

Abstract
Aims: The NF-E2 p45-related factor 2 (Nrf2) signaling pathway regulates the cellular antioxidant response and activation of Nrf2 has recently been shown to limit tissue damage from exposure to environmental toxicants, including As(III). In an attempt to identify improved molecular agents for systemic protection against environmental insults, we have focused on the identification of novel medicinal plant-derived Nrf2 activators. Results: Tanshinones [tanshinone I (T-I), tanshinone IIA, dihydrotanshinone, cryptotanshinone], phenanthrenequinone-based redox therapeutics derived from the medicinal herb Salvia miltiorrhiza, have been tested as experimental therapeutics for Nrf2-dependent cytoprotection. Using a dual luciferase reporter assay overexpressing wild-type or mutant Kelch-like ECH-associated protein-1 (Keap1), we demonstrate that T-I is a potent Keap1-C151-dependent Nrf2 activator that stabilizes Nrf2 by hindering its ubiquitination. In human bronchial epithelial cells exposed to As(III), T-I displays pronounced cytoprotective activity with upregulation of Nrf2-orchestrated gene expression. In Nrf2 wild-type mice, systemic administration of T-I attenuates As(III) induced inflammatory lung damage, a protective effect not observed in Nrf2 knockout mice. Innovation: Tanshinones have been identified as a novel class of Nrf2-inducers for antioxidant tissue protection in an in vivo As(III) inhalation model, that is relevant to low doses of environmental exposure. Conclusion: T-I represents a prototype Nrf2-activator that displays cytoprotective activity upon systemic administration targeting lung damage originating from environmental insults. T-I based Nrf2-directed systemic intervention may provide therapeutic benefit in protecting other organs against environmental insults. Antioxid. Redox Signal. 19, 1647–1661.
Introduction
Inorganic As(III) is a ubiquitous environmental contaminant found in water and dust that poses a considerable threat to human health worldwide (2, 3, 5, 6). Among various organ systems, the lung has been identified as a major target organ for As(III)-induced acute and chronic toxicities (8, 9, 16, 22, 40). Indeed, a strong association between As(III) exposure and increased incidence of pulmonary malfunctions (such as chronic cough, bronchitis and shortness of breath) has been substantiated, and human epidemiological studies provide strong evidence in support of a carcinogenic activity of As(III) on the lung (38, 40, 41, 49). Therefore, an urgent need exists for the identification and development of protective strategies that limit As(III)-induced lung damage.
Innovation.
Tanshinones have been identified as a novel class of NF-E2 p45-related factor 2 (Nrf2)-inducers for antioxidant tissue protection in an in vivo As(III) inhalation model, that is relevant to low doses of environmental exposure.
Cumulative evidence suggests that the NF-E2 p45-related factor 2 (Nrf2) signaling pathway represents an important cellular defense system that suppresses tissue damage caused by acute or chronic exposure to various environmental toxicants, including solar ultraviolet radiation, xenobiotics, and heavy metals (10, 20, 42, 59). Enhanced sensitivity of Nrf2 knockout (Nrf2−/−) versus Nrf2 wild-type (Nrf2+/+) mice towards pulmonary damage from various toxicological challenges, including diesel exhaust particles, cigarette smoke, diquat, and As(III) has been documented (4, 24, 26, 43, 57). Our earlier experiments have demonstrated that stable knockdown of endogenous Nrf2 rendered cells more sensitive to As(III)-induced cell death (53). We and others have also demonstrated the interventional potential of diet-derived Nrf2 activators including sulforaphane (SF), lipoic acid, and cinnamaldehyde for the suppression of As(III) cytotoxicity through modulation of the Nrf2-dependent cellular defense mechanism (15, 23, 48, 53, 56). For example, SF has been shown to limit organ toxicity of As(III) in vivo when As(III) was administered through drinking water (25). More recently, we have shown that Nrf2 status determines the sensitivity of mice to As(III)-induced lung damage and that Nrf2 modulation using SF prevents inflammatory tissue damage in an in vivo As(III) inhalation model that is relevant to low environmental human exposure to As(III)-containing dusts (62).
The transcription factor Nrf2 is thought to control tissue damage from environmental insults by upregulating the expression of genes involved in antioxidant response and xenobiotic metabolism (20, 25, 26, 59). Nrf2 is negatively regulated by Kelch-like ECH-associated protein-1 (Keap1), which forms an E3 ubiquitin ligase complex with Cullin 3 (Cul3) and Rbx1 (61). Under normal conditions, cells maintain low constitutive levels of Nrf2 because the Keap1-E3 ubiquitin ligase complex constantly ubiquitinates Nrf2 and targets it for degradation by the 26S proteasome. When cells are exposed to exogenous stimulants, such as SF and tert-butylhydroquinone (tBHQ), the activity of the Keap1-E3 ubiquitin ligase complex is thought to be impaired due to modifications of critical cysteine residues in Keap1, particularly C151, leading to stabilization of Nrf2 (60). Nrf2 then translocates to the nucleus and binds to the antioxidant response element (ARE) in the promoter regions of cytoprotective genes (13, 17, 27, 50, 61). The Nrf2-target genes encode proteins with diverse cellular functions. For example, γ-glutamylcysteine synthetase (γ-GCS) and NAD(P)H quinone oxidoreductase (NQO1) are involved in the synthesis of glutathione (GSH) and redox homeostasis, respectively (7, 45, 46, 55). Conjugating enzymes, including glutathione S-transferases (GSTs) and UDP-glucuronosyltransferase, facilitate the removal of toxic and carcinogenic chemicals by increasing their solubility and excretion (19, 21, 28). Transporters, such as multidrug resistance proteins and p-glycoproteins, are important in the uptake and removal of xenobiotics (18, 37, 51, 58).
Interestingly, the “dark” side of Nrf2 has been recently revealed. Somatic mutations in either Keap1 or Nrf2 that disrupt Keap1-mediated negative regulation of Nrf2 have been identified in many types of cancer, which result in high constitutive levels of Nrf2. In addition, reduced expression of Keap1, due to hypermethylation or loss of heterozygosity, also leads to high basal levels of Nrf2 in certain cancers (32, 44). As(III) itself is an Nrf2 activator and has been shown to induce the Nrf2 pathway in many cell lines in vitro and in the liver, bladder, and lung when As(III) was administered through drinking water or inhaled particles in vivo (25, 33, 62). It is perplexing how could both beneficial chemopreventive compounds and harmful arsenicals induce Nrf2. Intriguingly, in our recent studies we uncovered a distinct mechanism of Nrf2 activation by As(III) at low environmentally relevant doses that is different from that of dietary chemopreventive compounds, such as SF and tBHQ (33, 54) (manuscript accepted to Molecular and Cellular Biology). Unlike SF and tBHQ that activate Nrf2 in a Keap1-C151 dependent, and p62-independent manner (canonical), As(III) activates Nrf2 in a noncanonical manner that is Keap1-C151 independent, and p62-dependent. Detailed studies indicate that As(III) causes an increase of autophagosomes where Keap1 and p62 accumulates, leading to inactivation of Keap1 and upregulation of Nrf2. More importantly, the noncanonical activation of Nrf2 by arsenic results in a prolonged activation of Nrf2, which mimics high constitutive levels of Nrf2 observed in certain cancer types, deemed the dark side of Nrf2 (33) (manuscript accepted to Molecular and Cellular Biology). It is likely that the prolonged activation of Nrf2 by arsenic underlies arsenic toxicity and carcinogenicity.
Elucidating the differences in the mode of Nrf2 activation between canonical versus noncanonical Nrf2 activators led us to hypothesize that Nrf2 activation by canonical Nrf2 activators is protective, whereas its activation by noncanonical Nrf2 activators is harmful. In addition, the canonical Nrf2 activators can alleviate arsenic-mediated toxic effects. In an attempt to identify improved canonical Nrf2 activators for systemic protection against As(III)-induced tissue damage we have focused on medicinal plant-derived Nrf2 activators. Here we demonstrate that tanshinone I (T-I), a phenanthrenequinone constituent of the Chinese medicinal herb Danshen (Salvia miltiorrhiza), is a potent Keap1-C151-dependent Nrf2 activator that stabilizes Nrf2 by hindering its ubiquitination. In human bronchial epithelial (HBE) cells exposed to As(III), T-I displayed pronounced cytoprotective activity with upregulation of Nrf2-orchestrated gene expression. In an in vivo As(III) inhalation model, T-I activated the Nrf2 signaling pathway attenuating As(III)-induced inflammation in lungs from Nrf2+/+ but not Nrf2−/− mice.
Results
Identification of tanshinones I and dihydrotanshinone as Nrf2-inducers
Employing a dual luciferase reporter assay with the mGST-ARE firefly luciferase plasmid reported previously, we measured the ability of four major tanshinones [T-I, dihydrotanshinone (DHT), tanshinone IIA (T-IIA), and cryptotanshinone (CT)] in inducing the transcriptional activity of Nrf2 (Fig. 1A). The transcriptional activity of Nrf2 was upregulated upon exposure to low micromolar concentrations that was most pronounced in response to T-I and DHT (Fig. 1B). Similarly, the protein level of Nrf2 was enhanced by tanshinone exposure (Fig. 1C). Again, T-I and DHT were the most active tanshinones causing upregulation of Nrf2 at the protein level similar to that induced by SF (Fig. 1C). Based on the activity of T-I in upregulating Nrf2 we focused our subsequent mechanistic and cytoprotection studies on T-I.
FIG. 1.

Nrf2 activation by tanshinones. (A) Chemical structures of T-I, DHT, T-IIA, and CT. (B) MDA-MB-231 cells transfected with expression vectors for mGST-ARE firefly luciferaseand renilla luciferase reporters were left untreated or treated with each of the indicated compounds for 16 h. Dual luciferase activities were measured. The experiment was repeated three times, with triplicate samples in each experiment. The data are expressed as means±SD (*p<0.05). (C) MDA-MB-231 cells were exposed to the indicated doses of tanshinones for4 h. SF (5 μM) treatment for 4 h was included as a positive control. Total cell lysates were subjected to immunoblot analysis. ARE, antioxidant response element; CT, cryptotanshinone; DHT, dihydrotanshinone; GST, glutathione S-transferase; Nrf2, NF-E2 p45-related factor 2; SF, sulforaphane; T-I, tanshinone I; T-IIA, tanshinone IIA; SD, standard deviation.
T-I activates Nrf2 in a C151-dependent manner and decreases its ubiquitination
Consistent with the result demonstrating upregulation of Nrf2 at the protein level in response to T-I treatment (Fig. 1C), the transcriptional activity of Nrf2 is also induced by T-I in a dose-dependent manner (Fig. 2A). At doses ≥4 μM a significant induction of luciferase activity was observed that was five fold over control at 20 μM, at which dose there was no observable toxicity (Fig. 2A).
FIG. 2.

T-I activates Nrf2 by decreasing Nrf2 ubiquitination and increasing Nrf2 stability in a Keap1-C151-dependent manner. (A) A dual luciferase reporter assay was performed after cotransfected MDA-MB-231 cells were treated with the indicated concentration of T-I for 16 h. The data are expressed as means±SD (*p<0.05). (B) MDA-MB-231 cells were cotransfected with the plasmids encoding the indicated proteins. Cells were then treated with either SF (5 μM) or T-I (5 μM) along with MG132 (10 μM) for 4 h before cell lysates were collected for ubiquitination assay. Anti-Nrf2 immunoprecipitates were analyzed by immunoblot with anti-HA antibodies for detection of ubiquitin-conjugated Nrf2. (C) Cells were either left untreated or treated with T-I (5 μM) for 4 h. Cycloheximide (50 μM) was added and cells were lysed at the indicated time points. Cell lysates were subjected to immunoblot analysis using anti-Nrf2 and anti-β-actin antibodies. The intensity of the bands was quantified using Quantity One software and plotted against the time after cycloheximide treatment. (D) The ARE luciferase reporter gene assay was performed in the same way as described in (A), except that Keap1-C151S, rather than Keap1-WT, was cotransfected in half of the samples. In addition, a Keap1-siRNA against the 3′ untranslated region was cotransfected to suppress endogenous Keap1. The transfected cells were then treated with T-I (5 μM), As(III) (10 μM), SF (5 μM), or tBHQ (50 μM) for 16 h, before the measurement of firefly and renilla luciferase activities. Data were expressed as means±SD (*p<0.05 control vs. compound treatment; #p<0.05 Keap1-WT vs. Keap1-C151S). An aliquot of cell lysates was used for immunoblot analysis. Keap1, Kelch-like ECH-associated protein-1; tBHQ, tert-butylhydroquinone.
Next, the mechanism of Nrf2 activation by T-I was investigated. As demonstrated previously, established Nrf2 inducers, such as SF and tBHQ cause Nrf2 activation through inhibition of the Keap1-mediated ubiquitination of Nrf2 (60). Therefore, an in vivo ubiquitination assay was performed using MDA-MB-231 cells cotransfected with expression vectors for Nrf2, Keap1, and HA-ubiquitin, and were either left untreated or treated with SF or T-I. When cells were treated with T-I, ubiquitination of Nrf2 decreased compared to the untreated control (Fig. 2B). As expected, SF also decreased Nrf2 ubiquitination (Fig. 2B). Next, the half-life of endogenous Nrf2 protein was determined. Cycloheximide was added to untreated or T-I-treated cells for the indicated time. The protein level of Nrf2 was detected by immunoblot analysis (Fig. 2C, upper panel). The intensity of the Nrf2 band was measured and plotted against the time after addition of cycloheximide and the half-life of Nrf2 was calculated (Fig. 2C, lower panel). The half-life of Nrf2 under untreated conditions was 14.6 min; however, after T-I treatment, the half-life of Nrf2 increased to 31.9 min (Fig. 2C). These results indicate that T-I activates the Nrf2-dependent response by decreasing its ubiquitination and thus, stabilizing Nrf2 at the protein level.
Previously, we reported that the critical cysteine residue at amino acid 151 (C151) in Keap1 is required for activation of Nrf2 by SF or tBHQ. We also have shown that As(III)-induced Nrf2 activation occurs independent of Keap1 (C151) (54, 60). Therefore, to determine the specific mechanism by which T-I activates the Nrf2 pathway, we investigated whether T-I-mediated Nrf2 activation was also dependent on C151 (Fig. 2D). A specific Keap1-siRNA targeting the 3′ untranslated region was included during transfection to knockdown the expression of endogenous Keap1. Cells were then cotransfected with expression vectors for either Keap1-WT or Keap1-C151 along with ARE-firefly luciferase and renila luciferase. Before measurement of dual luciferase, cells were treated with As(III), SF, and tBHQ for 16 h. Pronounced enhancement of Nrf2 activity was observed with all treatments [T-I, As(III), SF, tBHQ] when Keap1 wild-type (Keap1-WT) was cotransfected (Fig. 2D, upper panel; black bars). In contrast, upon expression of the mutated Keap1-C151S, Nrf2 activation by T-I, SF, and tBHQ was dramatically blunted, but As(III) was still able to activate Nrf2 (Fig. 2D, upper panel; gray bars). This is consistent with a C151-dependent (T-I, SF, tBHQ) or independent [As(III)] mechanism of activation. Immunoblot analysis of cell lysates further confirmed the results obtained from the luciferase assay. For cells transfected with Keap1-WT, all treatments increased the protein level of Nrf2 (Fig. 2D, lower panel). When Keap1-C151S was transfected into cells, only As(III) increased Nrf2; however, induction of Nrf2 by T-I, SF, and tBHQ was blocked (Fig. 2D, lower panel). These results demonstrate for the first time that T-I is a canonical Nrf2 inducer and it activates the Nrf2 pathway through the critical C151 sensor residue in Keap1.
T-I activates Nrf2 and its downstream genes in HBE cells and protects HBE cells from As(III)-induced oxidative stress and cytotoxicity
Since the lung is the major target organ for arsenic-mediated toxicity and carcinogenicity, the ability of T-I to activate the Nrf2 signaling pathway was tested in HBE cells. When cells were exposed to T-I for 4 h, endogenous Nrf2 protein levels increased in a dose-dependent manner (Fig. 3A). Similar to SF, T-I had no effect on Keap1 protein levels (Fig. 3A). Next, the time dependent induction of Nrf2 by T-I was conducted using two doses (Fig. 3B). A significant increase in Nrf2 protein level was observed as early as 2 h and persisted up to 12 h at both doses (Fig. 3B). Again, there was no effect on Keap1 protein levels. Nrf2 protein levels returned to basal levels by 24 h (Fig. 3B), but protein levels of two Nrf2 downstream target genes, NQO1 and γ-GCS, displayed sustained upregulation up to 48 h (Fig. 3B). Consistent with the notion that T-I activates the Nrf2 pathway by stabilizing Nrf2 proteins, the mRNA level of Nrf2 did not change upon treatment with T-I or SF (Fig. 3C). However, mRNA levels of NQO1 and GCLM, were significantly induced by T-I treatment and the fold of their induction was comparable to that elicited by SF (Fig. 3C). Taken together these results indicate that T-I activates expression of Nrf2 target genes by upregulation of Nrf2 at the protein level in lung epithelial cells.
FIG. 3.

T-I activates the Nrf2 pathway. (A) HBE cells were treated with the indicated doses of T-I for 4 h. Cell lysates were subjected to immunoblot analysis. (B) HBE cells were treated with T-I (5 or 10 μM) for the indicated duration. Cell lysates were subjected to immunoblot analysis. SF was included as a positive control in both (A) and (B). (C) HBE cells were treated with T-I (5 or 10 μM) for 16 h and mRNA was extracted. The relative mRNA levels of Nrf2, NQO1, and GCLM were then determined by quantitative real-time RT-PCR. Data are expressed as means±SD (*p<0.05). HBE, human bronchial epithelial; NQO1, NAD(P)H quinone oxidoreductase; RT-PCR, reverse transcription–polymerase chain reaction.
Next, we examined the feasibility of using T-I for cytoprotection against As(III)-mediated toxicity. First, the cytotoxicity of T-I was tested in HBE cells treated with several doses of T-I for 28 h. Ninety percent of the cells remained viable after 20 μM T-I treatment (Fig. 4A). Therefore, a nontoxic dose (5 μM) was chosen for the subsequent protection assays. Reactive oxygen species (ROS) and cell viability were measured after HBE cells were challenged by As(III) with or without T-I pretreatment. As(III) treatment increased ROS levels, whereas T-I itself did not enhance ROS (Fig. 4B, top panel). Cotreatment with As(III) and T-I suppressed As(III)-induced ROS levels (Fig. 4B, top panel). However, this T-I-mediated suppression of ROS was not observed in cells where Nrf2 expression was blocked by Nrf2-siRNA, indicating that the protection conferred by T-I is Nrf2-dependent (Fig. 4B, bottom panel). For the cytotoxicity protection assay, cells were exposed to several doses of As(III) in combination with either dimethyl sulfoxide (DMSO), 5 μM T-I, or 1.25 μM SF (a dose effective in inducing Nrf2 in HBE cells, data not shown) and cell viability was measured after 48 h. Remarkably, T-I and SF treatment improved cell viability in response to As(III) treatment (Fig. 4C). Taken together, these data indicate that T-I is able to maintain the cellular redox balance upon As(III) challenge and protects cells against As(III)-induced cell death.
FIG. 4.

T-I protects HBE cells against As(III) cytotoxicity. (A) Cell viability was measured in HBE cells treated with several doses of T-I at 28 h. (B) HBE cells were transfected with either control-siRNA or Nrf2-siRNA for 24 h. HBE cells were then pretreated with DMSO or T-I (5 μM) for 4 h before the treatment with As(III) (10 μM) for an additional 24 h. DCF-based fluorescence was measured using flow cytometry. Data were expressed as means±SD (*p<0.05 DMSO group vs. As(III)-treated group; #p<0.05 As(III)-treated group vs. As(III)+T-I-treated group). (C) Cells were left untreated or pretreated with 5 μM T-I for 24 h before addition of the indicated dose of As(III). Cell viability was measured 48 h after addition of As(III). Data are expressed as means±SD (*p<0.05 DMSO group vs. T-I-treated group; #p<0.05 DMSO group vs. SF-treated group). DMSO, dimethyl sulfoxide.
T-I activates the Nrf2 signaling pathway and attenuates As(III)-induced inflammation in lungs from Nrf2+/+ but not Nrf2−/− mice
To further explore the potential cytoprotective activity of T-I in a relevant animal model, we tested T-I in an in vivo As(III) inhalation model recently established by our team. First, a pilot study was carried out to test the best treatment regimen (dose and injection frequency) that results in repeated activation of Nrf2-dependent response. We demonstrated that systemic delivery of T-I (10 mg/kg, i.p., every 48 h) is effective in upregulating pulmonary protein levels of Nrf2 and Nrf2 target genes (γ-GCS and NQO1), in Nrf2+/+ but not from Nrf2−/− mice, as measured by both immunohistochemistry (IHC) (Fig. 5A) and immunoblot analysis (Fig. 5B). Next, feasibility of T-I-based tissue protection was examined. Mice were given either corn oil (control) or T-I (10 mg/kg, dissolved in corn oil) through i.p. injection every other day, while breathing in As(III)-containing dust for 15 consecutive days. Hematoxylin and eosin (HE) staining revealed infiltration of inflammatory cells and alveolar septal thickening in the lungs of both Nrf2+/+ and Nrf2−/− mice exposed to dust containing As(III) (Fig. 6A, HE panel). T-I injection did not affect the lungs of mice in either genotype (Fig. 6A, HE panel). Importantly, in mice exposed to As(III) and cotreated with T-I, pulmonary pathological alterations were reduced in Nrf2+/+ mice, whereas no improvement was observed in Nrf2−/− lungs (Fig. 6A, HE panel). Furthermore, IHC analysis for 8-hydroxy-2′-deoxyguanosine (8-OHdG) was performed to detect As(III)-induced oxidative DNA damage. As(III) enhanced 8-OHdG staining in both Nrf2+/+ and Nrf2−/− mice (Fig. 6A, 8-OHdG panel). In contrast, T-I itself did not have any effect; rather cotreatment with T-I and As(III) suppressed 8-OHdG staining in Nrf2+/+, but not in Nrf2−/−mice (Fig. 6A, 8-OHdG panel). These results indicate that T-I protects against arsenic-mediated pulmonary damage through activation of the Nrf2 pathway. To further support the data obtained, analysis of bronchoalveolar lavage (BAL) fluid for total and distinct inflammatory cell types was conducted. As(III) increased inflammatory cell infiltration as assessed by the number of total cells as well as the number of macrophages, neutrophils, or lymphocytes in both Nrf2+/+ and Nrf2−/− mice (Fig. 6B). T-I administration in unchallenged mice did not change any of the inflammatory cell numbers in either genotype. However, T-I treatment decreased As(III)-induced inflammatory cell infiltration (total, macrophages, lymphocytes) only in Nrf2+/+ mice, but not in Nrf2−/− mice (Fig. 6B). A similar effect was observed with neutrophils but did not reach to the level of statistical significance (Fig. 6B). These data suggest that T-I can decrease pulmonary inflammatory response associated with As(III) exposure through activation of the Nrf2-dependent defensive mechanism.
FIG. 5.
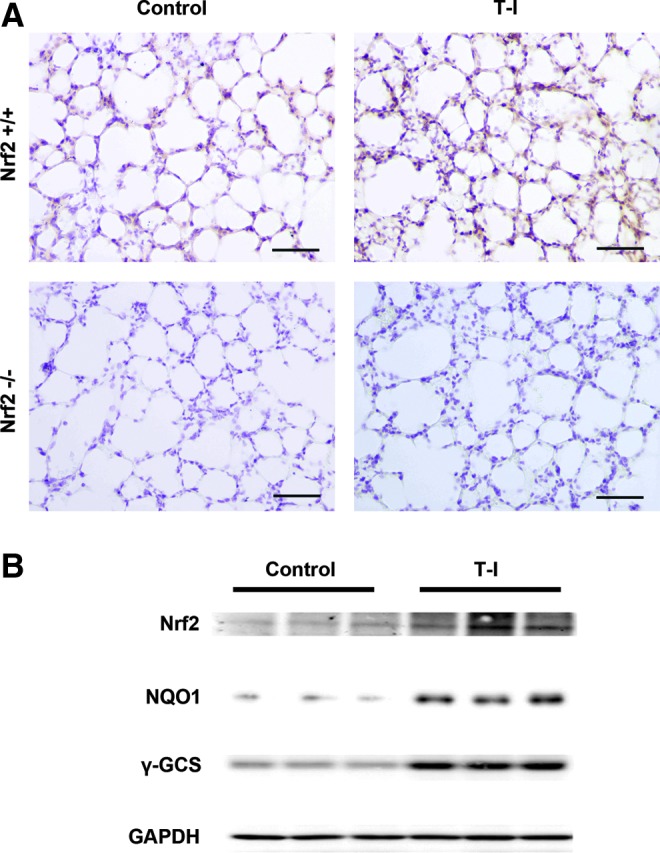
Pulmonary Nrf2 activation by systemic administration of T-I. Nrf2+/+ and Nrf2−/− mice were i.p. injected with 10 mg/kg T-I. Mice were sacrificed at 48 h postinjection and the lung was isolated. (A) Lung tissue sections were subjected to IHC analysis with anti-Nrf2 antibodies (n=3 in each group; one representative tissue section is shown per group). Scale bar: 100 μm. (B) Lung tissue lysates form Nrf2+/+ mice were subjected to immunoblot analysis with the indicated antibodies. Each lane contains a lung tissue sample from individual mice. Note: immunoblot analysis results were not included with tissues from Nrf2−/− mice because low expression levels of Nrf2, NQO1, and γ-GCS. IHC, immunohistochemistry; γ-GCS, γ-glutamylcysteine synthetase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 6.

T-I attenuates As(III)-induced pathological alterations and inflammatory cell infiltration in lungs from Nrf2+/+ but not Nrf2−/− mice. Nrf2+/+ and Nrf2−/− mice received systemic delivery of corn oil or T-I (10 mg/kg, i.p., every 48 h) for 15 days. During these 15 days, mice were also exposed to As(III)-containing dusts for 30 min everyday. (A) HE staining and IHC of 8-OHdG of lavaged lung tissue sections from Nrf2+/+ and Nrf2−/− mice. A representative image of the lung tissue from each group is shown. (B) Cell differential analysis was performed on the BAL cells from each mouse. After staining, at least 200 cells were counted under a microscope. The absolute number of total cells, macrophages, neutrophils, or lymphocytes was plotted. Results are expressed as means±SD (n=5) (*p<0.05 DMSO group vs. As(III)-administrated group; #p<0.05 As(III)-administrated group vs. As(III)+T-I-administrated group). BAL, bronchoalveolar lavage; HE, hematoxylin and eosin; 8-OHdG, 8-hydroxy-2′-deoxyguanosine. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
As expected, immunoblot analysis confirmed increased protein levels of Nrf2, NQO1, γ-GCS in the lungs of Nrf2+/+ mice exposed to the single or combined action of T-I and As(III) at the time of tissue collection at 15 day post-treatment (Fig. 7A). On the other hand, protein levels of Nrf2, NQO1, and γ-GCS were not affected by any of the treatments in Nrf2−/− mice (data not shown). Since NF-κB is a common signaling pathway regulating inflammatory response, protein levels of phosphorylated-p65 (p-p65) versus p65 were assessed as a measurement of NF-κB activation in both Nrf2+/+ and Nrf2−/− mice. A significant increase in p-p65 protein level but not p65 was observed in the lung of both Nrf2+/+ and Nrf2−/−mice when treated with As(III) (Fig. 7A). T-I alone did not affect p-p65 and p65 protein levels in both genotypes (Fig. 7A). When Nrf2+/+ and Nrf2−/− mice were treated with both T-I and As(III), p-p65 protein levels slightly decreased when compared to the As(III) alone treated group in the lungs of Nrf2+/+ mice but not Nrf2−/− mice. Independent ELISA-based analysis with interleukin (IL)-6 and TNF-α confirmed the As(III)-induced activation of the NF-κB signaling pathway and its attenuation by T-I administration that only occurred in Nrf2+/+; however, suppression of IL-6 levels by T-I was not statistically significant (Fig. 7B). Participation of CD4+ T cells, including Th1 and Th2 cells, in the As(III)-mediated inflammation response has recently been revealed (11, 12, 62). Therefore, we employed real-time reverse transcription–polymerase chain reaction (RT-PCR) to detect the mRNA levels of Th2 cytokines (IL-13 and IL-4) and Th1 cytokines (IL-2 and interferon gamma [IFNγ]) (Fig. 8). Indeed, As(III) upregulated IL-13 and IL-4 mRNA levels in both Nrf2+/+ and Nrf2−/− mice, and T-I treatment alone did not affect any of the cytokines. However, when As(III) was administered together with T-I, the As(III)-mediated increase in IL-13 was suppressed only in Nrf2+/+ lungs (Fig. 8). For IL-4, a similar effect was observed but did not reach the level of statistical significance (Fig. 8). Since IL-13 stimulates airway fibrosis largely through activation of transforming growth factor beta (TGF-β) (31, 34), the mRNA level of TGF-β was also assessed. Indeed, TGF-β mRNA levels increased when treated with As(III), which was suppressed by T-I coadministration in Nrf2+/+ mice only, but did not reach the level of statistical significance (Fig. 8). Conversely, As(III) decreased IL-2 and IFNγ mRNA levels in both Nrf2+/+ and Nrf2−/− lungs (Fig. 8), and T-I treatment alone did not affect IL-2 and IFNγ. Again, when treated in conjunction with As(III), T-I restored mRNA levels of IL-2 and IFNγ in Nrf2+/+ mice, but not in Nrf2−/− mice (Fig. 8). Furthermore, monocyte chemoattractant protein-1 (MCP-1) mRNA levels increased in response to As(III) in both genotypes. However, T-I treatment suppressed the As(III)-mediated response only in Nrf2+/+mice (Fig. 8). This result agrees with the enhanced inflammatory cell infiltration in response to As(III) exposure and its Nrf2-dependent suppression by T-I as observed by BAL cell counting (Fig. 6B). Collectively, these results suggest that As(III)-induced pulmonary inflammatory pathology and pathogenic Th2 cytokine response can be suppressed by systemic delivery of the novel Nrf2 activator T-I.
FIG. 7.

T-I activates the Nrf2 signaling pathway attenuating As(III)-induced inflammation in lungs from Nrf2+/+ but not Nrf2−/− mice. (A) Lung tissue lysates (three mice per group) were subjected to immunoblot analysis (left panel). The intensity of bands was quantified and the relative value of p65/p65 was plotted (right panel). (B) The amount of IL-6 and TGF-β in the BAL fluid was measured by ELISA. Results are expressed as means±SD (n=5) (*p<0.05 control group vs. As(III)-administrated group; #p<0.05 As(III) group vs. As(III)+T-I-administrated group). IL, interleukin; TGF-β, transforming growth factor beta.
FIG. 8.

T-I restores As(III)-induced immuno-proinflammatory cytokine production in lungs from Nrf2+/+ but not Nrf2−/− mice. Relative mRNA expression of IL-13, IL-4, TGF-β, IL-2, IFNγ, and MCP-1 was measured by real-time RT-PCR. mRNAs extracted from three mice per group were used to run RT-PCR in duplicate and the mean±SD was calculated (n=3) (*p<0.05 control group vs. As(III)-administrated group; #p<0.05 As(III) group vs. As(III)+T-I-administrated group). MCP-1, monocyte chemoattractant protein-1; IFNγ, interferon gamma.
Discussion
Environmental and occupational exposure to As(III) represents an unresolved major public health concern affecting large populations on a global scale. An urgent need exists for the development of strategies that prevent or limit tissue damage and pathologies associated with acute and chronic As(III) exposure. Tanshinones, phenanthrenequinone constituents of the Chinese medicinal herb Danshen (S. miltiorrhiza), have recently emerged as potent antioxidant, anti-inflammatory, and cytoprotective factors showing efficacy in cancer (14, 35), cardiovascular disease (29, 30), ischemia reperfusion injury (1), and hepatic fibrosis (52).
By comparing the ability of four major tanshinones in inducing Nrf2 activity, we identified that both T-I and DHT are Nrf2 activators. Since T-I elicits less cytotoxicity than DHT, T-I was further characterized for its mechanistic action of Nrf2 activation and for its potential therapeutic application. Our experiments demonstrate for the first time that T-I induces the Nrf2-dependent response primarily by hindering its ubiquitination and degradation of Nrf2, and thus, stabilizing Nrf2 protein levels in a Keap1-C151-dependent manner (Figs. 1 and 2). According to this result, T-I is a canonical Nrf2 activator. More importantly, the potential therapeutic use of T-I as a novel Nrf2 inducer has not been reported until now. Unlike many experimental Nrf2-inducers with undefined or unfavorable pharmacokinetic profiles, tanshinones are investigational drugs currently in advanced stages of clinical development in human patients with cardiovascular diseases and other indications (clinicTrials.gov: NCT01452477; NCT01637675). Therefore, in this study we investigated the therapeutic potential of T-I in protecting against arsenic toxicity using both HBE cells in vitro and a whole animal inhalation model relevant to human low doses of environmental exposure to arsenic in vivo.
In HBE cells exposed to As(III), T-I upregulated the Nrf2-orchestrated gene expression and displayed pronounced cytoprotective activity, an effect associated with pharmacological activation of Nrf2 by T-I (Figs. 3 and 4). T-I conferred cellular protection by maintaining cellular redox homeostasis under As(III) challenge and thus, enhancing cell viability (Fig. 4). Coordinated induction of the Nrf2-mediated cellular defense response, including upregulation of cellular antioxidant, phase II detoxifying enzymes, transporters and many other cytoprotective proteins, may contribute to the T-I-mediated protection observed in response to As(III) exposure.
In an in vivo As(III) dust inhalation model conducted in Nrf2+/+ and Nrf2−/− mice, preventive efficacy of systemic administration of T-I was only observed in Nrf2+/+ mice, consistent with the notion that T-I-mediated tissue protection is derived from activation of the Nrf2 pathway (Fig. 6). The results obtained using HE staining, 8-OHdG IHC and BAL cell counting indicate that inhaled As(III) dust caused similar severity of pulmonary oxidative damage and inflammation (except lymphocyte infiltration shown in Fig. 6B, lymphocyte panel) in both Nrf2+/+ and Nrf2−/− mice as judged by infiltration of inflammatory cells and alveolar septal thickening (Fig. 6). These results are coherent with our previous study demonstrating that in mice given drinking water contaminated with As(III), lung inflammation occurred irrespective of Nrf2 genotype (25). The reason that Nrf2+/+ mice did not show reduced inflammation compared to Nrf2−/− mice may be due to the extremely low basal level of Nrf2 in the lung. However, administration of T-I attenuated inflammatory events in the lung of Nrf2+/+ mice without showing therapeutic effects in Nrf2−/− mice. Similarly, protein levels of p-p65, an inflammatory marker indicative of NF-κB activation, also increased upon As(III) exposure irrespective of Nrf2 status, but was suppressed by T-I administration only in Nrf2+/+ mice (Fig. 7). These results strongly suggest that activation of Nrf2 by T-I plays a critical role in protecting lungs from As(III)-induced inflammation and toxicity.
Th2 polarized immune response is implicated in the pathology of allergic diseases (39). When assessing the potential participation of CD4+ T cells, including Th1 and Th2 cells, in As(III)-induced lung inflammation using real-time RT-PCR-based profiling of cytokine expression, we observed an As(III)-induced Th2 polarized cytokine response as evidenced from the upregulation of IL-13 and IL-4 mRNA in lung tissue (Fig. 8). In addition to IL-13 and IL-4, proinflammatory cytokines, including TGF-β and MCP-1 were also increased by As(III) inhalation. In contrast, other proinflammatory cytokines, such as Th1 cytokines (IL-2 and IFNγ) displayed an As(III)-induced decrease, changes that were observed in response to As(III) exposure irrespective of Nrf2 status. This observation suggests that the inflammatory response to As(III) is not Nrf2-dependent. However, when mice were treated with T-I in combination with As(III) exposure, the mRNA levels of all cytokines were restored in Nrf2+/+ mice only (Fig. 8), suggesting that Nrf2 plays a protective role against the As(III)-induced inflammatory response by acting as an immune modulator. However, the detailed mechanisms by which Nrf2 modulates the immune response require further investigation.
There is mounting evidence suggesting that As(III) induces ROS by depleting GSH or by damaging mitochondria (33, 47). Therefore, it was proposed that As(III) may indirectly activate the Nrf2 pathway by increasing ROS (36). However, the doses used in these earlier experiments showing generation of ROS in response to As(III) are extremely high. On the other hand, low environmentally relevant doses of arsenic do not seem to cause detectable changes in the intracellular redox status (33). Therefore, it is unlikely that the Nrf2-mediated protection against arsenic toxicity solely relies on the Nrf2-dependent antioxidant response that neutralizes ROS. Recently, we found that at low concentrations, arsenic is able to block autophagic flux, resulting in accumulation of autophagosomes where Keap1 and p62 proteins aggregate. Nrf2 activation by canonical Nrf2 activators, such as SF and tBHQ completely reversed autophagosomal accumulation in response to As(III), suggesting that canonical Nrf2 activators can be used to block As(III)-mediated autophagy deregulation (manuscript accepted to Molecular and Cellular Biology). The possible mechanisms of Nrf2 activator-mediated protection against arsenic toxicity are still not clear, although many compounds, such as SF, CA, tBHQ, and lipoic acid have been shown to protect against arsenic toxicity both in vitro and in vivo (33). Shinkai et al. demonstrated that activation of Nrf2 by SF reduced intracellular accumulation of As(III), and thus, reduced As(III) toxicity (48). The authors have suggested that upregulation of certain Nrf2 target genes, such as γ-GCS, GST isoforms and MRP1 enhance the excretion of arsenic (48). Therefore, it is highly possible that T-I alleviates the arsenic effect by reducing the intracellular concentration of arsenic though reduced uptake and enhanced export, which requires further investigation.
Taken together, our experiments demonstrate the feasibility of preventing As(III)-induced lung inflammation by systemic administration of T-I, a novel canonical Nrf2 activator characterized for the first time in this study. The T-I-mediated intervention may also prove to be efficacious for other types of environmental insults and may also confer protection against tissue damage in other organs.
Materials and Methods
Chemicals and cell culture
T-I and As(III) (NaAsO2) were purchased from Sigma, and SF was purchased from Santa Cruz. Human bronchial epithelium cells 16HBE14o (HBE) were obtained from California Pacific Medical Center, San Francisco. HBE cells were grown in Eagle's minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS; Atlanta Biological), 5% L-glutamine, and 0.1% gentamycin (Invitrogen). Human MDA-MB-231 breast carcinoma cells, purchased from ATCC, were cultured in MEM supplemented with 10% FBS, 5% L-glutamine, 0.1% gentamycin, 2 mM HEPES and 6 ng/ml bovine insulin (Invitrogen). All mammalian cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Transfection of siRNA, cDNA, and luciferase reporter gene assay
Transfection of cDNA was performed using Lipofectamine Plus (Invitrogen) and HiPerfect was used for transfection of siRNA, both were used according to the manufacturer's instructions. Nrf2-siRNA (SI03187289) and Keap1-siRNA (SI03246439) were purchased from Qiagen. Activation of Nrf2-dependent transcriptional activity by test compounds was examined as previously published (54). MDA-MB-231 cells were transfected with the mGST-ARE firefly luciferase reporter plasmid together with expression plasmids for Nrf2, Keap1, and renilla luciferase, an internal control, using Lipofectamine Plus (Invitrogen) according to the manufacturer's instructions. At 24 h post-transfection, cells were treated with the test compounds for 16 h before cell lysis for analysis of reporter gene activity. Reporter assays were performed using Promega dual-luciferase reporter gene assay system. All samples were run in triplicate for each experiment and the data represent the means of three independent experiments.
Cell viability
As(III)-induced toxicity was measured by functional impairment of the mitochondria using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma). Approximately 5×104 HBE cells per well were seeded in a 96-well plate and pretreated with DMSO, 5 μM T-I or 1.25 μM SF for 24 h. Cells were then cotreated with the indicated concentrations of As(III) (up to 20 μM) for 48 h. Twenty microliters of 2 mg/ml MTT was directly added to the cells. After incubation (37°C, 0.5–3 h), the plate was centrifuged and the medium was removed by aspiration. One hundred microliters of isopropanol/HCl was added to each well and was shaken at room temperature to dissolve the crystals. Absorbance was measured at 570 nm using the Synergy 2 Multi-Mode Microplate Reader (Biotek). All samples were run in triplicate for each experiment and the data represent the means of three independent experiments.
ROS detection
Cells were pretreated with DMSO (control) or 5 μM T-I for 4 h before the treatment with10 μM As(III) foran additional 24 h. Cells were then washed with phosphate-buffered saline (PBS) and fresh medium containing 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (Sigma; 10 μg/ml final concentration) was added. Plates were incubated for 20–60 min at 37°C. Cells were washed twice with PBS, trypsinized, washed again with PBS, and resuspended in PBS to ∼106 cells per ml. Fluorescence was measured using flow cytometry with excitation at 488 nm and emission at 515–545 nm. All steps were handled in the dark.
Antibodies, immunoblot analysis, ubiquitination assay, and protein half-life
Antibodies for Nrf2, Keap1, NQO1, γ-GCS and β-actin were purchased from Santa Cruz. Cells were harvested in sample buffer (50 mM Tris-HCl [pH 6.8], 2% sodium dodecyl sulfate [SDS], 10% glycerol, 100 mM dithiothreitol (DTT), and 0.1% bromophenol blue). After sonication, cell lysates were electrophoresed through an SDS-polyacrylamide gel and subjected to immunoblot analysis. For ubiquitination assay, MDA-MB-231 cells were cotransfected with expression vectors for HA-tagged ubiquitin, Nrf2 and Keap1, cells were treated with either 5 μM SF or T-Ialongwith 10 μM MG132 for 4 h. Cells were harvested in buffer containing 2% SDS, 150 mM NaCl, 10 mM Tris-HCl (pH 8.0), and 1 mM DTT and immediately boiled. The lysates were then diluted fivefold in buffer lacking SDS and incubated with an anti-Nrf2 antibody. Immunoprecipitated proteins were analyzed by immunoblot with an antibody against the HA epitope (Santa Cruz). To measure the half-life of Nrf2, MDA-MB-231 cells were either left untreated or treated with 5 μM T-I for 4 h. 50 μM cycloheximide was added to block protein synthesis. Total cell lysates were collected at different time points and subjected to immunoblot analysis with an anti-Nrf2 antibody. The relative intensity of the bands was quantified using the ChemiDoc CRS gel documentation system and Quantity One software from BioRad.
mRNA extraction and real-time RT-PCR
RT-PCR was done on total mRNA extracted from cells using TRIzol (Invitrogen). Equal amounts of mRNA were used to generate cDNA using the Transcriptor First Strand cDNA synthesis kit purchased from Roche. RT-PCR procedures and primer sequences of Nrf2, NQO1, GCLM, and GAPDH were described previously and the LightCycler 480 system was used (Roche) (50).
Quantification of cDNA amount for mIL-13, mIL-4, mIL-2, mIFNγ, mTGF-β, mMCP-1, and mβ-actin in each tissue sample was performed with KAPA SYBR FAST qPCR Kit (Kapa Biosystems). All primer sets were designed with Primer 3 online free software. And the primers were synthesized by Sigma.
mIL-13: forward (caagaccagactcccctgtg) and reverse (aggccatgcaatatcctctg);
mIL-4: forward (ccaaggtgcttcgcatattt) and reverse (atcgaaaagcccgaaagagt);
mIL-2: forward (aagctctacagcggaagcac) and reverse (atcctggggagtttcaggtt);
mIFNγ: forward (actggcaaaaggatggtgac) and reverse (gctgatggcctgattgtctt);
mTGF-β: forward (gactctccacctgcaagacc) and reverse (gactggcgagccttagtttg);
mMCP-1: forward (cccaatgagtaggctggaga) and reverse (tctggacccattccttcttg);
mβ-actin: forward (aaggccaaccgtgaaaagat) and reverse (gtggtacgaccagaggcatac).
The real-time PCR conditions used were the following: one cycle of initial denaturation (95°C for 3 min), 40 cycles of amplification (95°C for 10 s, 60°C for 20 s and 72°C for 5 s), melting curve (95°C for 5 s, 65°C for 1 min and 97°C continuous), and a cooling period (40°C for 30 s). Mean crossing point (Cp) values and standard deviations (SD) were determined. Cp values were normalized to the respective crossing point values of the mβ-actin reference gene. Data are presented as a fold change in gene expression compared to the control group. All reporter gene and RT-PCR analysis were repeated in three independent experiments and in duplicates. Data are all shown as means±SD.
Animals and treatments
Nrf2+/+ and Nrf2−/− mice were obtained by breeding Nrf2 heterozygous mice. All animals received water and food ad libitum. Eight-week-old mice were used for the experiment. Nrf2+/+ and Nrf2−/− mice were randomly allocated into four groups (n=5 per group): (i) control (corn oil); (ii) T-I(10 mg/kg, dissolved in corn oil); (iii) As(III); (iv) As(III)+T-I; T-I was administrated through intraperitoneal (i.p.) injection every other day for 15 days. During these 15 days mice were also exposed to 4.8 mg/m3 of the synthetic dust containing 10% As(III) for 30 min/day (62). All 40 mice survived As(III)-dust exposure and/or T-I injections. The dose of T-I was tested initially in a pilot study to ensure Nrf2 was activated up to 48 h after i.p. injection.
BAL and lung tissue collection
After treatment, mice were euthanized and lungs were isolated by carefully opening the thoracic cavity. BAL fluid was obtained by lavaging the lung with 0.5 ml PBS three times. The BAL fluid was centrifuged at 1500 rpm for 8 min at 4°C. Cell pellets were pooled, washed, and resuspended in PBS. Total cell counts were determined using standard hematologic procedures (63). Cytospins of BAL cells were prepared and slides were stained with a HEMA3 STAT PACK kit (Fisher Scientific Company). Macrophages, neutrophils, and lymphocytes were identified using the standard morphologic criteria. A minimal of 200 cells was examined. The means±SD were obtained by analyzing three batches of BAL fluid, each from individual mice in the same group. The supernatant of the first injection was stored at −80°C until used for an ELISA. Lungs were then collected and divided into two parts: one part was frozen in liquid nitrogen for total RNA extraction and protein analysis. The other part was fixed in 10% buffered formalin to be embedded in paraffin and cut into 4 μm sections for histological and immunochemical analyses.
HE staining and IHC
Tissue sections were stained with HE for pathological examination. IHC analysis was performed as previously described (25). A monoclonal antibody for 8-OHdG was purchased from Trevigen. Briefly, antigen retrieval of formalin-fixed paraffin-embedded tissue sections was carried out by microwave heating for 7 min at the highest setting to allow the retrieval solution to boil. Next, the sections were microwaved for 10 min at the lowest setting to maintain the retrieval solution at the boiling temperature. The retrieval solution contains 1×TBS with 0.1% Tween 20 (TBS-T) in 1 mol/L sodium citrate. After antigen retrieval, tissue sections were exposed to 3.5 M HCl for 15 min at room temperature and washed in TBS-T. Subsequently, tissue sections were treated with 0.3% peroxidase to quench endogenous peroxidase activity. Tissue sections were incubated with 5% normal goat serum for 30 min followed by 2 h incubation with an Nrf2 antibody at 1:100 dilution at room temperature. Sections were then incubated with a biotinylated goat anti-rabbit secondary antibody for 1 h. The ABC kit (Vector Laboratories) was then used according to the manufacturer's instructions. Finally, tissue sections were developed for 30 s using the 3,3′-diaminobenzidine staining kit (Dako), and counterstained with hematoxylin.
ELISA of cytokines in BAL fluid
The ELISA was purchased from eBiosciences and used according to the manufacturer's instructions. Briefly, the plate was coated with 100 μl capture antibody in coating buffer per well and incubated overnight at room temperature. The plate was washed with 250 μl wash buffer, blocked with 200 μl of the assay diluents, and incubated at room temperature for 1 h. A 100 μl of the BAL fluid was added and incubated at room temperature for 2 h. One hundred microliters detection antibody was then added to each well and incubated for 1 h at room temperature. Subsequently, 100 μl avidin-HRP was added and the plate was incubated for 30 min at room temperature. One hundred microliters of the substrate solution was added to each well and incubated for 15 min at room temperature and then 50 μl of the stop solution was added to stop the reaction. The plate was then read at 450 nm and analyzed. The ELISA was performed in triplicate. Serial dilutions of standards were also used to obtain a standard curve.
Statistics
Results are presented as the mean±SD of at least three independent experiments performed in duplicate or triplicate each. Statistical tests were performed using SPSS 10.0. Unpaired Student's t-tests were used to compare the means of two groups. One-way analysis of variance was applied to compare the means of three or more groups. p<0.05 was considered to be significant.
Abbreviations Used
- 8-OHdG
8-hydroxy-2′-deoxyguanosine
- ARE
antioxidant response element
- As(III)
arsenic
- BAL
bronchoalveolar lavage
- CT
cryptotanshinone
- Cul3
Cullin 3
- DHT
dihydrotanshinone
- DMSO
dimethyl sulfoxide.
- DTT
dithiothreitol
- FBS
fetal bovine serum
- GAPDH
glyceride-3-phosphate dehydrogenase
- GSH
glutathione
- GSTs
glutathione S-transferases
- H2DCFDA
2′,7′-dichlorodihydrofluorescein diacetate
- HBE
human bronchial epithelial
- HE
hematoxylin and eosin
- IFNγ
interferon gamma
- IHC
immunohistochemistry
- IL
interleukin
- Keap1
Kelch-like ECH-associated protein-1
- MCP-1
monocyte chemoattractant protein-1
- MEM
minimal Eagle's medium
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NQO1
NAD(P)H quinone oxidoreductase
- Nrf2
NF-E2 p45-related factor 2
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- RT-PCR
reverse transcription–polymerase chain reaction
- SD
standard deviation
- SDS
sodium dodecyl sulfate
- SF
sulforaphane
- tBHQ
tert-butylhydroquinone
- TGF-β
transforming growth factor beta
- T-I
tanshinone I
- T-IIA
tanshinone IIA
- γ-GCS
γ-glutamylcysteine synthetase
Acknowledgments
This work was supported by NIH grants 2R01 ES015010 and R01 CA154377 to D.D.Z., 1R03CA167580 to G.T.W., P42ES004940 to R.C.L., and P30ES006694, a center grant.
Author Disclosure Statement
The authors declare no conflicts of interest.
References
- 1.Adams JD. Wang R. Yang J. Lien EJ. Preclinical and clinical examinations of Salvia miltiorrhiza and its tanshinones in ischemic conditions. Chin Med. 2006;1:3. doi: 10.1186/1749-8546-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aelion CM. Davis HT. Lawson AB. Cai B. McDermott S. Associations of estimated residential soil arsenic and lead concentrations and community-level environmental measures with mother-child health conditions in South Carolina. Health Place. 2012;18:774–781. doi: 10.1016/j.healthplace.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anawar HM. Garcia-Sanchez A. Hossain MN. Akter S. Evaluation of health risk and arsenic levels in vegetables sold in markets of Dhaka (Bangladesh) and Salamanca (Spain) by hydride generation atomic absorption spectroscopy. Bull Environ Contam Toxicol. 2012;89:620–625. doi: 10.1007/s00128-012-0692-x. [DOI] [PubMed] [Google Scholar]
- 4.Aoki Y. Sato H. Nishimura N. Takahashi S. Itoh K. Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol. 2001;173:154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- 5.Argos M. Ahsan H. Graziano JH. Arsenic and human health: epidemiologic progress and public health implications. Rev Environ Health. 2012;0:1–5. doi: 10.1515/reveh-2012-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrd DM. Roegner ML. Griffiths JC. Lamm SH. Grumski KS. Wilson R. Lai S. Carcinogenic risks of inorganic arsenic in perspective. Int Arch Occup Environ Health. 1996;68:484–494. doi: 10.1007/BF00377874. [DOI] [PubMed] [Google Scholar]
- 7.Chan JY. Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 8.Chen CJ. Chen CW. Wu MM. Kuo TL. Cancer potential in liver, lung, bladder and kidney due to ingested inorganic arsenic in drinking water. Br J Cancer. 1992;66:888–892. doi: 10.1038/bjc.1992.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CL. Hsu LI. Chiou HY. Hsueh YM. Chen SY. Wu MM. Chen CJ. Ingested arsenic, cigarette smoking, and lung cancer risk: a follow-up study in arseniasis-endemic areas in Taiwan. JAMA. 2004;292:2984–2990. doi: 10.1001/jama.292.24.2984. [DOI] [PubMed] [Google Scholar]
- 10.Cho HY. Kleeberger SR. Nrf2 protects against airway disorders. Toxicol Appl Pharmacol. 2010;244:43–56. doi: 10.1016/j.taap.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 11.Cho Y. Ahn KH. Back MJ. Choi JM. Ji JE. Won JH. Fu Z. Jang JM. Kim DK. Age-related effects of sodium arsenite on splenocyte proliferation and Th1/Th2 cytokine production. Arch Pharm Res. 2012;35:375–382. doi: 10.1007/s12272-012-0219-3. [DOI] [PubMed] [Google Scholar]
- 12.Conde P. Acosta-Saavedra LC. Goytia-Acevedo RC. Calderon-Aranda ES. Sodium arsenite-induced inhibition of cell proliferation is related to inhibition of IL-2 mRNA expression in mouse activated T cells. Arch Toxicol. 2007;81:251–259. doi: 10.1007/s00204-006-0152-7. [DOI] [PubMed] [Google Scholar]
- 13.Cullinan SB. Gordan JD. Jin J. Harper JW. Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong Y. Morris-Natschke SL. Lee KH. Biosynthesis, total syntheses, and antitumor activity of tanshinones and their analogs as potential therapeutic agents. Nat Prod Rep. 2011;28:529–542. doi: 10.1039/c0np00035c. [DOI] [PubMed] [Google Scholar]
- 15.Du Y. Villeneuve NF. Wang XJ. Sun Z. Chen W. Li J. Lou H. Wong PK. Zhang DD. Oridonin confers protection against arsenic-induced toxicity through activation of the Nrf2-mediated defensive response. Environ Health Perspect. 2008;116:1154–1161. doi: 10.1289/ehp.11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferreccio C. Gonzalez C. Milosavjlevic V. Marshall G. Sancha AM. Smith AH. Lung cancer and arsenic concentrations in drinking water in Chile. Epidemiology. 2000;11:673–679. doi: 10.1097/00001648-200011000-00010. [DOI] [PubMed] [Google Scholar]
- 17.Furukawa M. Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashi A. Suzuki H. Itoh K. Yamamoto M. Sugiyama Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem Biophys Res Commun. 2003;310:824–829. doi: 10.1016/j.bbrc.2003.09.086. [DOI] [PubMed] [Google Scholar]
- 19.Hayes JD. Flanagan JU. Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 20.Hayes JD. McMahon M. Chowdhry S. Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the keap1-nrf2 pathway. Antioxid Redox Signal. 2010;13:1713–1748. doi: 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- 21.Hjelle JJ. Hazelton GA. Klaassen CD. Increased UDP-glucuronosyltransferase activity and UDP-glucuronic acid concentration in the small intestine of butylated hydroxyanisole-treated mice. Drug Metab Dispos. 1985;13:68–70. [PubMed] [Google Scholar]
- 22.Hopenhayn-Rich C. Biggs ML. Smith AH. Lung and kidney cancer mortality associated with arsenic in drinking water in Cordoba, Argentina. Int J Epidemiol. 1998;27:561–569. doi: 10.1093/ije/27.4.561. [DOI] [PubMed] [Google Scholar]
- 23.Huerta-Olvera SG. Macias-Barragan J. Ramos-Marquez ME. Armendariz-Borunda J. Diaz-Barriga F. Siller-Lopez F. Alpha-lipoic acid regulates heme oxygenase gene expression and nuclear Nrf2 activation as a mechanism of protection against arsenic exposure in HepG2 cells. Environ Toxicol Pharmacol. 2010;29:144–149. doi: 10.1016/j.etap.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Iizuka T. Ishii Y. Itoh K. Kiwamoto T. Kimura T. Matsuno Y. Morishima Y. Hegab AE. Homma S. Nomura A. Sakamoto T. Shimura M. Yoshida A. Yamamoto M. Sekizawa K. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells. 2005;10:1113–1125. doi: 10.1111/j.1365-2443.2005.00905.x. [DOI] [PubMed] [Google Scholar]
- 25.Jiang T. Huang Z. Chan JY. Zhang DD. Nrf2 protects against As(III)-induced damage in mouse liver and bladder. Toxicol Appl Pharmacol. 2009;240:8–14. doi: 10.1016/j.taap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kensler TW. Wakabayashi N. Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi A. Ohta T. Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273–286. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi M. Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Lam FF. Yeung JH. Chan KM. Or PM. Dihydrotanshinone, a lipophilic component of Salvia miltiorrhiza (danshen), relaxes rat coronary artery by inhibition of calcium channels. J Ethnopharmacol. 2008;119:318–321. doi: 10.1016/j.jep.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 30.Lam FF. Yeung JH. Chan KM. Or PM. Mechanisms of the dilator action of cryptotanshinone on rat coronary artery. Eur J Pharmacol. 2008;578:253–260. doi: 10.1016/j.ejphar.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 31.Lanone S. Zheng T. Zhu Z. Liu W. Lee CG. Ma B. Chen Q. Homer RJ. Wang J. Rabach LA. Rabach ME. Shipley JM. Shapiro SD. Senior RM. Elias JA. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and −12 in IL-13-induced inflammation and remodeling. J Clin Invest. 2002;110:463–474. doi: 10.1172/JCI14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lau A. Villeneuve NF. Sun Z. Wong PK. Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58:262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lau A. Whitman SA. Jaramillo MC. Zhang DD. Arsenic-mediated activation of the Nrf2-Keap1 antioxidant pathway. J Biochem Mol Toxicol. 2012;27:99–105. doi: 10.1002/jbt.21463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee DK. Park SH. Yi Y. Choi SG. Lee C. Parks WT. Cho H. de Caestecker MP. Shaul Y. Roberts AB. Kim SJ. The hepatitis B virus encoded oncoprotein pX amplifies TGF-beta family signaling through direct interaction with Smad4: potential mechanism of hepatitis B virus-induced liver fibrosis. Genes Dev. 2001;15:455–466. doi: 10.1101/gad.856201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee WY. Cheung CC. Liu KW. Fung KP. Wong J. Lai PB. Yeung JH. Cytotoxic effects of tanshinones from Salvia miltiorrhiza on doxorubicin-resistant human liver cancer cells. J Nat Prod. 2010;73:854–859. doi: 10.1021/np900792p. [DOI] [PubMed] [Google Scholar]
- 36.Li B. Li X. Zhu B. Zhang X. Wang Y. Xu Y. Wang H. Hou Y. Zheng Q. Sun G. Sodium arsenite induced reactive oxygen species generation, nuclear factor (erythroid-2 related) factor 2 activation, heme oxygenase-1 expression, and glutathione elevation in Chang human hepatocytes. Environ Toxicol. 2011 doi: 10.1002/tox.20731. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 37.Maher JM. Cheng X. Slitt AL. Dieter MZ. Klaassen CD. Induction of the multidrug resistance-associated protein family of transporters by chemical activators of receptor-mediated pathways in mouse liver. Drug Metab Dispos. 2005;33:956–962. doi: 10.1124/dmd.105.003798. [DOI] [PubMed] [Google Scholar]
- 38.Mazumder DN. Haque R. Ghosh N. De BK. Santra A. Chakraborti D. Smith AH. Arsenic in drinking water and the prevalence of respiratory effects in West Bengal, India. Int J Epidemiol. 2000;29:1047–1052. doi: 10.1093/ije/29.6.1047. [DOI] [PubMed] [Google Scholar]
- 39.Minnicozzi M. Sawyer RT. Fenton MJ. Innate immunity in allergic disease. Immunol Rev. 2011;242:106–127. doi: 10.1111/j.1600-065X.2011.01025.x. [DOI] [PubMed] [Google Scholar]
- 40.Putila JJ. Guo NL. Association of arsenic exposure with lung cancer incidence rates in the United States. PLoS One. 2011;6:e25886. doi: 10.1371/journal.pone.0025886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radon K. Schulze A. Strien R. Ehrenstein V. Praml G. Nowak D. [Prevalence of respiratory symptoms and diseases in neighbours of large-scale farming in Northern Germany] Pneumologie. 2005;59:897–900. doi: 10.1055/s-2005-915572. [DOI] [PubMed] [Google Scholar]
- 42.Ramos-Gomez M. Kwak MK. Dolan PM. Itoh K. Yamamoto M. Talalay P. Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rangasamy T. Cho CY. Thimmulappa RK. Zhen L. Srisuma SS. Kensler TW. Yamamoto M. Petrache I. Tuder RM. Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren D. Villeneuve NF. Jiang T. Wu T. Lau A. Toppin HA. Zhang DD. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108:1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ross D. Kepa JK. Winski SL. Beall HD. Anwar A. Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact. 2000;129:77–97. doi: 10.1016/s0009-2797(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 46.Sasaki H. Sato H. Kuriyama-Matsumura K. Sato K. Maebara K. Wang H. Tamba M. Itoh K. Yamamoto M. Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- 47.Sciandrello G. Mauro M. Catanzaro I. Saverini M. Caradonna F. Barbata G. Long-lasting genomic instability following arsenite exposure in mammalian cells: the role of reactive oxygen species. Environ Mol Mutagen. 2011;52:562–568. doi: 10.1002/em.20657. [DOI] [PubMed] [Google Scholar]
- 48.Shinkai Y. Sumi D. Fukami I. Ishii T. Kumagai Y. Sulforaphane, an activator of Nrf2, suppresses cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. FEBS Lett. 2006;580:1771–1774. doi: 10.1016/j.febslet.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 49.Smith AH. Goycolea M. Haque R. Biggs ML. Marked increase in bladder and lung cancer mortality in a region of Northern Chile due to arsenic in drinking water. Am J Epidemiol. 1998;147:660–669. doi: 10.1093/oxfordjournals.aje.a009507. [DOI] [PubMed] [Google Scholar]
- 50.Sun Z. Zhang S. Chan JY. Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vernhet L. Seite MP. Allain N. Guillouzo A. Fardel O. Arsenic induces expression of the multidrug resistance-associated protein 2 (MRP2) gene in primary rat and human hepatocytes. J Pharmacol Exp Ther. 2001;298:234–239. [PubMed] [Google Scholar]
- 52.Wang X. Morris-Natschke SL. Lee KH. New developments in the chemistry and biology of the bioactive constituents of Tanshen. Med Res Rev. 2007;27:133–148. doi: 10.1002/med.20077. [DOI] [PubMed] [Google Scholar]
- 53.Wang XJ. Sun Z. Chen W. Eblin KE. Gandolfi JA. Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol Appl Pharmacol. 2007;225:206–213. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang XJ. Sun Z. Chen W. Li Y. Villeneuve NF. Zhang DD. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: enhanced Keap1-Cul3 interaction. Toxicol Appl Pharmacol. 2008;230:383–389. doi: 10.1016/j.taap.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wild AC. Moinova HR. Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 56.Wondrak GT. Villeneuve NF. Lamore SD. Bause AS. Jiang T. Zhang DD. The cinnamon-derived dietary factor cinnamic aldehyde activates the Nrf2-dependent antioxidant response in human epithelial colon cells. Molecules. 2010;15:3338–3355. doi: 10.3390/molecules15053338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu KC. Zhang Y. Klaassen CD. Nrf2 protects against diquat-induced liver and lung injury. Free Radic Res. 2012;46:1220–1229. doi: 10.3109/10715762.2012.700709. [DOI] [PubMed] [Google Scholar]
- 58.Xu C. Li CY. Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28:249–268. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]
- 59.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 60.Zhang DD. Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang DD. Lo SC. Cross JV. Templeton DJ. Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng Y. Tao S. Lian F. Chau BT. Chen J. Sun G. Fang D. Lantz RC. Zhang DD. Sulforaphane prevents pulmonary damage in response to inhaled arsenic by activating the Nrf2-defense response. Toxicol Appl Pharmacol. 2012;265:292–299. doi: 10.1016/j.taap.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ziora D. Mazur B. Grzanka P. Niepsuj G. Oklek K. [BAL from two different lung segments indicated by high resolution computed tomography (HRCT) in patients with sarcoidosis. II. The role of T gamma delta lymphocytes (T gamma delta)] Pneumonol Alergol Pol. 1999;67:435–442. [PubMed] [Google Scholar]