

Abstract
Mutations of p53 are remarkably rare in acute promyelocytic leukemias (APLs). Here, we demonstrate that the APL-associated fusion proteins PML–RAR and PLZF-RAR directly inhibit p53, allowing leukemic blasts to evade p53-dependent cancer surveillance pathways. PML–RAR causes deacetylation and degradation of p53, resulting in repression of p53 transcriptional activity, and protection from p53-dependent responses to genotoxic stress. These phenomena are dependent on the expression of wild-type PML, acting as a bridge between p53 and PML–RAR. Recruitment of histone deacetylase (HDAC) to p53 and inhibition of p53 activity were abrogated by conditions that either inactivate HDACs or trigger HDAC release from the fusion protein, implicating recruitment of HDAC by PML–RAR as the mechanism underlying p53 inhibition.
Keywords: histone deacetylase, p53, PML, PML–RAR, promyelocytic leukemia
Introduction
Acute myeloid leukemias (AMLs) are associated with chromosomal translocations resulting in the formation of fusion proteins (Rabbitts, 1991, 1994; Look, 1997). Fusion proteins, however, are not sufficient to induce leukemogenesis (Westervelt and Ley, 1999): a ‘two-hit' model hypothesizes that they induce a ‘pre-leukemic' state, which favors the clonal occurrence of secondary genetic lesions required for the overt leukemic phenotype (Westervelt and Ley, 1999; He et al, 2000; Kogan et al, 2001).
In acute promyelocytic leukemia (APL), PML (or—in a few cases—PLZF) is found in a reciprocal translocation with the retinoic acid (RA) receptor alpha (RAR), resulting in the fusion protein PML–RAR (or PLZF–RAR: Lin et al, 1999; Minucci and Pelicci, 1999). RARs repress transcription of RA target genes, due to recruitment of histone deacetylases (HDACs). Physiological concentrations of RA cause dissociation of the HDAC complex and transcriptional activation of RA target genes (Minucci and Pelicci, 1999). APL blasts are characterized by a differentiation block at the promyelocytic stage, caused by aberrant recruitment of HDACs by PML–RAR (or PLZF–RAR) in the presence of physiological concentrations of RA. In turn, HDAC recruitment causes inappropriate transcriptional repression of RAR target genes. In the case of PML–RAR, altered recruitment of HDAC is due to oligomerization of the fusion protein through the PML moiety (Minucci et al, 2001). Pharmacological concentrations of RA cause dissociation of the PML–RAR/HDAC complex and degradation of the fusion protein, restoring the normal differentiation pathway (Minucci et al, 2001). The clinical treatment of APL patients with RA is, indeed, a paradigm for ‘differentiation therapy' (Warrell et al, 1993; Altucci and Gronemeyer, 2001).
PML is required for p53 acetylation, stabilizing the complex between p53 and the p53 acetylases p300 and CBP (Ferbeyre et al, 2000; Fogal et al, 2000; Pearson et al, 2000; Gottifredi and Prives, 2001; Pearson and Pelicci, 2001; Prives and Manley, 2001).
The tumor suppressor p53 controls genomic stability (Ko and Prives, 1996; Levine, 1997; Vogelstein et al, 2000; Ryan et al, 2001). p53 is maintained at a low level by constitutive degradation, mainly due to association with the MDM2 protein (Prives, 1998). Upon stress, p53 undergoes post-translational modifications (phosphorylation and acetylation), which lead to increased stability and transcriptional activity (Appella and Anderson, 2001; Prives and Manley, 2001). p53 regulates the expression of genes involved in the cellular stress response, mediating irreversible growth arrest or apoptosis (Levine, 1997; Vogelstein et al, 2000; Vousden, 2000; Ryan et al, 2001).
Paradoxically, mutations of p53 are exceedingly rare in APL (Longo et al, 1993; Trecca et al, 1994). Here, we show that PML–RAR inhibits p53 function by promoting its deacetylation and degradation, thereby allowing APL blasts to overcome p53-mediated oncogene surveillance pathways.
Results
PML–RAR inhibits p53-dependent stress responses
We used murine hematopoietic progenitors purified from bone marrow by depletion of cells expressing differentiation markers (lin- cells). Lin- represents, therefore, cells at early stages of differentiation, including true stem cells (Spangrude et al, 1988; Heimfeld et al, 1991; Morrison et al, 1997). One subpopulation of lin- cells, the colony-forming cells (CFCs: about 3–5% of the lin- cells at an early differentiative state), can be assayed for their ability to form colonies in semi-solid medium (methylcellulose). Lin- cells can be maintained ex vivo using high concentrations of cytokines. Since hematopoietic progenitors are more resistant to stress in the presence of cytokines (Quelle et al, 1998), we treated lin- cells with X-rays or hydrogen peroxide (H2O2) in the absence of cytokines, and observed a drastic decrease in the number of CFCs (Figure 1A). This stress response is p53-dependent, since it is nearly absent in p53 null lin- cells (Figure 1A).
Figure 1.
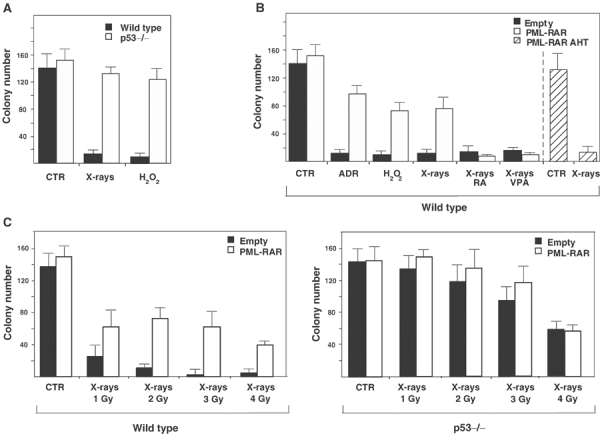
PML–RAR abrogates the p53-dependent loss of CFCs upon stress. (A) Lin- cells from wild-type (black bars) or p53−/− (white bars) mice were subjected to cytokine deprivation and exposed to X-rays (2 Gy), oxidative stress (100 μM H2O2), or left untreated (CTR). A total of 5000 cells were then plated in methylcellulose medium, in the presence of cytokines. After 8 days, colonies were counted. (B) Lin- cells were prepared from wild-type mice, and infected as indicated. Sorted cells underwent cytokine deprivation (CTR) and were then exposed to X-rays, adriamycin (ADR), or oxidative stress as indicated. X-ray treatment was performed in the absence or in the presence of RA (1 μM) or VPA (1 mM). Cells were then plated in methylcellulose medium. (C) After cytokine deprivation (CTR), wild-type and p53−/− cells were exposed to increasing doses of X-rays, and then plated in methylcellulose medium.
To express uniform levels of PML–RAR in lin- cells, we used high-titer retroviruses encoding the fusion protein and GFP as a selectable marker (Minucci et al, 2000, 2002). This strategy allows targeting of bona fide leukemia-initiating cells, since reinoculation of infected cells causes leukemia in more than 85% of the recipient mice (Minucci et al, 2002). PML–RAR expression markedly protected lin- cells against reduction in the number of CFCs following treatment with X-rays, hydrogen peroxide or adriamycin (Figure 1B). As the stress-induced reduction of CFCs is p53-dependent (Figure 1A), we hypothesized that PML–RAR might inhibit p53 function. We, therefore, expressed PML–RAR in p53 null lin- cells, and subjected the cells to an escalating X-ray dose (Figure 1C). At all doses, PML–RAR protected wild-type, lin- cells from loss of CFCs (Figure 1C, left panel). As expected, control p53 null lin- cells were almost completely resistant to low doses of X-rays, and still significantly protected at high doses (Figure 1C, right panel). PML–RAR expressing p53 null lin- cells showed a slightly larger number of colonies at lower doses of X-rays, which was not statistically significant (Figure 1C, right panel). Notably, PML–RAR was unable to protect p53 null cells from the loss of CFCs observed at higher X-ray doses (4 Gy), demonstrating that the protection by PML–RAR is largely—if not completely—due to inhibition of p53 (Figure 1C, right panel).
Treatment with pharmacological doses of RA dissociates the PML–RAR/HDAC complex, triggers PML–RAR degradation and induces differentiation of APL blasts (Minucci and Pelicci, 1999). Likewise, HDAC inhibitors release the differentiation block of PML–RAR-expressing cells, without inducing degradation of the fusion protein (Gottlicher et al, 2001). Mutation of three residues within the RAR moiety (AHT mutation) disrupts binding of PML–RAR to HDAC, and abrogates its ability to block differentiation (Grignani et al, 1998). We therefore analyzed the effect of RA, valproic acid (VPA, an HDAC inhibitor) and PML–RAR AHT. In the absence of stress, RA or VPA did not affect the number of CFCs (Supplementary Figure 1). As shown in Figure 1B, the protective effect of PML–RAR on CFCs was lost in the presence of RA or VPA, and PML–RAR AHT did not protect from loss of CFCs, although expressed at comparable levels to PML–RAR (Figure 1B and data not shown). These results demonstrate that PML–RAR protects CFCs from a p53-dependent stress response, in a manner that requires HDAC recruitment and activity.
HDAC-dependent inhibition of p53 transcriptional activity
To analyze the capacity of the fusion protein to repress the p53 transcriptional activity, transient transfection assays were performed in p53−/− murine embryo fibroblasts (MEFs), using transfected p53 and a reporter containing the physiologically p53-responsive Bax promoter. PML–RAR suppressed p53-mediated transactivation (Figure 2A). Identical results were obtained using other p53-responsive reporters (p21, mdm2: data not shown). PML–RAR did not repress transcription from p53-unrelated promoters (such as those responsive to GAL4-VP16 or p65 of the NF-kB complex: data not shown). In the presence of RA, or when harbouring the AHT mutation, PML–RAR lost its ability to repress p53 transactivation (Figure 2B), suggesting a requirement for HDAC recruitment. To extend these observations, we checked the effect of PML–RAR on the transcriptional activity of p53 in the context of endogenous promoters. We analyzed samples derived from mice that developed APL upon reinoculation of PML–RAR-expressing lin- cells (Minucci et al, 2002). Leukemic or normal mice were treated with X-rays, killed, and then spleens or bone marrows (with a strong infiltration of leukemic blasts) were analyzed for RNA levels of several p53 target genes by hybridization to a filter array. X-ray treatment caused a strong induction of the tested genes in normal mice, which was markedly or completely inhibited in the leukemic mice (Figure 2C and Supplementary Figure 2). In some cases, basal transcript levels were also downregulated in APL samples (Supplementary Figure 2). Real-time PCR analysis on selected genes showed, upon X-ray treatment, a two- to three-fold increase in the expression of Bax, Fas, MDM2 and Apaf-1 in normal spleen and marrow cells. In contrast, leukemic cells failed to display upregulation of these p53-target genes under the same experimental conditions (Figure 2D and data not shown).
Figure 2.
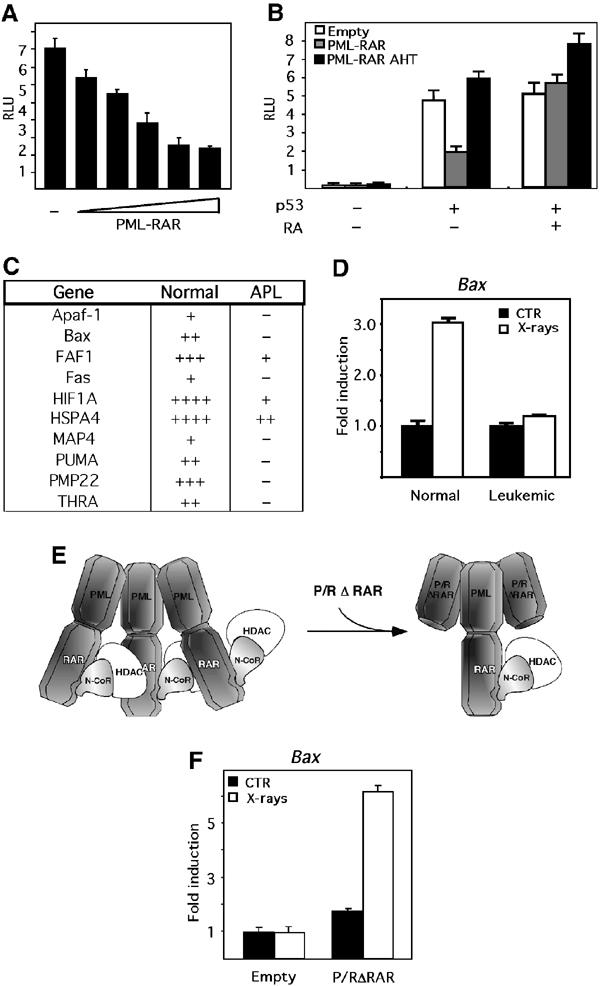
PML–RAR suppresses p53 transcriptional activity in a HDAC-dependent manner. (A) p53−/− MEFs were transfected with the reporter bax-luciferase, and increasing amounts of PML–RAR expression vector (50, 100, 150, 200 and 250 ng), in the presence of a p53 expression vector. Values were normalized against the activity of a co-transfected β-galactosidase-expressing plasmid. (B) p53−/− MEFs were transfected as in (A), with empty, PML–RAR- or PML–RAR AHT-expressing vectors, in the absence or presence of a p53-expressing vector. Where indicated, cells were treated with 1 μM RA. (C) Analysis of a ‘p53 pathway' Superarray filter was performed on RNA samples derived from normal and APL mice, prior/after X-ray treatment in vivo; the table shows the extent of induction measured in normal and APL mice. (D) Normal and leukemic spleen cells were left untreated (CTR), or treated with X-rays, and mRNA levels of p53 transcriptional targets (Bax, Fas, Apaf-1, MDM2) were checked by quantitative PCR (Q-PCR) analysis. Fas, MDM2 and Apaf-1 showed identical results. (E) Scheme for the use of P/RΔRAR as a dominant-negative PML–RAR. (F) APL blasts were infected with the empty vector, or with a P/RΔRAR expression vector, and GFP-positive cells were sorted. Cytokine-deprived cells were left untreated or X-ray irradiated, and then analyzed for Bax, fas, MDM2 and Apaf-1 expression. Fas, MDM2 and Apaf-1 gave identical results.
Overt leukemia represents the final stage of a series of genetic or epigenetic alterations triggered by the fusion protein. The inhibition of p53 observed in leukemic blasts could, therefore, not depend directly on PML–RAR expression (Di Croce et al, 2002). To clarify this issue, we used a dominant-negative PML–RAR, which lacks the RAR moiety and retains the PML portion (P/RΔRAR: see Figure 2E). This mutant forms stable complexes with PML–RAR (through its oligomerization domain), and reverts the differentiation block induced by PML–RAR (data not shown), suggesting that it might inhibit oligomerization and aberrant HDAC recruitment by the fusion protein (Minucci et al, 2000). We used P/RΔRAR to inhibit PML–RAR in the leukemic blasts. X-rays induced a marked upregulation of Bax (or fas, MDM2, Apaf-1) in the APL blasts expressing P/RΔRAR, as opposed to cells transduced with the empty vector, thereby showing that PML–RAR expression is required to maintain p53 in an inactive state in APL blasts (Figure 2F and data not shown).
PML–RAR destabilizes p53
We next wanted to determine the mechanisms through which PML–RAR antagonizes p53. To investigate whether the fusion protein exerts an effect on p53 stability in vivo, we measured endogenous p53 levels in samples derived from wild-type or PML–RAR leukemic mice. Western blot (Figure 3A) and immunohistochemistry (Figure 3B) analyses revealed markedly reduced basal levels of p53 in samples derived from spleens and bone marrow of leukemic mice, as compared to wild-type mice (Figure 3A and B and data not shown). In vivo X-ray treatment stabilized p53 in the wild-type samples (Figure 3A and B), whereas it had no effect on the leukemic samples (note that the Western blots of Figure 3A, right panels, were overloaded with the leukemic samples).
Figure 3.
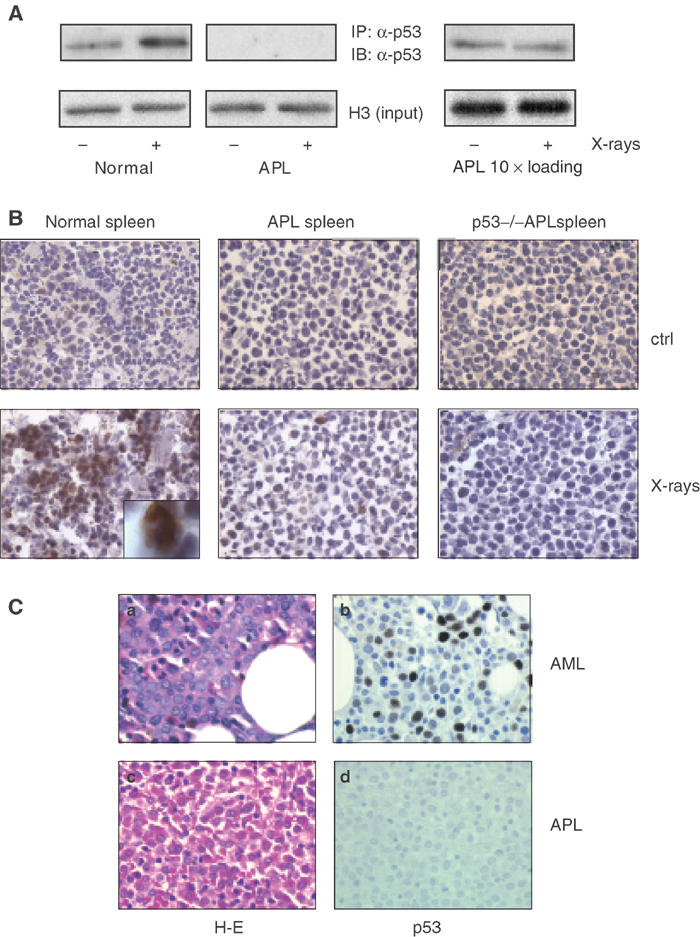
APL blasts have low levels of p53 and show no stabilization of p53 after X-ray treatment. (A) Normal and leukemic mice were given whole-body irradiation (9 Gy) or left untreated as indicated. The animals were killed 6 h later, spleen cells were isolated and nuclear extracts were prepared. The analysis was performed by immunoprecipitation (IP) followed by immunoblotting (IB) as indicated. Inputs were normalized according to histone H3 levels verified by immunoblotting prior to IP. Note that the starting material for the IP from leukemic samples was 10-fold that of normal samples due to the low expression of p53 in leukemic blasts. (B) Frozen spleen sections of normal and leukemic animals treated with X rays (or untreated mice as control) were stained with an anti-p53 antibody. An immature, wild-type myeloid precursor (by morphological criteria) showing strong p53 inducibility upon X-ray treatment is shown at a higher magnification in the inset. P53−/− spleens are shown as a negative control for p53 expression. (C) Immunohistochemistry analysis of p53 levels in human APL and non-APL samples. A representative case of AML (M2), diffusely immunoreactive for p53, is shown in the upper part of the panel. A case of APL, unreactive for p53, is shown in the lower part of the panel (40 × original magnification). (a and c) Hematoxylin and eosin, (b and d), hematoxylin counterstain.
PML–RAR blocks leukemic cells at an early stage of myeloid differentiation. As the inducibility of p53 at an early stage of hematopoiesis has not been tested previously in vivo, the lack of p53 expression and induction observed in leukemic cells could be the result of the block of differentiation at a nonpermissive state for p53 induction. The inset in Figure 3B, however, shows that p53 can be strongly induced in early myeloid precursors in normal spleen, showing that the lack of p53 induction in APL cells is due to the direct activity of the fusion protein (Figure 3A and B).
To correlate these results with the human disease, we measured p53 levels in APL patients. Expression of wild-type or mutant p53 can be detected by immunohistochemistry (IHC) in up to 65% of human AMLs (Seliger et al, 1996; Invernizzi et al, 2001). In a random sampling of 40 non-APL AML cases, we confirmed that approximately 50% of the samples expressed p53. The results are based mainly on IHC analysis; in a few cases, we performed Western blot analysis, which confirmed our findings in IHC (data not shown). In all, 10/40 AML cases (25%) expressed p53 at good levels (i.e., readily detectable by IHC in >50% of the blast cells, or showing a clear band in Western blot analysis using 50 μg of protein extract), 8/40 (20%) expressed p53 at good levels, but in a minority of the cells (10–35% of blast cells), and 22/40 (55%) did not express p53.
In 15/15 APL cases, we could not detect p53 by immunohistochemical analysis (Figure 3C, P=0.006), suggesting that downregulation of p53 is a constant feature of human APL blasts, and that it may explain the impairment of the p53 transcriptional response in PML–RAR-expressing cells.
PML–RAR inhibits p53 by triggering its proteasome-mediated, MDM2-dependent degradation
We next studied the roles of the proteasome and MDM2 in PML–RAR-triggered degradation of p53. Treatment of APL mice in vivo with the proteasome inhibitor epoxomicin led to a clear stabilization of p53, especially following X-ray treatment (Figure 4A, lanes 7–8). As MDM2 targets proteasome-dependent degradation of p53 (Prives, 1998), we checked the requirement of this oncogene in PML–RAR-triggered degradation of p53. We analyzed this phenomenon in a model system amenable to experimental manipulation. We used U937 hematopoietic progenitor cells, which express PML–RAR under a zinc-inducible promoter (PR9 cells; Grignani et al, 1998) and lack functional p53. These cells were engineered to express the p53-estrogen receptor chimera (p53ER), a p53 conditional allele activated by 4-hydroxytamoxifen (OHT) treatment (Moroni et al, 2001). In the absence of zinc (i.e., in cells not expressing PML–RAR), activation of p53 by OHT treatment provoked accumulation of G1 cells and a strong decrease of cells incorporating bromo-deoxyuridine (Supplementary Figure 3A). In agreement with these effects, OHT treatment stimulated p21 expression, which was further enhanced by X-rays (Supplementary Figure 3B). Induction of PML–RAR prevented both G1 arrest and p21 induction, showing that PML–RAR antagonizes p53 in this system (Supplementary Figure 3A–B). In addition, induction of PML–RAR led to a marked decrease in the level of p53ER, while addition of the proteasome inhibitor MG132 restored normal levels of p53 in the presence of PML–RAR (data not shown). This artificial model system thus fully recapitulates the observations previously described for primary APL cells. Expression in PR9-p53ER cells of the physiological inhibitor of MDM2, p19/ARF (Sherr, 2001), completely inhibited the proteasome-mediated degradation of p53ER triggered by PML–RAR (Figure 4B). Additionally, PML–RAR did not affect p53 protein levels in MDM2−/−/p53−/− MEFs transiently transfected with expression vectors for PML–RAR and p53, unless MDM2 was also co-transfected in limiting concentrations (to minimize the degradation of p53 by overexpression of MDM2 alone; see Figure 4C). Taken together, these results indicate that PML–RAR inhibits p53 by triggering its proteasome-mediated, MDM2-dependent degradation.
Figure 4.
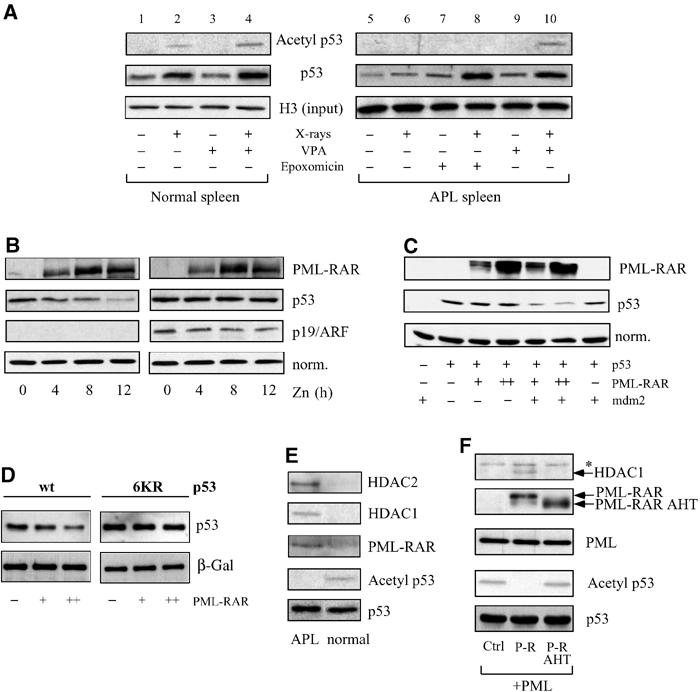
PML–RAR leads to HDAC-dependent deacetylation and to mdm-2-mediated degradation of p53. (A) Normal and leukemic animals were treated in vivo with the HDAC inhibitor VPA (400 mg/kg i.p.) or with the proteasome inhibitor epoxomicin (3 mg/kg i.p.) as indicated, and then subjected to X-ray treatment (9 Gy). Nuclear extracts were prepared from the spleens of these animals and analyzed by immunoprecipitation against p53 and immunoblotting against p53 or acetyl-p53. Input normalization for the immunoprecipitation was performed with an anti-histone H3 antibody; starting material for the immunoprecipitation from leukemic samples was 10-fold that of normal samples due to the low expression of p53 in leukemic blasts. (B) PR9-p53ER cells were infected with the empty vector or a p19/ARF vector, treated with Zn and OHT, and then analyzed for p53ER levels. (C) Mdm2−/− p53−/− MEFs were transfected with the indicated constructs and then analyzed for p53 levels. (D) p53−/− MEFs were transfected with increasing amounts of a PML–RAR expression vector in the presence of a wild-type or 6KR p53-expressing vector. Lysates were normalized for the levels of a co-transfected β-galactosidase, and then analyzed for p53 levels. (E) Nuclear extracts derived from X-ray-treated normal and leukemic spleens were normalized for p53 amounts, immunoprecipitated with antibodies against p53 and analyzed by immunoblot using antibodies against p53, acetylated p53, RAR, HDAC1 and HDAC2, as indicated in the figure. (F) Phoenix cells were transfected with expression vectors for PML-IV, and co-transfected with PML–RAR or PML–RAR AHT (or with the empty vector). Equal amounts of input lysates were analyzed by immunoblot to check for total p53 levels (not shown), and then immunoprecipitated with an anti-p53-specific antibody followed by immunoblotting using antibodies against p53, PML (to detect PML–RAR, PML–RAR AHT and PML), HDAC1 and acetylated p53. Asterisk, a nonspecific band not co-migrating with HDAC1.
HDAC-associated PML–RAR leads to p53 deacetylation
The stability and transcriptional activity of p53 are regulated by post-translational modifications, including phosphorylation and acetylation (Appella and Anderson, 2001; Prives and Manley, 2001; Ryan et al, 2001). Acetylation plays a positive role in the accumulation of the p53 protein in response to stresses (Ito et al, 2001) and wild-type PML modulates p53 acetylation (Pearson et al, 2000; Gottifredi and Prives, 2001; Pearson and Pelicci, 2001; Prives and Manley, 2001). Normal and APL mice were treated in vivo with X-rays, and nuclear extracts from spleens and bone marrow were analyzed for p53 levels and acetylation by immunoprecipitation followed by Western blotting. PML–RAR expression prevented p53 stabilization following X-ray treatment, and no detectable p53 acetylation was observed, prior to or after X-ray treatment (Figure 4A, lanes 5 and 6; Figure 4D, where levels of immunoprecipitated p53 are normalized from normal and APL samples, to directly compare the fraction of acetylated p53/total p53). We induced stabilization of p53 in APL mice, by in vivo treatment with the proteasome inhibitor epoxomicin. Under this artificial experimental condition, levels of p53 after X-rays were comparable to those obtained in the wild-type mice in the absence of proteasome inhibition, yet no acetylation of p53 was detected (Figure 4A, lanes 2 and 8). Similar results were obtained in PR9-p53ER cells (Supplementary Figure 4B), and in Phoenix cells, which contain wild-type p53 (Deppert et al, 1987; data not shown), and that are defective in the pathway leading to p53 degradation (see below). In these cells, PML–RAR expression induced deacetylation—but not destabilization— of p53 (Figure 4F). Together, these results suggest that PML/RAR acts directly on p53 acetylation.
Degradation of p53 by MDM2 is regulated by acetylation of p53, and p53 mutants that cannot undergo acetylation/deacetylation cannot be degraded by MDM2 (Ito et al, 2001). As MDM2 cooperates with PML–RAR in triggering p53 degradation, we checked whether similar mutations would affect degradation of p53 by PML–RAR. We transfected a p53 mutant in lysine 382 (p53 K382R) in p53−/− MEFs, in the presence of PML–RAR. The point mutation K382R reduces, but does not abolish, the sensitivity of p53 to PML–RAR-mediated degradation (data not shown). To test whether other acetylation sites of p53 are involved, we used a p53 construct carrying mutations in the known acetylation sites (p536KR). Strikingly, the stability of this mutant is essentially unaffected by PML–RAR (Figure 4D), similar to what was observed for MDM2 (Ito et al, 2001). This result links tightly the acetylation dynamics of p53 to the observed effect of PML–RAR on its stability.
To investigate whether the effect of PML–RAR on p53 acetylation depends on its ability to recruit HDAC, we measured the effects of in vivo treatment of normal and APL mice with X-rays in the presence of VPA. VPA enhanced both p53 acetylation and protein levels after X-ray treatment in normal mice (Figure 4A, lane 4). Notably, in APL mice VPA induced stabilization of p53 at a level similar to that observed in the APL mice treated with the proteasome inhibitor epoxomicin (Figure 4A, lanes 8 and 10). In this case, however, p53 acetylation was comparable to that measured in normal mice, directly linking p53 acetylation and stability (Figure 4A, lanes 4 and 10). Likewise, trichostatin A (TSA), a class I and II HDAC inhibitor, or RA, which dissociates the PML–RAR/HDAC complex, induced stabilization and blocked deacetylation of p53 by PML–RAR in PR9-p53ER cells (Supplementary Figure 4A and B). Consistently, the PML–RAR AHT mutant, which is defective for HDAC recruitment, was unable to deacetylate p53 in Phoenix cells (Figure 4F). These data suggest that PML–RAR exerts its effect on p53 when complexed to functional HDACs. Confirming previous reports, we found that PML–RAR immunoprecipitates from blast cells contain both HDAC1 and HDAC2 (data not shown). Finally, we investigated whether PML–RAR favors the formation of a stable p53–HADC complex in vivo. Strikingly, in cells derived from APL mice, p53 (in its deacetylated state) was found in complexes with both PML–RAR and HDAC1 or HDAC2 (Figure 4E).
Identical results were obtained in Phoenix cells transfected with PML–RAR. As the results obtained parallel completely those obtained in more ‘physiological' systems, we decided to use Phoenix cells, although they express viral proteins that are responsible for the downregulation of the p53 pathway. In cells transfected with the PML–RAR AHT mutant, we could not detect a stable p53–HDAC1 complex (Figure 4F). Taken together, these results support a model whereby deacetylation of p53 is triggered by the association of PML–RAR/HDAC with p53, resulting in reduced stability and transcriptional activity of p53.
PML–RAR requires the product of the wild-type PML allele to inhibit p53
PML associates with p53 directly, through the carboxy-terminal region of the PML-IV isoform (Fogal et al, 2000). This region is lost in the PML–RAR fusion protein, that—unlike PML-IV—binds poorly to p53 in vitro (data not shown; Guo et al, 2000). As PML–RAR forms stable complexes with wild-type PML, it could associate with p53 indirectly, through wild-type PML. To this end, we measured the extent of PML–RAR/p53 complex formation in Phoenix cells, which express limiting amounts of endogenous PML (data not shown), in the absence or presence of overexpressed PML. Overexpression of PML did not affect the levels of PML–RAR (Supplementary Figure 5). As shown in Figure 5A, PML–RAR (and PML–RAR AHT) were immunoprecipitated by anti-p53 antibodies at a much higher efficiency when co-expressed with wild-type PML. It appears, therefore, that PML–RAR and p53 can form a stable complex in vivo in the presence of PML. Recruitment of HDAC1 by p53, and deacetylation of p53 by PML–RAR, was only observed in the presence of co-transfected PML-IV (Figure 4F). The above results suggest that wild-type PML is indispensable for the ability of PML–RAR to inhibit p53. Indeed, p53 showed a slightly decreased transcriptional activity in PML−/− cells (Figure 5B). Expression of PML–RAR led to suppression of p53 activity in wild-type (two- to three-fold repression), but not in PML−/−MEFs, even following transfection of five-fold higher amounts of expression vector (Figure 5B). As a control, MDM2 was able to lower p53 transcriptional activity in both cell types (data not shown). Reintroduction of PML into PML null fibroblasts resulted in the restoration of PML–RAR's ability to suppress p53, conclusively showing that wild-type PML is required for the inhibitory effect of PML–RAR on p53 (Figure 5B).
Figure 5.
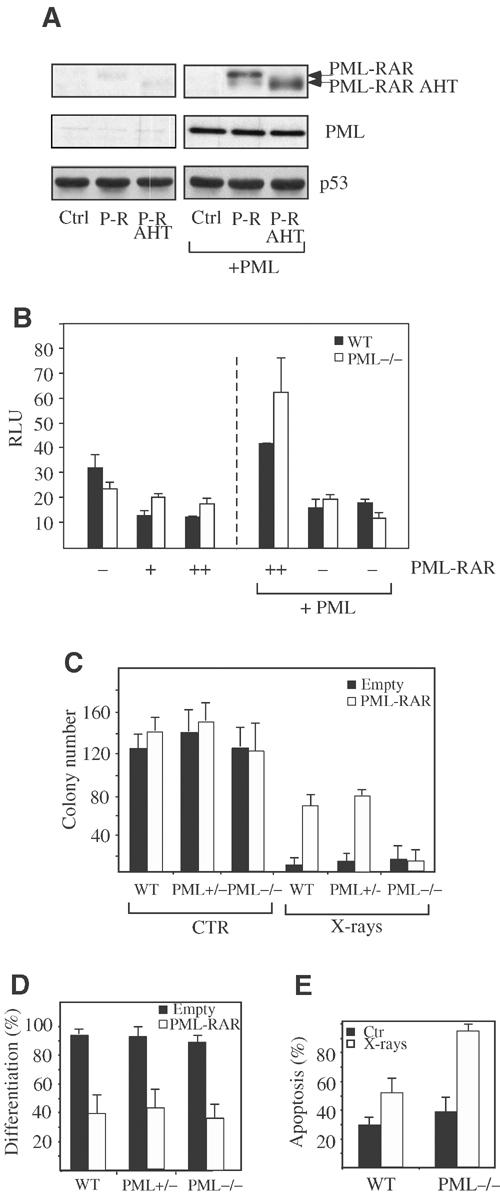
PML–RAR requires wild-type PML to inhibit p53. (A) Phoenix cells were transfected with expression vectors for PML–RAR or PML–RAR AHT (or with the empty vector), in the absence—left panels—or in the presence—right panels—of PML-IV. Input lysates were immunoprecipitated with an anti-p53-specific antibody, followed by immunoblotting using antibodies against p53 and PML (to detect PML–RAR, PML–RAR AHT and PML). (B) Wild-type and PML−/− MEFs were transiently transfected with the p53-responsive pGL13 reporter, in the presence of the indicated expression vectors. (C) Lin- cells prepared from wild-type, PML+/− and PML−/− mice were transduced with a PML–RAR expression vector, or with an empty control vector and sorted. After cytokine deprivation (CTR), cells were left untreated or exposed to X-rays. Cells were then plated in methylcellulose medium. After 8 days, colonies were counted. (D) Lin- cells from wild-type, PML+/− and PML−/− mice were transduced with a PML–RAR expression vector, or with an empty control vector. Sorted cells were plated in methylcellulose medium, and colonies were analyzed for the presence of the myeloid differentiation markers Mac1 and GR1. (E) Spleen cells from leukemic mice (wild type or PML−/−) were subjected to cytokine deprivation, and then treated with X-rays. After 24 h, apoptosis was measured.
We next investigated the biological effects of PML–RAR expression in PML−/− cells. The degree of protection from X-ray treatment observed in PML−/− was significantly weaker than in p53−/− cells, at all doses of X-rays tested (Figure 5C and data not shown). Strikingly, PML–RAR expression did not result in further protection from loss of CFCs in PML−/− cells, in contrast to the protective effect on wild-type controls and PML+/− cells (Figure 5C). The lack of a PML–RAR response was not due to a general loss of PML–RAR activity in the absence of PML, as PML–RAR inhibited myeloid differentiation of PML−/− cells as efficiently as in wild-type and PML+/− cells (Figure 5D).
From these results, it would be predicted that APL blasts derived from the re-injection of PML–RAR-expressing cells into PML−/− mice would be more sensitive to X-rays than APL blasts derived from wild-type mice. As shown in Figure 5E, 24 h after irradiation, PML−/− APL blasts underwent more than 80% apoptosis, whereas PML+/+ APL blasts displayed only half this amount. Taken together, these results demonstrate that PML is essential for the ability of PML–RAR to inhibit p53 function.
Degradation of p53 induced by other AML fusion proteins
We investigated whether the results obtained with PML–RAR can be extended to other APL variants. Strikingly, PLZF-RAR inhibits p53-dependent transcription and leads to degradation of p53 (Figure 6A and B). APL fusion proteins may therefore generally act through direct inhibition of p53 through degradation. In our study, we included AML1-ETO since an alternative mechanism for inhibition of the p53 pathway has been described for this fusion protein (causing a different form of AML), acting through transcriptional repression of the p14(ARF) locus (Linggi et al, 2002). AML1-ETO does not inhibit p53 directly in our transient transfection assays (Figure 6), consistent with its indirect suppression of p53. We believe, therefore, that distinct AML fusion proteins (in tumors that maintain wild-type p53 sequence) may have been selected for distinct mechanisms of p53 inactivation, in any case leading to functional impairment of the pathway.
Figure 6.
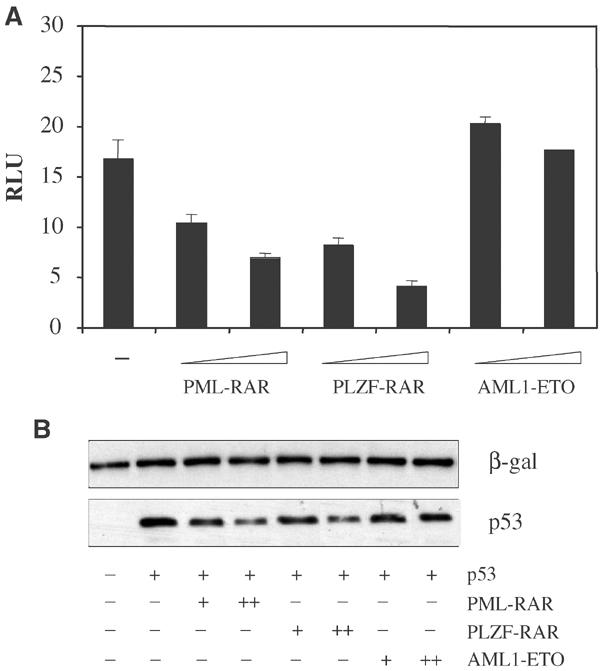
PLZF-RAR destabilizes p53. (A) p53−/− MEFs were transfected with the reporter bax-luciferase, and increasing amounts of PML–RAR, PLZF-RAR and AML1-ETO expression vectors, in the presence of a p53 expression vector. (B) p53−/− MEFs were transfected with increasing amounts of PML–RAR, PLZF-RAR and AML1-ETO expression vectors, in the presence of a p53-expressing vector. Lysates were normalized for the levels of a co-transfected β-galactosidase, and then analyzed for p53 levels.
Discussion
Mutations in p53 are the most frequent genetic alterations in human cancer; they are, however, extremely rare in APL cells, most of which express wild-type p53 (Longo et al, 1993; Trecca et al, 1994). From the results presented here, the lack of p53 mutations can easily be explained by the lack of selective pressure favoring their occurrence, given the functional inactivation of p53 mediated by PML–RAR and other rare APL fusion proteins (PLZF-RAR).
APL cells (as most tumor cells) exhibit two main biological phenotypes: (i) arrest of differentiation and (ii) enhanced survival upon stress (Grignani et al, 1994). Arrest of differentiation was known to occur via deregulation of RAR target genes (Minucci et al, 2001). We now show that PML–RAR directly achieves enhanced survival against genotoxic stresses, by inhibition of the PML–p53 pathway. PML–RAR functions, therefore, as a true bifunctional protein, by deregulating both RAR and PML-dependent pathways (Figure 7). The biological effects mark the ‘pre-leukemic' phase of the disease, and are present as soon as the fusion protein is expressed. The clonality of the leukemia may be explained by the stochastic nature of the consequences of the genomic instability established by p53 inhibition, and the following ‘second' (or multiple) hit(s). Despite the fact that the chromosomal translocation t(15;17), which yields the PML–RAR fusion protein, represents a unique cytogenetic abnormality in the majority of APL patients, almost 40% of APL cases show additional karyotypic anomalies, suggesting that indeed human APL results from combined genetic mutations (Berger et al, 1991; Hiorns et al, 1997; Zimonjic et al, 2000; Kogan et al, 2001; Le Beau et al, 2002).
Figure 7.
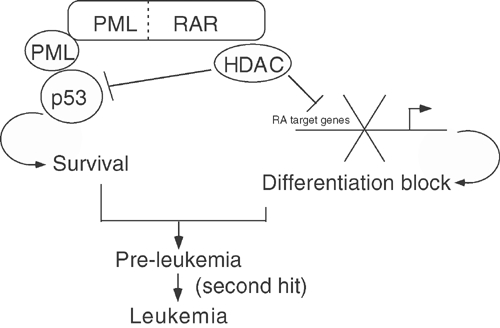
PML–RAR acts as an HDAC-dependent inhibitor of pathways regulated by wild-type PML and RAR. PML–RAR acts as a bifunctional protein: (i) it inhibits the transcription of RA target genes in the presence of physiological concentrations of ligand, thus blocking myeloid differentiation; and (ii) it inhibits p53 function (in a manner requiring wild-type PML), allowing enhanced survival of APL blasts. Both differentiation block and enhanced survival are mediated by recruitment of HDAC by the fusion protein.
Our finding that inhibition of p53 is achieved by exploiting the capacity of PML–RAR to associate with the product of the remaining, wild-type PML allele describes a novel mechanism of oncogenic activation. Intriguingly, the biological phenotypes observed in PML null cells, or upon expression of PML–RAR in wild-type cells, are not completely overlapping. The absence of PML leads to impairment rather than abrogation of the p53 response (Pearson and Pelicci, 2001). In contrast, PML–RAR-expressing cells show higher resistance to the p53-dependent stress response, and this effect depends on the presence of wild-type PML. These results suggest that PML–RAR acts as a ‘gain-of-function' mutation, rather than by simple inactivation of PML.
Our results show that expression of PML–RAR in PML−/− cells may still result in leukemias (due to enhanced resistance to stress in PML−/− cells, which may compensate for the lack of active p53 inhibition by the fusion protein in the absence of PML), but that the leukemic blasts are less resistant to stress than those arising from wild-type cells. Our results lead to the surprising conclusion that there could be a selective advantage in maintenance of wild-type PML expression in APL blasts, which may become especially evident under conditions of stress. Analysis of PML–RAR effects in wild-type and PML−/− cells supports this hypothesis, since APL blasts derived from wild-type cells are more resistant to stress (Figure 5E).
We have demonstrated that recruitment of HDACs by a leukemic fusion protein represents a novel mechanism enabling leukemic cells to overcome p53 action. The mechanism through which the fusion protein achieves this effect—inhibition of p53 acetylation, and subsequent alteration of p53 stability/activity–demonstrates the role that alterations in p53 post-translational modifications may play in cancer. Recruitment of HDAC by PML–RAR, followed by p53 degradation, resembles the capacity of MDM2 to recruit HDAC1 and then destabilize p53 physiologically (Ito et al, 2001).
It appears, therefore, that PML–RAR (through its capacity to further recruit HDAC1/2 to the PML–p53 complex) reinforces the action of MDM2: in support of this model, MDM2 is required for p53 degradation by the fusion protein. Recent results suggest functional and physical interactions among wild-type PML amd mdm-2 (Louria-Hayon et al, 2003; Wei et al, 2003). PML–RAR would therefore be potentially able to interact (directly, or via PML) with mdm-2 itself. Our results obtained with the PML–RAR AHT mutant (unable to recruit HDAC, and unable to alter p53 acetylation/function) exclude that (in the absence of direct HDAC recruitment by PML–RAR) this potential interaction would be sufficient to trigger mdm-2 activity on p53.
Notably, our results underline the importance of further studies of p53 post-translational modifications in other forms of leukemia and tumors characterized by a low frequency of p53 mutations (Appella and Anderson, 2001; Soussi and Béroud, 2001).
Interestingly, whereas recent results suggest that the continued expression of PML–RAR is not necessarily required for maintaining repression of PML–RAR target genes due to epigenetic changes (Di Croce et al, 2002), the results presented here show that PML–RAR is continuously inhibiting the function of p53 (Figure 2F). Therefore, blocking PML–RAR function may at any time be sufficient to restore relevant pathways for sensitivity to pharmacological treatment.
The dual phenotypic effect of the fusion protein is reached by a unifying molecular mechanism: recruitment of HDAC, either to the promoter of RA target genes, or to the PML/p53 complex. HDACs therefore represent an ideal candidate for blocking the action of the fusion protein. Having established p53 acetylation as a relevant oncogenic target, our results allow the definition of a new mechanism of action by HDACi, aimed at restoring normal levels of p53 acetylation. These direct effects of HDACi on an oncogenic transcription factor (PML–RAR) add to the more general, previously described effects of HDACi on cancer cells, and lend further support to their use in cancer therapy (Altucci and Gronemeyer, 2001; Marks et al, 2001; Minucci et al, 2001).
Experimental procedures
Plasmids and antibodies
Retroviral vectors for PML and PML–RAR have been described (Minucci et al, 2000). PINCO P/RΔRAR contains the PML region maintained in PML–RAR (Grignani et al, 1996). p53ER was obtained by fusing p53 cDNA with the estrogen-receptor ligand-binding domain (Littlewood et al, 1995; Pearson et al, 2000; Moroni et al, 2001). MDM2-Luc has been described (Barak et al, 1993).
The monoclonal DO-1 (Santa Cruz) antibody was used to detect p53 in PR9 and Phoenix cells, while the polyclonal 393-fl (Santa Cruz) antibody was used on primary murine cells. Immunostaining of bone marrow biopsies from APL or AML patients was performed using DO7 monoclonal antibodies (Novocastra); polyclonal CM5 (Novocastra) antibody was used for immunostaining of murine spleen sections. Acetylation at lysine 382 was detected using a polyclonal serum (Sakaguchi et al, 1998).
Preparation of lin- cells
Lin- cells were purified from the bone marrow, infected and then sorted as described (Minucci et al, 2000). In PML–RAR AHT transduction experiments, the GFP-positive cells with the highest mean fluorescence were sorted, since we observed a lower level of expression of PML–RAR AHT compared to PML–RAR. The mean GFP fluorescence signal correlates with the levels of expression of the protein under study (Minucci et al, 2000).
Isolation of PR9-p53ER cells
PR9 cells were infected with either pBABE-Puro or pBABE-Puro/p53ER. Cells were selected by culture in 10 μg/ml puromycin. Individual clones were isolated by plating at limiting dilution, and tested for the expression of p53ER and growth suppression upon addition of OHT.
Assays of p53 transcriptional activity
Transient transfection assays in MEFs were performed as described (Pearson et al, 2000). Data are plotted as means plus standard deviations from four independent experiments.
Survival assays
Sorted cells or uninfected, control lin- cells were subjected to cytokine deprivation. Cells were then treated with X-rays (2 Gy), with H2O2 (0.1 mM in PBS for 20 min) or adriamycin (2 ng/ml). For analysis of CFCs, cells were plated 12 h after stress in methylcellulose medium containing the following cytokines: IL3 (20 ng/ml), IL6 (20 ng/ml), SCF (100 ng/ml), G-CSF (60 ng/ml), GM-CSF (20 ng/ml). 8–10 days after plating, colonies were counted. Data are plotted as means plus standard deviations from three independent experiments.
For spleen cells from leukemic mice, apoptosis was quantified 24 h after X-ray treatment by propidium iodide staining according to standard techniques.
Differentiation assays
Sorted cells or uninfected control lin- cells were plated (5000 cells/plate) in methylcellulose medium containing the following cytokines: IL3 (20 ng/ml), IL6 (20 ng/ml), SCF (100 ng/ml), G-CSF (60 ng/ml), GM-CSF (20 ng/ml). 8–10 days after plating, colonies were pooled and analyzed for the presence of Mac1 and GR1 differentiation markers as described (Minucci et al, 2000). Data are plotted as means plus standard deviations from three independent experiments.
Real-time PCR
Splenocytes or bone marrow cells from leukemic and nonleukemic animals were subjected to cytokine deprivation for 4 h and then treated with X-rays (2 Gy). 10 h after irradiation, RNA was extracted, reverse transcribed and the cDNA was used for quantitative PCR. The values displayed were normalized against GAPDH.
Immunohistochemistry
p53 expression was assayed by means of the avidin–biotin peroxidase complex (ABC) method, using the 3,3′-diaminobenzidine tetrahydrochloride chromogen in Bouin-fixed bone marrow biopsies from patients with APL and other FAB subtypes of AML at diagnosis. Immunohistochemical detection of p53 on frozen murine spleen sections was performed as described (Komarova et al, 1997).
Superarray
We used the ‘GEArray Q series Mouse p53 signalling Pathway Gene Array' filter of p53 targets according to the manufacturer's instructions (Superarray Bioscience Corporation, Cat. No. MM-027N-4).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
We acknowledge Pier Paolo Pandolfi and Kristian Helin for reagents, Rosangela Invernizzi, Mario Faretta, Silvia Soddu for discussions, Francesco Contegno, Giusy Bonizzi, Sabrina Giavara, Shin'ichi Saito for performing initial experiments and Francesco Marchesi, Giuseppina Mazzaro, Fabio Dalla Valle for technical help. This work has been funded by AIRC, FIRC, EEC, MIUR and Ministero della Sanitá grants to SM and PGP.
References
- Altucci L, Gronemeyer H (2001) The promise of retinoids to fight against cancer. Nat Rev Cancer 1: 181–193 [DOI] [PubMed] [Google Scholar]
- Appella E, Anderson CW (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 268: 2764–2772 [DOI] [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M (1993) mdm2 expression is induced by wild type p53 activity. EMBO J 12: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger R, Le Coniat M, Derre J, Vecchione D, Jonveaux P (1991) Cytogenetic studies in acute promyelocytic leukemia: a survey of secondary chromosomal abnormalities. Genes Chromosomes Cancer 3: 332–337 [DOI] [PubMed] [Google Scholar]
- Deppert W, Haug M, Steinmayer T (1987) Modulation of p53 protein expression during cellular transformation with simian virus 40. Mol Cell Biol 7: 4453–4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, Fuks F, Lo Coco F, Kouzarides T, Nervi C, Minucci S, Pelicci PG (2002) Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295: 1079–1082 [DOI] [PubMed] [Google Scholar]
- Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW (2000) PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 14: 2015–2027 [PMC free article] [PubMed] [Google Scholar]
- Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G (2000) Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J 19: 6185–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottifredi V, Prives C (2001) P53 and PML: new partners in tumor suppression. Trends Cell Biol 11: 184–187 [DOI] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20: 6969–6978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Lazar MA, Minucci S, Pelicci PG (1998) Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature 391: 815–818 [DOI] [PubMed] [Google Scholar]
- Grignani F, Fagioli M, Alcalay M, Longo L, Pandolfi PP, Donti E, Biondi A, Lo Coco F, Pelicci PG (1994) Acute promyelocytic leukemia: from genetics to treatment. Blood 83: 10–25 [PubMed] [Google Scholar]
- Grignani F, Testa U, Rogaia D, Ferrucci PF, Samoggia P, Pinto A, Aldinucci D, Gelmetti V, Fagioli M, Alcalay M, Seeler J, Nicoletti I, Peschle C, Pelicci PG (1996) Effects on differentiation by the promyelocytic leukemia PML/RARalpha protein depend on the fusion of the PML protein dimerization and RARalpha DNA binding domains. EMBO J 15: 4949–4958 [PMC free article] [PubMed] [Google Scholar]
- Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Paolo Pandolfi P (2000) The function of PML in p53-dependent apoptosis. Nat Cell Biol 2: 730–736 [DOI] [PubMed] [Google Scholar]
- He L, Bhaumik M, Tribioli C, Rego EM, Ivins S, Zelent A, Pandolfi PP (2000) Two critical hits for promyelocytic leukemia. Mol Cell 6: 1131–1141 [DOI] [PubMed] [Google Scholar]
- Heimfeld S, Hudak S, Weissman I, Rennick D (1991) The in vitro response of phenotypically defined mouse stem cells and myeloerythroid progenitors to single or multiple growth factors. Proc Natl Acad Sci USA 88: 9902–9906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiorns LR, Swansbury GJ, Mehta J, Min T, Dainton MG, Treleaven J, Powles RL, Catovsky D (1997) Additional chromosome abnormalities confer worse prognosis in acute promyelocytic leukaemia. Br J Haematol 96: 314–321 [DOI] [PubMed] [Google Scholar]
- Invernizzi R, Pecci A, Bellotti L, Ascari E (2001) Expression of p53, bcl-2 and ras oncoproteins and apoptosis levels in acute leukaemias and myelodysplastic syndromes. Leuk Lymphoma 42: 481–489 [DOI] [PubMed] [Google Scholar]
- Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 20: 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko LJ, Prives C (1996) p53: puzzle and paradigm. Genes Dev 10: 1054–1072 [DOI] [PubMed] [Google Scholar]
- Kogan SC, Brown DE, Shultz DB, Truong BT, Lallemand-Breitenbach V, Guillemin MC, Lagasse E, Weissman IL, Bishop JM (2001) BCL-2 cooperates with promyelocytic leukemia retinoic acid receptor alpha chimeric protein (PMLRARalpha) to block neutrophil differentiation and initiate acute leukemia. J Exp Med 193: 531–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova EA, Chernov MV, Franks R, Wang K, Armin G, Zelnick CR, Chin DM, Bacus SS, Stark GR, Gudkov AV (1997) Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. EMBO J 16: 1391–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Beau MM, Bitts S, Davis EM, Kogan SC (2002) Recurring chromosomal abnormalities in leukemia in PML-RARA transgenic mice parallel human acute promyelocytic leukemia. Blood 99: 2985–2991 [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 [DOI] [PubMed] [Google Scholar]
- Lin RJ, Egan DA, Evans RM (1999) Molecular genetics of acute promyelocytic leukemia. Trends Genet 15: 179–184 [DOI] [PubMed] [Google Scholar]
- Linggi B, Muller-Tidow C, van de Locht L, Hu M, Nip J, Serve H, Berdel WE, van der Reijden B, Quelle DE, Rowley JD, Cleveland J, Jansen JH, Pandolfi PP, Hiebert SW (2002) The t(8;21) fusion protein, AML1 ETO, specifically represses the transcription of the p14(ARF) tumor suppressor in acute myeloid leukemia. Nat Med 8: 743–750 [DOI] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI (1995) A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res 23: 1686–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo L, Trecca D, Biondi A, Lo Coco F, Grignani F, Maiolo AT, Pelicci PG, Neri A (1993) Frequency of RAS and p53 mutations in acute promyelocytic leukemias. Leuk Lymphoma 11: 405–410 [DOI] [PubMed] [Google Scholar]
- Look AT (1997) Oncogenic transcription factors in the human acute leukemias. Science 278: 1059–1064 [DOI] [PubMed] [Google Scholar]
- Louria-Hayon I, Grossman T, Sionov RV, Alsheich O, Pandolfi PP, Haupt Y (2003) PML protects p53 from Mdm2-mediated inhibition and degradation. J Biol Chem 278: 33134–33141 [DOI] [PubMed] [Google Scholar]
- Marks P, Rifkind R, Richon V, Breslow R, Miller T, Kelly W (2001) Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 1: 194–202 [DOI] [PubMed] [Google Scholar]
- Minucci S, Maccarana M, Cioce M, De Luca P, Gelmetti V, Segalla S, Di Croce L, Giavara S, Matteucci C, Gobbi A, Bianchini A, Colombo E, Schiavoni I, Badaracco G, Hu X, Lazar MA, Landsberger N, Nervi C, Pelicci PG (2000) Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol Cell 5: 811–820 [DOI] [PubMed] [Google Scholar]
- Minucci S, Monestiroli S, Giavara S, Ronzoni S, Marchesi F, Insinga A, Diverio D, Gasparini P, Capillo M, Colombo E, Matteucci C, Contegno F, Lo-Coco F, Scanziani E, Gobbi A, Pelicci PG (2002) PML-RAR induces promyelocytic leukemias with high efficiency following retroviral gene transfer into purified murine hematopoietic progenitors. Blood 100: 2989–2995 [DOI] [PubMed] [Google Scholar]
- Minucci S, Nervi C, Lo Coco F, Pelicci PG (2001) Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene 20: 3110–3115 [DOI] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG (1999) Retinoid receptors in health and disease: co-regulators and the chromatin connection. Semin Cell Dev Biol 10: 215–225 [DOI] [PubMed] [Google Scholar]
- Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 3: 552–558 [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Wandycz AM, Hemmati HD, Wright DE, Weissman IL (1997) Identification of a lineage of multipotent hematopoietic progenitors. Development 124: 1929–1939 [DOI] [PubMed] [Google Scholar]
- Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406: 207–210 [DOI] [PubMed] [Google Scholar]
- Pearson M, Pelicci PG (2001) PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene 20: 7250–7256 [DOI] [PubMed] [Google Scholar]
- Prives C (1998) Signaling to p53: breaking the MDM2-p53 circuit. Cell 95: 5–8 [DOI] [PubMed] [Google Scholar]
- Prives C, Manley J (2001) Why is p53 acetylated? Cell 107: 815–818 [DOI] [PubMed] [Google Scholar]
- Quelle FW, Wang J, Feng J, Wang D, Cleveland JL, Ihle JN, Zambetti GP (1998) Cytokine rescue of p53-dependent apoptosis and cell cycle arrest is mediated by distinct Jak kinase signaling pathways. Genes Dev 12: 1099–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbitts TH (1991) Translocations, master genes, and differences between the origins of acute and chronic leukemias. Cell 67: 641–644 [DOI] [PubMed] [Google Scholar]
- Rabbitts TH (1994) Chromosomal translocations in human cancer. Nature 372: 143–149 [DOI] [PubMed] [Google Scholar]
- Ryan KM, Phillips AC, Vousden KH (2001) Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol 13: 332–337 [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E (1998) DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev 12: 2831–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seliger B, Papadileris S, Vogel D, Hess G, Brendel C, Storkel S, Ortel J, Kolbe K, Huber C, Huhn D, Neubauer A (1996) Analysis of the p53 and MDM-2 gene in acute myeloid leukemia. Eur J Haematol 57: 230–240 [DOI] [PubMed] [Google Scholar]
- Sherr CJ (2001) The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2: 731–737 [DOI] [PubMed] [Google Scholar]
- Soussi T, Béroud C (2001) Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer 1: 233–239 [DOI] [PubMed] [Google Scholar]
- Spangrude GJ, Heimfeld S, Weissman IL (1988) Purification and characterization of mouse hematopoietic stem cells. Science 241: 58–62 [DOI] [PubMed] [Google Scholar]
- Trecca D, Longo L, Biondi A, Cro L, Calori R, Grignani F, Maiolo AT, Pelicci PG, Neri A (1994) Analysis of p53 gene mutations in acute myeloid leukemia. Am J Hematol 46: 304–309 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Vousden KH (2000) p53: death star. Cell 103: 691–694 [DOI] [PubMed] [Google Scholar]
- Warrell RP Jr, de The H, Wang ZY, Degos L (1993) Acute promyelocytic leukemia. N Engl J Med 329: 177–189 [DOI] [PubMed] [Google Scholar]
- Wei X, Yu ZK, Ramalingam A, Grossman SR, Yu JH, Bloch DB, Maki CG (2003) Physical and functional interactions between PML and MDM2. J Biol Chem 278: 29288–29297 [DOI] [PubMed] [Google Scholar]
- Westervelt P, Ley TJ (1999) Seed versus soil: the importance of the target cell for transgenic models of human leukemias. Blood 93: 2143–2148 [PubMed] [Google Scholar]
- Zimonjic DB, Pollock JL, Westervelt P, Popescu NC, Ley TJ (2000) Acquired, nonrandom chromosomal abnormalities associated with the development of acute promyelocytic leukemia in transgenic mice. Proc Natl Acad Sci USA 97: 13306–13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5