

Summary
Anti-Gal is the most abundant natural antibody in humans, constituting ∼ 1% of immunoglobulins. Anti-Gal is naturally produced also in apes and Old World monkeys. The ligand of anti-Gal is a carbohydrate antigen called the ‘α-gal epitope’ with the structure Galα1-3Galβ1-4GlcNAc-R. The α-gal epitope is present as a major carbohydrate antigen in non-primate mammals, prosimians and New World monkeys. Anti-Gal can contributes to several immunological pathogeneses. Anti-Gal IgE produced in some individuals causes allergies to meat and to the therapeutic monoclonal antibody cetuximab, all presenting α-gal epitopes. Aberrant expression of the α-gal epitope or of antigens mimicking it in humans may result in autoimmune processes, as in Graves' disease. α-Gal epitopes produced by Trypanosoma cruzi interact with anti-Gal and induce ‘autoimmune like’ inflammatory reactions in Chagas' disease. Anti-Gal IgM and IgG further mediate rejection of xenografts expressing α-gal epitopes. Because of its abundance, anti-Gal may be exploited for various clinical uses. It increases immunogenicity of microbial vaccines (e.g. influenza vaccine) presenting α-gal epitopes by targeting them for effective uptake by antigen-presenting cells. Tumour lesions are converted into vaccines against autologous tumour-associated antigens by intra-tumoral injection of α-gal glycolipids, which insert into tumour cell membranes. Anti-Gal binding to α-gal epitopes on tumour cells targets them for uptake by antigen-presenting cells. Accelerated wound healing is achieved by application of α-gal nanoparticles, which bind anti-Gal, activate complement, and recruit and activate macrophages that induce tissue regeneration. This therapy may be of further significance in regeneration of internally injured tissues such as ischaemic myocardium and injured nerves.
Keywords: anti-Gal, increased immunogenicity, tumour vaccine, wound healing, α-gal epitopes
Anti-Gal and its ligand the α-gal epitope in mammals
Anti-Gal is the most abundant antibody in humans, constituting ∼ 1% of immunoglobulins, and is found as IgG, IgM and IgA isotypes.1 It is continuously produced throughout life as an immunological response to antigenic stimulation by bacteria of the normal flora.2 As many as 1% of human B cells can produce anti-Gal,3 most of which are quiescent and only those along the gastrointestinal tract produce this antibody in response to continuous antigenic stimulation by gastrointestinal bacteria. Anti-Gal in humans is encoded by several heavy-chain genes primarily of the VH3 immunoglobulin gene family.4–5 The distribution of anti-Gal in mammals is unique in that it is found only in humans, apes and Old World monkeys (monkeys of Asia and Africa)6 (Table 1). In contrast, the rest of the mammals, including non-primate mammals, prosimians and New World monkeys (monkeys of South America) produce the ligand of anti-Gal, a carbohydrate antigen called the α-gal epitope with the structure Galα1-3Galβ1-4GlcNAc-R on carbohydrate chains of glycolipids (Fig. 1) and glycoproteins.6–9 The synthesis of α-gal epitopes in mammals is catalysed by the glycosylation enzyme α1,3galactosyltransferase (α1,3GT)9–12 (Table 1). The combining site of anti-Gal has a ‘pocket’ for the α-gal epitope with a size corresponding to the free trisaccharide Galα1-3Galβ1-4GlcNAc. This is suggested from studies on the affinity of this natural antibody to oligosaccharides.13 The affinity of anti-Gal to the free trisaccharide Galα1-3Galβ1-4GlcNAc is approximately sevenfold higher than that to the disaccharide Galα1-3Gal. The similarity in binding of anti-Gal to α-gal epitopes on carbohydrate chains of glycoproteins (mannose in the fourth position) and glycolipids (no mannose in the carbohydrate chains) further implies that the pocket size is limited to a trisaccharide length of the antigen.14
Reciprocal distribution of the natural anti-Gal antibody and of its ligand the α-gal epitope in mammals
| Mammalian group | α1,3galactosyltransferase | α-gal epitope (Galα1-3Galβ1-4GlcNAc-R) | Natural anti-Gal antibody |
|---|---|---|---|
| Non-primate mammals | + | + | − |
| Prosimians (lemurs) | + | + | − |
| New World monkeys | + | + | − |
| Old World monkeys | − | − | + |
| Apes | − | − | + |
| Humans | − | − | + |
Figure 1.

Insertion of α-gal glycolipids into cell membranes of injected tumours. α-Gal glycolipids are injected into solid tumours in the form of micelles in which the hydrophobic ceramide tails form the core of the micelle whereas the hydrophilic carbohydrate chains protrude into the surrounding aqueous environment. The glycolipids spontaneously insert into the outer phospholipid leaflet of tumour cell membranes because the hydrophobic tail of glycolipids is energetically much more stable when surrounded by the fatty acid tails of phospholipids in tumour cell membranes than within the micelle where they are surrounded by water molecules. This insertion results in presentation of multiple α-gal epitopes on the tumour cell membranes (described as phospholipid bilayer) and binding of the natural anti-Gal antibody molecules to these epitopes. The representative α-gal glycolipid has 10 carbohydrate units (ceramide decasaccharide) and two branches (antennae), each capped by an α-gal epitope marked by a dashed line rectangle.
All mammals synthesizing α-gal epitopes are immunotolerant to it and cannot produce anti-Gal. It is probable that anti-Gal tolerance is induced at the level of B-cell development in the bone marrow, by deletion or receptor editing. This is suggested from studies on B cells in individuals with blood group A or B antigens, which have structures that are similar to the α-gal epitope. No lymphocytes producing anti-blood group A or anti-B antibodies were detected among Epstein–Barr virus-transformed human B cells obtained from the blood of individuals with the corresponding blood group A or B as self-antigen.3 This suggests that B cells capable of producing antibodies to major self-carbohydrate antigens are deleted or undergo receptor editing in the bone marrow and do not reach the circulation.
The α1,3GT gene (also called GGTA1) was inactivated in ancestral Old World monkeys and apes at a period thought to be 20–28 million years ago.15–19 This evolutionary event was likely to be a catastrophic epidemiological event that correlates with the almost complete extinction of hominoids, as indicated in the fossil record.16 The inactivation of the α1,3GT gene in ancestral Old World monkeys and apes is a unique event in mammalian evolution because it was followed by production of large amounts of the natural anti-Gal antibody against the eliminated α-gal epitope.1,4 Anti-Gal could provide immune protection against pathogens endemic to the Old World land mass that were detrimental to primates and that expressed α-gal epitopes.9,15 Several pathogens, including enveloped viruses,21–24 bacteria2,25 and protozoa,27,28 were found to express α-gal epitopes and so can be destroyed by anti-Gal.
It is of interest to note that the initial discovery of anti-Gal was on red blood cells (RBC) of patients with β-thallassaemia,30 on human normal senescent RBC1–31 and on sickle cell anaemia RBC.32 It is possible that a cryptic antigen capable of binding anti-Gal is exposed on human RBC that are ∼ 120 days old, or on thalassaemia and sickle cell anaemia RBC where this antigen is exposed at a younger age of the RBC.1–32 The amount of this cryptic antigen on RBC is very low, resulting in the binding of only a few hundred anti-Gal IgG molecules and its molecular structure has yet to be characterized.
As detailed below, anti-Gal contributes to a number of pathological phenomena. Nevertheless, as this antibody is ubiquitous in humans, anti-Gal may be amenable to exploitation in a number of clinical settings, some of which are presented in this review.
Allergic reactions caused by anti-Gal IgE
Anti-Gal-producing B cells occasionally undergo isotype switch to produce anti-Gal IgE antibodies, which cause allergies by binding to α-gal epitopes.33–37 Such allergies were found in a small proportion of cancer patients treated with the monoclonal antibody cetuximab (anti-EGFR) which carries on its Fab α-gal epitopes.33 Anti-Gal IgE binding to the infused cetuximab induces a systemic allergic reaction that may be severe enough to result in a life-threatening anaphylactic shock.33 An isotype switch to anti-Gal IgE was observed in several individuals following a bite from the tick Amblyomma americanum, which transmits Lyme disease.34 Additional studies indicated that allergic reactions to meat are also associated with anti-Gal IgE antibodies interacting with α-gal epitopes in beef and pork.35,36 Such allergic reactions may be avoided by performing a skin test with nanoparticles that present multiple α-gal epitopes on glycolipids. Such nanoparticles interact with anti-Gal but do not activate T cells,38–39 so they may elicit a localized skin allergic response in the presence of anti-Gal IgE.
Anti-Gal and autoimmunity in Graves' disease
Aberrant expression of α-gal epitopes in human tissues may result in anti-Gal mediated autoimmune processes.40 Graves' disease in which autoantibodies constantly stimulate thyroid cells is an example of association between anti-Gal and autoimmunity. Patients with Graves' disease display elevated titres of anti-Gal IgG,41 raising the possibility that α-gal epitopes aberrantly expressed on thyrocytes induce activation of quiescent anti-Gal B cells. Anti-Gal was further found to mimic thyroid-stimulating hormone (TSH) function if α-gal epitopes are expressed on TSH receptors (TSHR). Anti-Gal purified from normal human serum binds in vitro to porcine thyrocytes and stimulates them to proliferate, produce cAMP and incorporate iodine into thyroglobulin in a manner similar to TSH-mediated stimulation of these thyrocytes because TSHR on porcine thyrocytes are glycoproteins that carry α-gal epitopes.42–43 By a similar mechanism, anti-Gal binds to recombinant porcine TSHR expressed on transfected mouse 3T3 fibroblasts (cells producing the α-gal epitopes) and stimulate these cells to produce cAMP in a manner similar to stimulation by TSH.42 However, anti-Gal does not bind and does not stimulate in vitro normal human thyrocytes as these cells lack α-gal epitopes.42
In contrast to normal human thyrocytes, cultured Graves' disease thyrocytes bind anti-Gal and are stimulated by this antibody to produce cAMP, and to proliferate and display iodine uptake as well as iodine incorporation into thyroglobulin.44 Moreover, specific depletion of anti-Gal antibody from the serum of patients with Graves' disease resulted in a decrease of ∼ 80% in the stimulatory effect of the serum on autologous thyrocytes.44 Together, these observations suggest that α-gal epitopes are aberrantly expressed on Graves' disease thyrocytes, through an as yet unknown mechanism. These epitopes elicit anti-Gal production above the normal level, and the binding of this antibody to the aberrantly expressed epitopes results in stimulation of these thyrocytes at a level that is higher than that of the anti-TSHR antibodies directed against the peptide epitopes on this receptor. It is not clear at present whether aberrant expression of the α1,3GT gene is feasible because of the insertion of a single base mutation in this gene.40 A report on an alternative galactosyltransferase in pig cells45 raises the possibility that a similar enzyme may be aberrantly active in Graves' disease thyrocytes. Alternatively, anti-Gal binding to these thyrocytes may be facilitated by an unknown cell surface molecule with a structure similar to that of the α-gal epitope (e.g. a peptide mimetic to the α-gal epitope), which stimulates production of anti-Gal and interacts with it, ultimately resulting in chronic thyrocyte stimulation. Peptides mimetic to the α-gal epitope were previously identified.46–47 Another possibility may be that the cryptic antigen binding anti-Gal on senescent normal RBC1–31 and on pathological RBC30–32 may undergo increased expression on Graves' disease thyrocytes, resulting in anti-Gal binding and propagation of the autoimmune process. Characterization of this antigen on senescent RBC will enable the evaluation of its expression on Graves' disease thyrocytes.
A marked increase in anti-Gal titre has also been reported in other autoimmune diseases including Henoch–Schönlein purpura, where the increase is primarily in anti-Gal IgA antibody48 and in Crohn's disease where the increase is mainly of anti-Gal IgG antibody.49 It is not clear at present whether anti-Gal contributes to the pathology of these diseases.
Anti-Gal mediated ‘autoimmune like’ phenomena in Chagas' disease
Chagas' disease caused by Trypanosoma cruzi is marked by 10–30-fold higher anti-Gal titres than in uninfected individuals.42,43 Accordingly, T. cruzi was found to present on its cell membrane multiple α-gal epitopes on glycoinositolphospholipids and lipophosphoglycans.27,28 Anti-Gal readily binds to these epitopes on T. cruzi and induces complement-mediated lysis of this pathogen, suggesting that anti-Gal contributes to protection against T. cruzi infections.50–54 However, T. cruzi parasites escaping anti-Gal protection penetrate various tissues. The parasite in cells is protected from destruction by anti-Gal. Intracellular parasites continue to produce and release glycoinositolphospholipids and lipophosphoglycans with α-gal epitopes, as shown in Vero cells infected with T. cruzi.55 These released glycoconjugates stimulate the immune system for continuous production of anti-Gal at high titres. Anti-Gal further interacts with these glycoconjugates resulting in extensive localized ‘autoimmune-like’ inflammatory reactions characterized by extensive infiltration of macrophages and lymphocytes and which result in cardiomyopathy, hepatomegaly and megacolon characteristic of acute and chronic Chagas' disease.54–56 The α-gal epitope was also found to be expressed on Trypanosoma brucei,57 Leishmania mexicana and Leishmania major glycoinositolphospholipids,28,29 so it is possible that anti-Gal also contributes to the chronic inflammations seen following infections with these protozoa.
A similar anti-Gal-mediated autoimmune-like process may result from the adhesion to cells of bacterial fragments expressing carbohydrate epitope resembling α-gal epitope structure.25,26 Adhesion of such antigens to cells in the gastrointestinal tract, urinary tract or other tissues may result in anti-Gal-induced destruction of the cells by complement or antibody-dependent cell-mediated cytotoxicity.
Anti-Gal as a barrier in xenotransplantation
Anti-Gal has been a major immune barrier in xenotransplantation, i.e. transplantation of organs and tissues from various animals, in particular from pigs, into humans. Xenotransplantation research is motivated by the paucity of organ allografts available for transplantation. Pig cells, however, express very high numbers of α-gal epitopes (several million epitopes/cell).9–59 Anti-Gal in xenograft recipients binds to α-gal epitopes on the endothelial cells of xenografts and induces their complement-mediated cytolysis followed by platelet aggregation, occlusion of small blood vessels, collapse of the vascular bed and hyperacute rejection of the xenograft within 0·5–12 hr.60–64 This immunological barrier was recently overcome by the generation of α1,3GT knockout pigs (GT-KO pigs), which lack α-gal epitopes.65–66 Heart and kidney xenografts from these GT-KO pigs transplanted into monkeys were found to survive for from several weeks to several months67,68 and even up to 6 months in one monkey recipient of pig heart.70 The ultimate rejection of such xenografts lacking α-gal epitopes is associated with the production of many antibodies designated anti-non gal antibodies against multiple immunogenic pig proteins.67–71 Prevention of anti-non gal antibody production will require the development of tolerance-inducing methods that selectively prevent the immune response to the multiple pig xenoproteins without significantly affecting the immune protection against various pathogens. Another issue that has to be resolved in xenotransplantation is the effect of pig endogenous retrovirus in human recipients. This virus was shown to infect in vitro human fibroblasts co-incubated with pig cells.72 As the genome of this retrovirus can be incorporated in various domains of the recipient's genome, a white paper has been published that recommends that before large studies on xenotransplantation of porcine xenografts in humans, the study of pig endogenous retrovirus genome incorporation should be performed in xenograft recipients.73 If such incorporation is observed, then it should be elucidated whether it has any detrimental outcomes.73
The issues of anti-Gal and anti-non gal response to a xenograft implant are not only of future relevance in xenotransplantation, but are of present relevance in patients with a porcine heart valve. There is an inverse correlation between the age of the recipient and the rate at which the implanted valve function is deteriorating due to calcification, which seems to be caused by binding of antibodies to the valve.74 The observation that in porcine valve recipients there is elevation of anti-Gal antibody titre as an immune response to the α-gal epitopes on the porcine valve75 raised the possibility that this antibody may contribute to the calcification of the valve. Indeed, valve specimens from GT-KO pigs pre-incubated in human serum (i.e. valves not binding anti-Gal) displayed significantly less calcification following subcutaneous implantation in rats than valves from wild-type pig implants pre-incubated in human serum (i.e. valves binding anti-Gal to their α-gal epitopes).76–77 It remains to be determined whether in the absence of α-gal epitopes on porcine valve implants (i.e. valve implants from GT-KO pigs), the deterioration of the implant in young recipients will be reduced in comparison to valves from wild-type pigs, or is this deterioration also associated with extensive production of anti-non-gal antibodies.
Anti-Gal-mediated enhancement of viral vaccines immunogenicity by targeting to antigen-presenting cells
The presence of anti-Gal in large amounts in humans provides a unique opportunity for exploiting this antibody in several clinical settings. One versatile clinical application is to enhance immunogenicity of viral and other microbial vaccines as well as cancer vaccines by targeting vaccines for effective uptake by antigen-presenting cells (APC) such as dendritic cells and macrophages, provided that the vaccine expresses α-gal epitopes.78 Studies were performed in anti-Gal producing α1,3GT knockout mice (GT-KO mice) with ovalbumin as a vaccine model. If the vaccine expresses α-gal epitopes, anti-Gal forms immune complexes with it. These immune complexes are effectively internalized by APC via interaction between the Fc portion of immunocomplexed anti-Gal and Fcγ receptors (FcγR) on the APC.79 These APC transport the vaccine to regional lymph nodes where they present the vaccinating peptides on MHC molecules for the activation of vaccine-specific T cells.79
Increased immunogenicity of viral vaccines by in situ immunocomplexing with anti-Gal was demonstrated with influenza virus vaccine and gp120 vaccine of HIV engineered to express α-gal epitopes.80,81 Vaccination with inactivated influenza virus processed by the use of recombinant α1,3GT to express multiple α-gal epitopes elicited a 100-fold higher specific CD4+ T-cell response and anti-influenza virus antibody response as well as approximately sixfold higher CD8+ T-cell response than vaccination with virus lacking α-gal epitopes.80 Moreover, GT-KO mice immunized with influenza virus vaccine-presenting α-gal epitopes were ∼ 11-fold more resistant to challenge with live virus than GT-KO mice immunized with virus lacking α-gal epitopes.80 A similar increase in immunogenicity was observed with gp120 recombinant protein81 or with a fusion protein of gp120 and p24 of HIV on which α-gal epitopes were synthesized by the use of recombinant α1,3GT.82 As gp120 has 13–16 N-linked carbohydrate chains of the complex type on which α-gal epitopes can be synthesized with recombinant α1,3GT, it may serve as an effective platform for targeting proteins lacking carbohydrate chains (e.g. core and matrix proteins of HIV) to APC by formation of immune complexes between the fusion protein and anti-Gal.82
Conversion of tumours into vaccines by intra-tumoral injection of α-gal glycolipids
Anti-Gal may be exploited for eliciting a protective immune response against micrometastases by enhancing the immunogenicity of tumour-associated antigens (TAA) on autologous tumour cells. Many of the TAA are autologous TAA unique to each cancer patient and are generated by multiple coding mutations as a result of genomic instability.83,84 Some of these mutations confer a growth advantage to tumour cells (driver mutations) whereas others have no effect on growth (passenger mutations). The autologous TAA may serve as target for active immunotherapy against metastatic cells.
The immune system in humans has the potential to mount a protective immune response against the autologous TAA. This is evidenced by the correlation between the extent of CD8+ T cells and T helper type 1 memory T-cell infiltration into tumours and improved prognosis in malignancies such as melanoma, ovarian carcinoma and colorectal carcinoma.86–89 However, in a large proportion of cancer patients no T cells are found in resected tumours because tumours evolve to evade recognition by APC, so no anti-TAA immune response is elicited in the pre-clinical stage.
Identification of the multiple autologous TAA in individual patients and their synthesis for vaccination purposes are not feasible at present. Therefore, the tumour itself is a practical source for vaccinating autologous TAA. Immunogenicity of the undefined autologous TAA can be increased by anti-Gal-mediated in situ targeting of tumour cells within tumour lesions to APC.78 It was assumed that anti-Gal binding to tumour cells manipulated to express α-gal epitopes results in effective uptake of these cells by APC via Fc/FcγR interaction between the Fc portion of anti-Gal coating tumour cells and FcγR on APC, thereby converting the tumour itself into a vaccine (Fig. 2). Accordingly, studies performed in GT-KO mice producing anti-Gal and using the tumour model of B16 mouse melanoma, which lacks α-gal epitopes, have demonstrated increased immunogenicity of melanoma tumour antigens or of pancreatic carcinoma MUC1 tumour antigen after induction of α-gal epitope expression on the tumour cells.90–93
Figure 2.
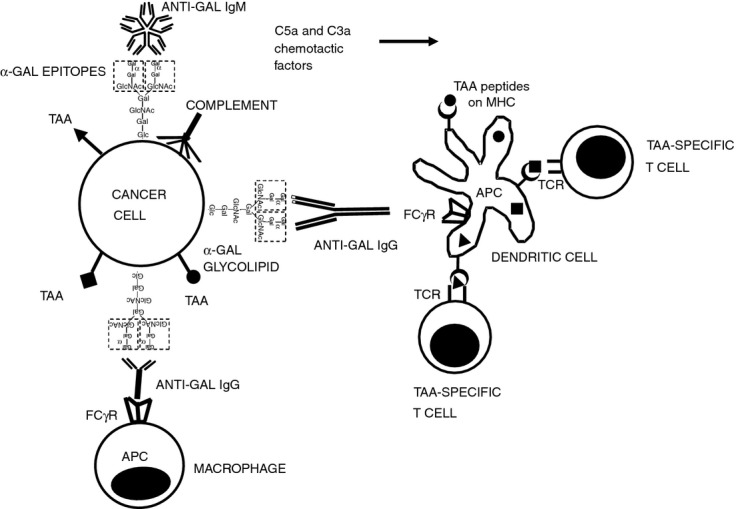
Targeting of tumour cells to antigen-presenting cell (APC) by the natural anti-Gal antibody. Following insertion of injected α-gal glycolipids into tumour cell membranes both anti-Gal IgM and IgG antibodies (released from ruptured capillaries) bind to α-gal epitopes on the tumour cells. Binding of anti-Gal IgM results in effective complement activation and generation of the chemotactic complement cleavage peptides C5a and C3a, which induce rapid migration of APC (dendritic cells and macrophages) into the treated tumour. Following anti-Gal IgG binding to the α-gal epitopes on tumour cells (i.e. opsonization), its Fc portion interacts with Fcγ receptor (FcγR) on APC. This interaction stimulates APC to internalize intact or lysed tumour cells and the tumour-associated antigens (TAA) they carry. Internalized TAA are transported by the APC to regional lymph nodes, processed and various immunogenic TAA peptides (•, ▪, ▴) are presented by the APC in association with class I and class II MHC molecules. These immunogenic peptides interact with the corresponding T-cell receptors (TCR) and activate the multiple TAA-specific cytotoxic and helper T-cell clones that mediate a protective anti-tumour immune response.
Whereas extensive expression of α-gal epitopes on vaccinating tumour cells can be achieved by in vitro stable transfection of the cells with the α1,3GT gene90–93 or by in vitro synthesis of α-gal epitopes on tumour cells with recombinant α1,3GT,94–95 such methods are not effective in inducing in situ expression of these epitopes in tumour lesions to convert them into autologous tumour vaccines. In situ expression of α-gal epitopes on tumour cells can be achieved by intra-tumoral injection of α-gal glycolipids.96 These glycolipids can be obtained in relatively large amounts from rabbit RBC membranes where they comprise the majority of glycolipids.96 α-Gal glycolipids include a hydrophobic ceramide ‘tail’ and a hydrophilic carbohydrate chain linked to the ceramide and carrying one or several carbohydrate branches capped by α-gal epitopes. These glycolipids are purified from rabbit RBC by chloroform : methanol extraction and removal of phospholipids and cholesterol.96 Purified α-gal glycolipids dissolve in water as micelles (Fig. 1). When injected into subcutaneous B16 melanoma lesions growing in GT-KO mice, the α-gal glycolipids in the micelles insert spontaneously into tumour cell membranes. This insertion occurs because the hydrophobic lipid ‘tail’ of α-gal glycolipids is energetically much more stable when surrounded by cell membrane phospholipids than in micelles surrounded by water (Fig. 1). The multiple α-gal epitopes of the inserted α-gal glycolipids bind anti-Gal. This interaction activates complement that induces lysis of some of the tumour cells and generates chemotactic complement cleavage peptides as C5a and C3a. These chemotactic factors induce rapid and extensive recruitment of dendritic cells and macrophages into the injected tumour.96–97 The recruited APC further internalize anti-Gal-coated tumour cells via Fc/FcγR interaction (Fig. 2). Within ∼ 2 weeks post-injection, APC in GT-KO mice transport the internalized melanoma TAA to regional lymph nodes where they present TAA peptides and activate tumour-specific CD8+ and CD4+ T cells.96–98 This immune response results in protection by CD8+ T cells against distant micrometastases and is potent enough to overcome the immunosuppressive effect of regulatory T cells.98 No immune tolerance was broken as treated mice displayed no indication of autoimmunity to normal tissue antigens and no vitiligo.96,97 Lack of autoimmunity to normal tissue antigens was also observed in GT-KO mice immunized with B16 melanoma engineered to express α-gal epitopes by stable transfection with α1,3GT cDNA.90–99
Of note is the report that generation of complement C5a in a tumour microenvironment enhances tumour growth by suppressing the anti-tumour CD8+ T-cell-mediated response.100 The immunotherapy by intra-tumoral injection of α-gal glycolipids is unlikely to be affected by this process as it elicits an anti-tumour CD8+ protective immune response in the draining lymph nodes and spleen, rather than within the treated tumour.98 Nevertheless, a proportion of the tumour cells with inserted α-gal glycolipids are destroyed within the injected lesions by complement-mediated cytotoxicity and by antibody-dependent cell-mediated cytotoxicity following anti-Gal binding to the α-gal epitopes de novo expressed on the tumour cells in the treated tumour.96–97
Immunotherapy by intra-tumoral injection of α-gal glycolipids in cancer patients
Immunotherapy by intra-tumoral injection with α-gal glycolipids was studied for safety in a Phase I dose escalation trial in patients with malignant solid tumours at advanced stages of disease.101 Studies were approved by the Food and Drugs Administration (IND12946) and the Institutional Review Board. Informed consent was received from participating patients. Intra-tumoral injection of 0·1, 1 and 10 mg α-gal glycolipids per tumour was found to be safe and no patients developed clinical signs of toxicity, or any indication for autoimmunity.101 Some patients with tumours such as renal carcinoma and pancreatic adenocarcinoma appeared to have an unexpectedly long survival after injection whereas other patients did not.101
Immunotherapy with α-gal glycolipids may be of particular interest in cancer patients before the surgical removal of their primary tumour, i.e. in neoadjuvant immunotherapy. Intra-tumoral injection of α-gal glycolipids in cancer patients shortly after diagnosis, i.e. 2–4 weeks before resection, may convert the treated tumour into a temporary vaccine that ‘educates’ the immune system to recognize and destroy metastatic cells presenting autologous TAA. This treatment may be suitable in patients with mammary, colon or pancreatic carcinoma as well as other internal solid tumours that can be injected by imaging. Because of immunological memory, the immune response elicited in the period between diagnosis and resection may protect against micrometastases long after the primary tumour was removed.
Acceleration of wound healing and tissue regeneration by α-gal nanoparticles
Anti-Gal can be exploited also for accelerating the healing of wounds and burns.39,102Wound healing requires the local recruitment and activation of macrophages, which orchestrate this process by secreting various cytokines that direct the regeneration of the injured tissue.104,105 Accelerated macrophage recruitment and activation can be achieved by application of α-gal nanoparticles to wounds. These nanoparticles are submicroscopic liposomes prepared from α-gal glycolipids, phospholipids and cholesterol, all extracted from rabbit RBC membranes.39,102 When applied into injuries, α-gal nanoparticles effectively bind the natural anti-Gal antibody that ‘leaks’ from ruptured capillaries and activate the complement cascade, which generates chemotactic cleavage complement peptides such as C5a and C3a (Fig. 3). These chemotactic peptides induce rapid and extensive migration of macrophages into the treated wound, observed already within 24 hr post-treatment.39 The recruited macrophages are further activated following the binding of the Fc portion of anti-Gal coating α-gal nanoparticles via FcγR on the macrophages (Fig. 3). The activated macrophages produce cytokines such as vascular endothelial growth factor, interleukin-1, platelet-derived growth factor and colony-stimulating factor that promote healing of injuries.39 As the α-gal glycolipids are surrounded in the nanoparticle wall by phospholipids they are energetically stable and do not insert into cell membranes in treated wounds.
Figure 3.
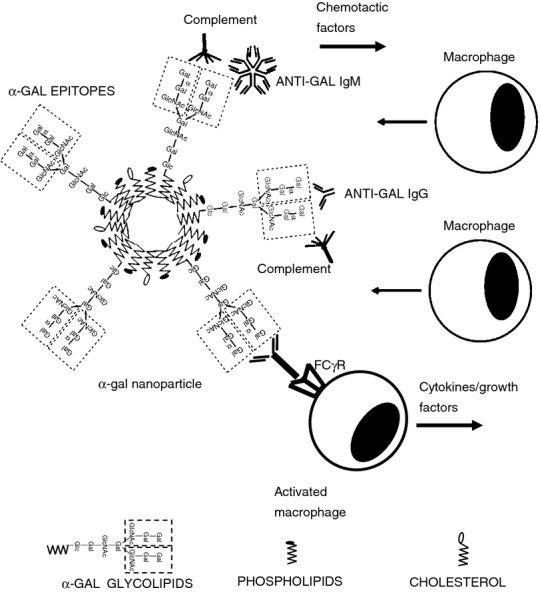
Acceleration of wound healing by α-gal nanoparticles applied to wounds. Binding of the natural anti-Gal antibody to α-gal epitopes on α-gal nanoparticles activates the complement system within the treated wound. The resulting complement cleavage chemotactic factors induce rapid recruitment of macrophages into the wound. The recruited macrophages undergo activation as a result of interaction between the Fc ‘tails’ of anti-Gal immunocomplexed to the α-gal nanoparticles with Fcγ receptor (FcγR) on macrophages. The activated macrophages produce and secrete cytokines/growth factors that accelerate healing and may recruit stem cells. Symbols for α-gal glycolipids, phospholipids and cholesterol are illustrated at the bottom of the figure.
The α-gal nanoparticle treatment was found to decrease wound and burn healing time in GT-KO mice by 50–70% in comparison with physiological healing time.39–102 Moreover, anti-Gal-mediated accelerated healing of the wound is associated with rapid restoration of normal skin architecture, thereby avoiding fibrosis and scar formation.39 In anti-Gal-producing GT-KO pigs the healing time of a full-thickness excisional skin wound (20 × 20 mm) was found to decrease by 40% following topical application of α-gal nanoparticles in comparison to saline-treated wounds on the same animal.103
It is of interest to note that whereas the natural anti-phospholipid antibody ‘anti-phosphatidyl-choline (anti-PC) IgM Ab’ was reported to be an anti-inflammatory antibody,107 the natural anti-Gal antibody may be a pro-inflammatory as well as anti-inflammatory antibody, depending on the type of cells or particles presenting the α-gal epitope. If this epitope is expressed on cells (e.g. xenograft cells or tumour cells with inserted α-gal glycolipids), the interaction with anti-Gal may lead to cell lysis and a pro-inflammatory response. However, if the α-gal epitope is expressed on nanoparticles and there is no insertion into cells because of the stability of the α-gal glycolipid molecules surrounded by phospholipids, then anti-Gal/α-gal epitope interaction results in recruitment and activation of macrophages into pro-healing activity, as observed in the wound-healing studies.39,102
Future directions on regeneration of ischaemic myocardium and of injured nerves
Repair and regeneration of skin wounds and of internal injuries are mediated by similar mechanisms so it may be possible that acceleration of injured tissue repair and regeneration by α-gal nanoparticles can be achieved also in internal injuries such as in ischaemic myocardium and in injured nerves. After myocardial infarction, as in wound healing, macrophages migrate to the injured myocardium, debride it of dead cells and secrete cytokines/growth factors that recruit stem cells. The recruited stem cells receive cues from the adjacent healthy cells, the microenvironment and the extracellular matrix, to differentiate into cardiomyocytes that regenerate the tissue and restore its physiological activity.108,109 Myocardium with limited ischaemic damage may display spontaneous regeneration by this mechanism. However, in more extensive ischaemic damage, migration of macrophages into injured myocardium and the recruitment of stem cells are processes that are too slow to prevent irreversible fibrosis and scar formation, which is the default mechanism for tissue repair. It may be possible that post-infarction fibrosis can be reduced or prevented by injection of α-gal nanoparticles into the injured cardiac muscle via a catheter, shortly after the ischaemia event (i.e. before the onset of myocardium fibrosis). Injected α-gal nanoparticles will bind anti-Gal and induce rapid chemotactic migration of macrophages, which will be activated and may further recruit stem cells from the circulation or from uninjured adjacent myocardium. It may further be possible that the recruited stem cells are guided by the microenvironment and extracellular matrix to differentiate into cardiomyocytes that repopulate the ischaemic myocardium and restore its biological activity before the onset of fibrosis.
Activated macrophages are also pivotal in regeneration of severed nerves, as in spinal cord injury. Regeneration of nerves requires regrowth of multiple sprouts from injured axons. These sprouts ‘attempt’ to reconnect across the lesion and grow into the distal axonal tube of the damaged neurons. This axonal sprout growth depends on growth factors such as vascular endothelial growth factor secreted by macrophages migrating to the injury site and inducing local angiogenesis.111 If this growth of axonal sprouts is delayed because of insufficient recruitment of macrophages into the injury site, the ongoing fibrosis process will irreversibly prevent regeneration of the injured nerve. α-Gal nanoparticles applied to the injury site will bind anti-Gal and may induce rapid macrophage migration and activation for the secretion of vascular endothelial growth factor, vascularization and hence growth of many axonal sprouts. The resulting growth of multiple axonal sprouts may enable regrowth of the axons into the distal portion of the axonal tubes and ultimately induce regeneration of the injured nerve.
It should be stressed that the proposed treatments for regeneration of ischaemic myocardium and injured nerve by application of α-gal nanoparticles are theoretical possibilities at present. The assumption that such regeneration will precede the default fibrosis processes may be studied in the experimental mammalian animal models that produce anti-Gal such as GT-KO mouse or GT-KO pig which were found to be instrumental in the study of wound healing, or in anti-Gal-producing monkeys, which have been essential in evaluating the role of anti-Gal as a barrier for xenotransplantation.
Conclusions
The natural anti-Gal antibody, which is the most abundant natural antibody in humans, is involved in a number of detrimental processes that may result in allergic, autoimmune and ‘autoimmune-like’ pathogeneses. Because of its abundance in humans, anti-Gal may be exploited in increasing immunogenicity of microbial vaccines and of autologous tumour vaccines expressing α-gal epitopes by targeting vaccines to APC. Application of α-gal nanoparticles to wounds further accelerates wound healing by rapid recruitment and activation of macrophages. Additional basic and clinical research exploiting this unique natural antibody may lead to numerous beneficial clinical applications that are, as yet, undefined.
Disclosures
The author has no conflict of interests.
References
- Galili U, Rachmilewitz EA, Peleg A, Flechner I. A unique natural human IgG antibody with anti-α-galactosyl specificity. J Exp Med. 1984;160:1519–31. doi: 10.1084/jem.160.5.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffis JM. Interaction between human natural anti-α-galactosyl immunoglobulin G and bacteria of the human flora. Infect Immun. 1988;56:1730–7. doi: 10.1128/iai.56.7.1730-1737.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili U, Anaraki F, Thall A, Hill-Black C, Radic M. One percent of circulating B lymphocytes are capable of producing the natural anti-Gal antibody. Blood. 1993;82:2485–93. [PubMed] [Google Scholar]
- Wang L, Radic MZ, Galili U. Human anti-Gal heavy chain genes: preferential use of VH3 and the presence of somatic mutations. J Immunol. 1995;155:1276–85. [PubMed] [Google Scholar]
- Kearns-Jonker M, Swensson J, Ghiuzeli C, et al. The human antibody response to porcine xenoantigens is encoded by IGHV3–11 and IGHV3–74 IgVH germline progenitors. J Immunol. 1999;163:4399–412. [PubMed] [Google Scholar]
- Galili U, Clark MR, Shohet SB, Buehler J, Macher BA. Evolutionary relationship between the anti-Gal antibody and the Galα1→3Gal epitope in primates. Proc Natl Acad Sci USA. 1987;84:1369–73. doi: 10.1073/pnas.84.5.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili U, Macher BA, Buehler J, Shohet SB. Human natural anti-α-galactosyl IgG. II. The specific recognition of α(1→3)-linked galactose residues. J Exp Med. 1985;162:573–82. doi: 10.1084/jem.162.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro RG, Bhoyroo VD. Occurrence of α-d-galactosyl residues in the thyroglobulin from several species. Localization in the saccharide chains of the complex carbohydrate units. J Biol Chem. 1984;259:9858–66. [PubMed] [Google Scholar]
- Galili U, Shohet SB, Kobrin E, Stults CLM, Macher BA. Man, apes, and Old World monkeys differ from other mammals in the expression of α-galactosyl epitopes on nucleated cells. J Biol Chem. 1988;263:17755–62. [PubMed] [Google Scholar]
- Basu M, Basu S. Enzymatic synthesis of blood group related pentaglycosyl ceramide by an α-galactosyltransferase. J Biol Chem. 1973;248:1700–6. [PubMed] [Google Scholar]
- Blake DD, Goldstein IJ. An α-d-galactosyltransferase in Ehrlich ascites tumor cells. Biosynthesis and characterization of a trisaccharide (α-d-galacto(1–3)-N-acetyllactosamine) J Biol Chem. 1981;256:5387–93. [PubMed] [Google Scholar]
- Betteridge A, Watkins WM. Two α-3-d galactosyltransferases in rabbit stomach mucosa with different acceptor substrate specificities. Eur J Biochem. 1983;132:29–35. doi: 10.1111/j.1432-1033.1983.tb07321.x. [DOI] [PubMed] [Google Scholar]
- Galili U, Matta KL. Inhibition of anti-Gal IgG binding to porcine endothelial cells by synthetic oligosaccharides. Transplantation. 1996;62:356–62. doi: 10.1097/00007890-199607270-00018. [DOI] [PubMed] [Google Scholar]
- Galili U. Evolution and pathophysiology of the human natural anti-Gal antibody. Springer Semin Immunopathol. 1993;15:155–71. doi: 10.1007/BF00201098. [DOI] [PubMed] [Google Scholar]
- Galili U, Swanson K. Gene sequences suggest inactivation of α1-3 galactosyltransferase in catarrhines after the divergence of apes from monkeys. Proc Natl Acad Sci USA. 1991;88:7401–4. doi: 10.1073/pnas.88.16.7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili U, Andrews P. Suppression of α-galactosyl epitopes synthesis and production of the natural anti-Gal antibody: a major evolutionary event in ancestral Old World primates. J Hum Evol. 1995;29:433–42. [Google Scholar]
- Joziasse DH, Shaper JH, Jabs EW, Shaper NL. Characterization of an α1-3-galactosyltransferase homologue on human chromosome 12 that is organized as a processed pseudogene. J Biol Chem. 1991;266:6991–8. [PubMed] [Google Scholar]
- Koike C, Fung JJ, Geller DA, et al. Molecular basis of evolutionary loss of the α1,3-galactosyltransferase gene in higher primates. J Biol Chem. 2002;277:10114–20. doi: 10.1074/jbc.M110527200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike C, Uddin M, Wildman DE, Gray EA, Trucco M, Starzl TE, Goodman M. Functionally important glycosyltransferase gain and loss during catarrhine primate emergence. Proc Natl Acad Sci USA. 2007;104:559–64. doi: 10.1073/pnas.0610012104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teranishi K, Manez R, Awwad M, Cooper DK. Anti-Gal α1-3Gal IgM and IgG antibody levels in sera of humans and old world non-human primates. Xenotransplantation. 2002;9:48–154. doi: 10.1034/j.1399-3089.2002.1o058.x. [DOI] [PubMed] [Google Scholar]
- Repik PM, Strizki JM, Galili U. Differential host dependent expression of α-galactosyl epitopes on viral glycoproteins: a study of Eastern equine encephalitis virus as a model. J Gen Virol. 1994;75:1177–81. doi: 10.1099/0022-1317-75-5-1177. [DOI] [PubMed] [Google Scholar]
- Galili U, Repik PM, Anaraki F, Mozdzanowska K, Washko G, Gerhard W. Enhancement of antigen presentation of influenza virus hemagglutinin by the natural human anti-Gal antibody. Vaccine. 1996;14:321–8. doi: 10.1016/0264-410x(95)00189-8. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Porter CD, Strahan KM, Preece AF, Gustafsson K, Cosset FL, Weiss RA, Collins MK. Sensitization of cells and retroviruses to human serum by (α1-3) galactosyltransferase. Nature. 1996;379:85–8. doi: 10.1038/379085a0. [DOI] [PubMed] [Google Scholar]
- Welsh RM, O'Donnell CL, Reed DJ, Rother RP. Evaluation of the Galα1-3Gal epitope as a host modification factor eliciting natural humoral immunity to enveloped viruses. J Virol. 1998;72:4650–6. doi: 10.1128/jvi.72.6.4650-4656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Cai L, Wu B, Li L, Xiao Z, Cheng J, Wang PG. The wciN gene encodes an α-1,3-galactosyltransferase involved in the biosynthesis of the capsule repeating unit of Streptococcus pneumoniae serotype 6B. Biochemistry. 2012;51:5804–10. doi: 10.1021/bi300640b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posekany KJ, Pittman HK, Bradfield JF, Haisch CE, Verbanac KM. Induction of cytolytic anti-Gal antibodies in α-1,3-galactosyltransferase gene knockout mice by oral inoculation with Escherichia coli O86:B7 bacteria. Infect Immun. 2002;70:6215–22. doi: 10.1128/IAI.70.11.6215-6222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida IC, Ferguson MA, Schenkman S, Travassos LR. Lytic anti-α-galactosyl antibodies from patients with chronic Chagas' disease recognize novel O-linked oligosaccharides on mucin-like glycosyl-phosphatidylinositol-anchored glycoproteins of Trypanosoma cruzi. Biochem J. 1994;304:793–802. doi: 10.1042/bj3040793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider P, Schnur LF, Jaffe CL, Ferguson MA, McConville MJ. Glycoinositol-phospholipid profiles of four serotypically distinct Old World Leishmania strains. Biochem J. 1994;304:603–9. doi: 10.1042/bj3040603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila JL, Rojas M, Galili U. Immunogenic Galα1-3Gal carbohydrate epitopes are present on pathogenic American Trypanosoma and Leishmania. J Immunol. 1989;142:2828–34. [PubMed] [Google Scholar]
- Galili U, Korkesh A, Kahane I, Rachmilewitz A. Demonstration of a natural anti-galactosyl IgG antibody on thalassemic red blood cells. Blood. 1983;61:1258–64. [PubMed] [Google Scholar]
- Galili U, Flechner I, Kniszinski A, Danon D, Rachmilewitz EA. The natural anti-α-galactosyl IgG on human normal senescent red blood cells. Br J Haematol. 1986;62:317–24. doi: 10.1111/j.1365-2141.1986.tb02935.x. [DOI] [PubMed] [Google Scholar]
- Galili U, Clark MR, Shohet SB. Excessive binding of the natural anti-α-galactosyl IgG to sickle red cells may contribute to extravascular cell destruction. J Clin Invest. 1986;77:27–33. doi: 10.1172/JCI112286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-α-1,3-galactose. N Engl J Med. 2008;358:1109–17. doi: 10.1056/NEJMoa074943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commins SP, James HR, Kelly LA, et al. The relevance of tick bites to the production of IgE antibodies to the mammalian oligosaccharide galactose-α-1,3-galactose. J Allergy Clin Immunol. 2011;127:1286–93. doi: 10.1016/j.jaci.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commins SP, Satinover SM, Hosen J, Mozena J, Borish L, Lewis BD, Woodfolk JA, Platts-Mills TA. Delayed anaphylaxis, angioedema, or urticaria after consumption of red meat in patients with IgE antibodies specific for galactose-α-1,3-galactose. J Allergy Clin Immunol. 2009;123:426–33. doi: 10.1016/j.jaci.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisset M, Richard C, Astier C, et al. Anaphylaxis to pork kidney is related to IgE antibodies specific for galactose-α-1,3-galactose. Allergy. 2012;67:699–704. doi: 10.1111/j.1398-9995.2012.02799.x. [DOI] [PubMed] [Google Scholar]
- Nuñez R, Carballada F, Gonzalez-Quintela A, Gomez-Rial J, Boquete M, Vidal C. Delayed mammalian meat-induced anaphylaxis due to galactose-α-1,3-galactose in 5 European patients. J Allergy Clin Immunol. 2011;128:1122–4. doi: 10.1016/j.jaci.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Tanemura M, Yin D, Chong AS, Galili U. Differential immune response to α-gal epitopes on xenografts and allografts: implications for accommodation in xenotransplantation. J Clin Invest. 2000;105:301–10. doi: 10.1172/JCI7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigglesworth K, Racki WJ, Mishra R, Szomolany-Tsuda E, Greiner DL, Galili U. Rapid recruitment and activation of macrophages by anti-Gal/α-gal liposome interaction accelerates wound healing. J Immunol. 2011;186:4422–32. doi: 10.4049/jimmunol.1002324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili U. Abnormal expression of α-galactosyl epitopes in man: a trigger for autoimmune processes? Lancet. 1989;2:358–61. doi: 10.1016/s0140-6736(89)90539-4. [DOI] [PubMed] [Google Scholar]
- Etienne-Decerf J, Malaise M, Mahieu P, Winand R. Elevated anti-α-galactosyl antibody titres. A marker of progression in autoimmune thyroid disorders and in endocrine ophthalmopathy? Acta Endocrinol. 1987;115:67–74. doi: 10.1530/acta.0.1150067. [DOI] [PubMed] [Google Scholar]
- Winand RJ, Anaraki F, Etienne-Decerf J, Galili U. Xenogeneic thyroid-stimulating hormone-like activity of the human natural anti-Gal antibody. Interaction of anti-Gal with porcine thyrocytes and with recombinant human thyroid-stimulating hormone receptors expressed on mouse cells. J Immunol. 1993;151:3923–34. [PubMed] [Google Scholar]
- Thall A, Etienne-Decerf J, Winand RJ, Galili U. The α-galactosyl epitope on mammalian thyroid cells. Acta Endocrinol. 1991;124:692–9. doi: 10.1530/acta.0.1240692. [DOI] [PubMed] [Google Scholar]
- Winand RJ, Winand-Devigne J, Meurisse M, Galili U. Specific stimulation of Graves' disease thyrocytes by the natural anti-Gal antibody from normal and autologous serum. J Immunol. 1994;153:1386–95. [PubMed] [Google Scholar]
- Sharma A, Naziruddin B, Cui MJ, et al. Pig cells that lack the gene for α1-3 galactosyltransferase express low levels of the gal antigen. Transplantation. 2003;75:430–6. doi: 10.1097/01.TP.0000053615.98201.77. [DOI] [PubMed] [Google Scholar]
- Sandrin MS, Vaughan HA, Xing PX, McKenzie IF. Natural human anti-Gal α(1,3)Gal antibodies react with human mucin peptides. Glycoconj J. 1997;14:97–105. doi: 10.1023/a:1018521217276. [DOI] [PubMed] [Google Scholar]
- Lang J, Zhan J, Xu L, Yan Z. Identification of peptide mimetics of xenoreactive α-Gal antigenic epitope by phage display. Biochem Biophys Res Commun. 2006;344:214–20. doi: 10.1016/j.bbrc.2006.03.112. [DOI] [PubMed] [Google Scholar]
- Davin JC, Malaise M, Foidart J, Mahieu P. Anti-α-galactosyl antibodies and immune complexes in children with Henoch–Schönlein purpura or IgA nephropathy. Kidney Int. 1987;31:1132–9. doi: 10.1038/ki.1987.119. [DOI] [PubMed] [Google Scholar]
- D'Alessandro M, Mariani P, Lomanto D, Bachetoni A, Speranza V. Alterations in serum anti-α-galactosyl antibodies in patients with Crohn's disease and ulcerative colitis. Clin Immunol. 2002;103:63–8. doi: 10.1006/clim.2001.5180. [DOI] [PubMed] [Google Scholar]
- Towbin H, Rosenfelder G, Wieslander J, et al. Circulating antibodies to mouse laminin in Chagas disease, American cutaneous leishmaniasis, and normal individuals recognize terminal galactosyl(α1-3)-galactose epitopes. J Exp Med. 1987;66:419–32. doi: 10.1084/jem.166.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani SR, Travassos LR. Anti-α-galactosyl antibodies in chagasic patients. Possible biological significance. Braz J Med Biol Res. 1988;21:1275–86. [PubMed] [Google Scholar]
- Almeida IC, Milani SR, Gorin PA, Travassos LR. Complement-mediated lysis of Trypanosoma cruzi trypomastigotes by human anti-α-galactosyl antibodies. J Immunol. 1991;146:2394–400. [PubMed] [Google Scholar]
- Gazzinelli RT, Pereira ME, Romanha A, Gazzinelli G, Brener Z. Direct lysis of Trypanosoma cruzi: a novel effector mechanism of protection mediated by human anti-gal antibodies. Parasite Immunol. 1991;13:345–56. doi: 10.1111/j.1365-3024.1991.tb00288.x. [DOI] [PubMed] [Google Scholar]
- Travassos LR, Almeida IC. Carbohydrate immunity in American trypanosomiasis. Springer Semin Immunopathol. 1993;15:183–204. doi: 10.1007/BF00201100. [DOI] [PubMed] [Google Scholar]
- Souto-Padron T, Almeida IC, de Souza W, Travassos LR. Distribution of α-galactosyl epitopes on Trypanosoma cruzi trypomastigotes and amastigotes forms for infected Vero cells detected by chagasic antibodies. J Eukaryot Microbiol. 1994;41:47–54. doi: 10.1111/j.1550-7408.1994.tb05933.x. [DOI] [PubMed] [Google Scholar]
- Avila JL. α-Galactosyl-bearing epitopes as potent immunogens in Chagas' disease and leishmaniasis. Subcell Biochem. 1999;32:173–213. doi: 10.1007/978-1-4615-4771-6_8. [DOI] [PubMed] [Google Scholar]
- Ramasamy R, Field MC. Terminal galactosylation of glycoconjugates in Plasmodium falciparum asexual blood stages and Trypanosoma brucei bloodstream trypomastigotes. Exp Parasitol. 2012;130:314–20. doi: 10.1016/j.exppara.2012.02.017. [DOI] [PubMed] [Google Scholar]
- Lüderitz O, Simmons DA, Westphal G. The immunochemistry of Salmonella chemotype VI O-antigens. The structure of oligosaccharides from Salmonella group U (o 43) lipopolysaccharides. Biochem J. 1965;97:820–6. doi: 10.1042/bj0970820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanemura M, Maruyama S, Galili U. Differential expression of α-gal epitopes (Galα1-3Galß1-4GlcNAc-R) on pig and mouse organs. Transplantation. 2000;69:187–90. doi: 10.1097/00007890-200001150-00034. [DOI] [PubMed] [Google Scholar]
- Galili U. Interaction of the natural anti-Gal antibody with α-galactosyl epitopes: a major obstacle for xenotranplantation in humans. Immunol Today. 1993;14:480–2. doi: 10.1016/0167-5699(93)90261-i. [DOI] [PubMed] [Google Scholar]
- Good AH, Cooper DK, Malcolm AJ, et al. Identification of carbohydrate structures which bind human anti-porcine antibodies: implication for discordant xenografting in man. Transplant Proc. 1992;24:559–62. [PubMed] [Google Scholar]
- Sandrin M, Vaughan HA, Dabkowski PL, McKenzie IFC. Anti-pig IgM antibodies in human serum react predominantly with Galα1-3Gal epitopes. Proc Natl Acad Sci USA. 1993;90:11391–5. doi: 10.1073/pnas.90.23.11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BH, Cotterell AH, McCurry KR, et al. Cardiac xenografts between primate species provide evidence of the α-galactosyl determinant in hyperacute rejection. J Immunol. 1994;154:5500–10. [PubMed] [Google Scholar]
- Xu Y, Lorf T, Sablinski T, et al. Removal of anti-porcine natural antibodies from human and nonhuman primate plasma in vitro and in vivo by a Galα1-3Galβ1-4Glc-R immunoaffinity column. Transplantation. 1998;65:172–9. doi: 10.1097/00007890-199801270-00005. [DOI] [PubMed] [Google Scholar]
- Lai L, Kolber-Simonds D, Park KW, et al. Production of α-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science. 2002;295:1089–92. doi: 10.1126/science.1068228. [DOI] [PubMed] [Google Scholar]
- Phelps CJ, Koike C, Vaught TD, et al. Production of α1,3-galactosyltransferase-deficient pigs. Science. 2003;299:411–4. doi: 10.1126/science.1078942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwaki K, Tseng L, Dor FJ, et al. Heart transplantation in baboons using α1,3-galactosyltransferase gene-knockout pigs as donors: initial experience. Nat Med. 2005;11:29–31. doi: 10.1038/nm1171. [DOI] [PubMed] [Google Scholar]
- Yamada K, Yazawa K, Shimizu A, et al. Marked prolongation of porcine renal xenograft survival in baboons through the use of α1,3-galactosyltransferase gene-knockout donors and the cotransplantation of vascularized thymic tissue. Nat Med. 2005;11:32–4. doi: 10.1038/nm1172. [DOI] [PubMed] [Google Scholar]
- Chen G, Qian H, Starzl H, et al. Acute rejection is associated with antibodies to non-Gal antigens in baboons using Gal-knockout pig kidneys. Nat Med. 2005;11:1295–8. doi: 10.1038/nm1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YL, Kuwaki K, Dor FJ, et al. α1,3-Galactosyltransferase gene-knockout pig heart transplantation in baboons with survival approaching 6 months. Transplantation. 2005;80:493–500. doi: 10.1097/01.tp.0000181397.41143.fa. [DOI] [PubMed] [Google Scholar]
- Galili U. Induced anti-non gal antibodies in human xenograft recipients. Transplantation. 2012;93:11–6. doi: 10.1097/TP.0b013e31823be870. [DOI] [PubMed] [Google Scholar]
- Patience C, Takeuchi Y, Weiss RA. Infection of human cells by an endogenous retrovirus of pigs. Nat Med. 1997;3:282–6. doi: 10.1038/nm0397-282. [DOI] [PubMed] [Google Scholar]
- Cooper DK, Keogh AM, Brink J, et al. Report of the Xenotransplantation Advisory Committee of the International Society for Heart and Lung Transplantation: the present status of xenotransplantation and its potential role in the treatment of end-stage cardiac and pulmonary diseases. J Heart Lung Transplant. 2000;19:1125–65. doi: 10.1016/s1053-2498(00)00224-2. [DOI] [PubMed] [Google Scholar]
- Jamieson WR, Rosado LJ, Munro AI, et al. Carpentier–Edwards standard porcine bioprosthesis: primary tissue failure (structural valve deterioration) by age groups. Ann Thorac Surg. 1988;46:155–62. doi: 10.1016/s0003-4975(10)65888-2. [DOI] [PubMed] [Google Scholar]
- Konakci KZ, Bohle B, Blumer R, et al. α-Gal on bioprosthesis: xenograft immune response in cardiac surgery. Eur J Clin Invest. 2005;35:17–23. doi: 10.1111/j.1365-2362.2005.01441.x. [DOI] [PubMed] [Google Scholar]
- Lila N, McGregor CG, Carpentier S, et al. Gal knockout pig pericardium: new source of material for heart valve bioprostheses. J Heart Lung Transplant. 2010;29:538–43. doi: 10.1016/j.healun.2009.10.007. [DOI] [PubMed] [Google Scholar]
- McGregor CG, Carpentier A, Lila N, Logan JS, Byrne GW. Cardiac xenotransplantation technology provides materials for improved bioprosthetic heart valves. J Thorac Cardiovasc Surg. 2011;141:269–75. doi: 10.1016/j.jtcvs.2010.08.064. [DOI] [PubMed] [Google Scholar]
- Galili G, LaTemple DC. The natural anti-Gal antibody as a universal augmenter of autologous tumor vaccine immunogenicity. Immunol Today. 1997;18:281–5. doi: 10.1016/s0167-5699(97)80024-2. [DOI] [PubMed] [Google Scholar]
- Abdel-Motal UM, Wigglesworth K, Galili U. Mechanism for increased immunogenicity of vaccines that form in vivo immune complexes with the natural anti-Gal antibody. Vaccine. 2009;27:3072–82. doi: 10.1016/j.vaccine.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Abdel-Motal UM, Guay HM, Wigglesworth K, Welsh RM, Galili U. Increased immunogenicity of influenza virus vaccine by anti-Gal mediated targeting to antigen presenting cells. J Virol. 2007;81:9131–41. doi: 10.1128/JVI.00647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Motal UM, Wang S, Lu S, Wigglesworth K, Galili U. Increased immunogenicity of human immunodeficiency virus gp120 engineered to express Galα1-3Galβ1-4GlcNAc-R epitopes. J Virol. 2006;80:6943–51. doi: 10.1128/JVI.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Motal UM, Wang S, Lu S, Wigglesworth K, Galili U. Increased immunogenicity of HIV-1 p24 and gp120 following immunization with gp120/p24 fusion protein vaccine expressing α-gal epitopes. Vaccine. 2010;28:1758–65. doi: 10.1016/j.vaccine.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–24. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, van Loo P, Wedge DC, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Wick M, Schreiber H. Unique tumor antigens redefined as mutant tumor-specific antigens. Semin Immunol. 1996;8:289–93. doi: 10.1006/smim.1996.0037. [DOI] [PubMed] [Google Scholar]
- thor Straten P, Becker JC, Guldberg P, Zeuthen J. In situ T cells in melanoma. Cancer Immunol Immunother. 1999;48:386–95. doi: 10.1007/s002620050591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- Mlecnik B, Tosolini M, Kirilovsky A, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;29:610–8. doi: 10.1200/JCO.2010.30.5425. [DOI] [PubMed] [Google Scholar]
- LaTemple DC, Abrams JT, Zhang SU, Galili U. Increased immunogenicity of tumor vaccines complexed with anti-Gal: studies in knock out mice for α1,3galactosyltranferase. Cancer Res. 1999;59:3417–23. [PubMed] [Google Scholar]
- Deriy L, Ogawa H, Gao G, Galili U. In vivo targeting of vaccinating tumor cells to antigen presenting cells by a gene therapy method with adenovirus containing the α1,3galactosyltransferase gene”. Cancer Gene Ther. 2005;12:528–39. doi: 10.1038/sj.cgt.7700812. [DOI] [PubMed] [Google Scholar]
- Rossi GR, Mautino MR, Unfer RC, et al. Effective treatment of preexisting melanoma with whole cell vaccines expressing α(1,3)-galactosyl epitopes. Cancer Res. 2005;65:10555–61. doi: 10.1158/0008-5472.CAN-05-0627. [DOI] [PubMed] [Google Scholar]
- Deguchi T, Tanemura M, Miyoshi E, et al. Increased immunogenicity of tumor-associated antigen, mucin 1, engineered to express α-gal epitopes: a novel approach to immunotherapy in pancreatic cancer. Cancer Res. 2010;70:5259–69. doi: 10.1158/0008-5472.CAN-09-4313. [DOI] [PubMed] [Google Scholar]
- LaTemple DC, Henion TR, Anaraki F, Galili U. Synthesis of α-galactosyl epitopes by recombinant α1,3galactosyltransferase for opsonization of human tumor cell vaccines by anti-Gal. Cancer Res. 1996;56:3069–74. [PubMed] [Google Scholar]
- Manches O, Plumas J, Lui L, et al. Anti-Gal mediated targeting of human B lymphoma cells to antigen-presenting cells: a potential method for immunotherapy with autologous tumor cells. Haematologica. 2005;90:625–34. [PubMed] [Google Scholar]
- Galili U, Wigglesworth K, Abdel-Motal UM. Intratumoral injection of α-gal glycolipids induces xenograft-like destruction and conversion of lesions into endogenous vaccines. J Immunol. 2007;178:4676–87. doi: 10.4049/jimmunol.178.7.4676. [DOI] [PubMed] [Google Scholar]
- Galili U, Albertini M, Sondel P, Wigglesworth K, Sullivan M, Whalen G. In situ conversion of melanoma lesions into autologous vaccine by intratumoral injections of α-gal glycolipids. Cancers. 2010;2:773–93. doi: 10.3390/cancers2020773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Motal UM, Wigglesworth K, Galili U. Intratumoral injection of α-gal glycolipids induces a protective anti-tumor T cell response which overcomes Treg activity. Cancer Immunol Immunother. 2009;58:1545–56. doi: 10.1007/s00262-009-0662-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi GR, Unfer RC, Seregina T, Link CJ. Complete protection against melanoma in absence of autoimmune depigmentation after rejection of melanoma cells expressing α(1,3)galactosyl epitopes. Cancer Immunol Immunother. 2005;54:999–1009. doi: 10.1007/s00262-005-0667-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewski MM, DeAngelis RA, Benencia F, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–35. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen GF, Sullivan M, Piperdi B, Wasseff W, Galili U. Cancer immunotherapy by intratumoral injection of α-gal glycolipids. Anticancer Res. 2012;32:3861–8. [PubMed] [Google Scholar]
- Galili U, Wigglesworth K, Abdel-Motal UM. Accelerated healing of skin burns by anti-Gal/α-gal liposomes interaction. Burns. 2010;36:239–51. doi: 10.1016/j.burns.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Hurwitz Z, Ignotz R, Lalikos J, Galili U. Accelerated porcine wound healing with α-Gal nanoparticles. Plast Reconstr Surg. 2012;129:242–51. doi: 10.1097/PRS.0b013e31823aebb1. [DOI] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;41:738–40. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005;15:599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–21. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Elkon KB, Silverman GJ. Naturally occurring autoantibodies to apoptotic cells. Adv Exp Med Biol. 2012;750:14–26. doi: 10.1007/978-1-4614-3461-0_2. [DOI] [PubMed] [Google Scholar]
- Minatoguchi S, Takemura G, Chen XH, et al. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation. 2004;109:2572–80. doi: 10.1161/01.CIR.0000129770.93985.3E. [DOI] [PubMed] [Google Scholar]
- Dewald O, Zymek P, Winkelmann K, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–9. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- Yano T, Miura T, Whittaker P, et al. Macrophage colony-stimulating factor treatment after myocardial infarction attenuates left ventricular dysfunction by accelerating infarct repair. J Am Coll Cardiol. 2006;47:626–34. doi: 10.1016/j.jacc.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Dray C, Rougon G, Debarbieux F. Qunatitative analysis by in vivo imaging of the dynamics of vascular and axonal networks in injured mouse spinal cord. Proc Natl Acad Sci USA. 2009;106:9459–64. doi: 10.1073/pnas.0900222106. [DOI] [PMC free article] [PubMed] [Google Scholar]