterozygous individual with a significant probability of being lost in subsequent generations of diploid individuals by drift. A cell with 1% mutant mtDNAs within a cell of 5000 mtDNAs would harbor 50 mutant mtDNAs. Because cytokinesis divides the cell in half, low-frequency mtDNA alleles are much less likely to be lost in successive generations than nDNA alleles. Finally, the mitochondria and mtDNAs within the cell are not autonomous units. Rather, they function as a collective population through repeated fission and fusion and sharing of all their gene products. Subtle alterations in the mtDNA sequence and heteroplasmy level can have significant effects on cellular energy production, REDOX regulation, ROS production, Ca2+ regulation, mtDNA copy number, mitochondrial fission and fusion, mtDNA replication and turnover, etc. For example, 20% of normal mtDNAs within a cell can mask the respiratory-deficient phenotypic of 80% 3243A>G mutant mtDNAs (Yoneda et al. 1994). Also, low-frequency deleterious mtDNAs can be retained in a cell lineage for many generations without adversely affecting the cellular phenotype. An ND5 gene frameshift mutation, which was found to be homoplasmic in a human oncocytoma tumor, was present at low-heteroplasmy levels in the normal tissues of the patient and his two sisters. Thus, this deleterious mutation was silently transmitted through the maternal lineage (Gasparre et al. 2008). Hence, classical concepts of genotype–phenotype associations and their interaction with selection are violated by mtDNA genetics.
In conclusion, the formulations of Wonnapinij and associates (Wonnapinij et al. 2008, 2010) have provided a significant improvement for the mathematical analyses of mtDNA heteroplasmy variance in cells and offspring. However, the WCS-K tool is less powerful at detecting the role of selection on mtDNA mutation inheritance.
Heteroplasmic mtDNA Mutation Segregation in Somatic Cells and Tissues
Heteroplasmic mtDNAs also segregate in somatic cells and tissues and this greatly complicates phenotypic predictions in patients. Although it is standard practice to test for mutant nDNA genes in patient blood, the mtDNA heteroplasmy level in blood cells can be quite different from that of brain, heart, muscle, or kidney, the organs most susceptible to life-threatening mitochondrial disease.
The first reports of directional segregation of mtDNA heteroplasmy in mouse and human somatic cells appeared in the 1970s. In these experiments, the fate of the mtDNAs was monitored in replicating somatic cell hybrids and cybrids via the mtDNA chloramphenicol (CAP)-resistance (CAPR) or -sensitivity (CAPS) genetic markers. CAPR is the product of mutations in the mtDNA 16S rRNA gene (Blanc et al. 1981a,b). When CAPR and CAPS cells of the same species and same cell lineage (mouse L cell to mouse L cell, human HeLa cells to human HeLa cells) were fused, the mixture of CAPR and CAPS mtDNAs was retained for extended periods and seemed to segregate stochastically. By contrast, when cell lines of the same species but different origin (mouse L cell to mouse RAG cells, human HeLa cells to human WiL2 [WAL2] or HT1080 cells) were fused, then one of the two mtDNAs was directionally and rapidly lost. In the case of cybrids prepared between enucleated CAPR HeLa cells and WAL2A cells, the heteroplasmy could only be retained by continuous CAP selection (Wallace et al. 1976, 1977; Bunn et al. 1977; Wallace 1981, 1982, 1986). The directionality of mtDNA segregation was even more extreme when cells of different species were fused. When CAPR human cells were fused to CAPS mouse cells and the hybrids selected in CAP, the surviving hybrids retained the human mtDNA and all of the human chromosomes, and lost all of the mouse mtDNA and segregated the mouse chromosomes. This is the opposite of the well-established mouse–human hybrid segregation pattern (Wallace et al. 1976; Giles et al. 1980).
When clinically relevant mtDNA mutations became available, these mutants were also introduced into cultured cells. In six different cybrids lines heteroplasmic for the MELAS tRNALeu(UUR) 3243A>G mutation, the mutant 3243A>G mtDNA progressively increased in frequency. Only cell lines that were predominantly homoplasmic were stable, with the exception of one cell line, which maintained a stable 30% 3243A>G mutant heteroplasmy (Yoneda et al. 1992).
Segregation of Human Somatic Cell mtDNA Mutations
Shortly after it was discovered that mtDNA deletions accumulate in muscle of patients with mitochondrial myopathy (Holt et al. 1988), it was found that mtDNA deletions were progressively lost from blood cells in CPEO and Kearn Sayre patients, though they become progressively enriched in muscle (Shoffner et al. 1989). Moreover, mtDNA deletions in humans were rarely transmitted through the female germline (Shoffner et al. 1989), though mtDNA duplications can be maternally transmitted (Ballinger et al. 1992, 1994).
The absence of mtDNA deletions in blood cells when they are prevalent in postmitotic tissues led to the hypothesis that severely deleterious mtDNA mutations might be inhibiting bone marrow stem cell replication. Those stem cells that spontaneously segregated the deleted mtDNA would then have a proliferative advantage and repopulate the bone marrow.
The MELAS tRNALeu(UUR) 3243A>G mutation, like mtDNA deletions, commonly presents as CPEO and is also progressively lost from the blood cells, though not as rapidly as mtDNA deletions. Comparison of the 3243A>G heteroplasmy levels for 13 patients tested in white blood cells, oral mucosa cells, and urinary tract epithelial cells consistently showed that the blood cell mutant level was lowest and the urinary epithelial cells heteroplasmy was highest. For one patient the levels were 20%, 30%, and 65% and for another were 30%, 50%, and 80%. Because of their consistently high mutation heteroplasmy level, urinary epithelial cells are considered the best surrogate for postmitotic tissues when testing for 3243>G heteroplasmy levels of the body (Monnot et al. 2011).
The level of the 3243A>G mutation progressively declines in blood cells over the life of the individual. In a longitudinal analysis of 34 individuals carrying the 3243A>G mutation, 17 symptomatic and 17 carriers all showed an age-related decline in blood mutant heteroplasmy levels. For the symptomatic subjects the heteroplasmy level declined 0.534% per year, while for the asymptomatic carriers the heteroplasmy level declined 0.215% per year. At the same time, the symptomatic individual's phenotypes became worse while the asymptomatic individuals remained asymptomatic (Mehrazin et al. 2009). A mathematical model was developed for the decline of 3243A>G heteroplasmy in blood and compared to longitudinal data that has been collected on the blood cell heteroplasmy levels. The model and the data indicate that the loss of heteroplasmy is an exponential function of the form: Mage-corrected = m(t)eSt, where m(t) is the average mutational level as a function of time and S = 0.020 ± 0.003 (1/year) (Rajasimha et al. 2008).
While the percentage of the mtDNA tRNALeu(UUR) 3243A>G mutation heteroplasmy declines with age in blood, the heteroplasmy level of the MERRF tRNALys 8344A>G mutation does not (Rajasimha et al. 2008). Hence, different mtDNA mutations show different tissue-specific stabilities indicating that a variety of physiological processes must modulate the fate of heteroplasmic variants in tissues.
Segregation of Mouse Somatic Cell mtDNA Mutations
The original Jenuth et al. 1996 manuscript reported that germline segregation of mouse NZB-BALB heteroplasmy was random. However, in a subsequent paper these authors reported that NZB-BALB mtDNAs segregated directionally with age in different tissues (Jenuth et al. 1997). Over the life of an NZB-BALB heteroplasmic animal, the kidney and liver progressively increased the NZB mtDNA, while the blood and spleen cells accumulated the BALB mtDNA (Jenuth et al. 1997). The heteroplasmy of the tail, cerebral cortex, gastrocnemius muscle, heart ventricle, and lung were reported to remain relatively stable (Jenuth et al. 1997).
In a similar experiment involving mouse NZB and 129-mtDNA heteroplasmy, the directional segregation of liver and kidney toward NZB mtDNAs and of spleen toward 129 mtDNAs was confirmed. In addition, it was found that seminal vesicles, ovaries, and pancreas also segregated toward 129 mtDNAs. However, tail, brain, lung, and heart maintained relatively constant heteroplasmy (Sharpley et al. 2012). Hence, striking differences exist between germline and somatic cell mtDNA heteroplasmy segregation. Because the somatic tissue cells contain thousands of mtDNAs, this segregation is unlikely to be caused by a bottleneck involving a dramatic reduction in mtDNA copy number. Rather, this must involve a process by which either some mtDNAs are selectively replicated or eliminated or certain mitochondria are differentially propagated.
In contrast to the NZB/BALB mouse nuclei (Jenuth et al. 1997), it was discovered that the Mus musculus castaneous (CAST/Ei) mouse nucleus does not cause the tissue-specific segregation of the NZB and BALB mtDNAs. This discovery provided the opportunity to map genetic loci involved in the mtDNA sorting process. Heteroplasmic female Mus musculus mice were crossed with male Mus musculus castaneous (CAST/Ei) mice and the tissue-specific segregation of the NZB-BALB mtDNAs was found to be inherited as a simple Mendelian recessive trait. Using F2 intercross animals, Battersby, Shoubridge, and associates mapped three quantitative trait loci (QTLs). One locus on chromosome 5 at D5mit25, designated Smdq1 (segregation of mitochondrial DNA QTL#1) accounted for 35% of the variance in the heteroplasmy segregation phenotype of liver. A second locus found on chromosome 2 (Smdg2) accounted for 16% of the heteroplasmy variance in the kidney. A third locus on chromosome 6 (Smdq3) accounted for 20% of the variance for the spleen (Battersby et al. 2003). Analysis of the maximal oxygen consumption rates of the liver mitochondria of NZB-BALB heteroplasmic animals with NZB mtDNAs levels between 0% and 91%–97% on the BALB background revealed no significant difference. In the liver, the rate of enrichment of the NZB mtDNAs was constant with age with a selective advantage over BALB of ∼14% per replication cycle. No difference was found in the relative incorporation rates of BrdU of the two mtDNAs. Hence, the advantage of the NZB mtDNA did not appear to be caused by increased replication rate. Also, partial hepatectomy followed by liver lobe regeneration did not affect the NZB-BALB mtDNA ratio. Surprisingly, however, when hepatocytes were explanted from heteroplasmic mouse livers, the majority of the in vitro cultures reversed the segregation direction, losing the NZB mtDNA while enriching the BALB mtDNAs. Therefore, the authors surmised that direction and mechanism of segregation was not the result of differential mitochondrial physiology or mtDNA replication rate and thus might depend on mitochondrial maintenance or mitochondrial turnover (Battersby and Shoubridge 2001).
To determine whether the directional segregation of the NZB mtDNAs in hematopoietic tissues was the result of immune surveillance, NZB-BALB heteroplasmic females were crossed onto nuclear backgrounds that lacked the Tap1, β2m, or Rag1 genes. Absence of these genes should impair the presentation and recognition of mtDNA-coded peptides. However, the kinetics of selection for the BALB mtDNA was unaltered indicating that segregation was not the result of presentation of mitochondrially encoded peptides by the major histocompatibility locus (MHC) (Battersby et al. 2005).
Fine mapping of the chromosome 6 (Smdq3) locus led to the identification of the Gimap3 locus (GTPase of immunity-associated protein 3), which proved to be mutant in CAST/Ei relative to BALB/c, with the CAST/Ei gene being differentially spliced. The Gimap3 gene has two AUG start codons in exons 3 and 4 with a stop codon in exon 4 upstream of AUG start codon. When all five exons are spliced together as in BALB the second AUG is used to initiate the polypeptide. In CAST/Ei, a G to A transition in the splice acceptor site of exon 4 results in the deletion of exon 4. This results in the first AUG being used resulting in the addition of 58 amino acids to the amino terminus of the protein without affecting the carboxy-terminal transmembrane domain. Although the mtDNA copy number was not different between the two Gimap3 alleles, the overexpression of the CAST/Ei allele in heteroplasmic mice slowed the mtDNA segregation rate (Jokinen et al. 2010).
Mitochondrial Physiological Control of Somatic and Germ Cell mtDNA Segregation
It has been suggested that in hematopoietic tissues the selection is age-dependent and proportional to the initial heteroplasmy level. This implies that selection occurs at the organelle level. Because Gimap3 is a mitochondrial outer membrane protein, it might participate in mitochondrial partitioning or sorting. Gimap3 is part of a vertebrate-specific gene family and the protein is anchored to the mitochondrial outer membrane by its carboxyl terminus. It has been reported to be important in T-cell survival and development. One speculation is that “Gimap3 could act as a scaffold on mitochondrial membranes to remodel mitochondria in response to specific stimuli” (Jokinen et al. 2011; Jokinen and Battersby 2013).
Regulation of mtDNA Heteroplasmy by tRNAArg 9821 insA-Generated ROS
The discovery that Gimap3 regulates mtDNA segregation in hematopoietic cells does not seem to be the basis for the enrichment of the NZB mtDNAs in either liver or kidney, each of which is modulated by a different genetic locus. It has been proposed that because the rate of mtDNA selection remains constant in the liver with age and is independent of the initial heteroplasmy level selection for the NZB mtDNA may act at the mtDNA level (Jokinen et al. 2011; Jokinen and Battersby 2013). This proposal corresponds to the discovery of an important mtDNA sequence difference between NZB versus BALB or 129 mtDNAs. NZB and NIH3T3 mtDNAs differ from BALB and 129 mtDNAs by having an additional A in the length of a homopolymer run of As in the DHU loop of tRNAArg gene at nt 9821 (tRNAArg 9821insA). This same variant has been found to be enriched in heteroplasmic cultured mouse L cells (Fan et al. 2012). Extensive biochemical analysis of mouse L929 ρ0 cell cybrids that harbor either NIH3T3 or NZB mtDNAs revealed that they have the same mitochondrial respiration rate as cells with fewer As. However, the similarity in respiration rate proved to be the result of a compensatory 1.6- to 1.9-fold increase in mtDNA copy number in the tRNAArg 9821 insA cells. This increased mtDNA copy number was induced by increased mitochondrial ROS production associated with the tRNAArg 9821 insA allele. When the increased ROS production was neutralized by antioxidants, the mtDNA copy number declined and mitochondrial respiratory chain function was reduced. The increased ROS also partially inactivated the mitochondrial α-ketoglutarate dehydrogenase and aconitase enzymes thus reducing the efficiency of the tricarboxylic acid cycle and the tRNAArg 8921 insA cells had a reduced capacity to grow in galactose (Moreno-Loshuertos et al. 2006).
Because an alteration in a mitochondrial tRNA would be expected to inhibit mitochondrial protein synthesis, this could impede the synthesis of mitochondrial OXPHOS enzymes, inhibit electron transport, and result in increased ROS production. Hence, a major factor in the enrichment of the NZB mtDNAs in liver could be increased ROS production as a result of tRNAArg 9821 insA variant-induced partial mitochondrial protein synthesis defect (Fan et al. 2012). The increased ROS would increase the mtDNA copy number and this effect would become increasingly predominant as the proportion of NZB mtDNAs increased in the tissues.
Battersby and Shoubridge have argued that the tRNAArg 9821 insA could not be the basis of the NZB segregation because they were able to map two different chromosomal loci in the BALB/c CAST/Ei crosses that abolished NZB mtDNA directional segregation (Battersby and Shoubridge 2007). However, the gene(s) responsible for the liver and kidney replicative advantage of the NZB mtDNAs could be nDNA genes involved in regulating mitochondrial ROS production and/or REDOX regulation.
A ROS-driven mtDNA copy-number hypothesis could also explain the directional germline segregation of the NZB mtDNAs in NZB-129 heteroplasmic mice. Assuming that the female germline cells have mitochondria with a limited number of mtDNAs and fusion and fission are inhibited, then each mitochondrion would act as a semi-autonomous unit. Those PGCs and proto-oocytes with higher percentages of NZB mtDNAs would generate increased ROS that would damage the mitochondria and mtDNAs resulting in preferential loss of the germline cells with the higher NZB mtDNAs. This effect would increase until the percentage of NZB mtDNAs reached a sufficiently high level that the increased ROS signaled to the nucleus to increase the mtDNA copy number. The biochemical difference between the predominant 129 and NZB cells would then be negated permitting the very high NZB lineages to segregate toward homoplasmic NZB.
In contrast to germline cells, somatic tissue cells and cultured cells have mitochondria that contain multiple nucleoids and mitochondria that actively fuse within a cell mixing their contents and gene products. In this case, the mitochondrial polysomes of a heteroplasmic somatic cell can translate both mtDNAs and mRNAs and can complement each other in trans. This trans complementation of mtDNAs in heteroplasmic cells was first demonstrated in somatic cell hybrids and cybrids harboring CAPR and CAPS mtDNAs linked to different polypeptide electrophoretic forms of the ND3 gene, MVI versus MVII. In heteroplasmic cells in which the mtDNA translation products were differentially labeled by growth in 35S-methionine in the presence of cytosolic ribosome inhibitor, emetine, both MVI and MVII were equally labeled. When CAP was added, both MVI and MVII continued to be labeled. This means that the CAPR ribosomes had to be translating the MV mRNA from the CAPS mtDNA (Oliver and Wallace 1982; Oliver et al. 1983).
In the case of somatic tissues harboring NZB mtDNAs with the tRNAArg 9821insA allele mixed with either BALB or 129 mtDNAs, the ribosomes would use both mutant and normal tRNAs in proportion to the mtDNA heteroplasmy. Because inhibition of polysome elongation would be limited by the “slowest” ribosome, the presence of even a small proportion of the inhibitory NZB mtDNA tRNAArg 9821 insA would impede protein synthesis, reduce mitochondrial OXPHOS, and increase ROS production. The increased ROS production would then drive an increase in mtDNA copy number. The resulting repeated cycles of amplification of the mtDNAs within NZB mtDNA-containing cells could result in the progressive enrichment of the NZB mtDNAs. Hence, the tRNAArg 9821 insA allele could account for the progressive increase in NZB mtDNAs in liver and kidney of NZB and BALB or 129 heteroplasmic mice.
This hypothesis would also be consistent with the dramatic reversal of NZB segregation seen for NZB-BALB heteroplasmic hepatocytes when grown in culture in high glucose medium (Battersby and Shoubridge 2001). The high glucose would fully reduce the electron transport chain of the cells resulting in increased ROS production regardless of the haplotype combination. The increased mtDNA copy-number effect would be neutralized permitting the more energetically efficient BALB mtDNA to take over.
If mitochondrial ROS production is important in the replicative segregation of NZB versus BALB or 129 mtDNA heteroplasmy animals, then one might anticipate that antioxidant defenses might also be relevant in regulating their segregation. The degree of enrichment for the NZB mtDNA in liver and kidney and against NZB mtDNA in the germline could then be influenced by mouse strain-specific genetic variation. One potentially relevant nDNA genetic variant is the loss of the nicotinamide nucleotide transhydrogenase gene (Nnt) on chromosome 13 in the Jackson Laboratory strain C57BL/6 J (Toye et al. 2005; Huang et al. 2006; Fielder et al. 2012). NNT is a mitochondrial inner membrane protein, which uses the potential energy of the mitochondrial electrochemical gradient to transfer reducing equivalents from NADH to NADPH. The higher reducing potential of NADPH is required for the reduction of mitochondrial lipid peroxides and of H2O2 to H2O via the glutathione peroxidases. NADPH is also essential for the regulation of multiple enzymes and transcription factors via oxidation-reduction of thiol-disulfides (Wallace 2012). Because the Jackson Laboratory strain of C57BL/6 J, which lacks the Nnt gene was used in the NZB-129 heteroplasmy studies, it is possible that mitochondrial ROS production caused by the NZB tRNAArg 9821 insA allele was enhanced on this strain background resulting in enhanced selection against PGCs or primary or mature oocytes with significant levels of the NZB mtDNA and increased ROS production.
Regulation of mtDNA Heteroplasmy by Mitochondrial Fusion and Fission
Other mitochondrial quality assurance mechanisms such as mitochondrial dynamics and mitochondrial autophagy (mitophagy) might also be important in mtDNA heteroplasmy segregation (Youle and van der Bliek 2012; Jokinen and Battersby 2013). The mitochondria in somatic tissues and cultured cells are highly dynamic undergoing a frequent fusion and fission cycle. Perturbations of mitochondrial fission and fusion have been shown to affect the segregation of heteroplasmic mtDNAs. Mitochondrial fission is initiated by Drp1, a member of the dynamin protein family. Mitochondrial fusion is initiated by the Mfn1 and Mfn2 proteins that mediate the fusion of the outer mitochondrial membranes and OpaI, which mediates the fusion of the mitochondrial inner membranes (Youle and van der Bliek 2012). In a muscle-derived rhabdomyosarcoma cultured cell line carrying 80% tRNALeu(UUR) 3243A>G mtDNAs, RNAi knockdown of the mitochondrial fusion Drp1 and hFis1 gene mRNAs resulted in an average 13% and 11% increase in the mutant mtDNA over the wild type, respectively. By contrast, RNAi knockdown of Opa1 mRNA did not affect the mutant heteroplasmy level. Hence, in this replicating cell line, increased mitochondrial fusion was permissive for retention of mutant mtDNAs (Malena et al. 2009).
In mice in which the mitochondrial fusion genes, Mfn1 and Mfn2, were knocked down in skeletal muscle via floxed Mfn1 and Mfn2 genes and the muscle-specific MLC1f promoter-driven Cre, profound mtDNA depletion ensued. At 7 to 8 weeks, the double mutant mice exhibited hyperproliferation of muscle mitochondria, mtDNA depletion to ∼250 mtDNAs/nucleus versus ∼3500 mtDNA/nucleus in normal mouse muscle, a 5-fold increase in mtDNA point mutations, and a 14-fold increase in mtDNA deletions. Comparison of the mtDNA deletion levels at 8–13 weeks revealed an 80-fold increase in mtDNA deletions in Mfn−/−, Mfn2+/− mice (2.3 × 10−5/genome versus 2.8 × 10−7/genome). When the homozygous mtDNA polymerase γ mutator locus, PolgAD257A, was combined with the Mfn−/− mutation, the animals manifested a profound OXPHOS defect with complex I activity (Chen et al. 2010). Hence, both mtDNA fission and fusion are important in maintaining mtDNA integrity and the segregation of mtDNAs within cells and tissues.
Regulation of mtDNA Heteroplasmy by Inter-mtDNA Complementation
Mitochodnrial fission and fusion permit mtDNA mutant complementation within cells. The mtDNAs are contained in nucleoids and the distribution of mtDNAs within nucleoids and the partitioning of nucleoids into daughter mitochondria could be important in heteroplasmy segregation. In nucleoids, the mtDNAs are complexed with numerous copies of the mtDNA-packaging protein and transcription factor, TFAM. Knockdown of TFAM in HeLa cells perturbs the distribution of mtDNAs into daughter nucleoids and mitochondria (Kasashima et al. 2011). The number of mtDNAs per nucleoid has been estimated to be as low as one (∼1.4) (Kukat et al. 2011) and between two and 10 (Gilkerson et al. 2008; Poe et al. 2010; Rebelo et al. 2011). If mtDNA nucleoids contain more than one mtDNA and nucleoids could exchange mtDNAs, then nucleoids could be heteroplasmic, which would significantly affect the dynamics of mtDNA segregation.
To determine whether nucleoids are heteroplasmic, two cell lines each harboring a homoplasmic, nonoverlapping mtDNA deletion that deleted one or more tRNAs (FLPΔ nt 7846-9748 and CWΔ nt 10155-15945) were fused into somatic cell hybrids. The homoplasmic mtDNA deletion cells each had fragmented mitochondria and were deficient in mitochondrial protein synthesis and respiration. However, in the hybrids containing the two deleted mtDNAs the mitochondrial were elongated and functional in both protein synthesis and respiration. By using hybridization probes that were internal to the two deletions and differentially fluorescently labeled, the presence of the two deleted mtDNAs were monitored. This revealed that the respiring hybrids harbored both mtDNAs in elongated mitochondria and most of the nucleoids hybridized to either one or the other probe, though in some cases the two hybridization probes seemed to overlap. When the hybrid cells were released from selection of respiratory sufficiency (uridine) within 12 d, 55% of the cells had segregated to pure CWΔ mtDNA and 42% had segregated to pure FLPΔ, with only 3% retaining both mtDNAs. As the mtDNAs segregated the nucleoids resolved into either one mtDNA or the other. This indicated that nucleoids do not exchange mtDNAs, and that the instances where the deleted mtDNAs colocalized in the hybrids was the result of two nucleoids being adjacent to each other and thus not resolved by light microscopy (Gilkerson and Schon 2008; Gilkerson et al. 2008).
Further evidence that the nucleoids are clones of a single mtDNA genotype have come from the analysis of cultured cells that were heteroplasmic for a 7522 nt deletion, ∼80% deletion. By fragmenting the mitochondria, sorting the pico-green-stained mitochondria in a cell sorter, and performing differential PCR amplification for the deleted and normal mtDNA, it was again concluded that each nucleoid contained only one type of mtDNA (Poe et al. 2010). Therefore, the current evidence favors the conclusion that individual nucleoids harbor only one type of mtDNA.
Regulation of mtDNA Heteroplasmy by Mitophagy
The mtDNAs continually replicate within somatic cells including postmitotic cells, with the excess mtDNAs being removed by mitophagy. Thus, mitophagy provides another mechanism by which heteroplasmic mtDNA mutations can be segregated. Mitophagy is preceded by mitochondrial fission, which is thought to be asymmetric such that one daughter mitochondrion is more energetically competent than the other. Mitophagy is then envisioned to preferentially degrade the more respiration-compromised mitochondrion. This process is mediated by Parkin and PTEN-induced kinase 1 (PINK1), encoded by the Park2 and Park6 loci, respectively. In healthy mitochondria, PINK1 is incorporated into the mitochondrion but is rapidly degraded by the mitochondrial PARL rhomboid protease. However, if the mitochondrial membrane potential is significantly reduced, PINK1 becomes stabilized in the mitochondrial outer membrane where it phosphorylates serines on target proteins. PINK1 phosphorylation results in the attraction of the cytosolic protein Parkin to the mitochondrial outer membrane. Parkin is an E3 ubiquitin ligase that ubiquitinates mitochondrial outer membrane target proteins (Narendra et al. 2008, 2009, 2010; Guo 2010; Youle and van der Bliek 2012). Recent evidence has implied that Mfn1, Mfn2, and Miro1 (a protein involved in mitochondrial transport along microtubules) are important Parkin ubiquitination targets. The Parkin modification of mitochondrial proteins is followed by the attraction of the p62/SQSTM1 protein to the mitochondrion, which commits the mitochondrion to LC3-II encapsulation, fusion of the resulting autophagosome with a lysosome, and degradation (Narendra et al. 2012).
The relevance of mitophagy to mtDNA heteroplasmy segregation was first demonstrated in Parkin overexpression experiments. Cybrid cells that were stably heteroplasmic for a high percentage of an mtDNA COI mutation (COXICA65) had a 55% reduction in mitochondrial inner membrane potential and showed an increase in Parkin-bound mitochondria. Partial inhibition of mitochondrial fusion with vMIA and of the electron transport chain with azide to reduce mitochondrial electron transport, increased the Parkin bound to the mitochondria. Overexpressed Parkin in these cells over a 60-d period resulted in the selective loss of the COI mutant mtDNAs. Hence, mitophagy is capable of modulating mtDNA heteroplasmy.
However, in a cell line heteroplasmic for an mtDNA cytochrome b mutation with a 37% reduction in mitochondrial membrane potential overexpression of Parkin did not result in a reduction in the percentage of mutant mtDNAs (Suen et al. 2010). Mitophagy has been found to be dependent on both Parkin expression and mTORC1 inhibition in cells homoplasmic for deleterious mtDNA mutations (Gilkerson et al. 2012).
Because of the multiplicity of cellular processes that can impinge on mtDNA heteroplasmy, it is not surprising that different mtDNA mutations show different directionality and kinetics of segregation in different tissues.
IMPLICATIONS FOR MEDICINE
Complexities of Genetic Counseling
The absolute nature of maternal inheritance of the mtDNA creates an intractable dilemma for women that harbor high levels of a deleterious mtDNA mutation. They have a high probability that most if not all of their children will develop devastating diseases. Homoplasmic mtDNA mutations such as those commonly associated with LHON will be transmitted to all of a woman's offspring, though fortunately the penetrance of these milder mtDNA disease mutations is incomplete. For more deleterious mtDNA mutations, the outcome can be much worse. In one published pedigree where the mother was heteroplasmic for the mtDNA tRNAThr 15923A>G mutation she conceived and lost seven pregnancies. Five of the conceptions were lost prenatally. One boy was born and died at 42 h and another girl was born and died at 56 h of severe OXPHOS defects (Fig. 6) (Yoon et al. 1993).
Figure 6.
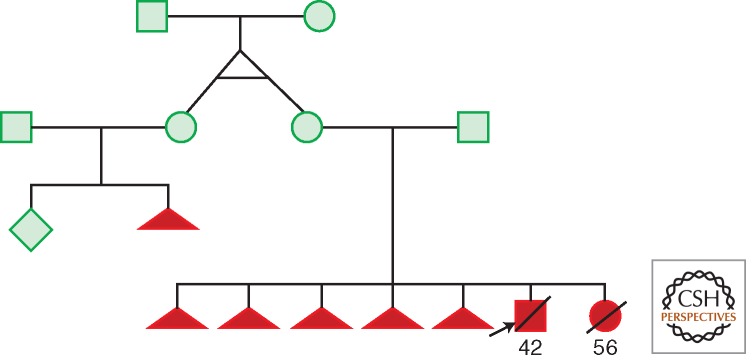
One published example of a germline lethal mtDNA mutation. A woman harboring a heteroplasmic mtDNA tRNAThr A15923G mutation lost five pregnancies in a row (triangles). The two surviving term infants died within 2 to 3 d. Postmortem mitochondrial analysis of one infant's skeletal muscle revealed that complex II + III and complex IV activities reduced to 6% and 5% of control and liver complex IV activity that was reduced 37% and complex II + III activity that was undetectable. The mother's twin sister had a similar reproductive history, yet both women were seemingly normal. (From Yoon et al. 1993; modified, with permission, from the authors.)
Even more common are women with subclinical levels of a pathogenic mutation such as the tRNALeu(UUR) 3243A>G or ATP6 8993T>G mutations but repeatedly transmit a high percentage of the deleterious mutant mtDNA to their offspring. Unfortunately, prenatal diagnostic procedures that have been effective in identifying fetuses with deleterious chromosomal gene mutations have proven to be relatively unreliable for diagnosing mtDNA diseases. These difficulties arise from the exclusively maternal inheritance of the mtDNA and the unpredictable nature of heteroplasmic mtDNA mutation segregation.
Obviously, all forms of prenatal diagnosis are useless for a woman harboring a homoplasmic mtDNA mutation because all of her fetuses will inherit her mutation. By contrast, heteroplasmic mtDNA mutations might be amenable to prenatal diagnosis if the woman was able to conceive a fetus with low levels of heteroplasmy. However, this can also be problematic because it is not certain that the fetal brain, heart, muscle, or kidney will have the same level of mtDNA mutation as found in the chorionic villus or amniocentesis sample. Furthermore, for most heteroplasmic mtDNA diseases, it is unclear what a “safe” level of heteroplasmy might be. Finally, some women might conceive multiple times and in every case the conceptus will have an unacceptably high level of mutant mtDNAs resulting in repeated fetal loss (Thorburn and Dahl 2001; Poulton and Bredenoord 2010).
Preimplantation Diagnosis
The ambiguities associated with chorionic villus sampling and amniocentesis diagnosis of mtDNA disease have led to investigation of the potential value of preimplantation genetic diagnosis (PGD). By determining the mtDNA heteroplasmic genotype of oocytes or embryos before fertilization or implantation, only those embryos with a desirable mtDNA genotype could be retained for implantation into the mother. Hence, many potential embryos could be screened with only the rare embryo with the optimal genotype permitted to go through gestation.
It would be particularly attractive if it were possible to remove the first polar body from an oocyte and determine its mtDNA genotype. Only those oocytes with the lowest polar body heteroplasmy level would then be utilized. Another possibility could be to fertilize the oocytes in vitro and then collect and genotype blastomeres or trophoblast cells to identify the low-heteroplasmy embryos.
The potential for PGD received significant initial support from a study of the mtDNA heteroplasmy genotypes of NZB-BALB mouse polar bodies, blastomeres, and embryos. In these studies, it was found that the mtDNA heteroplasmy level of the first polar body was within 0.1%–6.1% that of the rest of the embryo (R2 = 0.99), as compared to the lack of correlation between the mother's mtDNA genotype and that of her embryos (R2 = 0.32). Similarly, it was found that the mtDNA heteroplasmy of different blastomeres within the same embryo were also within 0%–6% of each other (Dean et al. 2003).
The positive results of the NZB-BALB heteroplasmy study were supported by studies on mice harboring the 4696 nt deleted mtDNA. Analysis of the second polar body following fertilization revealed that the polar body mtDNA genotype correlated with the percentage of deletion in the embryo with a coefficient of 0.95. Unfortunately, for mtDNA deletion mutations the benefits of PGD were compromised by the subsequent preferential replication of the deleted mtDNA during development. As mtDNA deletion-containing embryos developed, the percentage of deleted mtDNAs progressively increased, rising 17% during the 19 d after fertilization, 8% for the first 30 d after birth and before weaning, 8% for the first 100 d after weaning, and 6% for every subsequent 100 d. The maximum deletion level observed in any zygote was 78%, implying that the oogonia with higher levels of deletion did not yield oocytes. Ultimately, mtDNA deletion mice died of kidney failure, with the maximum deletion level observed in the kidneys being 90% (Sato et al. 2005). Because human mtDNA deletions have not been observed to be transmitted through the maternal lineage, the human female germ cells must have a lower tolerance for mitochondrial respiratory deficiency than the mouse (Wallace et al. 2013).
Based on the mouse polar body studies, sampling the mtDNA heteroplasmy levels of human meiosis I polar bodies would appear to be the most promising approach for PGD of heteroplasmic mtDNA diseases. Unfortunately, when human studies were conducted it was found that the mtDNA genotype of the first polar body might be very different from that of the embryo. The preimplantation embryos of three couples participating in assisted reproduction were studied, one each harboring the tRNALeu(UUR) 3243A>G MELAS mutation, the tRNALys 8344A>G MERRF mutation, and the ATP6 9185T>C cerebellar ataxia and tubulopathy mutation. The maternal heteroplasmy levels were 25%, 75%–95%, and 14%–25%, respectively, in tissues. In only half of the embryos (27 of 51) were the polar body genotypes within ±10% of that of the embryo. In those cases where the polar body was homoplasmic for the normal mtDNA the embryo was also homoplasmic normal (n = 7). However, for half of the embryos the polar body genotype was significantly different from that of the embryo. In cases where the embryo's heteroplasmy levels were very high, the polar body genotype was significantly lower. For polar bodies with >60% mutant the polar body genotype was on the average 3.5% ± 5% higher than the embryo. For polar body genotypes that were <60%, the polar body heteroplasmy was 11.8% ± 7.8% lower than the embryo. Hence, it was concluded that in humans, polar body biopsy would not provide a reliable preimplantation test for human mtDNA diseases (Gigarel et al. 2011).
The second possibility could be sampling one of the blastomeres of an early cleavage stage embryo. Studies of rhesus macaque oocytes having a 50:50 heteroplasmy generated by karyoplast–cytoplast fusion, the heteroplasmy levels among primate blastomeres might be very different. In quantifying the divergence in percentage heteroplasmy, the coefficient of variance increased from 17.7% to 25.0% to 30.9% and the range increased from 13.3% to 25.3% to 43.2% in two-, four-, and eight-cell embryos, respectively. By the eight-cell stage, some embryo blastomeres differed by as much as 10% to 80%. Furthermore, the variance in heteroplasmy level among oocytes, blastomeres, and offspring was sufficiently great as to render absolute predictions on the transmission of heteroplasmy among cells and across generations unreliable (Lee et al. 2012).
In contrast to the macaque reconstituted cell studies, analysis of 3243A>G heteroplasmy levels in human embryos revealed that heteroplasmy levels of sister blastomeres at the two-cell stage were similar as were the heteroplasmy levels of embryos at 3 versus 5 d. Fetal extraembryonic or embryonic tissues of 12 fetuses from seven carrier mothers were also found to be relatively consistent from gestational d 120 to term. The heteroplasmy levels of the tissues of four fetuses were also found to be relatively uniform: 74% ± 0.7% (three tissues), 42% ± 0.8% (five tissues), 71% ± 2% (five tissues), and 78% ± 0.9% (seven tissues). Comparison of placental versus fetal tissue were also reported to harbor similar heteroplasmy levels: two cases encompassed tissue 3243A>G frequencies of 74%–75% and 78%, while a third ranged from 57% to 42%. While the pooled average of heteroplasmy levels between chorionic villi and amniotic fluid samples showed <10% variation, the heteroplasmy levels of individual trophoblast or amniocyte cells was quite variable. Thus, while there is marked variability in different embryos from the same woman, the tissues within an embryo can be relatively uniform even though the individual cells within a tissue can vary considerably (Monnot et al. 2011)
Initial studies have suggested that preimplantation embryo blastomeres have relatively similar mtDNA heteroplasmy levels, in contrast to the macaque report. A 30-year-old female carrier with a 35% 3243A>G mtDNA mutation heteroplasmy requested PGD after having a daughter diagnosed at 2 years of age with MELAs syndrome and 84% 3243A>G mutant mtDNAs. Eight unfertilized oocytes displayed mutation loads ranging from 9% to 90% and eight arrested embryos encompassing cleavage and morula stages had mutation loads ranging from 7% to 91%. Comparison of blastomere genotypes from five cleavage-stage embryos with mean heteroplasmy levels of 24.8% to 85.5% revealed standard deviations ranging from 0.5% to 2.9% (Treff et al. 2012).
Multiple additional studies have been conducted on the potential of PGD for women that are heteroplasmic for the MELAS tRNALeu(UUR) 3243A>G and the NARP/Leigh syndrome ATP6 8993T>G mutations. In a study of three 8993T>G NARP and 18 3243A>G MELAS preimplantation embryos, the heteroplasmy levels were reported as stable among the different blastomeres. Another study of 10 8993T>G NARP and nine 3243A>G MELAS fetuses concluded that the various tissues had similar heteroplasmy levels and that the heteroplasmy levels did not change with gestational age. From this series, eight children carrying less than 30% mutant mtDNA in the prenatal period were born and appear healthy at 18 months to 9 years of age (Monnot et al. 2009b).
In a second series of 3243A>G and 8993T>G patients, 33 preimplantation embryos at 3 to 5 d and 25 fetuses at gestational age of 10 to 30 wk were analyzed from 16 unrelated carrier females. Twenty percent of the embryos lacked the mtDNA mutation. Of the 80% that had the mutation, the percentage of heteroplasmy did not differ significantly among blastomeres of the embryos. While the NARP embryos tended to have either <25% or >95% mutant mtDNAs, the MELAS embryos harbored a wide range of heteroplasmy levels from 0% to 80%. Among the fetuses, those harboring the NARP mutation tended to have either <30% or >65% mutant mtDNAs while the MELAS embryos again had a continuous distribution of heteroplasmy levels, though different tissue samples from the same embryo had similar heteroplasmy levels (Monnot et al. 2009a).
A third cohort of 8993T>G and 3243A>G patients concluded that for 8993T>G mutational loads fetuses with <60% mutant could have a “favorable” outcome, but for mutational loads of >90% there was a poor prognosis. By contrast the clinical correlation between genotype and phenotype for the 3243A>G mutation was significantly less clear. In general, it was stated that “assessment of mtDNA mutation load in chorionic villi, amniotic cells, or blastocytes … show that the mutation percentage is stable at different times or in different cells of one embryo” (de Die-Smulders and Smeets 2009).
PGC was performed on 13 couples harboring the 8993T>G heteroplasmy mutation encompassing 20 embryos and 123 blastomeres. Of the 20 embryos, five had detectable 8993T>G heteroplasmy. The heteroplasmy levels of the blastomeres of each embryo were quite similar with ranges of 0%–8%, 4%–15%, 4%–15%, 9%–20%, 9%–20%, 10%–12%, 11%–22%, 18%–21%, 10%–12%, and 18%–21%. In contrast to the similarity of heteroplasmy levels of blastomeres within an embryo, the heteroplasmy levels among embryos varied considerably. Interestingly, the heteroplasmy levels of individual lymphocytes from a subject with a 44.3% whole blood heteroplasmy proved to be highly variable, ranging from 11% to 70% (Tajima et al. 2007).
For a healthy woman with 35% of the 8993T>G mutation in her blood and who had a daughter who died at 2 years with an mtDNA mutation load of >95%, chorionic villus sampling of two subsequent pregnancies revealed >95% 8993T>G mutation and were terminated. The woman then underwent two rounds of in vitro fertilization in association with PGD. Fifty-nine cells from 10 embryos were analyzed and the mutation load found to be consistent within the blastomeres of the individual embryos. Ten embryos and seven oocytes that were tested, 12 (70%) had very high mutation loads. Of the 10 embryos, six had high mutation loads, three had 30%–40% heteroplasmy, but one had a heteroplasmy level of 2.4%. This embryo was implanted and the pregnancy resulted in a healthy girl with a cord blood mutation load of 4% (Thorburn et al. 2009).
Therefore, in contrast to the macaque studies, the heteroplasmy levels of different blastomeres of human embryos appear to have relatively similar mtDNA heteroplasmy levels. Hence, blastomere biopsy and mtDNA heteroplasmy analysis from eight-cell embryos may provide a reasonable estimate of the heteroplasmy levels in the remaining blastomeres and reflect the heteroplasmy levels in the ensuing pregnancy amniocytes and cord blood samples (Poulton and Bredenoord 2010).
While mtDNA analysis of blastomere biopsies appears to be a promising approach for determining the heteroplasmy level of preimplantation embryos, blastomere biopsy removes a significant proportion of the embryo. An alternative could be to biopsy extraembryonic cells in the trophectoderm of the blastocyst. In one published case, a 30-year-old woman harboring 35% 3243A>G requested PGD following birth of a MELAS syndrome daughter. Six developmentally competent blastocysts with mean heteroplasmy values ranging from 24% to 91% were sampled. Three to four trophectoderm cells were obtained and compared to one to three inner cell mass samples. The standard deviations of the independent samples from the various trophoblasts ranged from 2.1% to 5.0% and the means of the trophoblast and inner cell mass samples were all within 3% of each other. Based on this data, one developmentally competent embryo with trophoblast heteroplasmy levels of 12% was chosen for implantation. This resulted in the successful delivery of a child for which a buccal cell DNA analysis at 1 mo revealed a heteroplasmy level of 15%. Subsequent tests at 5 and 12 mo of buccal swab and urine sediment DNA heteroplasmy by a commercial laboratory reported <10% mutation load (Treff et al. 2012). However, subsequent follow-up of this case has revealed significant problems in the placenta, infant, and child, and a mutation load in the child's blood and urine of 42%–52% (MJ Falk, pers. comm.).
Hence, there are still important ambiguities in the results on the effectiveness of human PGD in identifying and avoiding heteroplasmic mtDNA disease mutations. One recently recognized variable that might be relevant is that different pathogenic mtDNA mutations behave differently in both tissues and preimplantation embryos. Following in vitro fertilization embryos with mtDNA mutant genotypes of 70% 3243A>G, 90% 8344A>G, and 54% 9185T>G were not adversely affected in preimplantation development. However, the mtDNA copy number of 3243A>G embryos was found to increase from the germinal vesicle stage to the blastocyts ∼2.7-fold, with the amount of the increase correlating with the percent heteroplasmy (R2 = 0.47). Hence, for the 3243G mutation, the mtDNA copy number appears to partially compensate for the mtDNA defect. By contrast, preimplantation embryos harboring the 8344A>G mutation did not show any increase in mtDNA copy number, even though the mtDNA mutation load of the 8344A>G embryos was higher than that of the 3243A>G embryos: oocyte and embryo heteroplasmy levels for the 8344A>G were 81% ± 15% and 68% ± 17% versus 37%±29% and 36% ± 30% for the 3243A>G. Thus, the 3243A>G mutation must generate different physiological signals than the 8344A>G mutation (Monnot et al. 2013). How such mutation-specific effects have affected the reliability of PGD remains to be determined.
Germline Gene Therapy
While the PGD by blastomere sampling seems promising for determining the heteroplasmy levels of the 8993T>G NARP/Leigh syndrome mutation and possibly the 3243A>G MELAS mutation, there are still major limitations of this method. First, a woman may not produce oocytes that have low levels of mutant mtDNA, and, without the appropriate oocyte, her in vitro fertilization regime could be unproductive. Second, while the heteroplasmic levels of the ATP6 8993T>G L126R mutation may be consistent throughout the embryo and fetus, the potential for intertissue differences in heteroplasmy levels for the 3243A>G mutation may be significantly higher. Third, PGD is useless for homoplasmic women. Because of these limitations, there is increasing interest in developing methods to simply replace the mutant mtDNAs in preimplantation embryos by pronuclear or spindle transfer.
One early attempt at altering the mtDNA genotype in human oocytes was ooplasmic transfer. In this technique ∼20% of the cytoplasm of an oocyte from a one woman is injected into an oocyte from another woman along with her husband's sperm (intracytoplasmic sperm injection, ICSI), and the embryos implanted into a woman's uterus. This technique was first used in an attempt to rejuvenate the oocytes of older women with the cytoplasm from the oocytes of younger women thus increasing their fertility. Multiple children have been born following application of this procedure and are alive today. Some of these children have been shown to be heteroplasmic for both the maternal oocyte mtDNA and the cytoplasmic donor mtDNA (Barritt et al. 2001a,b).
Given that it has been reported that the mixing of two different mouse mtDNAs within the same female germline can lead to offspring with neuro-psychiatric defects (Sharpley et al. 2012), concern has been raised that randomly mixing mtDNAs of different mtDNA lineages might be deleterious. Also, the addition of 20% normal mtDNAs would be unlikely to be sufficient to reduce the risk of the child's developing the mtDNA disease.
Rather than the transfer of mitochondria and mtDNAs, a more promising approach would be to isolate the nucleus from the oocyte or zygote of a woman harboring a deleterious mtDNA mutation and to transfer it into an enucleated oocyte or zygote from a woman with normal mtDNAs (Wallace 1987). As a model for pronuclear transfer, in mouse embryos for the heteroplasmic mtDNA 4696 nt deletion, the zygote nuclei were removed by micropipette from the mtDNA mutant zygotes following disaggregation of the cytoskeleton and the resulting karyoplast (plasma membrane bound pronuclei) fused to an enucleated oocyte by electroshock. About 6% of the mtDNA was carried along with the nucleus. Because the average mutant mtDNA in the karyoplast donor was 35%, this resulted in zygotes containing ∼2% deleted mtDNAs. Eleven mice were born and at weaning their tail heteroplasmy levels were found to be 6%–12% mutant, mean 11%. Hence, zygote nuclear transfer resulted in a significant reduction in the transmission of the deleted mtDNAs. Unfortunately, in the case of the mtDNA deletion, the deletion levels progressively increased over the next 300 d to 5%–44% mutant, mean 23% (Sato et al. 2005).
Pronuclear transfer technology has also been extended to human preimplantation embryos. Using uni-pronuclear or tri-pronuclear human embryos that otherwise would have been destroyed, the cytoskeleton was disrupted with cytochalasin B and nocodazole, the pronuclei pinched off into a karyoplast, the karyoplast placed under the zona pellucida of an enucleated recipient zygote, and the two cells fused together with inactivated viral envelope proteins. About 22% of the resulting reconstituted embryos developed to the eight-cell stage and of those that received two pronuclei, 8.3% developed to the blastocyst stage, ∼50% of the developmental potential of unmanipulated embryos. Using mtDNA control region polymorphisms to monitor the fate of the two mtDNAs, the mean mtDNA carryover along with the pronuclear karyoplast was 8.1% ± 7.6% (n = 8). Variation in the percentage of mutant mtDNAs in the different blastomeres within one eight-cell embryo was found to range from 5.1% to 30.2% and within another four-cell embryo to range from 8% to 39%. Further refinement of the karyoplast isolation technique resulted in nine embryos with an average carryover of karyoplast mtDNAs of 1.68% ± 1.81% (n = 9). Four of these embryos lacked detectable karyoplast mtDNA. Of those embryos that harbored karyoplast mtDNA the maximum donor mtDNA was found in a nine-cell embryo in which seven blastomeres lacked detectable donor mtDNAs, while the remaining two blastomeres harbored 6.1% and 11.4% donor mtDNAs (Craven et al. 2010).
Interestingly, the spread in blastomere heteroplasmy levels in the first round pronuclear transfer embryos (Craven et al. 2010) was reminiscent of the wide range of blastomere mtDNA heteroplasmy genotypes reported for the macaque embryo fusion experiments. This was much greater than the range of heteroplasmy levels observed in embryos generated from NZB/BALB heteroplasmic mice or in the blastomeres of heteroplasmic human preimplantation embryos. One possible explanation for this difference is that the oocyte cytoplasm may be relatively viscous resulting in the nonrandom partitioning of the mitochondria in different regions of the single-cell embryo. Depending on the angles of the initial cleavages, the mtDNAs of the karyoplast and the cytoplast could be asymmetrically distributed into daughter blastomeres resulting in differing heteroplasmy levels. By contrast, oocytes and embryos derived from heteroplasmic germline cells of the two mtDNAs could be more evenly distributed throughout the cytoplasm such that cleavages would generate blastomeres with similar mtDNA heteroplasmy levels.
One concern with the pronuclear transfer procedure is that it destroys an embryo. This concern has been overcome by transfer of the chromosomes from the prefertilization mtDNA mutant oocyte to an enucleated oocyte with normal mtDNA. In this case, however, the chromosomes of the mature oocyte are arrested in meiotic metaphase II. Hence, this method involves the physical transfer of the meiotic II spindle.
In the spindle transfer technique, the metaphase II (MII) spindle of the oocyte is visualized in the oocytes using the polarizing microscope and is removed by aspiration with a micropipette within a bleb of the ooycte cytoplasm and surrounded by the cell membrane. The spindle karyoplasts encompass ∼1.5% of the volume of an enucleated oocyte cytoplast. Because the spindle is relatively free of surrounding cytoplasm and mitochondria very little mtDNA is transferred. The karyoplast is treated with a Sendai virus-derived fusigen and placed in the perivitelline space of the cytoplast opposite the first polar body where it fuses. The resulting reconstituted oocyte can then be fertilized by intracytoplasmic sperm injection (ICSI).
This technology was first developed and applied to the oocytes of rhesus macaque monkeys. It resulted in preimplantation embryos that developed normally to generate pluripotent stem cells and when blastocysts of four- to eight-cell cleavage embryos were transferred into the reproductive tract of females, one pair of twins and two singleton infants were born. These four monkeys had the maternal chromosomes of the spindle donor but the mtDNA of the cytoplast donor. No spindle donor oocyte mtDNA was detected at a detection sensitivity of ∼3% (Tachibana et al. 2009). Three years after birth, the four-spindle transfer monkeys were phenotypically like control monkeys and showed no significant change in mtDNA carryover in blood and skin samples (Tachibana et al. 2013). Subsequent analysis revealed that the macaque karyoplasts carried 3538 ± 2310 mtDNAs while the cytoplast contributed 576,948 ± 209,069 mtDNA (n = 9) resulting in ∼0.6% heteroplasmy. Of 102 spindle transfer oocytes 62% developed into blastocyts. Two female embryos generated from spindle transfer oocytes were permitted to develop in utero for 135 d and then analyzed for mtDNA heteroplasmy. Of 24 oocytes isolated from the ovaries of the female fetuses 22 had no detectable spindle donor mtDNA while one from each fetus was heteroplasmic with 16% and 14% heteroplasmy, respectively (Lee et al. 2012).
One of the technological limitations of spindle transfer is that both the spindle donor karyoplast and the mtDNA donor cytoplast must be ready at the same time, requiring paired induction of ovulation of two women, which can be quite difficult. Studies on macaque oocytes have shown that the spindle donor oocyte can be frozen using a vitrification procedure. These oocytes can then be thawed, the spindle removed, and transferred to a fresh enucleated oocyte without diminution of fertilization. Blastocyst formation efficiency was 88% and 68% of that of controls. Vitrification of the cytoplasm donor oocytes, by contrast, blocked development (Tachibana et al. 2013). Hence, spindle transfer has the potential of eliminating most of the mutant mtDNAs from the maternal lineage, but low levels of heteroplasmy can still resurface during germline transmission.
To assess the feasibility of moving this technology into the clinic, spindle transplantation has been applied to human oocytes. Of 64 spindle-transfer oocytes, 94% survived ICSI and 75% formed pronuclei. However, in contrast to macaque results only about half (48%) formed two normal pronuclei and two polar bodies. The remainders were abnormal. Only 13% of the nonspindle transfer embryos were abnormal. Of the normal spindle transfer embryos, 76% went on to form blastocysts. From the 16 blastocysts, nine formed ESCs (56%) and these were indistinguishable from the controls. The mean karyoplast derived mtDNA levels were very low, 0.5% ± 04% for spindle transfer oocytes and embryos and 0.6% ± 0.9% for human ESCs (Tachibana et al. 2013).
In an independent study using parthenogenetically activated embryos that have undergone endoreduplication to establish a diploid karyotype, spindle transfer was also analyzed for its capacity to replace the mtDNA of the spindle donor mother. As before, the spindle was removed from the oocyte in a bleb of cytoplasm, the resulting karyoplast placed under the zona pellucida of a comparably enucleated oocyte, and the karyoplast fused to the cytoplast by either Sendai viral fusigen or electric shock. Sendai fusion proved preferable because electroshock was prone to induce premature oocyte activation. This study determined that the number of mtDNAs carried by the karyoplast was 1129 ± 785 or 0.36% of the total mtDNA in a metaphase II oocyte, which contains 311,146 ± 206,521 mtDNAs. An important innovation of this study was the realization that partial disaggregation of the karyoplast spindle complex by a 2-h exposure to room temperature or prior vitrification followed by partial reaggregation of the spindle at 37ºC significantly increased the efficiency of subsequent second polar body extrusion and embryo development. Several embryos were permitted to develop into blastocysts and from these ESC lines with normal karyotypes were derived. These were capable of differentiating into pancreatic cells, neurons, fibroblasts, and cardiomyocytes. Analysis of the mtDNA heteroplasmy level for all preimplantation embryos studied was 0.31% ± 0.27%. Of the three ESC lines that were generated, two lacked detectable karyoplast mtDNA while one had 2.79% ± 0.27% karyoplast mtDNA. The heteroplasmic ESC line subsequently lost the karyoplast mtDNA, the spindle donor mtDNAs becoming undetectable after passage 14 and remained so in 40 clones. Fibroblasts derived from one of the spindle transfer stem cell lines were converted back to induced pluripotent stem (iPS) cells, yet the mtDNAs from the karyoplast did not reappear. Finally, analysis of the mitochondrial respiratory complexes, respiration, and media acidification of one of the stem cell lines revealed that it was comparable to that of embryonic stem cells and iPS cells that had not undergone spindle transfer (Paull et al. 2013).
While both the pronuclear and spindle transfer techniques have been refined to transfer a minute amount of nuclear donor mtDNA, the possibility still remains that heteroplasmy can resurface in subsequent ESCs or maternal offspring (Paull et al. 2013). While low-level heteroplasmy of a pathogenic mutation would probably be masked by the predominance of normal mtDNAs, minimizing the risk of a clinical phenotype, there is a risk of incompatibility between the two mtDNA haplogroup lineages (Sharpley et al. 2012). This concern could be mitigated by screening potential oocyte mtDNA donor women for their mtDNA haplogroups. Then the mtDNA haplogroup of the woman harboring the deleterious mtDNA mutation could be matched with that of an oocyte donor; only matching mtDNA haplogroups would then be used.
Alternatives and Ethical Considerations
It has been argued that it may be unethical to manipulate human embryos in an effort to remove the risk of mtDNA disease. Certainly, all appropriate preclinical tests must be performed in an effort to reduce the risk for adverse outcomes in developing new human therapies. The question remains is human mtDNA germline gene therapy “ethical.” To answer this question, we must factor into our cost-benefit analysis the implications not only for society but also for the family and their prospective children. Women harboring severely pathogenic mtDNA mutations want to have healthy children that are free of pain and can live productive lives. Currently children with high heteroplasmy levels of severely deleterious mtDNA mutations experience the progressive loss of mental and physical capabilities, often experiencing unremitting discomfort and pain and many will ultimately progress to premature death. For such families, this can mean repeated medical crises, numerous emergency room admissions, devastating medical bills, the loss of time, affection, and resources for other family members, ending only by the death of the child. Therefore, if the society is to rule on the morality of helping women have healthy children, than the families who have suffered the ravages of mtDNA disease and personally paid the cost should be given a major voice in the decision.
ACKNOWLEDGMENTS
This work is supported by National Institutes of Health grants NS21328, NS070298, AG24373, and DK73691 and by Simons Foundation grant 205844 awarded to D.C.W.
REFERENCES
*Reference is also in this collection.
- Ashley MV, Laipis PJ, Hauswirth WW 1989. Rapid segregation of heteroplasmic bovine mitochondria. Nucleic Acids Res 17: 7325–7331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu GJ, Feigelson ED 2006. Astrostatistics: Goodness-of-fit and all that! Astronomical Data Analysis Software and Systems XV ASP Conference Series 351: 127 [Google Scholar]
- Ballinger SW, Shoffner JM, Hedaya EV, Trounce I, Polak MA, Koontz DA, Wallace DC 1992. Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nat Genet 1: 11–15 [DOI] [PubMed] [Google Scholar]
- Ballinger SW, Shoffner JM, Gebhart S, Koontz DA, Wallace DC 1994. Mitochondrial diabetes revisited. Nat Genet 7: 458–459 [DOI] [PubMed] [Google Scholar]
- Balloux F, Handley LJ, Jombart T, Liu H, Manica A 2009. Climate shaped the worldwide distribution of human mitochondrial DNA sequence variation. Proc R Soc B 276: 3447–3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barritt J, Willadsen S, Brenner C, Cohen J 2001a. Cytoplasmic transfer in assisted reproduction. Hum Reprod Update 7: 428–435 [DOI] [PubMed] [Google Scholar]
- Barritt JA, Brenner CA, Malter HE, Cohen J 2001b. Mitochondria in human offspring derived from ooplasmic transplantation. Hum Reprod 16: 513–516 [DOI] [PubMed] [Google Scholar]
- Battersby BJ, Shoubridge EA 2001. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum Mol Genet 10: 2469–2479 [DOI] [PubMed] [Google Scholar]
- Battersby BJ, Shoubridge EA 2007. Reactive oxygen species and the segregation of mtDNA sequence variants. Nat Genet 39: 571–572; author reply 572 [DOI] [PubMed] [Google Scholar]
- Battersby BJ, Loredo-Osti JC, Shoubridge EA 2003. Nuclear genetic control of mitochondrial DNA segregation. Nat Genet 33: 183–186 [DOI] [PubMed] [Google Scholar]
- Battersby BJ, Redpath ME, Shoubridge EA 2005. Mitochondrial DNA segregation in hematopoietic lineages does not depend on MHC presentation of mitochondrially encoded peptides. Hum Mol Genet 14: 2587–2594 [DOI] [PubMed] [Google Scholar]
- Baudouin SV, Saunders D, Tiangyou W, Elson JL, Poynter J, Pyle A, Keers S, Turnbull DM, Howell N, Chinnery PF 2005. Mitochondrial DNA and survival after sepsis: A prospective study. Lancet 366: 2118–2121 [DOI] [PubMed] [Google Scholar]
- Bergstrom CT, Pritchard J 1998. Germline bottlenecks and the evolutionary maintenance of mitochondrial genomes. Genetics 149: 2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc H, Adams CW, Wallace DC 1981a. Different nucleotide changes in the large rRNA gene of the mitochondrial DNA confer chloramphenicol resistance on two human cell lines. Nucleic Acids Res 9: 5785–5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc H, Wright CT, Bibb MJ, Wallace DC, Clayton DA 1981b. Mitochondrial DNA of chloramphenicol-resistant mouse cells contains a single nucleotide change in the region encoding the 3′ end of the large ribosomal RNA. Proc Natl Acad Sci 78: 3789–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blok RB, Gook DA, Thorburn DR, Dahl HH 1997. Skewed segregation of the mtDNA nt 8993 (T→G) mutation in human oocytes. Am J Hum Genet 60: 1495–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WM, Prager EM, Wan A, Wilson AC 1982. Mitochondrial DNA sequences in primates: Tempo and mode of evolution. J Mol Evol 18: 225–239 [DOI] [PubMed] [Google Scholar]
- Brown MD, Torroni A, Reckord CL, Wallace DC 1995. Phylogenetic analysis of Leber's hereditary optic neuropathy mitochondrial DNA's indicates multiple independent occurrences of the common mutations. Hum Mutat 6: 311–325 [DOI] [PubMed] [Google Scholar]
- Brown MD, Sun F, Wallace DC 1997. Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. Am J Hum Genet 60: 381–387 [PMC free article] [PubMed] [Google Scholar]
- Brown DT, Samuels DC, Michael EM, Turnbull DM, Chinnery PF 2001. Random genetic drift determines the level of mutant mtDNA in human primary oocytes. Am J Hum Genet 68: 533–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MD, Starikovskaya E, Derbeneva O, Hosseini S, Allen JC, Mikhailovskaya IE, Sukernik RI, Wallace DC 2002. The role of mtDNA background in disease expression: A new primary LHON mutation associated with Western Eurasian haplogroup J. Hum Genet 110: 130–138 [DOI] [PubMed] [Google Scholar]
- Bunn CL, Wallace DC, Eisenstadt JM 1974. Cytoplasmic inheritance of chloramphenicol resistance in mouse tissue culture cells. Proc Natl Acad Sci 71: 1681–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn CL, Wallace DC, Eisenstadt JM 1977. Mitotic segregation of cytoplasmic determinants for chloramphenicol resistance in mammalian cells. I: Fusions with mouse cell lines. Somatic Cell Genet 3: 71–92 [DOI] [PubMed] [Google Scholar]
- Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J, Yonekawa H 2007. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet 39: 386–390 [DOI] [PubMed] [Google Scholar]
- Cao L, Shitara H, Sugimoto M, Hayashi J, Abe K, Yonekawa H 2009. New evidence confirms that the mitochondrial bottleneck is generated without reduction of mitochondrial DNA content in early primordial germ cells of mice. PLoS Genet 5: e1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling PJ, Cree LM, Chinnery PF 2011. The implications of mitochondrial DNA copy number regulation during embryogenesis. Mitochondrion 11: 686–692 [DOI] [PubMed] [Google Scholar]
- Chapman R, Stephens J, Lansman R, Avise J 1982. Models of mitochondrial DNA transmission genetics and evolution in higher eucaryotes. Genet Res 40: 41–57 [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D 2010. Elements of evolutionary genetics. Roberts and Company, Greenwood Village, CO [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC 2010. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141: 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Elliott HR, Syed A, Rothwell PM 2010. Mitochondrial DNA haplogroups and risk of transient ischaemic attack and ischaemic stroke: A genetic association study. Lancet Neurol 9: 498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Elliott HR, Hudson G, Samuels DC, Relton CL 2012. Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol 41: 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomyn A 1996. Platelet-mediated transformation of human mitochondrial DNA-less cells. Methods Enzymol 264: 334–339 [DOI] [PubMed] [Google Scholar]
- Coskun PE, Wyrembak J, Derbereva O, Melkonian G, Doran E, Lott IT, Head E, Cotman CW, Wallace DC 2010. Systemic mitochondrial dysfunction and the etiology of Alzheimer's disease and Down syndrome dementia. J Alzheimers Dis 20: S293–S310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, et al. 2010. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 465: 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, Dahl HH, Chinnery PF 2008. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet 40: 249–254 [DOI] [PubMed] [Google Scholar]
- Dean NL, Battersby BJ, Ao A, Gosden RG, Tan SL, Shoubridge EA, Molnar MJ 2003. Prospect of preimplantation genetic diagnosis for heritable mitochondrial DNA diseases. Mol Hum Reprod 9: 631–638 [DOI] [PubMed] [Google Scholar]
- de Die-Smulders C, Smeets H 2009. The challenge of prenatal and preimplantation genetic diagnosis of mitochondrial disorders. European Human Genetics Conference, Abstract S09.3, Vienna Austria, May 23–26, 2009. Eur J Hum Genet 17: 9–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Stordeur E, Solignac M, Monnerot M, Mounolou JC 1989. The generation of transplasmic Drosophila simulans by cytoplasmic injection: Effects of segregation and selection on the perpetuation of mitochondrial DNA heteroplasmy. Mol Gen Genet 220: 127–132 [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF 2008. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet 83: 254–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Waymire K, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, MacGregor GR, Wallace DC 2008. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319: 958–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Lin CS, Potluri P, Procaccio V, Wallace DC 2012. MtDNA lineage analysis of mouse L cell lines reveals the accumulation of multiple mtDNA mutants and intermolecular recombination. Genes Dev 26: 384–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielder TJ, Yi CS, Masumi J, Waymire KG, Chen HW, Wang S, Shi KX, Wallace DC, MacGregor GR 2012. Comparison of male chimeric mice generated from microinjection of JM8.N4 embryonic stem cells into C57BL/6 J and C57BL/6NTac blastocysts. Transgenic Res 21: 1149–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyer C, Cree LM, Mourier A, Stewart JB, Koolmeister C, Milenkovic D, Wai T, Floros VI, Hagstrom E, Chatzidaki EE, et al. 2012. Variation in germline mtDNA heteroplasmy is determined prenatally but modified during subsequent transmission. Nat Genet 44: 1282–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuku N, Park KS, Yamada Y, Nishigaki Y, Cho YM, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, et al. 2007. Mitochondrial haplogroup N9a confers resistance against type 2 diabetes in Asians. Am J Hum Genet 80: 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparre G, Hervouet E, de Laplanche E, Demont J, Pennisi LF, Colombel M, Mege-Lechevallier F, Scoazec JY, Bonora E, Smeets R, et al. 2008. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum Mol Genet 17: 986–995 [DOI] [PubMed] [Google Scholar]
- Gigarel N, Hesters L, Samuels DC, Monnot S, Burlet P, Kerbrat V, Lamazou F, Benachi A, Frydman R, Feingold J, et al. 2011. Poor correlations in the levels of pathogenic mitochondrial DNA mutations in polar bodies versus oocytes and blastomeres in humans. Am J Hum Genet 88: 494–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles RE, Stroynowski I, Wallace DC 1980. Characterization of mitochondrial DNA in chloramphenicol-resistant interspecific hybrids and a cybrid. Somatic Cell Genet 6: 543–554 [DOI] [PubMed] [Google Scholar]
- Gilkerson RW, Schon EA 2008. Nucleoid autonomy: An underlying mechanism of mitochondrial genetics with therapeutic potential. Commun Integr Biol 1: 34–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson RW, Schon EA, Hernandez E, Davidson MM 2008. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J Cell Biol 181: 1117–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson RW, De Vries RL, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, Przedborski S, Schon EA 2012. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet 21: 978–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez R, O'Keeffe T, Chang L, Huebinger R, Minei J, Barber R 2009. Association of mitochondrial allele 4216C with increased risk for complicated sepsis and death after traumatic injury. J Trauma 66: 850–857; discussion 857–858 [DOI] [PubMed] [Google Scholar]
- Gomez-Duran A, Pacheu-Grau D, Lopez-Gallardo E, Diez-Sanchez C, Montoya J, Lopez-Perez MJ, Ruiz-Pesini E 2010. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum Mol Genet 19: 3343–3353 [DOI] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S 1990. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348: 651–653 [DOI] [PubMed] [Google Scholar]
- Goto H, Dickins B, Afgan E, Paul IM, Taylor J, Makova KD, Nekrutenko A 2011. Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study. Genome Biol 12: R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M 2010. What have we learned from Drosophila models of Parkinson's disease? Prog Brain Res 184: 3–16 [DOI] [PubMed] [Google Scholar]
- Hauswirth WW, Laipis PJ 1982. Mitochondrial DNA polymorphism in a maternal lineage of Holstein cows. Proc Natl Acad Sci 79: 4686–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Morgan-Hughes JA 1988. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331: 717–719 [DOI] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA 1990. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet 46: 428–433 [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ, Epstein CJ 2006. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet 15: 1187–1194 [DOI] [PubMed] [Google Scholar]
- Inoue K, Nakada K, Ogura A, Isobe K, Goto Y, Nonaka I, Hayashi J-I 2000. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet 26: 176–181 [DOI] [PubMed] [Google Scholar]
- Jansen RP, de Boer K 1998. The bottleneck: Mitochondrial imperatives in oogenesis and ovarian follicular fate. Mol Cell Endocrinol 145: 81–88 [DOI] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Fu K, Shoubridge EA 1996. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet 14: 146–151 [DOI] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Shoubridge EA 1997. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet 16: 93–95 [DOI] [PubMed] [Google Scholar]
- Ji F, Sharpley MS, Derbeneva O, Alves LS, Qian P, Wang Y, Chalkia D, Lvova M, Xu J, Yao W, et al. 2012. Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc Natl Acad Sci 109: 7391–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen R, Battersby BJ 2013. Insight into mammalian mitochondrial DNA segregation. Ann Med 45: 149–155 [DOI] [PubMed] [Google Scholar]
- Jokinen R, Marttinen P, Sandell HK, Manninen T, Teerenhovi H, Wai T, Teoli D, Loredo-Osti JC, Shoubridge EA, Battersby BJ 2010. Gimap3 regulates tissue-specific mitochondrial DNA segregation. PLoS Genet 6: e1001161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen R, Junnila H, Battersby BJ 2011. Gimap3: A foot-in-the-door to tissue-specific regulation of mitochondrial DNA genetics. Small GTPases 2: 31–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasashima K, Sumitani M, Endo H 2011. Human mitochondrial transcription factor A is required for the segregation of mitochondrial DNA in cultured cells. Exp Cell Res 317: 210–220 [DOI] [PubMed] [Google Scholar]
- Kazuno AA, Munakata K, Nagai T, Shimozono S, Tanaka M, Yoneda M, Kato N, Miyawaki A, Kato T 2006. Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PLoS Genet 2: e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrapko K 2008. Two ways to make an mtDNA bottleneck. Nat Genet 40: 134–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusnutdinova E, Gilyazova I, Ruiz-Pesini E, Derbeneva O, Khusainova R, Khidiyatova I, Magzhanov R, Wallace DC 2008. A mitochondrial etiology of neurodegenerative diseases: Evidence from Parkinson's disease. Ann NY Acad Sci 1147: 1–20 [DOI] [PubMed] [Google Scholar]
- Kimura M 1955. Solution of a process of random genetic drift with a continuous model. Proc Natl Acad Sci 41: 144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, Attardi G 1989. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 246: 500–503 [DOI] [PubMed] [Google Scholar]
- Koehler CM, Lindberg GL, Brown DR, Beitz DC, Freeman AE, Mayfield JE, Myers AM 1991. Replacement of bovine mitochondrial DNA by a sequence variant within one generation. Genetics 129: 247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman WJ, Willems PH, Smeitink JA 2012. Monogenic mitochondrial disorders. N Engl J Med 366: 1132–1141 [DOI] [PubMed] [Google Scholar]
- Krakauer DC, Mira A 1999. Mitochondria and germ-cell death. Nature 400: 125–126 [DOI] [PubMed] [Google Scholar]
- Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P 2011. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 32: 157–164 [DOI] [PubMed] [Google Scholar]
- Kukat C, Wurm CA, Spahr H, Falkenberg M, Larsson NG, Jakobs S 2011. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci 108: 13534–13539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Ma H, Juanes RC, Tachibana M, Sparman M, Woodward J, Ramsey C, Xu J, Kang EJ, Amato P, et al. 2012. Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell Rep 1: 506–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard WR, Sorensen MV, Galloway VA, Spencer GJ, Mosher MJ, Osipova L, Spitsyn VA 2002. Climatic influences on basal metabolic rates among circumpolar populations. Am J Human Biol 14: 609–620 [DOI] [PubMed] [Google Scholar]
- Liang M, Guan M, Zhao F, Zhou X, Yuan M, Tong Y, Yang L, Wei QP, Sun YH, Lu F, et al. 2009. Leber's hereditary optic neuropathy is associated with mitochondrial ND1 T3394C mutation. Biochem Biophys Res Commun 383: 286–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malena A, Loro E, Di Re M, Holt IJ, Vergani L 2009. Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum Mol Genet 18: 3407–3416 [DOI] [PubMed] [Google Scholar]
- Mehrazin M, Shanske S, Kaufmann P, Wei Y, Coku J, Engelstad K, Naini A, De Vivo DC, DiMauro S 2009. Longitudinal changes of mtDNA A3243G mutation load and level of functioning in MELAS. Am J Med Genet A 149A: 584–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini E, Mondragon-Palomino M, Procaccio V, Gaut B, Wallace DC 2006. Adaptive selection of mitochondrial complex I subunits during primate radiation. Gene 378: 11–18 [DOI] [PubMed] [Google Scholar]
- Mitchell P 1961. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191: 144–148 [DOI] [PubMed] [Google Scholar]
- MITOMAP 2012. A human mitochondrial genome database. http://www.mitomap.org [Google Scholar]
- Monnot S, Gigarel N, Frydman N, Burlet P, Gobin S, Sinico M, Bonniere M, Rio M, Rotig A, Frydman R, et al. 2009a. NARP and MELAS mutations differentially impact mitochondrial DNA segregation throughout human embryofetal development. http://www.ashg.org/2009meeting/abstracts/fulltext/f21163htm [Google Scholar]
- Monnot S, Gigarel N, Hesters L, Burlet P, Benachi A, Dumez Y, Tachdjian G, Rotig A, Frydman R, Munnich A, et al. 2009b. The challenge of prenatal and preimplantation genetic diagnosis of mitochondrial DNA disorders. European Human Genetics Conference, Abstract C07.4, Vienna Austria, May 23–26, 2009. Eur J Hum Genet 17: 24 [Google Scholar]
- Monnot S, Gigarel N, Samuels DC, Burlet P, Hesters L, Frydman N, Frydman R, Kerbrat V, Funalot B, Martinovic J, et al. 2011. Segregation of mtDNA throughout human embryofetal development: m.3243A>G as a model system. Hum Mutat 32: 116–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnot S, Samuels DC, Hesters L, Frydman N, Gigarel N, Burlet P, Kerbrat V, Lamazou F, Frydman R, Benachi A, et al. 2013. Mutation dependence of the mitochondrial DNA copy number in the first stages of human embryogenesis. Hum Mol Genet 22: 1867–1872 [DOI] [PubMed] [Google Scholar]
- Moreno-Loshuertos R, Acin-Perez R, Fernandez-Silva P, Movilla N, Perez-Martos A, Rodriguez de Cordoba S, Gallardo ME, Enriquez JA 2006. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat Genet 38: 1261–1268 [DOI] [PubMed] [Google Scholar]
- Nakada K, Sato A, Yoshida K, Morita T, Tanaka H, Inoue SI, Yonekawa H, Hayashi JI 2006. Mitochondria-related male infertility. Proc Natl Acad Sci 103: 15148–15153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ 2009. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 5: 706–708 [DOI] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Narendra D, Walker JE, Youle R 2012. Mitochondrial quality control mediated by PINK1 and Parkin: Links to parkinsonism. Cold Spring Harb Perspect Biol 4: a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckelmann N, Li K, Wade RP, Shuster R, Wallace DC 1987. cDNA sequence of a human skeletal muscle ADP/ATP translocator: lack of a leader peptide, divergence from a fibroblast translocator cDNA, and coevolution with mitochondrial DNA genes. Proc Natl Acad Sci 84: 7580–7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et al. 2012. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver NA, Wallace DC 1982. Assignment of two mitochondrially synthesized polypeptides to human mitochondrial DNA and their use in the study of intracellular mitochondrial interaction. Mol Cell Biol 2: 30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver NA, Greenberg BD, Wallace DC 1983. Assignment of a polymorphic polypeptide to the human mitochondrial DNA unidentified reading frame 3 gene by a new peptide mapping strategy. J Biol Chem 258: 5834–5839 [PubMed] [Google Scholar]
- Olivo PD, Van de Walle MJ, Laipis PJ, Hauswirth WW 1983. Nucleotide sequence evidence for rapid genotypic shifts in the bovine mitochondrial DNA D-loop. Nature 306: 400–402 [DOI] [PubMed] [Google Scholar]
- Ortiz RG, Newman NJ, Shoffner JM, Kaufman AE, Koontz DA, Wallace DC 1993. Variable retinal and neurologic manifestations in patients harboring the mitochondrial DNA 8993 mutation. Arch Ophthalmol 111: 1525–1530 [DOI] [PubMed] [Google Scholar]
- Paull D, Emmanuele V, Weiss KA, Treff N, Stewart L, Hua H, Zimmer M, Kahler DJ, Goland RS, Noggle SA, et al. 2013. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493: 632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne BA, Wilson IJ, Yu-Wai-Man P, Coxhead J, Deehan D, Horvath R, Taylor RW, Samuels DC, Santibanez-Koref M, Chinnery PF 2013. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet 22: 384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poe BG 3rd, Duffy CF, Greminger MA, Nelson BJ, Arriaga EA 2010. Detection of heteroplasmy in individual mitochondrial particles. Anal Bioanal Chem 397: 3397–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potluri P, Davila A, Ruiz-Pesini E, Mishmar D, O'Hearn S, Hancock S, Simon MC, Scheffler I, Wallace DC, Procaccio V 2009. A novel NDUFA1 mutation leads to a progressive mitochondrial complex I-specific neurodegenerative disease. Mol Genet Metab 96: 189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton J, Bredenoord AL 2010. Applying pre-implantation genetic diagnosis to mtDNA diseases: Implications of scientific advances. 174th ENMC International Workshop, Naarden, The Netherlands, March 19–21, 2010. Neuromuscul Disord 20: 559–563 [DOI] [PubMed] [Google Scholar]
- Rajasimha HK, Chinnery PF, Samuels DC 2008. Selection against pathogenic mtDNA mutations in a stem cell population leads to the loss of the 3243A→G mutation in blood. Am J Hum Genet 82: 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebelo AP, Dillon LM, Moraes CT 2011. Mitochondrial DNA transcription regulation and nucleoid organization. J Inherit Metab Dis 34: 941–951 [DOI] [PubMed] [Google Scholar]
- Rollins B, Martin MV, Sequeira PA, Moon EA, Morgan LZ, Watson SJ, Schatzberg A, Akil H, Myers RM, Jones EG, et al. 2009. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS ONE 4: e4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Wallace DC 2006. Evidence for adaptive selection acting on the tRNA and rRNA genes of the human mitochondrial DNA. Hum Mutat 27: 1072–1081 [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC 2004. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303: 223–226 [DOI] [PubMed] [Google Scholar]
- Sadun AA, Carelli V, Salomao SR, Berezovsky A, Quiros PA, Sadun F, DeNegri AM, Andrade R, Moraes M, Passos A, et al. 2003. Extensive investigation of a large Brazilian pedigree of 11778/haplogroup J Leber hereditary optic neuropathy. Am J Ophthalmol 136: 231–238 [DOI] [PubMed] [Google Scholar]
- Sadun AA, La Morgia C, Carelli V 2011. Leber's Hereditary Optic Neuropathy. Curr Treat Options Neurol 13: 109–117 [DOI] [PubMed] [Google Scholar]
- Samuels DC, Wonnapinij P, Cree LM, Chinnery PF 2010. Reassessing evidence for a postnatal mitochondrial genetic bottleneck. Nat Genet 42: 471–473 [DOI] [PubMed] [Google Scholar]
- Samuels DC, Wonnapinij P, Chinnery PF 2013. Preventing the transmission of pathogenic mitochondrial DNA mutations: Can we achieve long-term benefits from germ-line gene transfer? Hum Reprod 28: 554–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Kono T, Nakada K, Ishikawa K, Inoue S, Yonekawa H, Hayashi J 2005. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc Natl Acad Sci 102: 16765–16770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Nakada K, Shitara H, Kasahara A, Yonekawa H, Hayashi J 2007. Deletion-mutant mtDNA increases in somatic tissues but decreases in female germ cells with age. Genetics 177: 2031–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF 2004. The epidemiology of mitochondrial disorders–past, present and future. Biochim Biophys Acta 1659: 115–120 [DOI] [PubMed] [Google Scholar]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, Chinnery PF, Turnbull DM 2008. Prevalence of mitochondrial DNA disease in adults. Ann Neurol 63: 35–39 [DOI] [PubMed] [Google Scholar]
- Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA 2012. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci 109: 14508–14513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpley MS, Marciniak C, Eckel-Mahan K, McManus MJ, Crimi M, Waymire K, Lin CS, Masubuchi S, Friend N, Koike M, et al. 2012. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151: 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffner JM, Lott MT, Voljavec AS, Soueidan SA, Costigan DA, Wallace DC 1989. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc Natl Acad Sci 86: 7952–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC 1990. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 61: 931–937 [DOI] [PubMed] [Google Scholar]
- Snodgrass JJ, Leonard WR, Tarskaia LA, Alekseev VP, Krivoshapkin VG 2005. Basal metabolic rate in the Yakut (Sakha) of Siberia. Am J Human Biol 17: 155–172 [DOI] [PubMed] [Google Scholar]
- Snodgrass JJ, Leonard WR, Sorensen MV, Tarskaia LA, Mosher MJ 2008. The influence of basal metabolic rate on blood pressure among indigenous Siberians. Am J Phys Anthropol 137: 145–155 [DOI] [PubMed] [Google Scholar]
- Solignac M, Genermont J, Monnerot M, Mounolou JC 1984. Genetics of mitochondria in Drosophila mtDNA inheritance in heteroplasmic strains of Drosophila mauritiana. Mol Gen Genet 197: 183–188 [Google Scholar]
- Stephens M 1974. EDF statistics for goodness of fit and some comparisons. J Am Stat Assoc 69: 730–737 [Google Scholar]
- Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson NG 2008. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol 6: e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KA, Dubiner L, Simon M, Zaragoza M, Sengupta PP, Li P, Narula N, Dreike S, Platt J, Procaccio V, et al. 2013. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci 110: 3253–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ 2010. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci 107: 11835–11840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, Li Y, Ramsey C, Kolotushkina O, Mitalipov S 2009. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 461: 367–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner-Hedges R, Kang E, Lee HS, et al. 2013. Towards germline gene therapy of inherited mitochondrial diseases. Nature 493: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima H, Sueoka K, Moon SY, Nakabayashi A, Sakurai T, Murakoshi Y, Watanabe H, Iwata S, Hashiba T, Kato S, et al. 2007. The development of novel quantification assay for mitochondrial DNA heteroplasmy aimed at preimplantation genetic diagnosis of Leigh encephalopathy. J Assist Reprod Genet 24: 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Slatkin M 1983. Evolutionary dynamics of extranuclear genes. Genet Res 42: 257–268 [Google Scholar]
- Tatuch Y, Christodoulou J, Feigenbaum A, Clarke JTR, Wherret J, Smith C, Rudd N, Petrova-Benedict R, Robinson BH 1992. Heteroplasmic mtDNA mutation (T-G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am J Hum Genet 50: 852–858 [PMC free article] [PubMed] [Google Scholar]
- Thorburn DR, Dahl HH 2001. Mitochondrial disorders: Genetics, counseling, prenatal diagnosis and reproductive options. Am J Med Genet 106: 102–114 [DOI] [PubMed] [Google Scholar]
- Thorburn D, Wilton L, Stock-Myer S 2009. Healthy baby girl born following preimplantation genetic diagnosis for mitochondrial DNA m.8993T>G mutation. 11th International Congress of Inborn Errors of Metabolism, Abstract 117, Platform Presentation 3: Mitochondrial Disorders. Mol Genet Metab 98: 5–6 [Google Scholar]
- Torroni A, Petrozzi M, D'Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, et al. 1997. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet 60: 1107–1121 [PMC free article] [PubMed] [Google Scholar]
- Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, et al. 2005. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6 J mice. Diabetologia 48: 675–686 [DOI] [PubMed] [Google Scholar]
- Treff NR, Campos J, Tao X, Levy B, Ferry KM, Scott RT Jr 2012. Blastocyst preimplantation genetic diagnosis (PGD) of a mitochondrial DNA disorder. Fertil Steril 98: 1236–1240 [DOI] [PubMed] [Google Scholar]
- van den Ouweland JM, Lemkes HHP, Ruitenbeek W, Sandkjujl LA, deVijlder MF, Struyvenberg PAA, van de Kamp JJP, Maassen JA 1992. Mutation in mitochondrial tRNALeu(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet 1: 368–371 [DOI] [PubMed] [Google Scholar]
- Wai T, Teoli D, Shoubridge EA 2008. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat Genet 40: 1484–1488 [DOI] [PubMed] [Google Scholar]
- Wallace DC 1981. Assignment of the chloramphenicol resistance gene to mitochondrial deoxyribonucelic acid and analysis of its expression in cultured human cells. Mol Cell Biol 1: 697–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC 1982. Cytoplasmic inheritance of chloramphenicol resistance in mammalian cells. In Techniques in somatic cell genetics (ed. Shay JW), Chap. 12. pp. 159–187 Plenum, New York [Google Scholar]
- Wallace DC 1986. Mitotic segregation of mitochondrial DNAs in human cell hybrids and expression of chloramphenicol resistance. Somat Cell Mol Genet 12: 41–49 [DOI] [PubMed] [Google Scholar]
- Wallace DC 1987. Maternal genes: Mitochondrial diseases. In Medical and experimental mammalian genetics: A perspective (ed. McKusick VA, Roderick TH, Mori J, Paul MW), pp. 137–190 A.R. Liss, New York [Google Scholar]
- Wallace DC 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu Rev Genet 39: 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC 2007. Why do we have a maternally inherited mitochondrial DNA? Insights from Evolutionary Medicine. Annu Rev Biochem 76: 781–821 [DOI] [PubMed] [Google Scholar]
- Wallace DC 2008. Mitochondria as chi. Genetics 179: 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC 2011. Bioenergetic origins of complexity and disease. Cold Spring Harb Symp Quant Biol 76: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC 2012. Mitochondria and cancer. Nat Rev Cancer 12: 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC 2013a. Bioenergetics in human evolution and disease: Implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos Trans R Soc Lond B Biol Sci 10.1098/rstb.2012.0267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC 2013b. Mitochondrial bioenergetic etiology of disease. J Clin Invest 123: 1405–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W 2009. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev 23: 1714–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W 2010. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10: 12–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Bunn CL, Eisenstadt JM 1975. Cytoplasmic transfer of chloramphenicol resistance in human tissue culture cells. J Cell Biol 67: 174–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Pollack Y, Bunn CL, Eisenstadt JM 1976. Cytoplasmic inheritance in mammalian tissue culture cells. In Vitro 12: 758–776 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Bunn CL, Eisenstadt JM 1977. Mitotic segregation of cytoplasmic inherited genes for chloramphenicol resistance in mammalian cells. II: Fusions with human cell lines. Somatic Cell Genet 3: 93–119 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Ye JH, Neckelmann SN, Singh G, Webster KA, Greenberg BD 1987. Sequence analysis of cDNAs for the human and bovine ATP synthase b-subunit: Mitochondrial DNA genes sustain seventeen times more mutations. Curr Genet 12: 81–90 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK 1988a. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242: 1427–1430 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Zheng X, Lott MT, Shoffner JM, Hodge JA, Kelley RI, Epstein CM, Hopkins LC 1988b. Familial mitochondrial encephalomyopathy (MERRF): Genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 55: 601–610 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Brown MD, Lott MT 1999. Mitochondrial DNA variation in human evolution and disease. Gene 238: 211–230 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Fan W, Procaccio V 2010. Mitochondrial energetics and therapeutics. Annu Rev Path 5: 297–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Lott MT, Procaccio V 2013. Mitochondrial medicine: The mitochondrial biology and genetics of metabolic and degenerative diseases, cancer, and aging. In Emery and Rimoin's principles and practice of medical genetics (ed. Rimoin DL, Pyeritz RE, Korf BR). Churchill Livingstone, Philadelphia [Google Scholar]
- Wonnapinij P, Chinnery PF, Samuels DC 2008. The distribution of mitochondrial DNA heteroplasmy due to random genetic drift. Am J Hum Genet 83: 582–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnapinij P, Chinnery PF, Samuels DC 2010. Previous estimates of mitochondrial DNA mutation level variance did not account for sampling error: comparing the mtDNA genetic bottleneck in mice and humans. Am J Hum Genet 86: 540–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S 1969. Theory of gene frequencies. In Evolution and the genetics of populations, University of Chicago Press, Chicago [Google Scholar]
- Yoneda M, Miyatake TGA 1994. Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol Cell Biol 14: 2699–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda M, Chomyn A, Martinuzzi A, Hurko O, Attardi G 1992. Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc Natl Acad Sci 89: 11164–11168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KL, Ernst SG, Rasmussen C, Dooling EC, Aprille JR 1993. Mitochondrial disorder associated with newborn cardiopulmonary arrest. Pediatr Res 33: 433–440 [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM 2012. Mitochondrial fission, fusion, and stress. Science 337: 1062–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Itagaki K, Hauser CJ 2010a. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34: 55–559 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ 2010b. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou RR, Wang B, Wang J, Schatten H, Zhang YZ 2010. Is the mitochondrial cloud the selection machinery for preferentially transmitting wild-type mtDNA between generations? Rewinding Muller's ratchet efficiently. Curr Genet 56: 101–107 [DOI] [PubMed] [Google Scholar]