

Abstract
The genetic syndrome Fanconi anemia (FA) is characterized by aplastic anemia, cancer predisposition and hypersensitivity to DNA interstrand crosslinks (ICLs). FA proteins (FANCs) are thought to work in pathway(s) essential for dealing with crosslinked DNA. FANCs interact with other proteins involved in both DNA repair and S-phase checkpoint such as BRCA1, ATM and the RAD50/MRE11/NBS1 (RMN) complex. We deciphered the previously undefined pathway(s) leading to the ICLs-induced S-phase checkpoint and the role of FANCs in this process. We found that ICLs activate a branched pathway downstream of the ATR kinase: one branch depending on CHK1 activity and the other on the FANCs–RMN complex. The transient slow-down of DNA synthesis was abolished in cells lacking ATR, whereas CHK1-siRNA-treated cells, NBS1 or FA cells showed partial S-phase arrest. CHK1 RNAi in NBS1 or FA cells abolished the S-phase checkpoint, suggesting that CHK1 and FANCs/NBS1 proteins work on parallel pathways. Furthermore, we found that ICLs trigger ATR-dependent FANCD2 phosphorylation and FANCD2/ATR colocalization. This study demonstrates a novel relationship between the FA pathway(s) and the ATR kinase.
Keywords: DNA repair, Fanconi anemia, S-phase checkpoint
Introduction
To cope with DNA damage efficiently, cells activate a complex response that leads to DNA repair, cell cycle arrest and cell death. Failure to correctly activate and coordinate these different pathways involved in the response to DNA damage results in increased cell sensitivity to genotoxic stress and, in the long term, in an increased risk of cancer (Paulovich et al, 1997). Despite many recent findings concerning cell cycle checkpoints, their components and interactions, their relationships with other DNA damage response pathways remain poorly characterized. This is particularly true for DNA interstrand crosslinks (ICLs). ICLs are lesions introduced into DNA by many carcinogens and antitumoral drugs such as photoactivated psoralens, cisplatinum, mitomycin C (MMC) and derivatives of nitrogen mustard. ICLs covalently join both DNA strands, preventing their separation and blocking DNA and RNA polymerases. Because of this block during replication or transcription, ICLs are highly toxic for cells. Although still poorly defined, the repair of ICLs in mammalian cells is thought to be based on the combined intervention of nucleotide excision repair and recombination (De Silva et al, 2000; Dronkert and Kanaar, 2001; McHugh et al, 2001). Moreover, it has been proposed that ICL induction, at least by photoactivated psoralens, does not activate the G2 or the G1 checkpoint in human cells, suggesting that crosslinked DNA can be tolerated until they are encountered by a DNA replication fork (Akkari et al, 2000). Consequently, the S-phase checkpoint could have a major role as coordinator of the cellular response to ICLs. The S-phase checkpoint monitors progression through the S-phase, slowing down rapidly and transiently ongoing DNA synthesis in the presence of DNA lesions, allowing time to repair. The S-phase checkpoint response activated by ionizing radiation (IR) or triggered by DNA damage at the replication fork is more or less defined (Bartek and Lukas, 2001). How the S-phase checkpoint works in response to crosslinked DNA remains to be elucidated.
A particular class of human ICL hypersensitive mutants comprises cells carrying mutations in one of the Fanconi anemia (FA) genes. FA is a rare human autosomal recessive syndrome characterized by aplastic anemia, cancer predisposition, genetic instability, hypersensitivity to ICL agents and, to a lesser extent, IR (D'Andrea and Grompe, 1997, 2003; Ahmad et al, 2002). Eight responsible genes, FANCA, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9 and FANCL, have been cloned (Strathdee et al, 1992; Lo Ten Foe et al, 1996; de Winter et al, 1998, 2000a, 2000b; Timmers et al, 2001; Howlett et al, 2002; Meetei et al, 2003a). The FANC genes display little, if any, homology to each other or to other known genes. Although the functions of the FA proteins are still largely unknown, they have been shown to form a complex and are thought to work in a linear pathway that is essential for dealing with crosslinked DNA (D'Andrea and Grompe, 2003). Several studies have shown that the FA pathway is involved in additional networks controlling genome stability in human cells as demonstrated by the interaction between FA proteins and other caretaker gene products such as BRCA1, ATM, XP-F, BLM or the RAD50/MRE11/NBS1 (RMN) complex (Garcia-Higuera et al, 2001; Nakanishi et al, 2002; Pichierri et al, 2002; Taniguchi et al, 2002; Meetei et al, 2003b; Sridharan et al, 2003). These proteins are involved in DNA repair, S-phase checkpoint control and apoptosis, which are altered in FA (Dutrillaux et al, 1982; Rosselli et al, 1995; Escarceller et al, 1997; Thyagarajan and Campbell, 1997; Escarceller et al, 1998; Haneline et al, 1998; Guillouf et al, 1999; Centurion et al, 2000; Guillouf et al, 2000; Sala-Trepat et al, 2000). However, despite extensive analysis, the biochemical events under the control of the FA proteins remain elusive (Buchwald and Moustacchi, 1998; Ahmad et al, 2002).
In this work, we decided to decipher the yet undefined molecular pathway(s) involved in S-phase checkpoint activation by crosslinked DNA and to study the role, if any, of FANC proteins in that DNA damage inducible process. We show here that ICLs induce two pathways, both under the control of the ATR kinase, whose cooperation leads to S-phase checkpoint activation. The CHK1 and NBS1–FANCD2 pathways are activated by ATR, but control downstream independent events. We also demonstrate that the ATR–NBS1–FANCD2 checkpoint requires the core FANC complex (formed by the nuclear assembling of FANCA, FANCC, FANCG, FANCE, FANCF and FANCL) to assemble NBS1 or FANCD2 in colocalized nuclear foci. The formation of nuclear foci is independent of the ATR-induced NBS1 or FANCD2 phosphorylation.
Results
ATR kinase activates S-phase checkpoint in response to ICLs
The S-phase checkpoint is regulated by a complex network of proteins orchestrated by ATM or ATR protein kinases (Abraham, 2001). Loss of ATM activity by mutations in the ATM gene, which are responsible for ataxia telangiectasia (A-T) syndrome, leads to a defect in the S-phase checkpoint that is normally activated in response to IR. This defect is known as radioresistant DNA synthesis (RDS) and it is associated with radiosensitivity (Falck et al, 2002). Inactivation of the ATR kinase results in the impairment of the S-phase checkpoint function following replication fork stalling (Hekmat-Nejad et al, 2000; Heffernan et al, 2002; Costanzo et al, 2003).
To determine the kinase involved in the ICL-dependent inhibition of S-phase progression, we looked at the inhibition of DNA synthesis in wild-type (WT) cells, A-T derived cells, cells conditionally expressing a dominant-negative kinase-dead form of ATR (ATRkd) (Cliby et al, 1998) or ATR-siRNA-transfected cells. WT cells treated with ICL agents rapidly and transiently slow down DNA synthesis (Figure 1A). Similar to previously published data (Jaspers et al, 1982), ICL-dependent S-phase inhibition in A-T cells was similar to that observed in the ATM/ATR WT human MRC5 fibroblasts or GM3657 lymphoblasts (Figure 1A). On the contrary, the induction of the ATRkd protein by doxycyclin (ATRkd+Dox, Figure 1B) as well as the ectopic expression of ATR siRNA (Figure 1C) greatly impaired the cellular S-phase delay in response to photoactivated psoralen (8-MOP) (Figure 1A).
Figure 1.
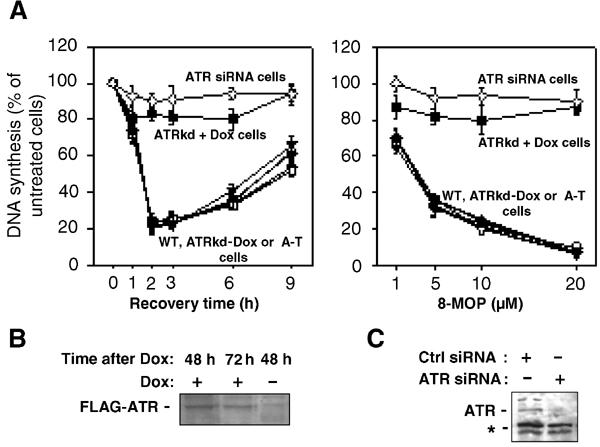
ATR is involved in the S-phase checkpoint induced by ICL agents. (A) Replicative DNA synthesis was assessed by 3H–T incorporation at various time points following exposure to 8-MOP (10 μM)+UVA (10 kJ/m2) (left panel) or 3 h post-treatment after various doses of 8-MOP photoactivated by UVA (10 kJ/m2) (right panel) in GM3657 (▾), MRC5 (•), ATM defective cells (□), ATRkd-Dox (○) control cells, ATRkd-expressing cells (▪), i.e. ATRkd+doxycyclin 48 h before crosslinking treatment, or HeLa cells transfected with ATR siRNA (▿) 48 h before the genotoxic treatment. (B) Induction by doxycyclin (1 μg/ml) of the Flag-tagged ATRkd protein in ATRkd-transfected cells as analyzed by Western blot with an anti-Flag antibody. (C) Inhibition of ATR expression by ATR-siRNA transfection. *Indicates aspecific bands recognized by the α-ATR antibody used.
These results indicate that the ATR kinase is necessary for the activation of the signaling pathway leading to inhibition of DNA synthesis in the presence of crosslinked DNA.
CHK1 and NBS1 are independent mediators of ATR signaling
ATR phosphorylates and activates CHK1 and NBS1, two main proteins involved in S-phase checkpoint activation (Abraham, 2001). We sought to determine the involvement of these proteins in ICL-dependent S-phase delay.
To analyze CHK1 involvement in S-phase checkpoint activation after ICL induction, we used siRNA transfection to silence CHK1 expression in HeLa cells (Figure 2A). As reported in Figure 2B, the absence of CHK1 protein in 8-MOP+UVA-treated cells led to a partial deficiency in the S-phase checkpoint. Cells derived from Nijmegen breakage syndrome (NBS) were then used to determine the involvement of NBS1 in ICL-dependent S-phase checkpoint. The ICL-dependent inhibition of DNA synthesis observed in NBS cells was comparable to that in CHK1-siRNA-transfected cells (Figure 2B). Since CHK1- and NBS1-deficient cells showed a similar defect in S-phase checkpoint activation, which is intermediate between ATR-proficient and ATR-deficient cells, we investigated whether CHK1 and NBS1 participate in the same or in separate pathways leading to ICL-dependent S-phase delay. NBS cells were transfected with CHK1 siRNA and DNA synthesis was monitored after ICL induction. Inhibition of CHK1 expression in cells deficient in NBS1 resulted in a total loss of the S-phase checkpoint (Figure 2B). In conclusion, blockage of CHK1- or NBS1-dependent events partially affects S-phase delay in response to ICLs, while concomitant inactivation of both CHK1 and NBS1 abolishes inhibition of DNA synthesis induced by ICL treatment, resulting in a loss of the S-phase checkpoint similar to that observed in ATRkd-expressing cells.
Figure 2.
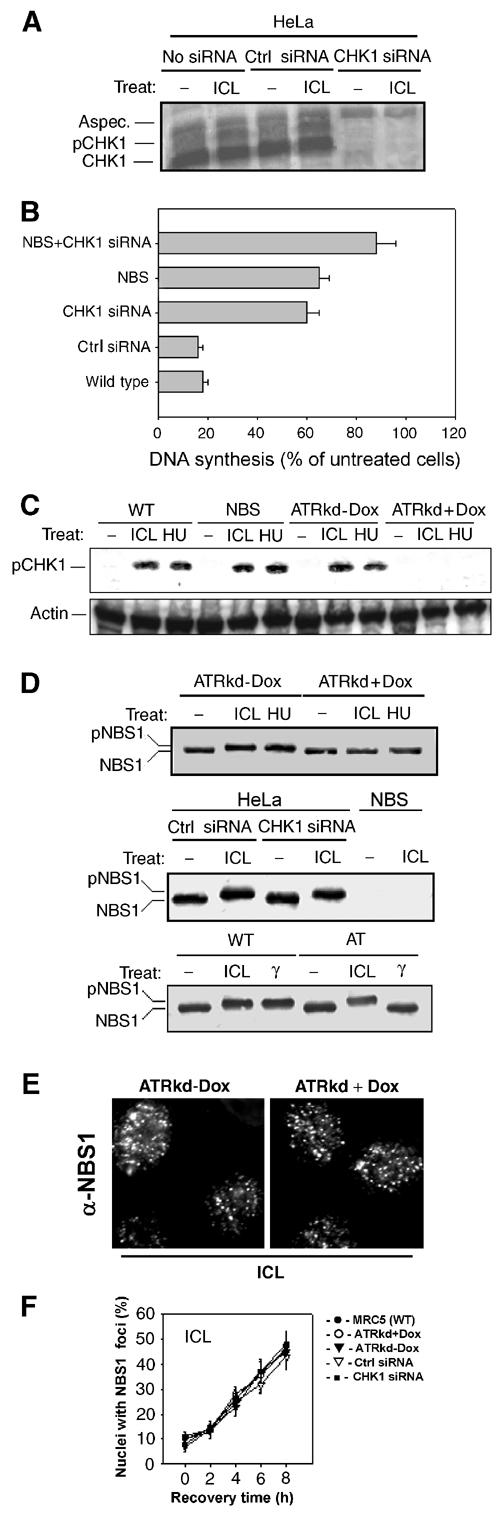
CHK1 and NBS1 are independently involved in ICL-dependent S-phase checkpoint downstream of ATR. (A) CHK1-siRNA transfection induces inhibition of CHK1 expression in HeLa cells. Protein extracts from mock-transfected (No siRNA), Ctrl siRNA or CHK1-siRNA-transfected cells were analyzed by Western blot with an anti-CHK1 antibody. (B) Replicative DNA synthesis in WT, NBS and/or CHK1-siRNA-inhibited cells 3 h after crosslinking treatment (10 μM 8-MOP+10 kJ/m2 UVA). (C) CHK1 phosphorylation in response to ICLs (10 μM 8-MOP+10 kJ/m2 UVA, 3 h recovery) or HU (2 mM, 6 h of treatment) exposure in WT, NBS, ATRkd-Dox or ATRkd-expressing cells (ATRkd+Dox) was assessed by Western blot with anti-phospho-CHK1 antibody directed against phospho-S345. (D) Analysis of NBS1 phosphorylation in response to ICLs (10 μM 8-MOP+10 kJ/m2 UVA, 3 h recovery) or HU (2 mM, 6 h of treatment) exposure in WT (ATRkd-Dox) and ATRkd-expressing (ATRkd+Dox) cells (first panel), in CHK1-siRNA-treated cells (middle panel) or in response to ICLs or IR (5 Gy) in control and A-T cells (lower panel), as observed by Western blot with an anti-NBS1 antibody. (E) Representative images showing ICL-induced NBS1 assembly in nuclear foci in cells expressing or not the inactive form of ATR. Images were taken 3 h after treatment with 8-MOP (10 μM+10 kJ/m2 UVA). (F) Quantification of NBS1 foci in WT, ATRkd-expressing and CHK1-siRNA-transfected cells following exposure to photoactivated 8-MOP.
To further extend our observations, we looked at CHK1 and NBS1 phosphorylation in response to ICLs since NBS1 and CHK1 phosphorylation are correlated with the activation of the S-phase (Abraham, 2001). As reported in Figure 2C, using an anti-phospho-CHK1-specific antibody, photoactivated psoralens induced CHK1 phosphorylation in all ATR WT expressing cells examined, i.e. MRC5, NBS and the uninduced ATRkd-Dox cells. In contrast, the pCHK1 band was lost in cells expressing the ATRkd protein (ATRkd+Dox) (Figure 2C). Similar to previous observations (Pichierri et al, 2002), ICL-dependent NBS1 phosphorylation was analyzed by the induction of a mobility shift of the band recognized by the NBS1-specific antibody. The mobility shift was clearly lost in ATRkd-expressing cells (ATRkd+Dox) but normally induced in the absence of CHK1 expression (Figure 2D). However, in A-T cells, NBS1 migration is clearly retarded following induction of ICLs while remaining unchanged in response to IR, suggesting that NBS1 phosphorylation following exposure to photoactivated psoralens is independent of ATM.
Even though ICL-dependent NBS1 phosphorylation was impaired in ATRkd-expressing cells, NBS1 subnuclear relocalization was similar in WT, ATRkd-expressing and CHK1-siRNA-transfected cells (Figure 2E and F), suggesting that NBS1 subnuclear assembly is independent of both ATR and CHK1.
All of these observations imply that two parallel pathways, both under the control of ATR kinase, are activated in response to ICLs and cooperate in the transient inhibition of DNA synthesis: one pathway dependent on CHK1 and the other dependent on NBS1 activation. Moreover, our data clearly indicate that activation of the ATR–NBS1 branch of the ICL S-phase checkpoint involves two molecular steps. Indeed, crosslinked DNA induces an ATR-independent NBS1 relocalization in nuclear foci and an ATR-dependent NBS1 phosphorylation.
Fanconi anemia proteins are involved in the ATR–NBS1 branch of the ICL-dependent S-phase checkpoint pathway
It has previously been reported that several of the FA complementation groups show a defect in the delay of S-phase progression in the presence of ICLs (Centurion et al, 2000; Sala-Trepat et al, 2000). We sought to understand how such a defect could be correlated to the data reported here. We analyzed inhibition of DNA synthesis in FA cells mutated in FANC core complex genes or mutated in FANCD2, the downstream effector of the FA pathway (Garcia-Higuera et al, 2001; D'Andrea and Grompe, 2003).
As reported in Figure 3A, all FA cell lines examined showed a reduced inhibition of DNA synthesis in response to 8-MOP+UVA treatment. The extent of the S-phase checkpoint defect in FA cells was similar in all of the FA complementation groups analyzed and was comparable to that observed in CHK1- or NBS1-deficient cells. This defect was reversed by the ectopic expression of the corresponding FA gene (Figure 3B). These results indicate that both FANC core complex and FANCD2 activities are involved in the ICL-dependent S-phase checkpoint control.
Figure 3.
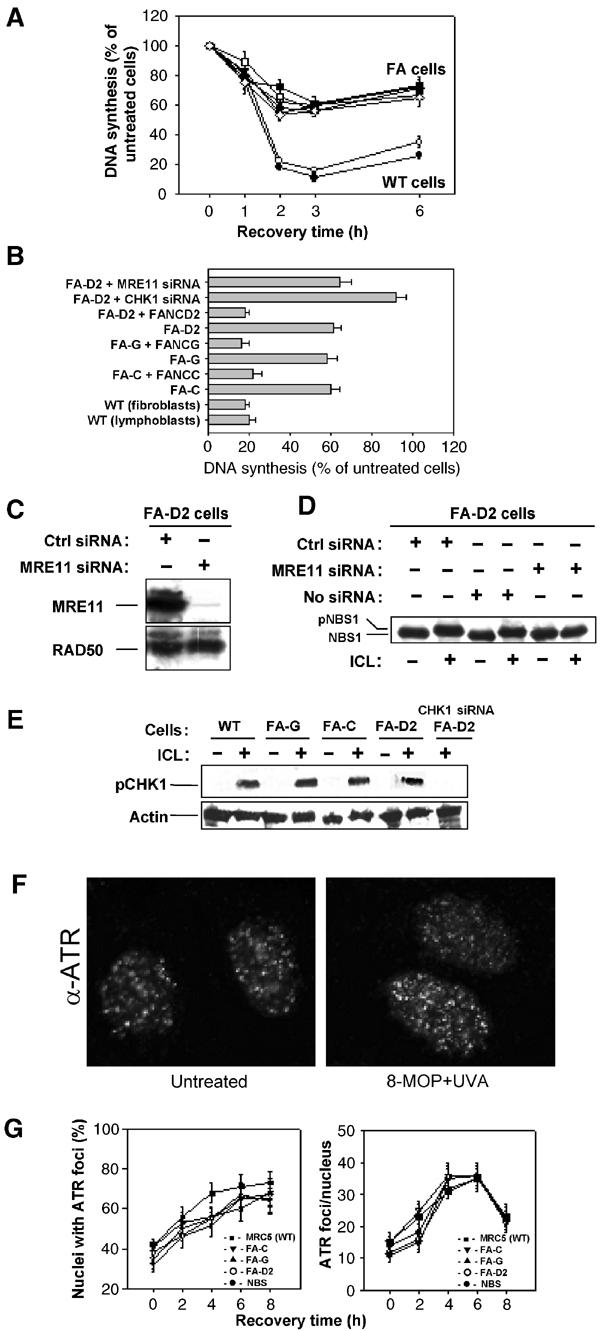
FA proteins are involved in S-phase checkpoint activation. (A) Replicative DNA synthesis was measured as a function of time after exposure to 10 μM 8-MOP+10 kJ/m2 UVA in WT, GM3657 (□, lymphoblasts), MRC5 (•, fibroblasts) and FA cells from A, C, G and D2 complementation groups. (B) Replicative DNA synthesis was measured 3 h after exposure to 10 μM 8-MOP+10 kJ/m2 UVA in WT, FA cells from C, G and D2 complementation groups, ectopically corrected FA cells or in FA-D2 cells transfected with CHK1 siRNA. (C) Inhibition of MRE11 expression by MRE11-siRNA transfection in FA cells. Equal loading of proteins was demonstrated by RAD50 immunoblotting. (D) Consequence of MRE11 interference on NBS1 phosphorylation following exposure to photoactivated psoralens. (E) Analysis of CHK1 phosphorylation in photoactivated (10 kJ/m2 UVA) 8-MOP (10 μM) treated FA cells by Western blot with a specific anti-phospho-CHK1 antibody directed against phospho-S345. (F, G) Formation and quantitation of ATR foci in response to 10 μM 8-MOP+10 kJ/m2 UVA treatment as evaluated by immunostaining of cells with an anti-ATR antibody in WT, NBS1 or FANC defective cells.
Our data (Figure 2) indicate the existence of an ATR–CHK1 and an ATR–NBS1 pathway in the ICL-dependent S-phase checkpoint. Moreover, we have recently reported that the integrity of the FANC core complex is specifically required for RAD50/MRE11/NBS1 relocalization and NBS1 phosphorylation in response to ICL agents (Pichierri et al, 2002, see also Figure 6C). These data, together with the recent results from D'Andrea's group demonstrating a functional link between FANCD2 and NBS1 (Nakanishi et al, 2002), strongly suggest that FANC proteins participate in the ATR–NBS1 branch of the ICL-dependent S-phase checkpoint. Accordingly, MRE11-siRNA transfection in FA-D2 cells, resulting in MRE11 depletion (Figure 3C) and loss of NBS1 phosphorylation (Figure 3D), did not modify the S-phase defect (Figure 3B, first histogram). Importantly, cells with a concomitant interference in both CHK1 and FANCD2 activity (by CHK1-siRNA transfection in FA-D2 cells) showed a complete loss of S-phase checkpoint activation (Figure 3B, second histogram), demonstrating that CHK1 is involved in a pathway different from FANCD2 to control the S-phase checkpoint. Furthermore, ICL-treated FA cells were able to phosphorylate CHK1 (Figure 3E). Since ICL-induced CHK1 phosphorylation was dependent on ATR activation (Figure 2C), our data also suggest that ATR could be normally activated in FA-deficient cells. Indeed, the formation of ATR nuclear foci following exposure to photoactivated psoralen was similar in WT, FA- and NBS1-deficient cells (Figure 3F and G).
Figure 6.
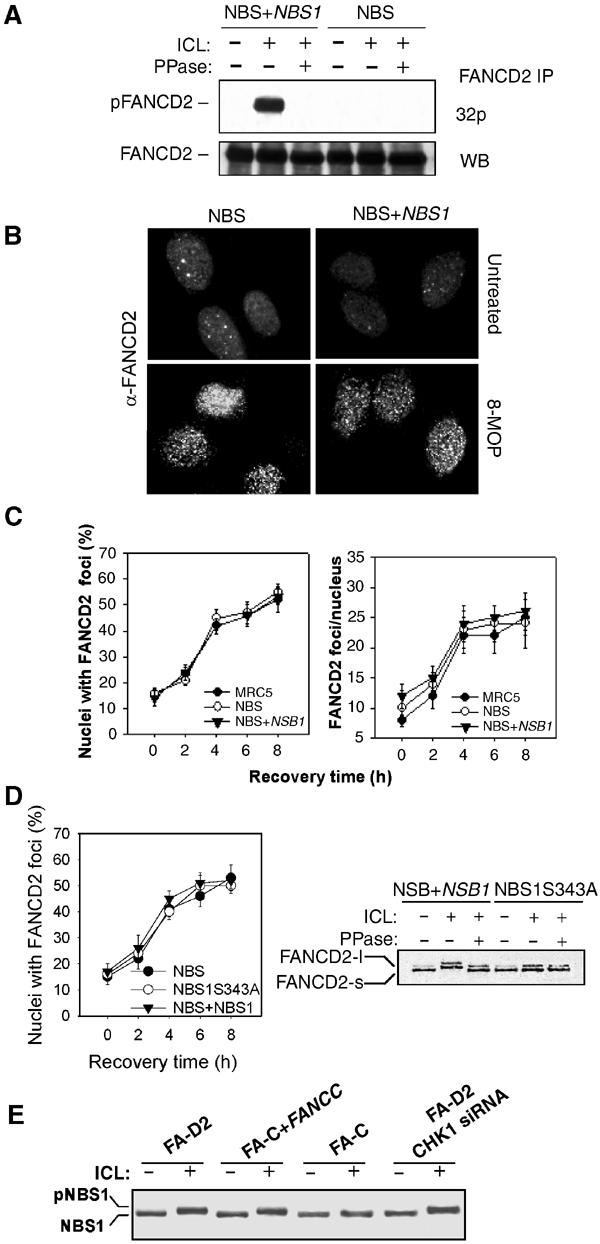
NBS1 is involved in FANCD2 phosphorylation. (A) FANCD2 phosphorylation was assessed in NBS and NBS-corrected cells 3 h after treatment with 8-MOP (10 μM)+UVA (10 kJ/m2). Radiolabeled cell extracts were analyzed by immunoprecipitation, phosphatase treatment, SDS–PAGE and autoradiography. Western blot with an anti-FANCD2 antibody demonstrated the equal loading of each slot. (B, C) FANCD2 foci formation and quantitation in NBS and NBS-corrected cells 6 h after treatment with 8-MOP (10 μM)+UVA (10 kJ/m2) as observed by immunostaining of the cells with an anti-FANCD2 antibody. (D) FANCD2 foci formation in the NBS1S343A cell line in response to 8-MOP (10 μM)+UVA (10 kJ/m2). (E) Phosphorylation of NBS1 analyzed by Western blot 3 h after ICL treatment (8-MOP (10 μM)+UVA (10 kJ/m2)) in FA-D2, FA-C, FA-C corrected cells or FA-D2 cells with CHK1 si RNA.
Together, our results demonstrate that FA proteins are involved in an S-phase checkpoint response that is independent of CHK1 phosphorylation and suggest that FA proteins participate in the ATR–NBS1 branch of the ICLs-dependent S-phase checkpoint.
FANCD2 is phosphorylated in response to photoactivated 8-MOP
FANCD2 protein exists as two isoforms, a faster migrating form (FANCD2-s) and a slower migrating monoubiquitinated form (FANCD2-l). Monoubiquitination of FANCD2 is induced by DNA damage, requires the FANC core complex to be observed and correlates with FANCD2 relocalization in discrete nuclear foci (Garcia-Higuera et al, 2001). Moreover, in response to IR, FANCD2 is also phosphorylated by ATM and this event is correlated with S-phase checkpoint activation (Taniguchi et al, 2002). Thus, to determine whether phosphorylation of FANCD2 could also be induced by ICLs, FANCD2 was immunoprecipitated from 32P metabolically labeled cells exposed to IR or ICLs. Immunoprecipitates were analyzed by SDS–PAGE followed by autoradiography (Figure 4A). A clear signal corresponding to FANCD2 was detected in immunoprecipitates from treated cells, and the signal was lost in immunoprecipitates exposed to phosphatase. The absence of signal in untreated cells validates the induced nature of the phosphorylation in DNA damaged cells. In conclusion, it appears that FANCD2 phosphorylation is induced by exposure to both IR and ICL agents.
Figure 4.
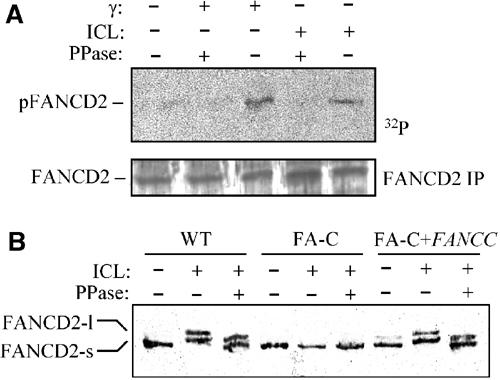
FANCD2 is phosphorylated after ICL treatment. (A) FANCD2 phosphorylation analyzed in HeLa cells exposed to 20 Gy of IR or 8-MOP (10 μM)+UVA (10 kJ/m2) and harvested 3 h later by immunoprecipitation of in vivo radiolabeled proteins, phosphatase treatment, SDS–PAGE and autoradiography. Western blot analysis with an anti-FANCD2 antibody demonstrates the equal loading of each slot. (B) FANCD2 phosphorylation was assessed in FA-C and FA-C corrected cells after treatment with 8-MOP (10 μM)+UVA (10 kJ/m2) by immunoprecipitation, phosphatase treatment and Western blot with an anti-FANCD2 antibody.
FANCD2 monoubiquitination is dependent on WT FANC core complex activity (Garcia-Higuera et al, 2001). We sought to determine whether ICL-induced FANCD2 phosphorylation also requires a functional FA pathway. As reported in Figure 4B, phosphatase treatment of FANCD2 immunoprecipitated from 8-MOP+UVA-treated FA-C cells did not modify the mobility of the FANCD2-S isoform in an SDS–PAGE gel. Ectopic expression of FANCC in FA-C cells restored both the presence of the monoubiquitinated FANCD2 isoform and the phosphorylation of the proteins, as demonstrated by the modification of the mobility of both FANCD2 isoforms caused by phosphatase treatment of the immunoprecipitates (Figure 4B).
Together, our data indicate that FANCD2 is phosphorylated in response to ICLs and that this modification relies upon the presence of a functional FA core complex.
FANCD2 is phosphorylated in an ATR–NBS1 pathway-dependent manner in response to photoactivated 8-MOP
Having demonstrated that the FA proteins are part of the ATR–NBS1 branch of the pathway controlling S-phase checkpoint activation in the presence of crosslinked DNA and that FANCD2 is phosphorylated in response to ICL induction, we sought to determine whether FANCD2 is phosphorylated in an ATR-dependent manner and whether NBS1 is involved.
We evaluated FANCD2 phosphorylation in ATM/ATR WT cells, in A-T derived cells and in cells with a disrupted ATR activity (ATRkd+Dox) by in vivo labeling of the protein with 32P, immunoprecipitation with anti-FANCD2 antibodies, SDS–PAGE analysis and autoradiography (Figure 5A). In response to IR or photoactivated 8-MOP, FANCD2 phosphorylation was observed in ATM/ATR WT cells. As previously described, in A-T cells, the IR-dependent FANCD2 phosphorylation was absent (Figure 5A, Taniguchi et al, 2002) but ICL-dependent FANCD2 phosphorylation was present. In contrast, in ATRkd-expressing cells, ICL-dependent FANCD2 phosphorylation was lost. Moreover, immunoprecipitated FANCD2 was recognized by a phospho-Ser/Thr-Gln showing that the protein is modified at a specific ATR phosphorylation site in response to ICL agent exposure (Figure 5B). Interestingly, similar to what has been observed following IR treatment of A-T cells (Taniguchi et al, 2002), unphosphorylated FANCD2 was able to aggregate in nuclear foci following exposure to ICLs (Figure 5C and D).
Figure 5.
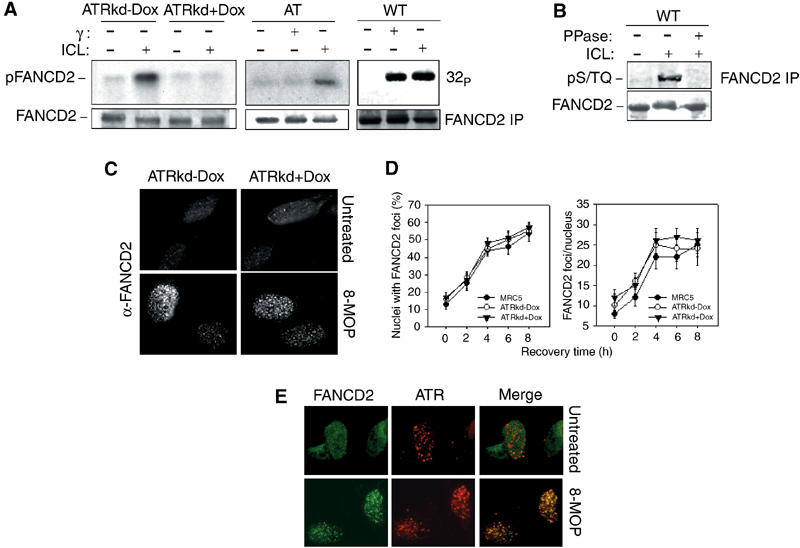
ATR is involved in ICL-dependent FANCD2 phosphorylation. (A) In vivo phosphoradiolabeled FANCD2 protein was immunoprecipitated from WT (HeLa), A-T derived, ATRkd-Dox or ATRkd-expressing (ATRkd+Dox) cells in response to 8-MOP (10 μM)+UVA (10 kJ/m2) or 20 Gy of IR. The FANCD2 immunoprecipitates were prepared 3 h after treatment and analyzed by SDS–PAGE and autoradiography. Western blotting with anti-FANCD2 antibodies is reported to show equal loading of the proteins into the gel. (B) FANCD2 was immunoprecipitated from ICL-treated HeLa cells and analyzed by an anti-pS/TQ specific antibody. (C, D) FANCD2 foci formation and quantitation in WT and ATR inactivated cells after ICL treatment (8-MOP (10 μM)+UVA (10 kJ/m2)) as observed by immunostaining of the cells 6 h after treatment with an anti-FANCD2 antibody. (C) Formation and colocalization of FANCD2 and ATR foci in response to 8-MOP (10 μM)+UVA (10 kJ/m2) exposure analyzed by immunostaining of cells with an anti-FANCD2 antibody (green) and an anti-ATR antibody (red). A representative nucleus obtained from cells fixed before and 6 h after treatment is displayed.
To determine if activated FANCD2 colocalizes with ATR, we carried out FANCD2 and ATR immunolabeling in WT cells following the generation of crosslinked DNA. As shown in Figure 5E, a clear and large colocalization of FANCD2 and ATR kinase was observed in ICL-treated cells.
Finally, we verified whether FANCD2 phosphorylation is related to NBS1 activity. To do this, we looked at FANCD2 phosphorylation and focus formation in NBS cells. As reported in Figure 6A, in the absence of NBS1, FANCD2 was not phosphorylated. Ectopic expression of WT NBS1 cDNA in NBS cells restored FANCD2 phosphorylation. In contrast, the ICL-dependent FANCD2 assembly in nuclear foci was observed in both NBS as well as in NBS-corrected cells (Figure 6B and C). The ICL-dependent FANCD2 phosphorylation, but not its subnuclear relocalization (Figure 6D), was also affected in an NBS cell line ectopically expressing the NBS1S343A mutant protein that is unable to be phosphorylated but capable of relocalizing with RAD50 and MRE11 in nuclear foci (Gatei et al, 2000). In contrast, FANCD2 or CHK1 appeared unnecessary for both NBS1 phosphorylation and relocalization in response to crosslinked DNA (Figure 6E and data not shown). Together, these observations suggest that NBS1 phosphorylation is required for FANCD2 phosphorylation, but is dispensable for its assembly in nuclear foci. In addition, our data suggest the existence of an ATR–NBS1–FANCD2 pathway, which is a part of the S-phase checkpoint in response to ICL DNA damage.
Discussion
In this study, we tackled the problem of S-phase checkpoint control in response to ICLs. We established that DNA damage induced by ICLs activates two parallel molecular pathways, both under the control of the ATR kinase, which cooperate to attain the full activation of the S-phase checkpoint. The first branch is represented by the ATR–CHK1 pathway and the second by the ATR–NBS1–FANCD2 pathway. Furthermore, our data show that the activation of the second branch requires three separate molecular steps: (i) a FANC core complex-dependent (Pichierri et al, 2002) and ATR-independent assembly of the RMN complex and of the FANCD2 protein in subnuclear foci; (ii) an ATR-mediated NBS1 phosphorylation; and (iii) an ATR-mediated/NBS1-dependent FANCD2 phosphorylation event. Our observations, combined with other data presented in the literature, can be summarized in the model reported in Figure 7.
Figure 7.
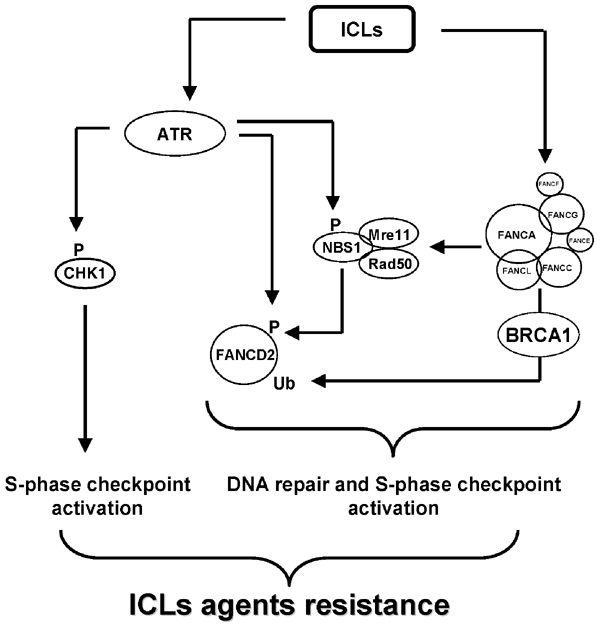
A model summarizing the links between the players involved in ICL-dependent S-phase checkpoint.
ATM and ATR kinases are two members of the phosphoinositol kinase family known to be at the top of a signal transduction cascade induced by DNA lesions which activate, by phosphorylation events, DNA repair, checkpoint and apoptotic proteins (Abraham, 2001). Mutations in ATM are responsible for the A-T syndrome, in which neuronal and immunological deficiencies are associated with cancer predisposition. ATM mutant cells are defective in DNA damage checkpoint, apoptosis and extremely sensitive to IR or agents causing DNA double-strand breaks (Lavin and Shiloh, 1997; Rotman and Shiloh, 1997; Shiloh, 1997). Less is known about ATR, essentially because of a lack of ATR mutant cells. Recently, a splice mutation in ATR has been shown to be associated with Seckel syndrome, an autosomal recessive disorder characterized by dwarfism and mental retardation (O'Driscoll et al, 2003). Although the cells carrying the ATR splice mutation showed a similar IR sensitivity to normal cells, a significant increase in cellular sensitivity to UV irradiation and the ICL-inducing agent MMC was observed (O'Driscoll et al, 2003). Additionally, previous studies using overexpression of a dominant kinase-deficient ATR protein (ATRkd), which abrogates WT activity, have demonstrated a defective cell cycle arrest after a block of replication fork progression. The results of this study suggest a role for ATR in activating the DNA damage checkpoint in response to other lesions than those on which ATM acts (Hekmat-Nejad et al, 2000; Abraham, 2001; Heffernan et al, 2002; Costanzo et al, 2003). In this work, we demonstrated that ATR is the kinase involved in the S-phase checkpoint activated by ICL treatment. Using a genetic approach, we have shown that, following its activation, ATR phosphorylates CHK1 and NBS1. Activation of these two parallel pathways allows full inhibition of DNA synthesis, resulting in S-phase delay. These observations are reminiscent of the IR-induced S-phase checkpoint, which involves ATM–CHK2–CDC25A and ATM–NBS1 parallel pathways (Falck et al, 2002). Further studies are needed to understand how ATR is assembled and activated in response to ICLs. Our data exclude a role for CHK1, NBS1 and FANC proteins as ATR activators. A recent work suggests that RPA could play a role in ATR activation (Zou and Elledge, 2003).
NBS1 is the product of the gene mutated in Nijmegen breakage syndrome. NBS1 mutations are associated with cancer predisposition, chromosome fragility, hypersensitivity to IR and RDS (Shiloh, 1997). Rare NBS patients present bone marrow failure and AML (Resnick et al, 2002). NBS1 associates with RAD50 and MRE11 in nuclear foci induced by the presence of DNA double-strand breaks. The RMN complex is thought to act in both DNA repair and S-phase checkpoint (Petrini, 1999). In response to DNA damage, NBS1 is phosphorylated at S343 in an ATM- or ATR-dependent manner, and this event is required for the S-phase checkpoint but is unnecessary for foci formation (Gatei et al, 2000; Lim et al, 2000; Wu et al, 2000; Zhao et al, 2000). Recently published work from our lab as well as D'Andrea's group has established a connection between the FA and RMN complex. We have reported that RMN relocalization in nuclear foci after ICL induction requires the FANC core complex activity (Pichierri et al, 2002), whereas D'Andrea's group has demonstrated an interaction between NBS1 and FANCD2 (Nakanishi et al, 2002). Moreover, the paper by Nakanishi and colleagues revealed that some NBS cell lines present a cellular sensitivity to MMC similar to that of FA cells, whereas chromosomal fragility in response to MMC was about 50% of that observed in FA cells. Interestingly, an S343A NBS1 mutant, which forms nuclear foci but lacks a phosphorylation site, did not show an MMC-dependent chromosome breakage sensitivity (Nakanishi et al, 2002). Together with the data presented here, it appears that NBS1 acts in the FANC pathway, between the FANC core complex and FANCD2. The FANC core complex is required for both NBS1 and FANCD2 relocalization in response to ICLs and NBS1 is required for FANCD2 phosphorylation. Surprisingly, in spite of the direct interaction and colocalization of NBS1 and FANCD2, their ability to form foci is mutually independent. What is the function of the NBS1/FANCD2 interaction? Although FANCD2 and ATR colocalized, FANCD2 foci formation is independent of ATR activity. Since phosphorylation of FANCD2 depends on both ATR and NBS1, we hypothesized that NBS1 acts as an adaptor for ATR-mediated FANCD2 phosphorylation in response to ICLs. It has been suggested that, in response to IR, NBS1 acts upstream of ATM, working as a sensor of DNA damage (Mirzoeva and Petrini, 2001), activating ATM which in turn phosphorylates NBS1, whose presence is required for downstream events, supporting a role as adaptor for NBS1. This proposed double sensor/adaptor role of NBS1 in response to IR seems to be inapplicable to ICL response. Indeed, NBS1 cannot be the sensor because its presence is not required for ATR activation and it needs the FANC core complex to be assembled in the RMN complex. Consequently, its role could be to position correctly FANCD2 in order to be phosphorylated by ATR.
Our observations, combined with several published data, lead to the proposal that the FANC pathway contributes to ICL resistance by two functions: one in the context of the ICL-dependent S-phase checkpoint and the other in the mechanisms of ICL repair (Figure 7). The RMN complex activity is relevant to both FANC activities, participating in both S-phase checkpoint (via NBS1) and DNA repair (via RAD50 and MRE11) mechanisms. How S-phase checkpoint and DNA repair respectively contribute to ICL sensitivity remains to be clarified, but recent data suggest that cellular hypersensitivity correlates better with an S-phase checkpoint defect and chromosomal fragility with a DNA repair defect (Nakanishi et al, 2002). Identification of FANCD2 downstream targets/interactors is of primary importance to determine how this protein, which plays a role in IR and ICL agent treatment, coordinates S-phase checkpoint and DNA repair.
Finally, this work extends previous data showing the participation of FANC proteins in a complicated network of proteins involved in the DNA damage response and whose mutations are associated to genomic instability and cancer, like ATM, BLM, XP-F, NBS1, BRCA1, MRE11 and NBS1. How the fine tuning of the relationship among these caretakers leads to the specific clinical and cellular characteristics of each syndrome remains to be resolved.
Materials and methods
Cell lines and genotoxic treatments
The following cell lines were used in this study: EBV-transformed lymphoblasts from FA-A (HSC-72), FA-C (HSC-536) and FA-D2 (GM16752) individuals; ectopically corrected FA-C lymphoblasts (FA-C+FANCC); the EBV-transformed lymphoblastoid cell line from a normal donor (WT), GM3657; SV40-transformed fibroblasts from FA-C (PD332), FA-G (PD352) and FA-D2 (PD20) individuals; ectopically corrected FA-G (FA-G+FANCG), FA-D2 (PD20-315) fibroblasts; SV40-transformed fibroblasts from a normal donor, MRC5; HeLa cells and SV40 fibroblasts conditionally overexpressing the dominant-negative inactive form of ATR (ATRkd). With the exception of the GM serie, which is from the Coriell Cell Repositories, all the FA cell lines were kindly provided by M Buchwald (Hospital for Sick Children, Toronto, Ontario, Canada)and BA Cox (Oregon Health and Science University Fanconi Anemia Cell Repository, Portland, USA). The ATRkd-expressing fibroblasts were kindly provided by S Handeli and SL Schreiber (Fred Hutchinson Cancer Center, Seattle, USA) and have been already described (Cliby et al, 1998). The NBS1S343A cell line was a gift from P Concannon (Gatei et al, 2000). All the cell lines were handled as described (Pichierri et al, 2002). Expression of the transgene in the ATRkd cells was achieved by the addition of 1 μM doxycyclin (Sigma, USA) for 48 h. ICL induction by activated psoralens was achieved by exposing cells to 10 μM 8-metoxypsoralen (8-MOP, Sigma, USA) for 20 min followed by 10 kJ/m2 of UVA. Replication arrest was induced by exposure to 2 mM hydroxyurea (HU, Sigma, USA) for the indicated times. Alternatively, cells were exposed to γ-rays from a 60Co source at a dose rate of 2 Gy/min. After treatment, cells were cultivated in complete medium at 37°C until the selected times at which they were processed.
Evaluation of DNA synthesis
To analyze DNA synthesis inhibition by ICL induction, cells were prelabeled with 14C-thymidine (50 nCi/ml) for 2 days. Cells were treated with 8-MOP+UVA and pulse-labeled with 3H–thymidine (10 μCi/ml) 15 min before harvesting. Cells were harvested and fixed with 70% methanol. Radioactivity was evaluated by scintillography. The measure of DNA synthesis was derived from resulting ratios of 3H to 14C counts per minute and expressed as a percentage of untreated cells.
Preparation of cell lysates, immunoprecipitation and Western blot analysis
Cells were lysed in RIPA buffer (PBS, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 50 mM NaCl) supplemented with protease and phosphatase inhibitors (10 μg/ml aprotinin, 10 μg/ml PMSF, 10 μg/ml leupeptin, 25 mM β-glycerophosphate, 1 mM Na orthovanadate, 1 mM NaF). For immunoprecipitation experiments, 2.5 × 107 cells for each experimental point were used and 2 mg of lysate prepared in RIPA buffer was precleared with protein A/G Sepharose beads, incubated overnight at 4°C with rabbit polyclonal anti-FANCD2 (6 μg; AbCam, UK) and incubated with 30 μl of protein A/G Sepharose beads for 2 h at 4°C. After extensive washing in RIPA buffer, proteins were eluted by boiling in 2 × electrophoresis sample buffer and subjected to SDS–PAGE followed by immunoblotting. For treatments with phosphatase, IPs were resuspended in phosphatase buffer and incubated in the presence or absence of 300 U λ-phosphatase for 1 h. The samples were boiled in 2 × electrophoresis sample buffer prior to loading onto the gel. Ponceau red staining of the blots was used to assess equal loading and transfer. Where indicated, WCE represents 5% of the input. For Western immunoblotting, 50 μg of whole-cell lysate was used for each experimental point. Blots were separately incubated for 1 h at RT with primary antibodies against CHK1 (1:200, Santa Cruz Biotechnologies, USA), pS345CHK1 (1:500, Cell Signalling Technologies, USA), NBS1 (1:10000, AbCam, USA), FANCD2 (1:400, Santa Cruz Biotechnologies, USA), pS/TQ (1:500, Cell Signalling, USA) and FLAGM2 (1:1000, Sigma, USA). Horseradish peroxidase-conjugated goat specie-specific secondary antibodies (Santa Cruz Biotechnologies, USA) were used at a dilution of 1:1000. Visualization of the signal was accomplished using ECL (Amersham, USA).
siRNA transfection
CHK1 expressions were knocked down by transfection with an siRNA duplex (Dharmacon, USA) directed against the sequence GCGTGCCGTAGACTGTCCA (CHK1) as described (Zhao et al, 2002). MRE11 expression was knocked down by transfection with a mix of three siRNAs directed against the following sequences of the MRE11 mRNA: CCTGCCTCGAGTTATTAAG; CTGCGAGTGGACTATAGTG; GATGCCATTGAGGAATTAG. ATR expression was knocked down using the smart pool ATR siRNA from Dharmacon. Each siRNA duplex (Dharmacon, USA) was resuspended in RNase-free water at a concentration of 20 μM and transfection was carried out using oligofectamine reagent (Invitrogen, USA) according to the manufacturer's protocol for adherent cells. As a control, an siRNA duplex directed against GFP was used. All the experiments were carried out 48 h after transfection when maximal inhibition of CHK1 expression was observed as analyzed by Western blot.
Immunofluorescence
Cells recovered at the indicated times were spread onto poly-L-lysine-coated slides, fixed for 10 min at RT in a 4% paraformaldehyde buffered solution and permeabilized with 0.5% Triton X-100 for 10 min at 4°C. Staining with rabbit polyclonal anti-ATR (Oncogene Research, USA), polyclonal anti-NBS1 (Oncogene Research, USA) and mouse monoclonal anti-FANCD2 (Santa Cruz Biotechnologies, USA) was performed overnight at 4°C in PBS/1% BSA, whereas species-specific fluorescein- or Texas Red-conjugated secondary antibodies (Jackson Immunoresearch, USA) were applied for 1 h at RT, followed by counterstaining for 5 min at RT with 0.5 μg/ml DAPI. Slides were mounted in Vectashield (Vector Labs, USA) and analyzed by fluorescence confocal microscopy. All the primary antibodies were used at a 1:300 dilution, whereas the secondary antibodies were employed at a 1:500 dilution. For the simultaneous visualization of ATR and FANCD2 foci, cells were fixed in −20°C methanol for 20 min followed by 10 min incubation in an ice-cold 1:1 methanol/acetone solution. Cell preparations were blocked for 1 h in 10% FBS/PBS, rinsed and incubated with primary antibodies for 1 h at 37°C. For each time point, at least 200 nuclei were examined and ATR or FANCD2 foci scored blindly at a magnification of 100 × using a fluorescence confocal microscope. Only nuclei showing >5 foci were considered as positive. Parallel samples, incubated either with the appropriate normal serum or only with the secondary antibody, confirmed that the observed fluorescence pattern was not attributable to artifacts.
32P in vivo labeling
In vivo labeling experiments used exponentially growing WT (HeLa, ATRkd), A-T or NBS cells. The cells were washed with phosphate-free DMEM medium, supplemented with 10% heat inactivated fetal calf serum and incubated for 30 min at 37°C in the same media. [32P]-orthophosphate (Amersham, USA) was added directly to the medium (0.6 mCi/ml). After 30 min incubation at 37°C, cells were either mock-treated or exposed to 8-MOP or γ-rays. The cells were lysed and FANCD2 immunoprecipitated as described above. The immunoprecipitated proteins were resolved by SDS–PAGE, analyzed by autoradiography and Western blotting.
Acknowledgments
The authors thank C Guillouf for a critical review of the manuscript and JA Dugas du Villard for excellent technical assistance. This work was supported by grants from Electricité de France (EDF), Association pour la Recherche sur le Cancer (ARC) and Fondation de France (FdF). PP was supported by a fellowship from ARC.
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Ahmad SI, Hanaoka F, Kirk SH (2002) Molecular biology of Fanconi anaemia—an old problem, a new insight. Bioessays 24: 439–448 [DOI] [PubMed] [Google Scholar]
- Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M (2000) DNA replication is required to elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol Cell Biol 20: 8283–8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2001) Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol 13: 738–747 [DOI] [PubMed] [Google Scholar]
- Buchwald M, Moustacchi E (1998) Is Fanconi anemia caused by a defect in the processing of DNA damage? Mutat Res 408: 75–90 [DOI] [PubMed] [Google Scholar]
- Centurion SA, Kuo HR, Lambert WC (2000) Damage-resistant DNA synthesis in Fanconi anemia cells treated with a DNA cross-linking agent. Exp Cell Res 260: 216–221 [DOI] [PubMed] [Google Scholar]
- Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH (1998) Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J 17: 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J (2003) An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11: 203–213 [DOI] [PubMed] [Google Scholar]
- D'Andrea AD, Grompe M (1997) Molecular biology of Fanconi anemia: implications for diagnosis and therapy. Blood 90: 1725–1736 [PubMed] [Google Scholar]
- D'Andrea AD, Grompe M (2003) The Fanconi anaemia/BRCA pathway. Nat Rev Cancer 3: 23–34 [DOI] [PubMed] [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol 20: 7980–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Winter JP, Leveille F, van Berkel CG, Rooimans MA, van Der Weel L, Steltenpool J, Demuth I, Morgan NV, Alon N, Bosnoyan-Collins L, Lightfoot J, Leegwater PA, Waisfisz Q, Komatsu K, Arwert F, Pronk JC, Mathew CG, Digweed M, Buchwald M, Joenje H (2000a) Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am J Hum Genet 67: 1306–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Winter JP, Rooimans MA, van Der Weel L, van Berkel CG, Alon N, Bosnoyan-Collins L, de Groot J, Zhi Y, Waisfisz Q, Pronk JC, Arwert F, Mathew CG, Scheper RJ, Hoatlin ME, Buchwald M, Joenje H (2000b) The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nat Genet 24: 15–16 [DOI] [PubMed] [Google Scholar]
- de Winter JP, Waisfisz Q, Rooimans MA, van Berkel CG, Bosnoyan-Collins L, Alon N, Carreau M, Bender O, Demuth I, Schindler D, Pronk JC, Arwert F, Hoehn H, Digweed M, Buchwald M, Joenje H (1998) The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat Genet 20: 281–283 [DOI] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R (2001) Repair of DNA interstrand cross-links. Mutat Res 486: 217–247 [DOI] [PubMed] [Google Scholar]
- Dutrillaux B, Aurias A, Dutrillaux AM, Buriot D, Prieur M (1982) The cell cycle of lymphocytes in Fanconi anemia. Hum Genet 62: 327–332 [DOI] [PubMed] [Google Scholar]
- Escarceller M, Buchwald M, Singleton BK, Jeggo PA, Jackson SP, Moustacchi E, Papadopoulo D (1998) Fanconi anemia C gene product plays a role in the fidelity of blunt DNA end-joining. J Mol Biol 279: 375–385 [DOI] [PubMed] [Google Scholar]
- Escarceller M, Rousset S, Moustacchi E, Papadopoulo D (1997) The fidelity of double strand breaks processing is impaired in complementation groups B and D of Fanconi anemia, a genetic instability syndrome. Somat Cell Mol Genet 23: 401–411 [DOI] [PubMed] [Google Scholar]
- Falck J, Petrini JH, Williams BR, Lukas J, Bartek J (2002) The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet 30: 290–294 [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262 [DOI] [PubMed] [Google Scholar]
- Gatei M, Young D, Cerosaletti KM, Desai-Mehta A, Spring K, Kozlov S, Lavin MF, Gatti RA, Concannon P, Khanna K (2000) ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat Genet 25: 115–119 [DOI] [PubMed] [Google Scholar]
- Guillouf C, Vit JP, Rosselli F (2000) Loss of the Fanconi anemia group C protein activity results in an inability to activate caspase-3 after ionizing radiation. Biochimie 82: 51–58 [DOI] [PubMed] [Google Scholar]
- Guillouf C, Wang TS, Liu J, Walsh CE, Poirier GG, Moustacchi E, Rosselli F (1999) Fanconi anemia C protein acts at a switch between apoptosis and necrosis in mitomycin C-induced cell death. Exp Cell Res 246: 384–394 [DOI] [PubMed] [Google Scholar]
- Haneline LS, Broxmeyer HE, Cooper S, Hangoc G, Carreau M, Buchwald M, Clapp DW (1998) Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac−/− mice. Blood 91: 4092–4098 [PubMed] [Google Scholar]
- Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, Kaufmann WK (2002) An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol 22: 8552–8561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Nejad M, You Z, Yee MC, Newport JW, Cimprich KA (2000) Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol 10: 1565–1573 [DOI] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD (2002) Biallelic inactivation of BRCA2 in Fanconi anemia. Science 13: 13. [DOI] [PubMed] [Google Scholar]
- Jaspers NG, de Wit J, Regulski MR, Bootsma D (1982) Abnormal regulation of DNA replication and increased lethality in ataxia telangiectasia cells exposed to carcinogenic agents. Cancer Res 42: 335–341 [PubMed] [Google Scholar]
- Lavin MF, Shiloh Y (1997) The genetic defect in ataxia-telangiectasia. Annu Rev Immunol 15: 177–202 [DOI] [PubMed] [Google Scholar]
- Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB (2000) ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 404: 613–617 [DOI] [PubMed] [Google Scholar]
- Lo Ten Foe JR, Rooimans MA, Bosnoyan-Collins L, Alon N, Wijker M, Parker L, Lightfoot J, Carreau M, Callen DF, Savoia A, Cheng NC, van Berkel CG, Strunk MH, Gille JJ, Pals G, Kruyt FA, Pronk JC, Arwert F, Buchwald M, Joenje H (1996) Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nat Genet 14: 320–323 [DOI] [PubMed] [Google Scholar]
- McHugh PJ, Spanswick VJ, Hartley JA (2001) Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol 2: 483–490 [DOI] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W (2003a) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 35: 165–170 [DOI] [PubMed] [Google Scholar]
- Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W (2003b) A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23: 3417–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva OK, Petrini JH (2001) DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol 21: 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D'Andrea AD (2002) Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol 4: 913–920 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Toczyski DP, Hartwell LH (1997) When checkpoints fail. Cell 88: 315–321 [DOI] [PubMed] [Google Scholar]
- Petrini JH (1999) The mammalian Mre11-Rad50-nbs1 protein complex: integration of functions in the cellular DNA-damage response. Am J Hum Genet 64: 1264–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P, Averbeck D, Rosselli F (2002) DNA cross-link-dependent RAD50/MRE11/NBS1 subnuclear assembly requires the Fanconi anemia C protein. Hum Mol Genet 11: 2531–2546 [DOI] [PubMed] [Google Scholar]
- Resnick IB, Kondratenko I, Togoev O, Vasserman N, Shagina I, Evgrafov O, Tverskaya S, Cerosaletti KM, Gatti RA, Concannon P (2002) Nijmegen breakage syndrome: clinical characteristics and mutation analysis in eight unrelated Russian families. J Pediatr 140: 355–361 [DOI] [PubMed] [Google Scholar]
- Rosselli F, Ridet A, Soussi T, Duchaud E, Alapetite C, Moustacchi E (1995) p53-dependent pathway of radio-induced apoptosis is altered in Fanconi anemia. Oncogene 10: 9–17 [PubMed] [Google Scholar]
- Rotman G, Shiloh Y (1997) The ATM gene and protein: possible roles in genome surveillance, checkpoint controls and cellular defence against oxidative stress. Cancer Surv 29: 285–304 [PubMed] [Google Scholar]
- Sala-Trepat M, Rouillard D, Escarceller M, Laquerbe A, Moustacchi E, Papadopoulo D (2000) Arrest of S-phase progression is impaired in Fanconi anemia cells. Exp Cell Res 260: 208–215 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (1997) Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet 31: 635–662 [DOI] [PubMed] [Google Scholar]
- Sridharan D, Brown M, Lambert WC, McMahon LW, Lambert MW (2003) Nonerythroid alphaII spectrin is required for recruitment of FANCA and XPF to nuclear foci induced by DNA interstrand cross-links. J Cell Sci 116: 823–835 [DOI] [PubMed] [Google Scholar]
- Strathdee CA, Gavish H, Shannon WR, Buchwald M (1992) Cloning of cDNAs for Fanconi's anaemia by functional complementation. Nature 358: 434. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D'Andrea AD (2002) Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell 109: 459–472 [DOI] [PubMed] [Google Scholar]
- Thyagarajan B, Campbell C (1997) Elevated homologous recombination activity in Fanconi anemia fibroblasts. J Biol Chem 272: 23328–23333 [DOI] [PubMed] [Google Scholar]
- Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox B, Olson S, D'Andrea AD, Moses R, Grompe M (2001) Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell 7: 241–248 [DOI] [PubMed] [Google Scholar]
- Wu X, Ranganathan V, Weisman DS, Heine WF, Ciccone DN, O'Neill TB, Crick KE, Pierce KA, Lane WS, Rathbun G, Livingston DM, Weaver DT (2000) ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature 405: 477–482 [DOI] [PubMed] [Google Scholar]
- Zhao H, Watkins JL, Piwnica-Worms H (2002) Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA 99: 14795–14800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC, Lin SC, Gerbino E, Song MH, Zdzienicka MZ, Gatti RA, Shay JW, Ziv Y, Shiloh Y, Lee EY (2000) Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature 405: 473–477 [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]