

Abstract
Phosphorylation of BCL-2 within an unstructured loop inhibits its antiapoptotic effect. We found that phosphorylated BCL-2 predominantly localized to the endoplasmic reticulum (ER) and tested whether phosphorylation would control its activity at this organelle, where Ca2+ dynamics serve as a critical control point for apoptosis. Phosphorylation greatly inhibits the ability of BCL-2 to lower [Ca2+]er and protect against Ca2+-dependent death stimuli. Cells expressing nonphosphorylatable BCL-2AAA exhibited increased leak of Ca2+ from the ER and further diminished steady-state [Ca2+]er stores when compared to cells expressing BCL-2wt. Consequently, when BCL-2 is phosphorylated, Ca2+ discharge from the ER is increased, with a secondary increase in mitochondrial Ca2+ uptake. We also demonstrate that phosphorylation of BCL-2 inhibits its binding to proapoptotic family members. This inhibitory mechanism manifested at the ER, where phosphorylated BCL-2 was unable to bind proapoptotic members. [Ca2+]er proved coordinate with the capacity of BCL-2 to bind proapoptotic BH3-only members, further integrating the apoptotic pathway and Ca2+ modulation. Unexpectedly, the regulation of ER Ca2+ dynamics is a principal avenue whereby BCL-2 phosphorylation alters susceptibility to apoptosis.
Keywords: apoptosis, BCL-2, calcium, endoplasmic reticulum, phosphorylation
Introduction
Apoptosis is a highly regulated pathway in mammalian cells in which the pro- and antiapoptotic proteins of the BCL-2 family constitute an important control point upstream of irreversible damage to cellular constituents including organelles (reviewed in Adams and Cory, 2001; Martinou and Green, 2001). The antiapoptotic members such as BCL-2 possess sequence conservation within four domains (BH1–4), and bind and sequester proapoptotic proteins. Proapoptotic BCL-2 family proteins can be subdivided into ‘multidomain' proteins such as BAX and BAK, which display homology within BH1–3 domains, and the ‘BH3-only' proteins, which only possess sequence homology within this amphipathic α-helix, which serves as the critical death domain. The ratio between the antiapoptotic and proapoptotic BCL-2 family members helps determine the susceptibility of cells to a death signal (Oltvai et al, 1993). Genetic and biochemical studies indicate that multidomain BAX and BAK function as a gateway to the intrinsic death pathway operating at both the mitochondria and endoplasmic reticulum (ER). The upstream BH3-only members respond to select death signals and subsequently trigger the conformational activation of BAX and BAK, inducing their intramembranous homo-oligomerization and permeabilization of the mitochondrial outer membrane (Wei et al, 2001). Released intermembranous proteins include cytochrome c, which complexes with Apaf 1 and caspase-9 to form a postmitochondrial apoptosome that activates effector caspases (Li et al, 1997). Conversely, BCL-2 can sequester activated BH3-only proteins, and thus inhibit the activation of BAX and BAK (Cheng et al, 2001).
In the absence of BAX and BAK, cells are resistant to a wide variety of death signals including agents that release Ca2+ from intracellular stores, such as the lipid second messengers arachidonic acid and C2-ceramide as well as oxidative stress. BAX and BAK also localize to the ER, and cells deficient in BAX, BAK have reduced resting [Ca2+]er, which accounts for their resistance to Ca2+-dependent death stimuli (Scorrano et al, 2003). Reciprocally, expression of the antiapoptotic protein BCL-2 protects cells from death by thapsigargin, an irreversible inhibitor of the sarcoplasmic–endoplasmic reticulum Ca2+ ATPase (SERCA) responsible for uptake of Ca2+ from the cytosol into the ER lumen (Lam et al, 1994). BCL-2 is also found at the ER (in addition to mitochondria and nuclear membrane), and its overexpression resulted in reduced [Ca2+]er, accompanied by an increased leak of Ca2+ from the ER stores (Foyouzi-Youssefi et al, 2000; Pinton et al, 2000). Several studies found that ER-targeted BCL-2 demonstrated selective protection against specific stimuli, suggesting that subcellular location may influence the antiapoptotic role of BCL-2 (Zhu et al, 1996; Wang et al, 2001). Organelle-specific correction of the ER or mitochondrial defect in BAX, BAK doubly deficient cells indicates that many intrinsic death signals require ER-released Ca2+ as well as mitochondrial-based BAX or BAK to restore fully apoptosis (Scorrano et al, 2003). Thus, regulation of ER calcium levels may prove to be a common general mechanism whereby cells adjust sensitivity to diverse death stimuli.
BCL-2 family proteins are frequently regulated by post-translational modifications that control their activity and conformation. Antiapoptotic BCL-2 is phosphorylated on specific residues within an unstructured loop in response to diverse stimuli including treatment with chemotherapeutic taxanes (Haldar et al, 1998; Ling et al, 1998; Scatena et al, 1998), survival factor addition (May et al, 1994; Poommipanit et al, 1999), or PKC activation (Ruvolo et al, 1998). In most instances, phosphorylation of BCL-2 has been associated with its inactivation, in that deletion of the loop region or mutation of select phosphorylation sites is reported to enhance the antiapoptotic activity of BCL-2 (Chang et al, 1997; Haldar et al, 1998; Srivastava et al, 1999; Yamamoto et al, 1999). However, in another cell system, phosphorylation is reported to augment the activity of BCL-2 to bryostatin-1 (May et al, 1994). As multiple kinases have been reported to phosphorylate BCL-2, the functional consequence of BCL-2 phosphorylation should be examined in the context of a given cell type and specific signal.
In cycling Jurkat cells, BCL-2 is normally phosphorylated at G2/M. Moreover, diverse agents that induce a G2/M cell cycle block including paclitaxel, vincristine, vinblastine, and nocodazole result in the phosphorylation of BCL-2. JNK kinase appears to be responsible for phosphorylation of Thr69, Ser70, and Ser87 within the unstructured loop. Mutation of these residues in the BCL-2AAA variant enhanced antiapoptotic activity in response to taxol and anti-Fas treatment (Yamamoto et al, 1999). Thus, phosphorylation can inactivate BCL-2 at G2/M, acting as a physiological means of regulating apoptotic susceptibility during the cell cycle. However, little is known concerning how the phosphorylation of BCL-2 actually affects susceptibility to cell death.
As the regulation of ER Ca2+ dynamics is a control point for apoptosis (Demaurex and Distelhorst, 2003; Orrenius et al, 2003), we asked whether the phosphorylation of BCL-2 might exert its influence on apoptosis by affecting the regulation of ER Ca2+. Unexpectedly, we found that phosphorylated BCL-2 predominantly localized to the ER, warranting further testing of this thesis. Data presented here identify a mechanism whereby phosphorylation of BCL-2 inhibits its antiapoptotic activity by altering Ca2+ dynamics at the ER.
Results
Nonphosphorylatable BCL-2AAA demonstrates enhanced protection against Ca2+-dependent death stimuli
Phosphorylation of BCL-2 has been noted to inhibit its antiapoptotic function (Chang et al, 1997; Haldar et al, 1998; Srivastava et al, 1999; Yamamoto et al, 1999). The microtubule-damaging agent paclitaxel arrests cells at G2/M resulting in phosphorylation of BCL-2, and Jurkat T cells expressing a nonphosphorylatable BCL-2AAA (T69A, S70A, S87A) were found to be more resistant to taxol-induced cell death (Yamamoto et al, 1999) (Figure 1A). To assess the spectrum of death stimuli to which nonphosphorylatable BCL-2 displays enhanced protection, we compared Jurkat T cells expressing comparable levels of BCL-2wt or BCL-2AAA with a control line bearing an empty neomycin selection vector (Vector) (Yamamoto et al, 1999). The BCL-2AAA-expressing cells proved more resistant than BCL-2wt-expressing cells to several intrinsic pathway death stimuli including thapsigargin, staurosporine, and etoposide, as well as extrinsic pathway anti-Fas antibody treatment (Figure 1A).
Figure 1.
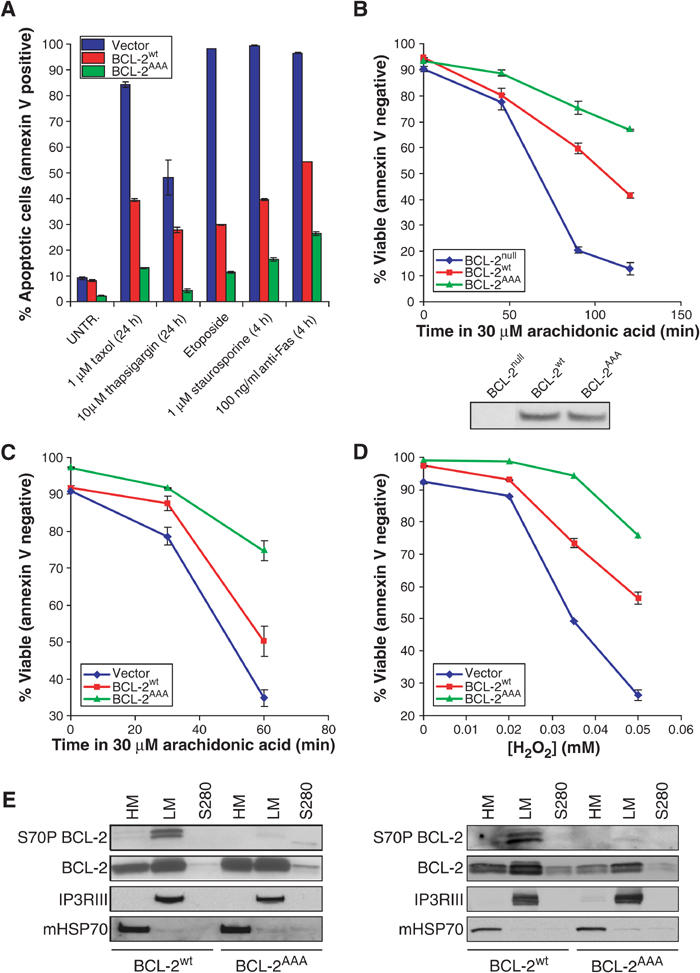
BCL-2AAA-expressing cells are more resistant than BCL-2wt-expressing cells to calcium-dependent death stimuli. (A) Jurkat cells expressing either neo vector, BCL-2wt, or BCL-2AAA were treated as indicated, stained with annexin V, and analyzed by FACS. (B) MEFs or (C) Jurkat lines were treated in medium without serum with 30 μM arachidonic acid for the indicated times. (D) Jurkat cells were treated in growth medium for 17 h with the indicated concentrations of H2O2. (E) Untreated Jurkat (left panel) or MEF (right panel) cells were fractionated as described in Materials and methods, and then run on SDS–PAGE and Western blotted with the indicated antibodies.
To extend these studies, we reconstituted BCL-2null murine embryonic fibroblast cell lines (MEFs) with comparable levels of stably expressed BCL-2wt or BCL-2AAA (Figure 1B). We then tested the response of the cell lines to the lipid second messenger arachidonic acid, in which apoptosis is heavily dependent on the release of Ca2+ from the ER followed by activation of mitochondrial permeability transition (Scorrano et al, 2001). BCL-2AAA, as compared to BCL-2wt, further improved survival of MEFs (Figure 1B) and Jurkat cells (Figure 1C) following exposure to 30 μM arachidonic acid. The oxidant H2O2 also induces a Ca2+-dependent death (Scorrano et al, 2003), and Jurkat cells expressing BCL-2AAA were significantly more protected than cells expressing BCL-2wt when treated with H2O2 (Figure 1D). This body of data suggests that phosphorylation of BCL-2 can substantially inhibit its ability to protect against Ca2+-dependent death stimuli.
Jurkat or reconstituted MEF cells expressing BCL-2wt or BCL-2AAA were subjected to subcellular fractionation, revealing that slower-migrating phosphorylated BCL-2 species localized principally to the light membrane fraction (enriched for ER) rather than the mitochondria-enriched heavy membrane fraction (Figure 1E). The predominant localization of phospho-BCL-2 to the ER suggests a testable thesis that phosphorylation of BCL-2 might control its activity at this organelle.
Nonphosphorylatable BCL-2AAA reduces the level of released Ca2+
An increasing body of evidence suggests that modulation of ER Ca2+ levels is an important mechanism by which BCL-2 family members can regulate the susceptibility of a cell to apoptosis (Nakamura et al, 2000; Orrenius et al, 2003; Scorrano et al, 2003). As BCL-2AAA demonstrated enhanced protection from Ca2+-dependent death stimuli, we investigated the Ca2+ dynamics of BCL-2null MEFs compared to those corrected with BCL-2AAA or BCL-2wt. Cells were treated with thapsigargin, which irreversibly inhibits the SERCA pump responsible for uptake of Ca2+ from the cytosol into the ER lumen. This results in the passive release of Ca2+ from ER stores and an increase in cytosolic Ca2+, [Ca2+]i, which we measured with the fluorescent dye Fura-2. The restoration of BCL-2wt protein lowered the [Ca2+]i of BCL-2null MEFs elicited by thapsigargin (Figures 2A and B). Of note, cells corrected with BCL-2AAA as compared to BCL-2wt demonstrated significantly further reduced [Ca2+]i in response to thapsigargin (P⩽0.05) (Figures 2A and B).
Figure 2.
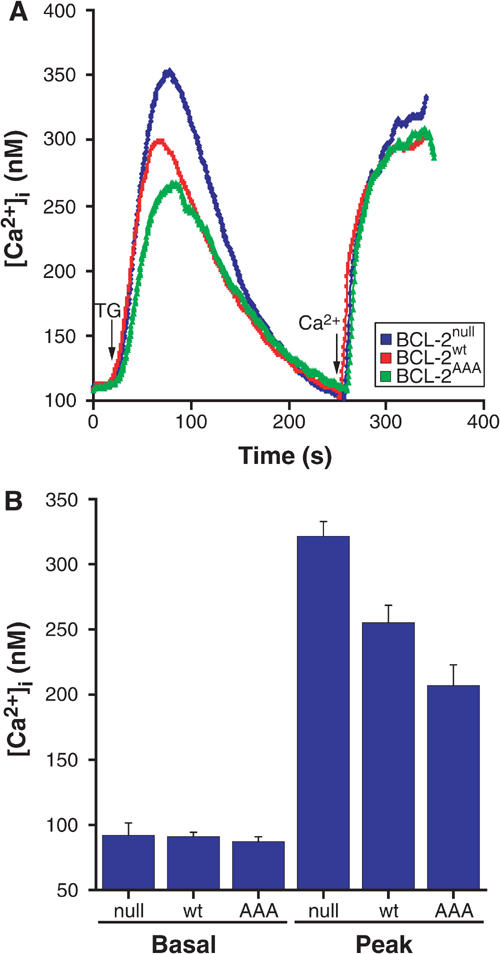
BCL-2wt lowers levels of releasable ER Ca2+, and nonphosphorylatable BCL-2AAA mutants further reduces levels. (A) MEF lines stably transfected with various BCL-2 constructs were loaded with the cytosolic calcium indicator Fura-2, and pulsed with 200 nM thapsigargin (TG) in the absence of extracellular calcium. After baseline was reached, calcium was added back to a final concentration of 2 mM free [Ca2+]. (B) For the curves shown in (A), basal and peak cytosolic levels of calcium were measured following TG-induced release of ER stores (average of 10 experiments).
The extent of capacitative Ca2+ entry was assessed by the addition of extracellular Ca2+. Prior studies reported that cells overexpressing BCL-2 displayed decreased capacitative Ca2+ entry (Foyouzi-Youssefi et al, 2000; Pinton et al, 2000). BCL-2null cells demonstrated greater capacitative Ca2+ entry than cells reconstituted with BCL-2wt (Figure 2A), indicating a role for BCL-2 at physiological levels. The capacitative entry was comparable for BCL-2AAA and BCL-2wt cells (Figure 2A).
Nonphosphorylatable BCL-2AAA expression results in diminished mitochondrial Ca2+ uptake
Mitochondria are closely juxtaposed to the ER, and modulate the Ca2+ response in various signaling pathways following release of Ca2+ from the ER (Hajnoczky et al, 1995; Rizzuto et al, 1998). In addition, mitochondrial Ca2+ uptake following ER discharge has been shown to contribute significantly to the susceptibility of a cell to Ca2+-dependent death stimuli (Bernardi, 1999). Therefore, we tested whether reconstitution of cells with BCL-2AAA, which alters [Ca2+]i following ER discharge, also influences the mitochondrial Ca2+ concentration, [Ca2+]m, as assessed in single cells by real-time fluorescence microscopy using the fluorescent Ca2+ indicator Rhod-2. Passive release of ER Ca2+ stores by thapsigargin was followed by a transient increase in [Ca2+]m. Real-time fluorescent images were acquired (Supplementary data, Movies 1–3), and fluorescence values of mitochondrial regions were quantitated. Ca2+ uptake by mitochondria was reduced in cells reconstituted with BCL-2wt compared to BCL-2null cells, and even further reduced with the expression of BCL-2AAA (Figure 3A). Quantitation of peak [Ca2+]m following thapsigargin (Figure 3B) revealed a 50% increase above the baseline for BCL-2null cells. Reconstitution with BCL-2wt reduced this response by approximately half (∼22% above baseline), and BCL-2AAA even further reduced Ca2+ uptake (∼13% above baseline). Thus, nonphosphorylatable BCL-2AAA demonstrates greater reduction of both intracellular Ca2+ (Figure 2B) and peak mitochondrial uptake of Ca2+ (Figure 3B) following release of ER stores, when compared to BCL-2wt protein.
Figure 3.
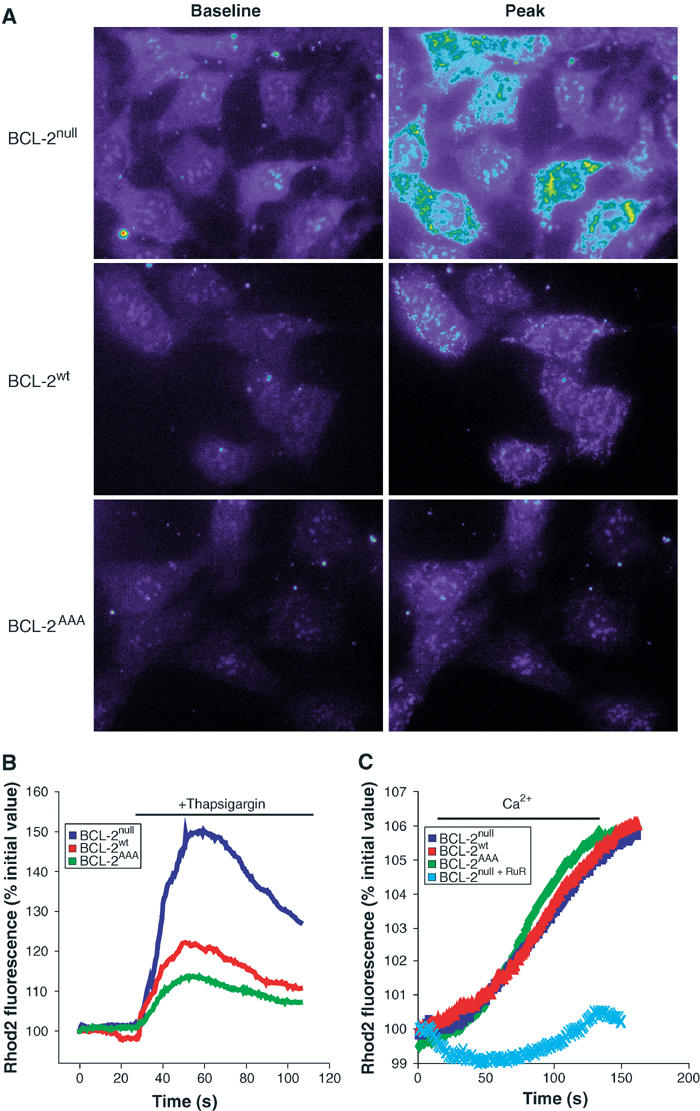
Mitochondria in BCL-2AAA-expressing cells take up lower levels of calcium following release from the ER. (A) MEF lines loaded with Rhod-2 were treated with 200 nM thapsigargin, and sequential images were taken. Baseline and peak fluorescence images were selected for comparison. Complete image sequences are given in Supplementary data, Movies 1–3. (B) For the images in (A), mitochondrial areas were selected, and normalized fluorescence intensity values were measured over time (see Materials and methods). (C) MEFs loaded with Rhod-2 were permeabilized with 0.001% digitonin and exposed to 14.5 μM Ca2+. Ruthenium red (100 μM), an inhibitor of the mitochondrial calcium uniporter, was added where indicated.
We next asked whether the alteration in peak [Ca2+]m observed in BCL-2AAA-expressing cells reflected a primary aberration in the capacity of mitochondria to uptake Ca2+, or was secondary to their lower [Ca2+]i following release of ER Ca2+ stores. We permeabilized the plasma membrane of Rhod-2-loaded cells with 0.001% digitonin and perfused the cells with buffers of fixed [Ca2+]. The kinetics and extent of Rhod-2 fluorescence were comparable in BCL-2null, BCL-2wt, and BCL-2AAA cells, and could be blocked by ruthenium red, an inhibitor of the mitochondrial calcium uniporter (Figure 3C). Thus, there is no inherent difference in mitochondrial Ca2+ handling in these cells, and the diminished level of [Ca2+]m in BCL-2AAA cells appears to be secondary to the lower [Ca2+]i to which the mitochondria are exposed.
Steady-state ER calcium levels are reduced in BCL-2AAA-expressing cells
In order to measure directly the concentration of Ca2+ within the ER, [Ca2+]er, we used aequorin, a calibratable Ca2+ photoprotein reporter that was selectively targeted to this subcellular location (erAEQ) (Pinton et al, 2000). We transfected erAEQ into BCL-2null, BCL-2wt, or BCL-2AAA MEFs, depleted [Ca2+]er to <20 μM, and then allowed them to refill by addition of Ca2+ to the perfusate. Luminescence of erAEQ indicated a marked reduction in the steady-state [Ca2+]er reached when BCL-2null MEFs (618±104 μM) were reconstituted with BCL-2wt (317±75 μM), which was even further reduced if reconstituted with BCL-2AAA (186±20 μM) (Figure 4A).
Figure 4.
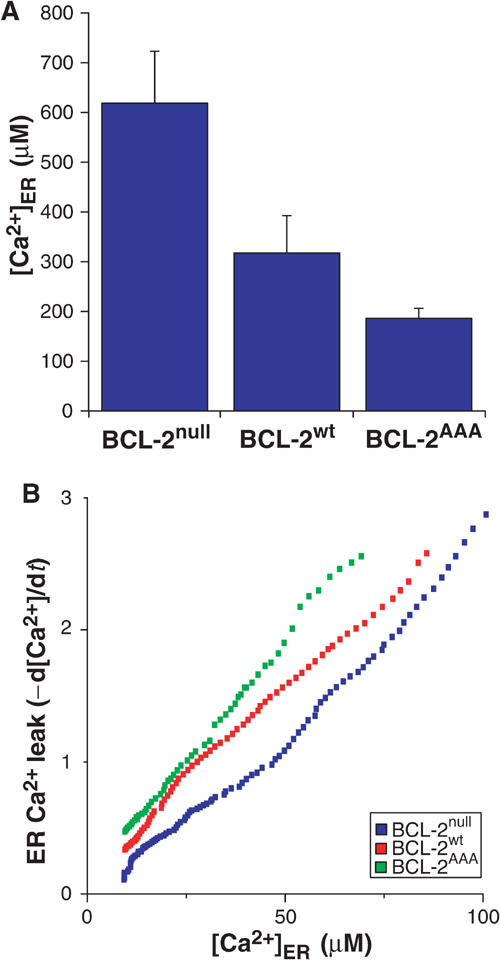
Cells expressing BCL-2AAA have a lower peak ER calcium levels, and higher calcium leak rate from ER. (A) Cells transfected with ER-targeted aequorin were calcium-depleted and processed as described in Materials and methods. While monitoring aequorin luminescence, calcium was added back to obtain the peak calcium content in ER. (B) At peak calcium, SERCA-mediated ER calcium uptake was inhibited with tBuBHQ, and the rate of leak was determined.
Steady-state [Ca2+]er reflects an equilibrium between active Ca2+ uptake and diffusion from the ER. Inhibition of the ER calcium pump SERCA with 2,5-di-(tert-butyl)-1,4-benzohydroquinone (tBuBHQ) enables the rate of leak of Ca2+ from the ER to be measured. After steady-state [Ca2+]er was determined, we added tBuBHQ to the cells, and calculated the change in [Ca2+]er over time as a measure of the rate of leak (d[Ca2+]er/dt). At each [Ca2+]er for which the rate of leak was measured, the stepwise decrease in steady-state [Ca2+]er (Figure 4A) correlated with an increased rate of Ca2+ leak from the ER (Figure 4B). These data revealed that reconstitution of BCL-2null cells with BCL-2wt increased the rate of Ca2+ leak, while the nonphosphorylatable BCL-2AAA enhanced the leak even further.
Phosphorylation of BCL-2 at the ER inhibits its binding to proapoptotic BCL-2 family proteins
It has been suggested that expression of antiapoptotic BCL-2 or BCL-XL may alter levels of various calcium handling proteins such as the ER Ca2+ uptake pump SERCA, calreticulin (Vanden Abeele et al, 2002), or IP3 receptor (Li et al, 2002). Therefore, we wished to test whether levels of such ER Ca2+ handling proteins might account for observed differences between cells expressing BCL-2AAA and BCL-2wt. In general, protein levels of the tested ER Ca2+ handling proteins including calreticulin, calcineurin, GRP 94, IP3RI, IP3RIII, and SERCA 2 appeared comparable in Jurkat cells or Jurkat with BCL-2wt or BCL-2AAA as well as MEFs that are BCL-2null or reconstituted with BCL-2wt or BCL-2AAA (Figure 5A).
Figure 5.
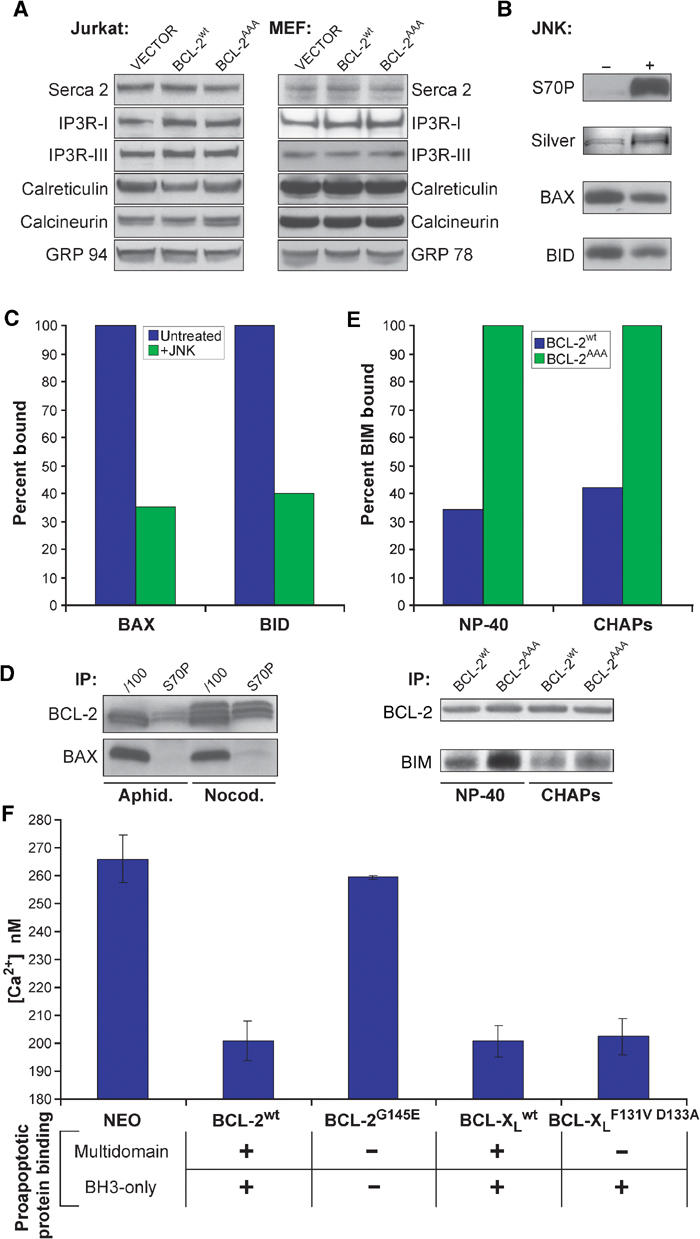
BCL-2 must bind to proapoptotic proteins to lower ER calcium, and this binding is prevented by phosphorylation. (A) RIPA extracts of Jurkat cell lines were analyzed for levels of calcium handling proteins. (B) GST-BCL-2 was treated with JNK kinase, run on SDS–PAGE, and either silver stained or Western blotted with anti-S70P antibody. (C) This protein was then mixed with purified BAX or BID protein in NP-40 buffer, captured with GSH beads, and association with BAX or BID was assessed by Western blot and quantitated by densitometry. (D) Jurkat cells were treated with aphidicolin or nocodazole for 16 h, and then whole-cell extracts were made in IP buffer, immunoprecipitated with the indicated antibody, and then assayed by Western blot. (E) Following subcellular fractionation, light membrane fractions from MEF lines were solubilized in NP-40 or CHAPS IP buffer, immunoprecipitated with anti-BCL-2 antibody, and assayed for associated BIM by Western blot. (F) Fl 5.12 lines expressing the indicated constructs were loaded with Fura-2, and peak calcium following thapsigargin treatment was measured.
We next tested whether phosphorylation of sites within the unstructured loop would affect another activity of BCL-2, its capacity to bind BH3 domains of proapoptotic members. Phosphorylation of recombinant GST-BCL-2wt by JNK substantially reduced its ability to bind recombinant multidomain BAX or the BH3-only protein BID (Figures 5B and C; ∼70% reduction). Thus, phosphorylation of BCL-2 in this purified system diminished its binding to both multidomain and BH3-only proapoptotic family members.
Immunoprecipitation (IP) of total BCL-2 from solubilized lysates of Jurkat cells expressing BCL-2wt resulted in co-precipitation of substantial amounts of BAX. However, when the phosphorylated fraction of BCL-2 was selectively immunoprecipitated with a BCL-2 S70-P-specific antibody, it did not co-precipitate BAX (Figure 5D). Of note, nocodazole-treated, G2/M-arrested cells had more phosphorylated BCL-2 than aphidicolin-treated, G1/S-arrested cells.
One principal role of BCL-2 in preventing apoptosis is to bind and sequester BH3-only proapoptotic proteins such as BIM, BID, and BAD (Cheng et al, 2001). As most phosphorylated BCL-2 resides at the ER (Figure 1E), we compared the ability of ER-localized BCL-2wt and BCL-2AAA to bind BH3-only proteins. The IP of nonphosphorylatable BCL-2AAA from ER-enriched light membrane fractions of MEFs co-precipitated substantially more BIM than did BCL-2wt protein (Figure 5E). This was noted when ER fractions were solubilized in either NP-40, which maximizes interactions, or in CHAPS, which does not induce conformational changes of BCL-2 members. While BIM was the predominant BH3-only protein of those tested, BID and BAD were also more highly associated with nonphosphorylated BCL-2. Together, this compilation of studies indicates that phosphorylation of ER-based BCL-2 can substantially reduce its binding to multidomain and BH3-only proapoptotic members.
Capacity of BCL-2 to bind proapoptotic BH3-only proteins correlates with ER Ca2+ levels
We next asked if the capacity of BCL-2 to lower steady-state [Ca2+]er was related to its ability to bind proapoptotic members. Fura-2 was used to measure Ca2+ release from the ER of Fl 5.12 cells expressing BCL-2wt or BCL-2G145E, a BCL-2 mutant that fails to bind either multidomain or BH3-only proapoptotic members (Yin et al, 1994). Strikingly, BCL-2G145E does not lower [Ca2+]i (Figure 5F), correlating with its inability to bind proapoptotic BCL-2 family proteins. Like BCL-2wt, BCL-XLwt also lowers [Ca2+]i (Figure 5F), and a portion of BCL-XL localizes to the ER-enriched light membrane fraction (Supplementary Figure 1). Of note, the BCL-XLF131VD133A mutant, which cannot bind BAX or BAK, but retains the ability to bind BH3-only proteins (Cheng et al, 2001), did substantially reduce [Ca2+]i (Figure 5F). Thus, both in vivo and in vitro, the capability of antiapoptotic BCL-2 or BCL-XL to bind BH3-only proteins is coordinate with the level of releasable ER Ca2+ stores.
Discussion
Here we address the mechanism by which phosphorylation of BCL-2 inhibits its antiapoptotic activity. The predominant localization of phosphorylated BCL-2 to the ER (Figure 1) provided the initial insight that prompted our assessment of the role for BCL-2 phosphorylation at this gateway to apoptosis. Specifically, we asked whether phosphorylation of BCL-2 would alter its regulation of ER Ca2+ dynamics.
We found that nonphosphorylatable BCL-2AAA displayed enhanced protection against arachidonic acid and oxidative stress, stimuli that depend on the release of ER Ca2+ stores (Figure 1). BCL-2AAA reduced steady-state [Ca2+]er even more profoundly than BCL-2wt, which appears to reflect a further increased rate of Ca2+ leak from the ER when BCL-2 cannot be phosphorylated (Figure 4). Consequently, diminished release of ER Ca2+ results in lower intracellular Ca2+ ([Ca2+]i) (Figure 2) and a secondary decrease in Ca2+ uptake by the mitochondria in cells expressing BCL-2AAA (Figure 3). However, there was no inherent defect in mitochondrial uptake of Ca2+ in BCL-2AAA cells when compared to BCL-2null or BCL-2wt expressing cells when mitochondria were exposed to Ca2+ (Figure 3). Thus, BCL-2 is more capable of reducing [Ca2+]er and blocking apoptosis when it is not phosphorylated. In this regard, phosphorylation of BCL-2 reduces protection from a variety of stimuli, including taxol and etoposide (Figure 1), which can also release ER Ca2+ (Ueoka and Yamaguchi, 1997; Srivastava et al, 1999; Kidd et al, 2002), likely reflecting the participation of ER Ca2+ release in many types of apoptotic death (Scorrano et al, 2003).
Phosphorylation of BCL-2 has been noted to reduce binding to BAX (Poommipanit et al, 1999). Our data indicate that phosphorylation modulates the binding of BCL-2 to BH3-only proteins such as BID and BIM as well as multidomain proapoptotic proteins such as BAX (Figure 5B–E). Of note, deamidation within the loop region of BCL-XL was recently shown to reduce its binding affinity for BIM (Deverman et al, 2002). Taken together, this suggests that post-translational modification of the loop region of antiapoptotic BCL-2 family members can regulate the binding between their pocket/groove and the BH3 domains of proapoptotic members. As in vitro phosphorylation of recombinant BCL-2 protein alters its binding to proapoptotic BAX or BID (Figure 5B and C), phosphorylation appears to be sufficient to induce an intramolecular conformational change that inhibits this activity.
Phosphorylation of BCL-2 has been reported in response to multiple stimuli, and apparently can be mediated by a variety of kinases, including Raf-1 (Blagosklonny et al, 1997), PKCα (Ruvolo et al, 1998), PKA (Srivastava et al, 1998), and JNK (Yamamoto et al, 1999). BCL-2 has also been noted to be phosphorylated following addition of growth factors (May et al, 1994; Poommipanit et al, 1999). In addition, v-cyclin/cdk6 can phosphorylate BCL-2 in U2OS and Cos-7 cells during G1/S (Ojala et al, 2000). Of note, we detected substantial phosphorylated BCL-2 in asynchronous cultures (Figure 1E) and even some phosphorylated BCL-2 in aphidicolin-arrested (G1/S) cells (Figure 5D). However, stress response kinases appear to be responsible for the robust phosphorylation of BCL-2 during cell cycle progression as a normal physiologic process that inactivates BCL-2 at G2/M (Yamamoto et al, 1999). JNK has been reported to be activated by an ER-resident complex following ER stress (Urano et al, 2000), consistent with our data. We and others have noted that the JNK pathway is also responsible for phosphorylation of BCL-2 on the same residues in the loop region in response to microtubule-damaging agents, which also arrest cells in G2/M (Haldar et al, 1998; Ling et al, 1998; Scatena et al, 1998). Given our results here, phosphorylation of BCL-2 during mitosis would increase [Ca2+]er, and could account for the increased G2/M susceptibility to apoptosis noted at this checkpoint (Yamamoto et al, 1999).
Both pro- and antiapoptotic BCL-2 family members reside at the ER and influence Ca2+ dynamics. A principal mechanism whereby antiapoptotic BCL-2 and BCL-XL block cell death is to bind and sequester proapoptotic BH3-only molecules (Cheng et al, 2001). This binding can occur at the ER (Germain et al, 2002; Thomenius et al, 2003), and as demonstrated here phosphorylated BCL-2 binds less BH3-only BIM or multidomain BAX proapoptotic protein (Figure 5). The BCL-XLF131VD133A mutant that does not bind BAX or BAK but still binds BH3-only molecules still reduced releasable stores of ER Ca2+ (Figure 5). Conversely, the BCL-2G145E mutant incapable of binding either BH3-only or multidomain proapoptotic molecules had lost its Ca2+ regulatory effect. This compilation of findings provides further support for a rheostat model in which the ratio of pro- to antiapoptotic BCL-2 family members also operates at the ER to coordinate Ca2+ dynamics. Together, our data indicate that the phosphorylation status of BCL-2 determines its capacity to bind BH3-only molecules and the releasable stores of [Ca2+]er. Overall, the control of ER Ca2+ dynamics appears to be a principal pathway by which the phosphorylation of BCL-2 regulates its antiapoptotic activity.
Materials and methods
Cell lines and plasmids
Fl 5.12 lines (Cheng et al, 2001), and Jurkat cells transfected with NEO-resistant vector, BCL-2wt, or BCL-2AAA (Yamamoto et al, 1999) have been described previously. MEFs were prepared from BCL-2null mice (Veis et al, 1993), transformed with SV40 genomic DNA, and then transfected with either human BCL-2wt or T69AS70AS87A=BCL-2AAA cloned into the MSCV puro vector (Clontech). Stable clones were selected in 1.3 μg/ml puromycin (Sigma), and grown in Iscove's modified Dulbecco's medium supplemented with minimal essential amino acids, L-glutamine, penicillin/streptomycin, and 10% fetal bovine serum. Clones with matched expression levels of BCL-2 were selected for further analysis.
Cell death assay
Jurkat or MEF cells were treated as described in the figure legends; taxol, thapsigargin, etoposide, and staurosporine were purchased from Sigma. Anti-Fas clone CH-11 is from Upstate. Arachidonic acid (Alexis Pharmaceuticals) was added to cells in RPMI without serum for the indicated times, and H2O2 (Sigma) was added directly to the growth medium. Annexin-V-cy-3 (Biovision) was used according to the manufacturer's protocol.
Fura-2 and Rhod-2 measurements
Fura-2-AM and Rhod-2-AM were purchased from Molecular Probes. Fura-2 was loaded into cells for 30 min at 37°C in Hanks balanced salt solution (HBSS) supplemented with 10% fetal bovine serum, and then cells were washed with PBS. For assay, cells were suspended in HBSS/10% FBS, and then extracellular calcium was chelated with the addition of 2 mM EGTA. Thapsigargin (200 nM; Sigma) was added to the medium to induce the release of calcium from the ER, and cytosolic Fura-2 fluorescence was assayed on a Perkin-Elmer LS-50B spectrofluorimeter, as described previously (Scorrano et al, 2003).
Rhod-2 assays were performed as described previously (Scorrano et al, 2003). Briefly, 105 cells were plated onto 25-mm round coverslips, and 24 h later cells were loaded with 4 μM Rhod-2-AM in HBSS for 3 h at 4°C. Coverslips were then mounted into the coverslip holder, washed with HBSS, and placed on the stage of a Nikon Eclipse TE200 inverted microscope equipped with a Perkin-Elmer Ultraview confocal scanner and a Hamamatsu Orca ER 12-bit CCD camera. Cells were excited using the 547 nm laser line and sequential images were taken every 500 ms with exposure times of 30 ms using a × 60 1.4 NA Plan Apo objective (Nikon) and Ultraview imaging suite (Perkin-Elmer). Images were then analyzed with Metafluor software (Universal Imaging). 20 areas of mitochondrial fluorescence for each experiment were identified in the peak fluorescent frame and the average intensity of each area was measured over the course of the experiment. These values were background subtracted, normalized for comparison, and averaged to give a curve representing the change in fluorescence over the course of the experiment. Curves representing the average of 10 experiments were compiled.
Intrinsic mitochondrial Ca2+ uptake ability was measured by exposing mitochondria in 0.001% digitonin-permeabilized cells to Ca2+-EGTA-buffered cytosolic solutions (Molecular Probes) with a final [Ca2+] of 14.5 μM.
Aequorin measurements
ER aequorin reconstitution, luminescence analysis, calibration of [Ca2+], and leak rate measurements were performed as described previously (Pinton et al, 2000; Scorrano et al, 2003). Briefly, cells were grown on 13 mM round glass coverslips, and transfected with the erAEQ construct using Fugene 6. After 24 h, luminal calcium was depleted for 10 min at 4°C in KRB (125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 5.5 mM glucose, and 20 mM Hepes pH 7.4)+1 mM EGTA, 10 μM ionomycin, and 100 μM tBuBHQ. Cells were washed twice in the same buffer without ionomycin but with 10% FCS, and then aequorin was reconstituted for 2 h at 4°C with 5 μM coelenterazine-N-AM (Molecular Probes) in KRB+1 mM EGTA and 100 μM tBuBHQ. Coverslips were then washed with KRB/EGTA/10% FCS, and placed in a KRB/EGTA-perfused, thermostated chamber in the luminometer. Luminescence of the coverslip was measured during addition of calcium to the perfusate (KRB+1 mM Ca2+). After [Ca2+]er reached steady state, ER Ca2+ leak measurements were performed by perfusing cells with KRB+EGTA and 100 μM tBuBHQ.
Immunoprecipitation, subcellular fractionation, and Western blotting
IPs and binding assays were conducted in either NP-40 buffer (0.2% NP-40, 150 mM KCl, 1 mM EGTA, 50 mM Hepes pH 7.5, 5 mM MgCl2, 50 mM NaF, 1 mM Na3VO4, 250 μM PMSF, and 5 μg/ml pepstatin) or CHAPS buffer (1% CHAPS, 100 mM KCl, 50 mM Hepes pH 7.5, 1 mM EGTA, 50 mM NaF, 1 mM Na3VO4, 250 μM PMSF, and 5 μg/ml pepstatin). IP was performed in 500 μl extract containing 750 μg protein, which was precleared by centrifugation at 16 000 g followed by exposure to 30 μl protein A/G beads (Santa Cruz). Cleared extracts were incubated with antibody for 2 hours, and captured on 30 μl beads for 1 hour, washed three times, and boiled in SDS sample buffer. Anti-human BCL-2 clone/100 antibody was purchased from Pharmingen, and N20 anti-BAX antibody was purchased from Santa Cruz. Anti-BID antibodies were described previously (Wang et al, 1996). Anti-S70P antibodies were generated in rabbits immunized against the following peptide (CRDPVARTpSPLQTP). For IP antibodies were used at 1 μg/100 μl, and for Western blotting antibodies were used at 1 μg/ml. Aphidicolin and nocodazole (Sigma) were added to cells for 16 h.
Subcellular fractionation was performed on 109 cells. Cells were washed 1 × with PBS, suspended in HIM buffer (200 mM mannitol, 70 mM sucrose, 10 mM HEPES pH 7.5, and 1 mM EGTA), and then disrupted using a polytron cell homogenizer (Kinematica). After a crude fraction was removed at 700 g, a heavy membrane fraction was removed at 7000 g for further purification on a percoll gradient (Danial et al, 2003). Light membrane fraction was separated from cytosolic fraction at 280 000 g. Proteins were either solubilized in NP-40 or CHAPS buffer for IP, or in RIPA buffer (1% NP-40, 1% Na-deoxycholate, 0.1% SDS, 150 mM NaCl, 10 mM HEPES pH 7.5, 2 mM EDTA, 50 mM NaF, 200 μM Na3VO4, 250 μM PMSF, and 5 μg/ml pepstatin) for Western blotting.
Methods for obtaining purified GST-BCL-2 (Letai et al, 2002), BAX (Suzuki et al, 2000), and BID (Zha et al, 2000) have been described previously. Purified JNK (Upstate) was incubated with GST-BCL-2 for 16 h at 30°C in a buffer containing 10 mM HEPES pH 7.5, 25 mM MgCl2, 1 mM EGTA, 1 mM ATP, and 250 μM PMSF.
Supplementary Material
Supplementary Figure 1BCL-XL is located in the ER-enriched light membrane fraction, as well as the cytosol and heavy membrane fraction.
Supplemental Figure
Supplementary data, Movie 1
Supplementary data, Movie 2
Supplementary data, Movie 3
Acknowledgments
We thank J Opferman and E Cheng for technical assistance and helpful discussions, K Yamamoto for design of the phospho-specific antibody, and E Smith for help with figure and manuscript preparation. MCB is sponsored by NIH training grant T32 CA09361. This work was supported in part by NIH grant R37CA50239. Research in LS lab is supported by Telethon (TCP012016).
References
- Adams JM, Cory S (2001) Life-or-death decisions by the Bcl-2 protein family. Trends Biochem Sci 26: 61–66 [DOI] [PubMed] [Google Scholar]
- Bernardi P (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79: 1127–1155 [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV, Giannakakou P, el-Deiry WS, Kingston DG, Higgs PI, Neckers L, Fojo T (1997) Raf-1/bcl-2 phosphorylation: a step from microtubule damage to cell death. Cancer Res 57: 130–135 [PubMed] [Google Scholar]
- Chang BS, Minn AJ, Muchmore SW, Fesik SW, Thompson CB (1997) Identification of a novel regulatory domain in Bcl-X(L) and Bcl-2. EMBO J 16: 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8: 705–711 [DOI] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang C-Y, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, Gygi SP, Korsmeyer SJ (2003) BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424: 952–956 [DOI] [PubMed] [Google Scholar]
- Demaurex N, Distelhorst C (2003) Cell biology. Apoptosis–the calcium connection. Science 300: 65–67 [DOI] [PubMed] [Google Scholar]
- Deverman BE, Cook BL, Manson SR, Niederhoff RA, Langer EM, Rosova I, Kulans LA, Fu X, Weinberg JS, Heinecke JW, Roth KA, Weintraub SJ (2002) Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell 111: 51–62 [DOI] [PubMed] [Google Scholar]
- Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N, Krause KH (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci USA 97: 5723–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Mathai JP, Shore GC (2002) BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J Biol Chem 277: 18053–18060 [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82: 415–424 [DOI] [PubMed] [Google Scholar]
- Haldar S, Basu A, Croce CM (1998) Serine-70 is one of the critical sites for drug-induced Bcl2 phosphorylation in cancer cells. Cancer Res 58: 1609–1615 [PubMed] [Google Scholar]
- Kidd JF, Pilkington MF, Schell MJ, Fogarty KE, Skepper JN, Taylor CW, Thorn P (2002) Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J Biol Chem 277: 6504–6510 [DOI] [PubMed] [Google Scholar]
- Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW (1994) Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci USA 91: 6569–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192 [DOI] [PubMed] [Google Scholar]
- Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB (2002) Bcl-X(L) affects Ca(2+) homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc Natl Acad Sci USA 99: 9830–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489 [DOI] [PubMed] [Google Scholar]
- Ling YH, Tornos C, Perez-Soler R (1998) Phosphorylation of Bcl-2 is a marker of M phase events and not a determinant of apoptosis. J Biol Chem 273: 18984–18991 [DOI] [PubMed] [Google Scholar]
- Martinou JC, Green DR (2001) Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2: 63–67 [DOI] [PubMed] [Google Scholar]
- May WS, Tyler PG, Ito T, Armstrong DK, Qatsha KA, Davidson NE (1994) Interleukin-3 and bryostatin-1 mediate hyperphosphorylation of BCL2 alpha in association with suppression of apoptosis. J Biol Chem 269: 26865–26870 [PubMed] [Google Scholar]
- Nakamura K, Bossy-Wetzel E, Burns K, Fadel MP, Lozyk M, Goping IS, Opas M, Bleackley RC, Green DR, Michalak M (2000) Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J Cell Biol 150: 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala PM, Yamamoto K, Castanos-Velez E, Biberfeld P, Korsmeyer SJ, Makela TP (2000) The apoptotic v-cyclin–CDK6 complex phosphorylates and inactivates Bcl-2. Nat Cell Biol 2: 819–825 [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609–619 [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol 4: 552–565 [DOI] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R (2000) Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 148: 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poommipanit PB, Chen B, Oltvai ZN (1999) Interleukin-3 induces the phosphorylation of a distinct fraction of bcl-2. J Biol Chem 274: 1033–1039 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766 [DOI] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, Carr BK, May WS (1998) A functional role for mitochondrial protein kinase Calpha in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem 273: 25436–25442 [DOI] [PubMed] [Google Scholar]
- Scatena CD, Stewart ZA, Mays D, Tang LJ, Keefer CJ, Leach SD, Pietenpol JA (1998) Mitotic phosphorylation of Bcl-2 during normal cell cycle progression and Taxol-induced growth arrest. J Biol Chem 273: 30777–30784 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Penzo D, Petronilli V, Pagano F, Bernardi P (2001) Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha aopototic signaling. J Biol Chem 276: 12035–12040 [DOI] [PubMed] [Google Scholar]
- Srivastava RK, Mi QS, Hardwick JM, Longo DL (1999) Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci USA 96: 3775–3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS, Longo DL (1998) Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase. Mol Cell Biol 18: 3509–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654 [DOI] [PubMed] [Google Scholar]
- Thomenius MJ, Wang NS, Reineks EZ, Wang Z, Distelhorst CW (2003) Bcl-2 on the endoplasmic reticulum regulates Bax activity by binding to BH3-only proteins. J Biol Chem 278: 6243–6250 [DOI] [PubMed] [Google Scholar]
- Ueoka S, Yamaguchi M (1997) Effect of apoptosis-related compounds on Ca2+ transport system in isolated rat liver nuclei. Mol Cell Biochem 166: 183–189 [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666 [DOI] [PubMed] [Google Scholar]
- Vanden Abeele F, Skryma R, Shuba Y, Van Coppenolle F, Slomianny C, Roudbaraki M, Mauroy B, Wuytack F, Prevarskaya N (2002) Bcl-2-dependent modulation of Ca(2+) homeostasis and store-operated channels in prostate cancer cells. Cancer Cell 1: 169–179 [DOI] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ (1993) Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75: 229–240 [DOI] [PubMed] [Google Scholar]
- Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ (1996) BID: a novel BH3 domain-only death agonist. Genes Dev 10: 2859–2869 [DOI] [PubMed] [Google Scholar]
- Wang NS, Unkila MT, Reineks EZ, Distelhorst CW (2001) Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J Biol Chem 276: 44117–44128 [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Ichijo H, Korsmeyer SJ (1999) BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 19: 8469–8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin XM, Oltvai ZN, Korsmeyer SJ (1994) BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369: 321–323 [DOI] [PubMed] [Google Scholar]
- Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ (2000) Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290: 1761–1765 [DOI] [PubMed] [Google Scholar]
- Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW (1996) Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J 15: 4130–4141 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1BCL-XL is located in the ER-enriched light membrane fraction, as well as the cytosol and heavy membrane fraction.
Supplemental Figure
Supplementary data, Movie 1
Supplementary data, Movie 2
Supplementary data, Movie 3