

Summary
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by multiple alterations affecting the normal function of immune cells, such as lymphocytes, dendritic cells (DCs) and monocytes. Although the understanding of autoimmunity has significantly increased, the breakthrough in effective therapies has been modest, making necessary the development of new therapeutic strategies. Here we propose that a new potential target for therapy is haem oxygenase-1 (HO-1), an enzyme that catalyses the degradation of the haem group into biliverdin, carbon monoxide (CO) and Fe2+. These products exhibit immunosuppressive and anti-inflammatory effects, which can contribute to improving tolerance during organ transplantation. Because HO-1 is highly expressed by immune cells involved in SLE pathogenesis, such as monocytes and DCs, we evaluated whether induction of HO-1 expression or the administration of CO could ameliorate disease in the FcγRIIb knockout (KO) mouse model for SLE. We found that CO administration decreased the expansion of CD11b+ cells, prevented the decline of regulatory T cells and reduced anti-histone antibodies observed in untreated FcγRIIb KO mice. Furthermore, CO-treated animals and HO-1 induction showed less kidney damage compared with untreated mice. These data suggest that HO-1 modulation and CO administration can ameliorate autoimmunity and prevent the lupus symptoms shown by FcγRIIb KO mice, highlighting HO-1 as a potential new target for autoimmune therapy.
Keywords: carbon monoxide, haem oxygenase-1, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the presence of autoantibodies against nuclear antigens, such as dsDNA, nucleosomes and ribonucleoproteins.1 Inflammatory systemic compromise is thought to be the consequence of immune complex deposition, which leads to inflammation and tissue damage through the activation of monocytes, macrophages and complement.2,3 Symptoms of SLE can be heterogeneous, varying from mild disease to life-threatening kidney injury. Most components of the immune system contribute to SLE pathogenesis, including elements of both the innate and adaptive immune response.5,6
Monocytes contribute to the initiation and perpetuation of the autoimmune response and organ damage observed during SLE.8 In a similar way, the impaired clearance of apoptotic fragments by monocytes is thought to allow the priming of self-reactive lymphocytes by nuclear autoantigens.8,9 Furthermore, the number of circulating monocytes expands in both people with SLE and in mice suffering from an SLE-like disorder. Expansion of monocytes is thought to be promoted by an unbalanced expression of activating/inhibitory Fcγ receptors.11–15 Further, lupus nephritis has been associated with tissue infiltration by monocytes expressing an inflammatory phenotype.16–17 Hence, an unbalanced expression of activator/inhibitory molecules on monocytes and dendritic cells (DCs) could contribute to autoimmune inflammation during SLE.
Although the understanding of autoimmunity has greatly increased, these new findings have not led to a significant breakthrough in the development of more efficient or safer therapies. Unfortunately, current available therapies for SLE are mainly based on unspecific immunosuppressive agents, such as corticoids.18
Haem oxygenase 1 (HO-1) is an enzyme that catalyses the degradation of the haem group into biliverdin, free iron and carbon monoxide (CO).19 Although enzymatic products derived from HO-1 activity, such as biliverdin and bilirubin, possess antioxidant and immunosuppressive activity,19 CO has been shown to down-modulate immune responses in a variety of pathogenic processes and it is thought to mediate most of the immunomodulatory effects attributed to HO-1.20–24 It remains to be determined whether the modulation of HO-1 activity could prevent the immune alterations that are characteristic of SLE. Only one report has shown that the administration of an HO-1 inducer reduces nephritis severity as well as anti-dsDNA antibody levels in the MRL-Faslpr mouse model of lupus.25 Importantly, HO-1-deficient mice, as well as individuals with HO-1 deficiency, display immunological alterations, such as chronic inflammation, that are thought to be mediated by myeloid immune cells.26–27 In this context, we have recently shown that patients with SLE display a decreased expression of HO-1 on peripheral blood monocytes, which might be related to SLE pathogenesis.28 These data suggest that HO-1 induction, as well as the anti-inflammatory capacity of CO as an HO-1 product, could be considered a potential therapy to improve SLE progression.
Here, we have evaluated the effects of HO-1 induction and CO exposure in the onset and development of autoimmunity, using FcγRIIb-deficient mice as an animal model for SLE. This strain of mice was chosen because an FcγRIIb deficiency leads to increased susceptibility to myeloid cell activation in response to stimulation with immune complexes.15,29 Remarkably, we found that FcγRIIb-deficient mice displayed a significant reduction in the expression of HO-1 in the spleen. In this organ these mice showed progressive immunological alterations such as the expansion of CD11b+ cells and the contraction of the CD4+ Foxp3+ cell [regulatory T (Treg) cell] population. When FcγRIIb-deficient mice were exposed to CO, the expansion of CD11b+ cells was partially prevented and the frequency of Treg cells was restored. This was not seen for CoPP-treated mice. In addition, CO treatment decreased anti-histone IgG levels in FcγRIIb-deficient mice. Importantly, HO-1 induction and exposure to CO ameliorated the renal damage leading to proteinuria. Taken together, these data suggest that CO treatment, and to a lesser extent HO-1 induction, may contribute to preventing the immunological alterations observed in lupus mice. Our results support the notion that HO-1 activity could play a crucial role in immune system homeostasis by inducing pleiotropic anti-inflammatory effects, which could be considered as a potential therapy target for SLE patients.
Materials and methods
Antibodies and reagents
FITC-conjugated anti-mouse HO-1 monoclonal antibody (clone HO-1-2) and anti-mouse HO-1 monoclonal antibody (clone HO-1-1) were purchased from Abcam (Cambridge, UK). Anti-mouse CD11c-phycoerythrin (PE)/FITC/allophycocyanin (APC) (clone HL3), anti-mouse CD11b-PE/FITC (clone M1/70), anti-mouse Gr1 (clone RB6-8C5), anti-mouse CD4-FITC/PE (clone GK1.5), anti-mouse CD4-Peridinin chlorophyll protein (PerCP) (clone H129.19), anti-mouse Foxp3-AlexaFluor 647, (clone MF3), anti-mouse CD25-APC, anti-mouse CD8-APC (clone 53-6.7), anti-mouse B220-FITC (clone RA3-6B2) and anti-mouse CD16/32-PE (clone 2.4G2) were all purchased from BD Biosciences (San Jose, CA). Anti-mouse CD25PE-Cy7 (clone PC61.5) was purchased from eBioscience (San Diego, CA). Anti-actin (clone C4) was purchased from Chemicon–Millipore (Billerica, MA).
Mice
C57BL/6J, 129SJ and B6;129S4-Fcgr2btm1Ttk/J mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All mice used in this study were sex-matched and age-matched in all the experiments. Female and male mice were used. The FcγRIIb−/− (Fcgr2b targeted mutagenesis) phenotype were confirmed by double staining with anti-mouse CD16/32-PE and anti-mouse B220-FITC. All mice were kept under specific pathogen-free conditions at the animal facility of the Pontificia Universidad Católica de Chile. All animal work was performed according to institutional guidelines and supervised by a veterinarian.
Assessment of urinary protein excretion
Proteinuria was estimated by examining fresh urine with Combur Test sticks for urinalysis (Roche, Basel, Switzerland) using a scale of 0–3, where 0/trace = negative, 1 = 30 mg/dl, 2 = 100 mg/dl, and 3 = 500 mg/dl. Proteinuria scores > 2 were considered to represent moderate glomerulonephritis.
Assessment of antinuclear antibodies
Antinuclear antibody levels in serum were determined with HEp-2 cells in 12-well slides (BioRad, Hercules, CA). HEp-2 cells were incubated for 30 min with 1 : 40 diluted serum derived from FcγRIIb-deficient mice, followed by staining for 30 min with FITC-conjugated anti-mouse IgG (Invitrogen, Life Technologies, Carlsbad, CA).
CO exposure
Mice were exposed to compressed CO at a concentration equal to 250 ppm for 2 hr per day from weeks 12–15 to week 52.23 A CO analyser was used to measure CO levels in the chambers and maintain a controlled CO concentration. Anti-coagulated cardiac blood samples (0·4 ml) were taken immediately after CO exposure and carboxyhaemoglobin levels were measured using a haemoximeter (ROCHE cobas b 221Blood gas system, Mannheim, Germany). Carboxyhaemoglobin levels were 23–27% in all experiments using CO gas.
Pharmacological modulation of HO-1 expression
Either CoPP or tin protoporhyrin (SnPP) (Frontier Scientific, Logan, UT) was used either to induce HO-1 expression or to inhibit HO-1 activity, respectively. The CoPP and SnPP were dissolved in 0·1 m NaOH. Treatments consisted of a weekly intraperitoneal dose of CoPP (3·0 mg/kg)31 or SnPP (3·5 mg/kg)32 from 12–15 weeks of age to 52 weeks.
Flow cytometry
Spleen or inguinal lymph nodes from 1-year-old mice from different groups were harvested and minced in PBS supplemented with 10% fetal calf serum, until a homogeneous cell suspension was achieved. Cells were washed with PBS–1% BSA, re-suspended at 2 × 106 cells/ml (50 μl/tube) and incubated with FITC-, PE-, PerCP-, PE-Cy7- and APC-conjugated antibodies for 30 min at 4°. For HO-1 and Foxp3 intracellular staining, fixed cells were incubated with a FITC-conjugated anti-HO-1 or Alexa Fluor 647-conjugated anti-Foxp3 antibody in permeabilization buffer (PBS/BSA 3%–Saponin 0·5%) for 4 hr. Cells were washed with permeabilization buffer and analysed using a FACS Canto II flow cytometer (BD Biosciences).
Real-time RT-PCR
RNA was extracted from spleens and kidneys using Trizol (Invitrogen), according to the manufacturer's instructions. RT-PCR and cDNA synthesis were performed using random primers (ImProm-II; Promega, Madison, WI). Real-time PCR were carried out using a StepOne plus thermal cycler (Applied Biosystems, Foster City, CA). Briefly, cDNAs amplified out of total RNA were tested for HO-1 amplification using the following primers: mouse HO-1forward 5′-CCTCTGACGAAGTGACGCC-3′ and reverse 5′-CAGCCCCACCAAGTTCAAA-3′. PCR amplification of hypoxanthine-guanine phosphoribosyltransferase (HPRT) was used as an internal control using the following primers: mouse HPRT forward 5′-ATCCAGCAGGTCAGCAAAGA-3′ and reverse 5′-CGTGATTAGCGATGATGAACC-3′. To corroborate amplification specificity, PCR products were subjected to a melting curve program. Abundance of HO-1 mRNA was determined by relative expression to HPRT by the  method.
method.
ELISA
Serum samples from different groups of mice were obtained during the treatment and total IgG antibodies against DNA (Invitrogen) and histones (Calbiochem, EMD Millipore, Bellerica, MA) were quantified by ELISA. Briefly, ELISA plates were coated overnight at 4° with 2 µg/ml DNA or 2 µg/ml histones in PBS, washed and then blocked with PBS/BSA 10% for 2 hr at room temperature. After washing three times, serum samples were diluted in PBS/BSA 1% starting at 1 : 100 and incubated for 2 hr at room temperature. IgG was detected with a goat anti-mouse IgG antibody conjugated with horseradish peroxidase (Invitrogen). After washing three times, horseradish peroxidase substrate was added (3, 30, 5, 50-tetramethylbenzidine; Sigma, St Louis, MO) and optical density (OD) at 450 nm was measured on a microplate reader.
Western blot
Kidneys were harvested from CoPP 3 mg/kg-treated C57BL/6J mice after 48 hr. Kidney tissue was prepared for Western blot analysis as follows: 100 mg of tissue was lysed in radioimmunoprecipitation assay lysis buffer. The protein concentration of each sample was determined using the bicinchoninic acid assay (Thermo Scientific, Rockford, IL). Equal amounts of protein (20 µg) were loaded onto 12% SDS–polyacrylamide gels. After electrophoresis, proteins in the gel were transferred to a nitrocellulose membrane (HybondC-Extra; Amersham Biosciences, Piscataway, NJ). Membranes were blocked with 4% non-fat dried milk in PBS for 2 hr and incubated with anti-mouse HO-1 mAb (clone HO-1-1) or anti-actin antibody (clone C4) in 2% non-fat dried milk in PBS for 12 hr at 4°. Immunoreactive bands were detected using horseradish peroxidase-linked antibody against rabbit IgG and visualized with an enhanced chemiluminescence kit (Thermo Scientific).
Immunofluorescence
To evaluate CoPP-driven HO-1 induction in murine myeloid cells (see Supplementary material, Fig. S5), we produced CD11c+ CD11b+ bone marrow-derived DCs by using granulocyte–macrophage colony-stimulating factor (GM-CSF) differentiation culture as previously described.33 DCs were seeded on coverslips at day 0 and at day 5 after differentiation, cells were treated either with vehicle or with 50 µm CoPP for 2 hr, then extensively washed and incubated for 16 hr at 37°. Then, cells were washed and fixed with 2% p-formaldehyde for 10 min at 4°. Next, cells were washed, permeabilized with 0·05% saponin-PBS at room temperature and coverslips were transferred to a cold chamber over a hydrophobic surface (paraffin-coated). Coverslips were treated with 50 µl of a 1/200 mouse anti-HO-1 already dissolved in 0·05% saponin-PBS for 16 hr. Next, cells were extensively washed and stained with a 1/200 goat anti-mouse Alexa Fluor 488 for 3 hr at 4°. Cells were washed and mounted with DABCO for confocal microcopy analysis. HO-1-associated fluorescence intensity per cell was measured using ImageJ 1.47c software (NIH, Bethesda, MD).
Statistics
Data and statistical analyses were performed using Prism 5 software (Graph Pad Software, Inc., San Diego, CA). For statistical analyses, analyses of variance and Student's t-test were used. P-values < 0·05 were considered statistically significant. Flow cytometry data were analysed using FACS Diva software (BD Biosciences).
Results
HO-1 expression is decreased in the lupus mouse model
It has been previously shown that an HO-1 deficiency in mice may promote an inflammatory state.27 Hence, we decided to evaluate the levels of HO-1 mRNA in the spleens of 12-month-old FcγRIIb-deficient mice by RT-PCR. As shown in Fig. 1(a), HO-1 expression was significantly decreased in splenocytes from FcγRIIb-deficient mice compared with wild-type (WT) animals (Fig. 1a). To better assess HO-1 expression in immune cells, splenocytes from FcγRIIb-deficient and WT mice were stained with specific surface markers for monocytes, DCs and CD4+ T cells, in addition to anti-HO-1-specific antibodies, and analysed by FACS. Monocytes, CD4+ T cells and DCs from FcγRIIb-deficient mice all showed decreased expression of HO-1 compared with WT mice (Fig. 1b–d). To evaluate HO-1 levels in non-lymphoid tissues, we measured HO-1 expression in kidneys and lungs by real-time PCR. In contrast, in other tissues where HO-1 is abundant, such as the kidneys, or is scarce, such as the lung, it showed similar levels of HO-1 mRNA expression in FcγRIIb-deficient mice compared with WT mice (data not shown). These results suggest that HO-1 metabolism is mainly affected in immune cells from FcγRIIb-deficient mice.
Figure 1.

Expression of haem oxygenase 1 (HO-1) is reduced in FcγRIIb-deficient mice. Single cell suspensions of splenocytes from FcγRIIb-deficient and C57BL/6 wild type (WT) mice at 12 months were obtained. (a) Total mRNA was purified and HO-1 mRNA levels were evaluated by real-time PCR. *P < 0·05 by unpaired t-test. The data are presented as relative expression to HPRT by  mean ± SEM of three independent experiments with nmice/group = 5. (b–d). Spleen cell suspensions from FcγRIIb-deficient and WT mice were stained with specific anti-CD11c phycoerythrin (PE), -CD11b PE and -CD4 peridinin chlorophyll protein monoclonal antibodies, fixed, permeabilized, stained with an anti-HO-1 FITC monoclonal antibody, and evaluated by FACS. The data are presented as HO-1 mean fluorescence intensity (MFI) Geo mean expression ratio FcγRIIb-deficient/WT mice of CD11c+ cells (b), CD11b+ cells (c) and CD4+ cells (d). FcγRIIb−/− (FcγRIIb-deficient). *P < 0·05, **P < 0·01 by unpaired t-test.
mean ± SEM of three independent experiments with nmice/group = 5. (b–d). Spleen cell suspensions from FcγRIIb-deficient and WT mice were stained with specific anti-CD11c phycoerythrin (PE), -CD11b PE and -CD4 peridinin chlorophyll protein monoclonal antibodies, fixed, permeabilized, stained with an anti-HO-1 FITC monoclonal antibody, and evaluated by FACS. The data are presented as HO-1 mean fluorescence intensity (MFI) Geo mean expression ratio FcγRIIb-deficient/WT mice of CD11c+ cells (b), CD11b+ cells (c) and CD4+ cells (d). FcγRIIb−/− (FcγRIIb-deficient). *P < 0·05, **P < 0·01 by unpaired t-test.
CO reduces the expansion of CD11b+ cells in the spleen of lupus mice
Because monocytes and granulocytes play a relevant role in the pathogenesis of several autoimmune diseases by infiltrating tissues during inflammation, we evaluated the presence of CD11b+ cells during the progression of autoimmunity in FcγRIIb-deficient mice. Interestingly, we found an age-dependent expansion of CD11b+ cells in the spleens of FcγRIIb-deficient mice (Fig. 2a,b), which is consistent with the development of autoimmunity as indicated by the emergence of circulating anti-nuclear antibodies (see Supplementary material, Fig. S1). No major changes were seen for CD4+, CD8+, CD11c+ and B220+ cells from FcγRIIb-deficient mice compared with C57BL/6 or C57BL/6 × 129S WT mice (see Supplementary material, Fig. S2a–e). Next, whether exposure to CO could ameliorate the increase in CD11b+cells in FcγRIIb-deficient mice was evaluated. Accordingly, mice were exposed to 250 ppm CO for 2 hr daily for 9 months. To corroborate the efficacy of CO treatment, carboxyhaemoglobin levels were determined in peripheral blood. CO treatment resulted in 23–27% saturation of carboxyhaemoglobin after 2 hr of exposure (see Supplementary material, Fig. S3). Remarkably, CO treatment decreased the expansion of spleen CD11b+ cells in 1-year-old mice (Fig. 2d,e). The effect of CO treatment in this CD11b+ population was mainly observed in CD11b+ Gr1+ cells (Fig. 2f,h). However, although not statistically significant, CO treatment also caused a decrease in the CD11b+ Gr1− population in FcγRIIb-deficient mice when compared with untreated controls (Fig. 2g,h).
Figure 2.
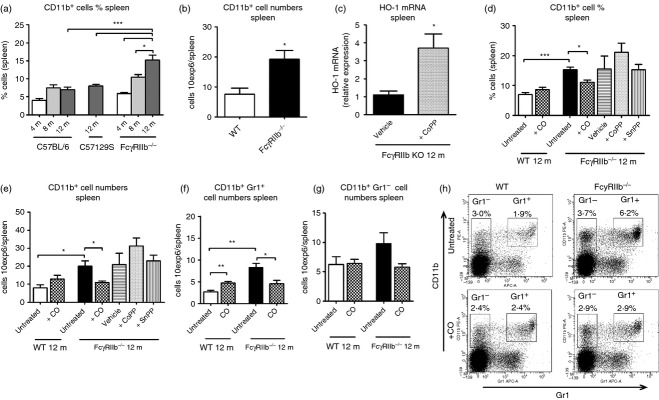
Study of CD11b+ cells in spleen from FcγRIIb-deficient mice. Spleen from FcγRIIb−/−, C57129S wild-type and C57BL/6 wild-type (WT) mice (n = 5/group) from 4 to 12 months-old were harvested and minced in PBS until a homogeneous cell suspensions were reached, then red blood cells were removed using hypotonic lysis buffer. (a) Spleen cell suspension from FcγRIIb-deficient (4–12 months), C57129S (12 months) and C57BL/6 (4–12 months) mice were stained with specific anti-CD11b phycoerythrin-conjugated monoclonal antibodies and evaluated by FACS. The graph represents the percentage of CD11b+ cells in spleens for three independent experiments. nmice/group = 6. *P < 0·05, ***P < 0·001 by one-way analysis of variance test. (b) Total cell counts from spleen cell suspension from FcγRIIb-deficient (12 months) and C57BL/6 (12 months) mice were measured by cell counting with Turk solution in a Neubauer chamber. The data are presented as cells × 106per spleen. *P < 0·05 by unpaired t-test. (c) Messenger RNA was purified from spleen cell suspension of cobalt protoporphyrin (CoPP) -treated FcγRIIb-deficient and untreated FcγRIIb-deficient mice (12 months), haem oxygenase 1 (HO-1) mRNA levels were evaluated by real-time PCR (CoPP administration from 12 to 52 week of age). *P < 0·05 by unpaired t-test. nmice/group = 5. (d–g) Spleen cell suspension from untreated and +CO groups of C57BL/6 (WT), and untreated, +CO, vehicle (intraperitoneal PBS, 500 µl), +CoPP (intraperitoneal 3·0 mg/kg in PBS, 500 µl), + tin protoporphyrin (SnPP) (intraperitoneal 3·5 mg/kg in PBS, 500 µl) of FcγRIIb-deficient mice were stained with specific anti-CD11b-phycoerythrin and anti-Gr1-allophycocyanin monoclonal antibodies and evaluated by FACS. All mice were killed at 12 months-old. Graphs represent the percentage (d) and total cell count (e) of CD11b+ cells, and total cell count of CD11b+ Gr1+ (f) and CD11b+ Gr1− (g) cells in spleens for two independent experiments. nmice/group = 7–8. ***P < 0·001, **P < 0·01 and *P < 0·05 by unpaired t-test, for untreated WT versus untreated FcγRIIb-deficient mice; untreated WT versus +CO WT; and untreated FcγRIIb-deficient mice untreated versus +CO FcγRIIb-deficient mice. (h) Representative dot plots of CD11b and Gr1 expression in spleens of untreated and CO-treated groups of C57BL/6 (WT) and FcγRIIb-deficient mice of 12 months.
Because HO-1 induction leads to CO production in cells, we evaluated whether CO effects could also be observed after treating 3-month-old FcγRIIb-deficient mice for 9 months with either CoPP (3 mg/kg) or SnPP (3·5 mg/kg); an HO-1 inductor and an HO-1 inhibitor, respectively. HO-1 induction in spleen, kidney and DCs was evaluated using RT-PCR (Fig. 2c), Western blot and confocal microscopy (see Supplementary material, Fig. S5). Unlike CO treatment, HO-1 induction by CoPP had no effect on the total number of splenic CD11b+ cells (Fig. 2d,e). As in CoPP-treated mice, FcγRIIb-deficient animals that received SnPP showed no changes in CD11b+ cells. Hence, CO exposure but not CoPP treatment was able to prevent CD11b+ cell expansion.
CO increases the frequency of regulatory T cells in the spleens of lupus mice
It has been reported that Treg cells are decreased in peripheral blood from patients with SLE and that they can show different alterations in their phenotype.34,35 Accordingly, the frequency of Treg cells in the spleens of FcγRIIb-deficient mice was determined by using flow cytometry. As shown in Fig. 3, an age-dependent reduction of CD4+ Foxp3+ cells was observed for FcγRIIb-deficient mice, which was not seen in WT mice (Fig. 3a,b). Frequency of Treg cells was also decreased in lymphatic nodes at 12 months in FcγRIIb-deficient mice compared with WT animals (Fig. 3c). Furthermore, Foxp3 mRNA expression was also decreased in the spleens of FcγRIIb-deficient mice compared with WT mice (Fig. 3d). On the contrary, no significant differences were observed between FcγRIIb-deficient and WT mice when the frequency of CD4+ CD25+ cells was determined in spleens (Fig. 3e).
Figure 3.
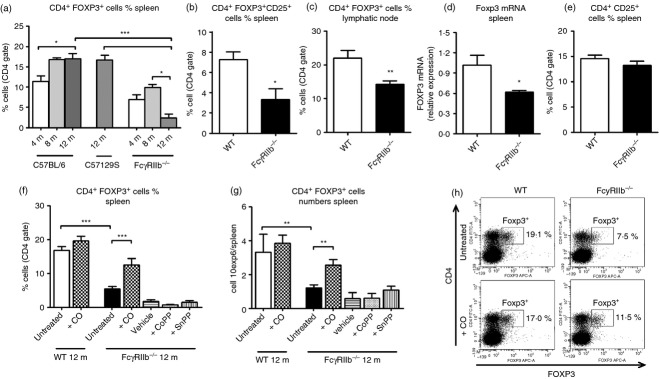
Study of FOXP3+ cells in spleen from FcγRIIb-deficient mice. (a) Spleen cell suspension from FcγRIIb-deficient (4–12 months), C57129S (12 months) and C57BL/6 (4–12 months) were stained with specific anti-CD4 FITC, fixed, permeabilized and stained with FOXP3 Alexa 647 monoclonal antibodies and evaluated by FACS. nmice/group = 6. *P < 0·05, ***P < 0·001 by one-way analysis of variance test. (b) Spleen cell suspensions from FcγRIIb-deficient (12 months) and C57BL/6 (12 months) mice were stained with anti-CD4 FITC, anti-CD25 phycoerythrin-Cy7, fixed, permeabilized and stained with FOXP3 Alexa 647 monoclonal antibodies and evaluated by FACS. nmice/group = 6. (c) Inguinal lymphatic nodes from FcγRIIb-deficient (12 months) and C57BL/6 (12 months) mice were harvested and minced until a cell suspension was reached. Then cells were stained with anti-CD4 FITC and anti-CD25 PE, fixed, permeabilized and incubated with anti-FOXP3 Alexa Fluor 647. (d) Messenger RNA was purified from spleen cell suspensions of wild-type (WT) and FcγRIIb-deficient mice (12 months), FOXP3 mRNA levels were evaluated by real-time PCR. *P < 0·05 by unpaired t-test. (e) Spleen cell suspensions from FcγRIIb-deficient (12 months) and C57BL/6 (12 months) mice were stained with anti-CD4 FITC, anti-CD25 phycoerythrin-Cy7 and analysed by FACS. (f–h) Spleen cell suspension from untreated and +CO groups of C57BL/6 (WT), and untreated, +CO, vehicle (intraperitoneal PBS, 500 µl), + cobalt protoporphyrin (CoPP) (intraperitoneal 3·0 mg/kg in PBS, 500 µl), + tin protoporphyrin (SnPP) (intraperitoneal 3·5 mg/kg in PBS, 500 µl) of FcγRIIb-deficient mice were stained with anti-CD4 FITC, fixed, permeabilized and stained with FOXP3 Alexa Fluor 647 monoclonal antibodies and evaluated by FACS. Graphs represent the percentage of CD4+ FOXP3+ (CD4 gated) cells (f) and the total cell count (h) of CD4+ FOXP3+ cells. nmice/group = 7–8. ***P < 0·001 and *P < 0·05 by unpaired t-test, untreated WT versus untreated FcγRIIb-deficient mice; untreated WT versus +CO WT; untreated FcγRIIb-deficient mice untreated versus +CO FcγRIIb-deficient mice. (g) Representative dot plots of CD4 and FOXP3 expression in spleens of untreated and treated CO groups of C57BL/6 (WT) and FcγRIIb-deficient mice of 12 months.
Whether CO, CoPP or SnPP treatment could restore CD4+ Foxp3+ cell numbers in the spleens of FcγRIIb-deficient mice was evaluated. As shown in Fig. 3, CO treatment indicated that the total number of CD4+ Foxp3+cells in FcγRIIb-deficient mice remained equivalent to that in WT mice (Fig. 3f–h). However, FcγRIIb-deficient animals treated with CoPP and SnPP showed no restoration of CD4+ Foxp3+ cells. Hence, CO exposure but not CoPP administration could prevent the contraction of the Foxp3+ cell population.
CO decreases autoantibody levels in lupus mice
The pathogenesis of SLE is intimately linked to the activation of auto-reactive B cells and the production of nephritogenic autoantibodies against dsDNA.37 Although FcγRIIb-deficient mice in the B6;129S4 genetic background show low levels of antinuclear antibodies, they develop mild renal symptoms. When IgG autoantibody levels were determined by ELISA, CO-treated animals exhibited decreased anti-histone but not anti-DNA autoantibody levels compared with untreated mice (Fig. 4a,b). CoPP treatment did not alter anti-histone or anti-DNA antibodies. On the contrary, in SnPP-treated mice anti-histone and anti-DNA antibody levels were both increased compared with control mice treated with vehicle. These findings suggest that CO treatment may ameliorate lupus renal damage by decreasing autoantibody levels. On the other hand, HO-1 inhibition by SnPP, as observed in HO-1 deficiency, may promote inflammation leading to autoantibody production and auoimmunity.27
Figure 4.
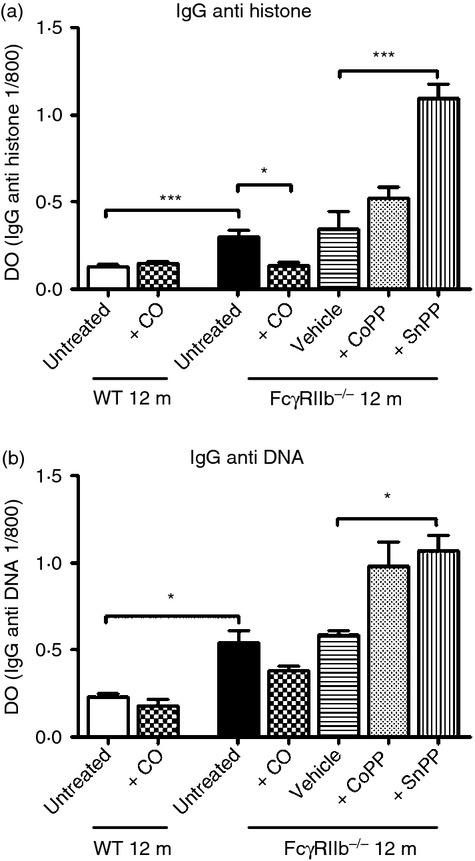
Therapeutic effects of CO and cobalt protoporphyrin (CoPP) treatment on autoantibody production in FcγRIIb-deficient mice. SerumIgG anti-dsDNA and IgG anti-histone antibody levels in wild-type (WT) and FcγRIIb-deficient mice treated with CO, vehicle, CoPP, tin protoporphyrin (SnPP) and untreated were determined by ELISA. (a) SerumIgG anti-histone antibody levels at 12 months of age from all groups studied. nmice/group = 7–8. (b) Serum IgG anti-DNA antibody levels at 12 months of age from all groups studied. nmice/group = 7–8. ***P < 0·001 and *P < 0·05 by unpaired t-test, untreated WT versus untreated FcγRIIb-deficient mice; untreated versus +CO FcγRIIb-deficient mice; vehicle versus + tin protoporphyrin (SnPP) FcγRIIb-deficient mice. The data shown are the mean ± SEM.
HO-1 induction and CO exposure ameliorate renal damage of lupus mice
To evaluate the therapeutic effect of HO-1 in renal manifestations, mice were treated with either CoPP or CO from week 12 to week 52 of age. The urinary protein levels were determined using reactive strips. As shown in Fig. 5, this weekly CoPP treatment or CO exposure delayed the onset of proteinuria and reduced the incidence of proteinuria at the end of the study when compared with untreated FcγRIIb-deficient mice (Fig. 5a).
Figure 5.

Therapeutic effects of CO and cobalt protoporphyrin (CoPP) treatment on proteinuria in FcγRIIb-deficient mice. Presence of urinary protein excretion was evaluated throughout the study (from 10 to 52 weeks) by examining fresh urine with Combur Test sticks for urinalysis (Roche). Urinary protein above 100 mg/dl was considered as proteinuria. (a) Presence of proteinuria in FcγRIIb-deficient mice treated with ♦ CO, ▴ CoPP or ▪ untreated over time. nmice/group = 7–8. The data shown are the Kaplan–Meier survival fractions profile. (b) Messenger RNA was purified from kidney from untreated and +CoPP FcγRIIb-deficient mice at 12 months of age. Messenger RNA haem oxygenase 1 (HO-1) levels were evaluated by real-time PCR. *P < 0·05 by unpaired t-test. The data are presented as relative expression of HO-1 mRNA to HPRT by 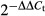 method. Mean ± SEM of two independent experiments with nmice/group = 4.
method. Mean ± SEM of two independent experiments with nmice/group = 4.
To determine whether CoPP treatment could induce HO-1 in kidney and so promote its effects locally, we quantified HO-1 mRNA levels in the kidney. As shown in Fig. 5, HO-1 was induced in the kidney of CoPP-treated mice, indicating that protection may be mediated locally (Fig. 5b). Therapeutic effects of CoPP may be related to a local decrease in inflammatory mediators, reactive oxygen species or cytoprotection. These results support the notion that HO-1 modulation could be exploited as a potential therapeutic target for lupus nephritis.
Discussion
Our results show that HO-1 expression is significantly reduced in the spleen of FcγRIIb-deficient mice compared with WT mice. These differences are specific for lymphoid tissue because no significant differences were found in kidneys or lungs. We also found immunological alterations, such as expansion of CD11b+ cells and the contraction of CD4+ Foxp3+ cells. To our knowledge this study is the first to report that CO exposure reduces immunological alterations seen in FcγRIIb-deficient mice, including prevention of CD11b+ cell expansion and the depletion of Treg cells. Furthermore, treatment with CO decreased histone-specific autoantibodies in FcγRIIb-deficient animals.
Although it has been reported that during acute inflammatory states there is an increase in HO-1 expression that may lead to an anti-inflammatory response, there are no reports evaluating HO-1 expression in murine models for lupus.38 Along these lines, we have previously reported for the first time that peripheral blood CD14+ cells from SLE patients showed a reduced expression of HO-1 on peripheral blood monocytes.28 Here, FcγRIIb-deficient mice (12 months old) showed a decreased HO-1 expression in spleen. Defective expression of FcγRIIb on immune cells may promote deregulated chronic activation via stimulatory FcγRs in response to immune complex unbalancing HO-1 homeostasis. Hence, FcγRIIb deficiency may affect HO-1 expression, so contributing to autoimmunity. The fact that HO-1 deficiency leads to chronic inflammation but not autoimmunity strongly suggests that an HO-1 deficiency is unlikely to directly cause autoimmunity.26–27 Further research is required to elucidate whether HO-1 deregulation may lead to an inflammatory condition or an exacerbated inflammatory response that could in turn contribute to the development of autoimmunity in susceptible hosts. This latter notion is supported by our recent observation that monocytes derived from SLE patients showed reduced HO-1 expression.28
As mentioned above, this is the first report showing that CO exposure modulates spleen CD11b+ cell expansion and Treg cell contraction. Expansion of CD11b+ cells, seen in the spleens of FcγRIIb-deficient mice, is most likely a result of the lack of FcγRIIb expression, which could lead to monocyte/granulocyte activation in response to immune complexes. In addition, this activation could promote a deregulated cytokine production, including GM-CSF, interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α), resulting in the subsequent inhibition of HO-1 expression.39,40 We found that CO exposure could restore the number of monocyte/granulocyte cells in the spleens of FcγRIIb-deficient mice to levels equivalent to WT mice. The effect of CO could be exerted by inhibiting signalling pathways triggered during FcγRIIb deficiency as it has been reported that CO could modulate p38 mitogen-activated protein kinase, mitogen-activated protein kinase kinase 3 and caspase 3 signalling.42 The nature of expanded splenic CD11b+ Gr1+ and CD11b+ Gr1− cell populations remains to be determined as several myeloid sub-populations have been described, including inflammatory monocytes, granulocytes and suppressor myeloid cells.43–44 However, it is remarkable that CO exposure could both limit the expansion of CD11b+ subpopulations to normal levels as well as maintaining Treg cell numbers.
Furthermore, the mechanism responsible for regulatory CD4+ Foxp3+ T-cell reduction in FcγRIIb-deficient mice has not yet been determined. One explanation may be that pro-inflammatory cytokine production by expanded CD11b+ cells may affect regulatory T-cell population homeostasis and survival. Based on recent reports, it is probable that monocytes may act as an important source of pro-inflammatory cytokines such as IL-6 and TNF-α, and that these cytokines, mainly IL-6, may affect Treg cell development and function.42,45 Moreover, it has been recently reported that CO can suppress the secretion of pro-inflammatory cytokines without altering IL-10 production by myeloid DCs.47 Hence, it is likely that by promoting IL-10 production, CO exposure would prevent monocyte expansion and activation, reducing the secretion of pro-inflammatory cytokines, such as IL-6, and sustaining the numbers of regulatory T cells.47 On the contrary, the effect of CO in the CD11b+ or CD4+ Foxp3+ cell populations could not be reproduced by CoPP treatment. This lack of response in CoPP-treated mice could have been because of differences in final CO concentrations in lymphoid tissues between our two approaches. As can be seen in the Supplementary material, Fig. S4, CoPP treatment failed to induce enough CO to be measured by blood carboxyhaemoglobin. It remains to be elucidated whether physiological CO levels derived from haem group degradation by HO-1 could play a relevant role in immune cell suppression. Another possible explanation for the reduced effect of CoPP on immune cells compared with CO therapy could be the occurrence of secondary side effects due to chronic HO-1 induction. The subsequent increase of haem degradation products after HO-1 induction may be harmful to cells, limiting the beneficial immunosuppressive effect of CoPP-induced CO release.48–49
Decreased IgG anti-histone levels and normal regulatory T-cell numbers following CO exposure may be secondarily associated with CD11b+ cell modulation. When the number of CD11b+ cells was decreased by CO, pro-inflammatory mediators may also be decreased, limiting autoantibody production and the contraction of the Treg cell population. The improvement of renal status in CO-treated mice may be associated with a decreased level of autoantibodies, as immune complex deposition in the kidney, which activates local monocyte populations. Consistent with this notion, it has been observed that hemin treatment prevented glomerulonephritis development in MRL lupus mice without modifying the total cell number in spleens.25 Similarly, we found that CoPP treatment ameliorates renal symptoms (urinary protein secretion) but failed to prevent CD11b+ cell expansion or Treg cell contraction. However, we could not see any reduction in anti-DNA or anti-histone antibodies in response to CoPP treatment, which indicates that the HO-1 modulation effect on proteinuria may be independent of a reduction in self-reactive IgG. Consistent with the importance of HO-1 for the modulation of the inflammatory response during lupus, inhibition of HO-1 by SnPP led to increased levels of anti-DNA and histone autoantibodies in FcγRIIb-deficient mice. As lupus nephritis is known to be partially mediated by nitric oxide production, it is possible that the underlying mechanism of CoPP in renal protection observed in our study could be associated with inhibition of local nitric oxide synthase in kidneys, whereas CO therapy would be mediated mainly by modulation of immune cells.25–50 It has been recently reported that biliverdin and bilirubin are able to protect mice from developing diabetic nephropathy by reducing NAD(P)H-dependent reactive oxygen species production. It is therefore possible that the local effect of HO-1 in the kidney in terms of protection from damage, could be mediated by an equivalent mechanism.51
To our knowledge, these are the first results characterizing the anti-inflammatory properties of CO in FcγRIIb-deficient mice suffering from a lupus-like syndrome. Our results suggest that an impaired immune cell function can be the result of reduced HO-1 expression in FcγRIIb-deficient cells, resulting in a bias to deregulated inflammation and autoimmunity. The anti-inflammatory effects of CO could be the result of a blockade of the expansion of immune cells, such as monocytes, which would in turn reduce inflammation. It remains to be determined whether CO exposure mediates anti-inflammatory effects in lupus through inhibition of production of pro-inflammatory cytokines such as IL-6, TNF-α or GM-CSF. Nonetheless, the data shown here suggest that HO-1 could work as a potent regulator of systemic homeostasis through pleiotropic mechanisms and that the understanding of these pathways is of therapeutic relevance in autoimmunity and inflammation research.
Acknowledgments
This work was supported by grants FONDECYT no 1070352, FONDECYT no 1050979, FONDECYT no 1040349, FONDECYT no 1100926, FONDECYT no 1110397, FONDECYT no 1100971, FONDECYT no 1110604, Proyecto de Inserción de Capital Humano Avanzado en la Academia 2011 No 791100015, Vicerrectoría de Investigación PUC Posdoctorado no 4/2010 and Millennium Institute on Immunology and Immunotherapy (P09-016-F). LJC is a Pew Latin American Fellow. We would like to thank Camila Schmidt for her skilful technical assistance. AMK is a Chaire De La Région Pays De La Loire De Chercheur Étranger D'excellence and the collaboration between AMK and IA is supported by a Grant ‘Nouvelles Equipes-nouvelles thématiques' from the La Région Pays De La Loire and by an INSERM CDD grant.
Glossary
- APC
allophycocyanin
- CO
carbon monoxide
- CoPP
cobalt protoporphyrin
- DCs
dendritic cells
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- HO-1
haem oxygenase 1
- IL-6
interleukin-6
- PE
phycoerythrin
- SLE
systemic lupus erythematosus
- SnPP
tin protoporphyrin
- TNF
tumour necrosis factor
- Tregs
regulatory T cells
- WT
wild-type
Disclosures
The authors declare that they have no financial or commercial conflict of interest.
Supporting Information
Figure S4. Blood carboxyhaemoglobin level after cobalt protoporphyrin (CoPP) treatment and CO exposure.
Figure S5. Haem oxygenase 1 (HO-1) protein induction by cobalt protoporphyrin (CoPP) determined by in vivo and in vitro assays.
References
- Rekvig OP, Nossent JC. Anti-double-stranded DNA antibodies, nucleosomes, and systemic lupus erythematosus: a time for new paradigms? Arthritis Rheum. 2003;48:300–12. doi: 10.1002/art.10739. [DOI] [PubMed] [Google Scholar]
- Brown EE, Edberg JC, Kimberly RP. Fc receptor genes and the systemic lupus erythematosus diathesis. Autoimmunity. 2007;40:567–81. doi: 10.1080/08916930701763710. [DOI] [PubMed] [Google Scholar]
- Anolik JH. B cell biology and dysfunction in SLE. Bull NYU Hosp Jt Dis. 2007;65:182–6. [PubMed] [Google Scholar]
- Cohen PLPL. T- and B-cell abnormalities in systemic lupus. J Invest Dermatol. 1993;100:69S–72S. doi: 10.1111/1523-1747.ep12355631. [DOI] [PubMed] [Google Scholar]
- Sanz I, Lee FE-H. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:326–37. doi: 10.1038/nrrheum.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnblom L, Elkon KB. Cytokines as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:339–47. doi: 10.1038/nrrheum.2010.64. [DOI] [PubMed] [Google Scholar]
- Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:317–25. doi: 10.1038/nrrheum.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsiari CG, Liossis S-NC, Sfikakis PP. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheum. 2010;39:491–503. doi: 10.1016/j.semarthrit.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Gaipl US, Munoz LE, Grossmayer G, et al. Clearance deficiency and systemic lupus erythematosus (SLE) J Autoimmun. 2007;28:114–21. doi: 10.1016/j.jaut.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Muñoz LE, Janko C, Grossmayer GE, et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1733–42. doi: 10.1002/art.24535. [DOI] [PubMed] [Google Scholar]
- Sullivan KE, Suriano A, Dietzmann K, Lin J, Goldman D, Petri MA. The TNFα locus is altered in monocytes from patients with systemic lupus erythematosus. Clin Immunol. 2007;123:74–81. doi: 10.1016/j.clim.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Vega N, Galindo-Rodríguez G, Bajaña S, Portales-Pérez D, Abud-Mendoza C, Sánchez-Torres C, González-Amaro R. Phenotypic analysis of IL-10-treated, monocyte-derived dendritic cells in patients with systemic lupus erythematosus. Scand J Immunol. 2006;64:668–76. doi: 10.1111/j.1365-3083.2006.01849.x. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Santiago-Raber M-L, Amano H, Amano E, Fossati-Jimack L, Moll T, Kotzin BL, Izui S. Contribution of NZB autoimmunity 2 to Y-linked autoimmune acceleration-induced monocytosis in association with murine systemic lupus. J Immunol. 2006;176:3240–7. doi: 10.4049/jimmunol.176.5.3240. [DOI] [PubMed] [Google Scholar]
- Müller M, Emmendörffer A, Lohmann-Matthes M-L. Expansion and high proliferate potential of the macrophage system throughout life time of lupus-prone NZB/W and MRL lpr/lpr mice. Lack of down-regulation of extramedullar macrophage proliferation in the postnatal period. Eur J Immunol. 1991;21:2211–7. doi: 10.1002/eji.1830210932. [DOI] [PubMed] [Google Scholar]
- Santiago-Raber M-L, Amano H, Amano E, et al. Fcγ receptor-dependent expansion of a hyperactive monocyte subset in lupus-prone mice. Arthritis Rheum. 2009;60:2408–17. doi: 10.1002/art.24787. [DOI] [PubMed] [Google Scholar]
- Kuroiwa T, Lee EG. Cellular interactions in the pathogenesis of lupus nephritis: the role of T cells and macrophages in the amplification of the inflammatory process in the kidney. Lupus. 1998;7:597–603. doi: 10.1191/096120398678920712. [DOI] [PubMed] [Google Scholar]
- Frosch M, Vogl T, Waldherr R, Sorg C, Sunderkötter C, Roth J. Expression of MRP8 and MRP14 by macrophages is a marker for severe forms of glomerulonephritis. J Leukoc Biol. 2004;75:198–206. doi: 10.1189/jlb.0203076. [DOI] [PubMed] [Google Scholar]
- Houssiau FA, Ginzler EM. Current treatment of lupus nephritis. Lupus. 2008;17:426–30. doi: 10.1177/0961203308090029. [DOI] [PubMed] [Google Scholar]
- Abraham NG, Kappas A. Pharmacological and clinical aspects of hemeoxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- Rémy S, Blancou P, Tesson L, et al. Carbon monoxide inhibits TLR-induced dendritic cell immunogenicity. J Immunol. 2009;182:1877–84. doi: 10.4049/jimmunol.0802436. [DOI] [PubMed] [Google Scholar]
- Simon T, Pogu S, Tardif V, et al. Carbon monoxide-treated dendritic cells decrease β1-integrin induction on CD8+ T cells and protect from type 1 diabetes. Eur J Immunol. 2013;43:209–18. doi: 10.1002/eji.201242684. [DOI] [PubMed] [Google Scholar]
- Listopad J, Asadullah K, Sievers C, Ritter T, Meisel C, Sabat R, Döcke W-D. Heme oxygenase-1 inhibits T cell-dependent skin inflammation and differentiation and function of antigen-presenting cells. Exp Dermatol. 2007;16:661–70. doi: 10.1111/j.1600-0625.2007.00581.x. [DOI] [PubMed] [Google Scholar]
- Chora ÂA, Fontoura P, Cunha A, et al. Hemeoxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clinl Invest. 2007;117:438–47. doi: 10.1172/JCI28844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau A, Hill M, Thébault P, et al. Tolerogenic dendritic cells actively inhibit T cells through heme oxygenase-1 in rodents and in nonhuman primates. FASEB J. 2009;23:3070–7. doi: 10.1096/fj.08-128173. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Takeno M, Iwasaki M, et al. Chemical induction of HO-1 suppresses lupus nephritis by reducing local iNOS expression and synthesis of anti-dsDNA antibody. Clin Exp Immunol. 2004;138:237–44. doi: 10.1111/j.1365-2249.2004.02594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancou P, Tardif V, Simon T, Rémy S, Carreño L, Kalergis A, Anegon I. Immunoregulatory Properties of Heme Oxygenase-1. In: Cuturi MC, Anegon I, editors. Suppression and Regulation of Immune Responses SE – 18. Vol. 677. NY: Humana Press; 2011. pp. 247–68. [DOI] [PubMed] [Google Scholar]
- Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, Agarwal A. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. 2004;165:1045–53. doi: 10.1016/S0002-9440(10)63365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrada AA, Llanos C, Mackern-Oberti JP, et al. Haemoxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology. 2012;136:414–24. doi: 10.1111/j.1365-2567.2012.03598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature. 1996;379:346–9. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcγ receptors on dendritic cells. J Exp Med. 2002;195:1653–9. doi: 10.1084/jem.20020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kim DH, Tsenovoy PL, et al. Treatment of obese diabetic mice with a hemeoxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57:1526–35. doi: 10.2337/db07-1764. [DOI] [PubMed] [Google Scholar]
- Cai Y, Cho G-S, Ju C, et al. Activated microglia are less vulnerable to hemin toxicity due to nitric oxide-dependent inhibition of JNK and p38 MAPK activation. J Immunol. 2011;187:1314–21. doi: 10.4049/jimmunol.1002925. [DOI] [PubMed] [Google Scholar]
- Riquelme SA, Bueno SM, Kalergis AM. IgG keeps virulent Salmonella from evading dendritic cell uptake. Immunology. 2012;136:291–305. doi: 10.1111/j.1365-2567.2012.03578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zhang X, Tang F, Zhu L, Liu Y. Reduction of forkhead box P3 levels in CD4+ CD25high T cells in patients with new-onset systemic lupus erythematosus. Clin Exp Immunol. 2008;153:182–7. doi: 10.1111/j.1365-2249.2008.03686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyara M, Amoura Z, Parizot C, et al. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol. 2005;175:8392–400. doi: 10.4049/jimmunol.175.12.8392. [DOI] [PubMed] [Google Scholar]
- Gerli R, Nocentini G, Alunno A, Bocci EB, Bianchini R, Bistoni O, Riccardi C. Identification of regulatory T cells in systemic lupus erythematosus. Autoimmun Rev. 2009;8:426–30. doi: 10.1016/j.autrev.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Muller S, Dieker J, Tincani A, Meroni PL. Pathogenic anti-nucleosome antibodies. Lupus. 2008;17:431–6. doi: 10.1177/0961203308090030. [DOI] [PubMed] [Google Scholar]
- Yachie A, Toma T, Mizuno K, Okamoto H, Shimura S, Ohta K, Kasahara Y, Koizumi S. Heme oxygenase-1 production by peripheral blood monocytes during acute inflammatory illnesses of children. Exp Biol Med. 2003;228:550–6. doi: 10.1177/15353702-0322805-26. [DOI] [PubMed] [Google Scholar]
- Liu B, Tan W, Barsoum A, et al. IL-17 is a potent synergistic factor with GM-CSF in mice in stimulating myelopoiesis, dendritic cell expansion, proliferation, and functional enhancement. Exp Hematol. 2010;38:877–84. doi: 10.1016/j.exphem.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Yoshimoto K, Tanaka M, Kojima M, et al. Regulatory mechanisms for the production of BAFF and IL-6 are impaired in monocytes of patients of primary Sjögren's syndrome. Arthritis Res Ther. 2011;13:R170. doi: 10.1186/ar3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hora K, Satriano JA, Santiago A, Mori T, Stanley ER, Shan Z, Schlondorff D. Receptors for IgG complexes activate synthesis of monocyte chemoattractant peptide 1 and colony-stimulating factor 1. Proc Natl Acad Sci U S A. 1992;89:1745–9. doi: 10.1073/pnas.89.5.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Alam J, Choi AMK. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribechini E, Greifenberg V, Sandwick S, Lutz M. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol. 2010;199:273–81. doi: 10.1007/s00430-010-0151-4. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Nakano M, Terabe F, et al. The influence of excessive IL-6 production in vivo on the development and function of Foxp3+ regulatory T cells. J Immunol. 2011;186:32–40. doi: 10.4049/jimmunol.0903314. [DOI] [PubMed] [Google Scholar]
- Chauveau C, Rémy S, Royer PJ, et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood. 2005;106:1694–702. doi: 10.1182/blood-2005-02-0494. [DOI] [PubMed] [Google Scholar]
- Galbraith RA, Kappas A. Regulation of food intake and body weight in rats by the synthetic heme analogue cobalt protoporphyrin. Am J Physiol. 1991;261:R1388–94. doi: 10.1152/ajpregu.1991.261.6.R1388. [DOI] [PubMed] [Google Scholar]
- Rosenberg DW, Drummond GS, Smith TJ. Depletion of cytochrome P-450 by thyroid hormone and cobalt-protoporphyrin IX in rat liver: evidence that susceptibility varies among forms of the hemeprotein. Pharmacology. 1995;51:254–62. doi: 10.1159/000139367. [DOI] [PubMed] [Google Scholar]
- Reilly CM, Farrelly LW, Viti D, et al. Modulation of renal disease in MRL/lpr mice by pharmacologic inhibition of inducible nitric oxide synthase. Kidney Int. 2002;61:839–46. doi: 10.1046/j.1523-1755.2002.00230.x. [DOI] [PubMed] [Google Scholar]
- Fujii M, Inoguchi T, Sasaki S, Maeda Y, Zheng J, Kobayashi K, Takayanagi R. Bilirubin and biliverdin protect rodents against diabetic nephropathy by downregulating NAD(P)H oxidase. Kidney Int. 2010;78:905–19. doi: 10.1038/ki.2010.265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S4. Blood carboxyhaemoglobin level after cobalt protoporphyrin (CoPP) treatment and CO exposure.
Figure S5. Haem oxygenase 1 (HO-1) protein induction by cobalt protoporphyrin (CoPP) determined by in vivo and in vitro assays.