

Abstract
The binding of eIF2–GTP–tRNAiMet ternary complex (TC) to 40S subunits is impaired in yeast prt1-1 (eIF3b) mutant extracts, but evidence is lacking that TC recruitment is a critical function of eIF3 in vivo. If TC binding was rate-limiting in prt1-1 cells, overexpressing TC should suppress the temperature-sensitive phenotype and GCN4 translation should be strongly derepressed in this mutant, but neither was observed. Rather, GCN4 translation is noninducible in prt1-1 cells, and genetic analysis indicates defective ribosomal scanning between the upstream open reading frames that mediate translational control. prt1-1 cells also show reduced utilization of a near-cognate start codon, implicating eIF3 in AUG selection. Using in vivo cross-linking, we observed accumulation of TC and mRNA/eIF4G on 40S subunits and a 48S ‘halfmer' in prt1-1 cells. Genetic evidence suggests that 40S–60S subunit joining is not rate-limiting in the prt1-1 mutant. Thus, eIF3b functions between 48S assembly and subunit joining to influence AUG recognition and reinitiation on GCN4 mRNA. Other mutations that disrupt eIF2–eIF3 contacts in the multifactor complex (MFC) diminished 40S-bound TC, indicating that MFC formation enhances 43S assembly in vivo.
Keywords: eIF3, GCN4 translational control, multifactor complex (MFC), PRT1, yeast
Introduction
Initiation of protein synthesis begins with the recruitment of initiator methionyl tRNA (Met-tRNAiMet) in a ternary complex (TC) with eIF2 and GTP to the 40S subunit, to form the 43S preinitiation complex. This is followed by recruitment of mRNA, prebound to the cap-binding complex eIF4F and the poly(A)-binding protein, to form the 48S preinitiation complex. The 43S complex scans the mRNA and AUG recognition triggers GTP hydrolysis in the TC, after which the 60S subunit joins and translation elongation begins. Following ejection of eIF2·GDP, the bound GDP is replaced with GTP by the guanine nucleotide exchange factor eIF2B in order to regenerate TC (Hershey and Merrick, 2000; Hinnebusch, 2000).
Mammalian eIF3 binds to the 40S ribosome and stimulates the binding of both TC and mRNA to 40S subunits in vitro (Hershey and Merrick, 2000; Hinnebusch, 2000). Yeast eIF3, consisting of the five subunits eIF3a/TIF32, eIF3b/PRT1, eIF3c/NIP1, eIF3i/TIF34 and eIF3g/TIF35, can restore binding of Met-tRNAiMet (Danaie et al, 1995; Phan et al, 1998) and mRNA (Phan et al, 2001) to 40S ribosomes in heat-inactivated extracts of the prt1-1 (eIF3b) mutant. Thus, yeast eIF3 performs two key functions ascribed to the mammalian factor. The recruitment of TC to the 40S subunit is also stimulated by eIF1 and eIF1A in vitro (Hershey and Merrick, 2000; Algire et al, 2002; Majumdar et al, 2003); however, the relative importance of these factors and of eIF3 for 43S formation in vivo is unclear.
In yeast, translational control of GCN4 mRNA is a sensitive in vivo indicator of the rate of TC binding to 40S ribosomes. GCN4 translation is activated by amino-acid starvation through a mechanism involving four upstream open reading frames (uORFs 1–4) in GCN4 mRNA. After translating uORF1, many 40S subunits remain attached to the mRNA and resume scanning; however, GCN4 translation is normally repressed because all of these 40S ribosomes rebind the TC before reaching uORF4, translate uORF4, and dissociate from the mRNA. Starvation leads to phosphorylation of eIF2α by GCN2, converting eIF2 from a substrate to a competitive inhibitor of eIF2B and reducing the concentration of TC (Hinnebusch, 1996). This allows ∼50% of the rescanning 40S ribosomes to rebind TC after bypassing uORF4 and reinitiate at GCN4 instead. gcn2Δ mutants fail to induce GCN4 in starved cells and have a Gcn− (general control nonderepressible) phenotype. Mutations in eIF2B that lower TC levels derepress GCN4 under nonstarvation conditions. This Gcd− (general control derepressed) phenotype does not require eIF2α phosphorylation and can be recognized in gcn2Δ cells (Hinnebusch, 1996).
We recently described the first mutation in eIF1A with a Gcd− phenotype, a deletion of the C-terminal 45 residues of the protein (Olsen et al, 2003). Importantly, overexpression of TC from a high-copy (hc) plasmid containing the genes for eIF2α, -β and -γ and tRNAiMet (hc TC) suppressed the Gcd− phenotype of this mutation. This showed that eIF1A is required for optimal TC binding to 40S subunits in vivo and implicated eIF1A in the reinitiation process on GCN4 mRNA. Mutations in eIF3 that reduce TC binding to 40S subunits should also produce a Gcd− phenotype, but none has been isolated.
In yeast, the TC is associated with eIF3, eIF1 and eIF5 in a multifactor complex (MFC) that can exist free of 40S ribosomes (Asano et al, 2000). eIF2 interacts indirectly with NIP1/eIF3c in a manner bridged by eIF5 and it also binds directly to the C-terminal domain (CTD) of TIF32/eIF3a (Asano et al, 2000, 2001; Valášek et al, 2002). The tif5-7A mutation in eIF5 disrupts the indirect contact between eIF2 and eIF3 and leads to temperature-sensitive (Ts−) growth in vivo and diminished TC recruitment in vitro (Asano et al, 1999, 2000). Reducing the direct contact between eIF2 and eIF3 by overexpressing a truncated TIF32 protein lacking the eIF2β-binding domain (hc-TIF32Δ6) confers a slow-growth (Slg−) phenotype that is partially suppressed by hc TC. Combining tif5-7A and hc-TIF32Δ6 produces a synthetic growth defect (Valášek et al, 2002), suggesting that the independent contacts between eIF2 and eIF3 in the MFC have additive effects on TC recruitment. However, the tif5-7A hc-TIF32Δ6 double mutant does not have a Gcd− phenotype, neither does it show reduced binding of TC to 40S subunits in cell extracts (Valášek et al, 2002). Thus, it appears that a reduction in TC recruitment is not the rate-limiting defect conferred by disruption of the MFC in this strain.
In an effort to implicate eIF3 directly in TC recruitment in vivo, we examined the effect of prt1-1 on the level of 43S complexes in mutant cells at the restrictive temperature. This mutation replaces Ser-518 with Phe, and does not affect eIF3 integrity (Phan et al, 2001). Surprisingly, we saw an accumulation of eIF2 on 40S subunits in extracts of heat-treated prt1-1 cells, and confirmed this result using a new technique for cross-linking preinitiation complexes in living cells. Consistent with the idea that 43S assembly is not rate-limiting in the prt1-1 mutant, its Ts− phenotype was not suppressed by hc TC and we observed a strong Gcn− phenotype that likely results from a delay in scanning by reinitiating ribosomes on GCN4 mRNA. Interestingly, prt1-1 also increases the probability of rejecting a non-AUG start codon, implicating eIF3 in AUG recognition. These findings fit well with the fact that an eIF3 ortholog is lacking in prokaryotes, where scanning does not occur and AUG recognition relies heavily on base-pairing between rRNA in the 30S subunit and mRNA.
Results
TC and mRNA binding to 40S subunits is not diminished in prt1-1 cells at the restrictive temperature
We asked whether incubating prt1-1 cells at the restrictive temperature would reduce the amount of eIF2 associated with 40S subunits, as predicted from a defect in 40S binding by the TC. Incubating prt1-1 cells for 20 min at 37°C produced a run-off of polysomes and accumulation of 80S monosomes (Figure 1A, right panel), indicating a severe reduction in translation initiation (Hartwell and McLaughlin, 1968). Substantial amounts of eIF3, eIF2, eIF1 and eIF5 (components of the MFC), and eIF1A were found in the fractions containing free 40S subunits in the wild-type (WT) extracts (Figure 1B, left panel). Unexpectedly, there was an even greater proportion of eIF2 subunits in the 40S fraction from prt1-1 cells (Figure 1B, right panel), at odds with the previous finding that prt1-1 impairs TC binding to 40S subunits in heat-treated extracts (Danaie et al, 1995; Phan et al, 1998).
Figure 1.
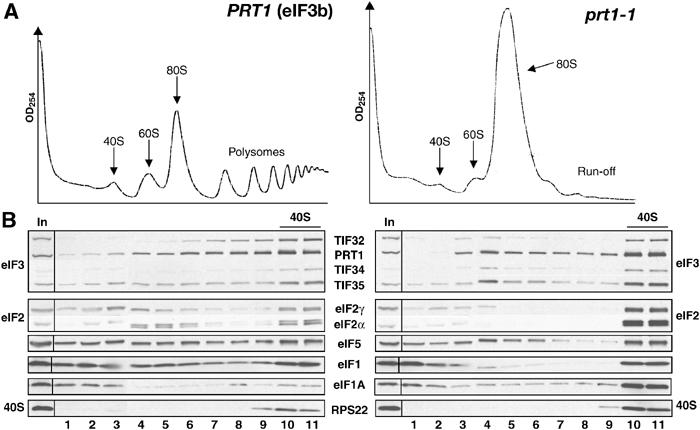
eIF2 remains bound to 40S subunits in extracts of prt1-1 cells incubated at the nonpermissive temperature. (A) Isogenic PRT1 (H2879) and prt1-1 (H1676) cells were grown in YPD medium at 25°C and treated for 20 min at 37°C. Cyclohexamide was added to 50 μg/ml just prior to harvesting and WCEs prepared with heparin (200 μg/ml) in the breaking buffer were separated on a 4.5–45% sucrose gradient by centrifugation at 39 000 r.p.m. for 2.5 h. The gradients were collected and scanned at 254 nm to visualize the ribosomal species. (B) WCEs described in (A) were separated on a 7.5–30% sucrose gradient by centrifugation at 41 000 r.p.m. for 5 h. Proteins were subjected to Western analysis using antibodies against the proteins listed between the blots. An aliquot of each WCE was analyzed in parallel (In, input).
Owing to this discrepancy, we considered the possibility that the results in Figure 1 were an artifact of extract preparation. The binding of MFC components to 40S subunits during sedimentation through sucrose gradients requires heparin in the extraction buffer (Asano et al, 2000), and it was possible that heparin suppressed a defect in eIF2 binding in the prt1-1 extract. To address this possibility, we developed a protocol for cross-linking 43S–48S complexes in vivo by formaldehyde (HCHO) treatment of living cells that eliminated the need for heparin. Cyclohexamide is also omitted because the cross-linking prevents polysome run-off in the extracts (data not shown).
Substantial proportions of eIF2, eIF3 and eIF1A cosedimented with the free 40S subunits in the whole-cell extracts (WCEs) from cross-linked WT cells (Figure 2B, left panel), although smaller proportions of 40S-bound eIF5 and eIF1 were observed compared to the conventional protocol (Figure 1B). A significant proportion of eIF4G also cosedimented with 40S subunits (Figure 2B, left panel), whereas this factor is undetectable in the 40S fraction of WCEs prepared with heparin (data not shown). Total RNA was extracted from each fraction and probed for Met-tRNAiMet and RPL41A mRNA by Northern analysis. The short length of this mRNA, 340 nt, ensures that free mRNP complexes sediment more slowly than 40S subunits. As expected, both tRNAiMet and RPL41A mRNA peaked in the 40S fraction of cross-linked WT cells (Figure 2C, left panel).
Figure 2.
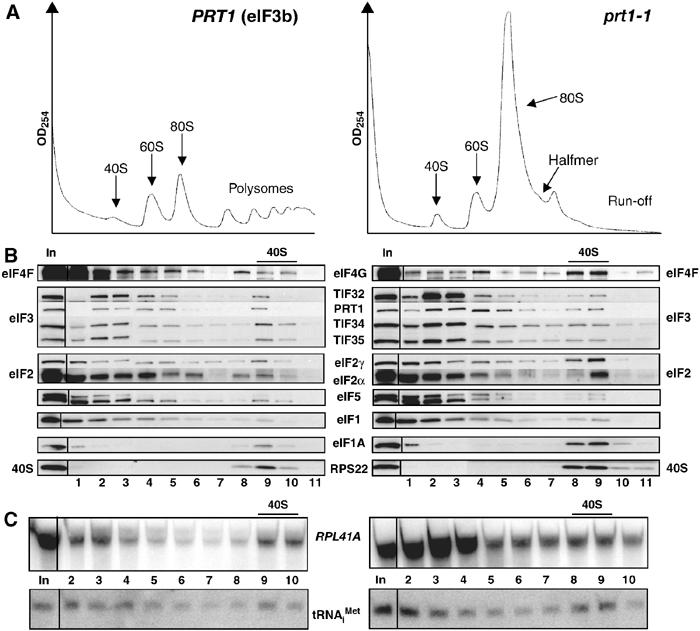
TC and mRNA remain bound to 40S subunits in HCHO-treated prt1-1 cells at the nonpermissive temperature. PRT1 (H2879) and prt1-1 (H1676) cells were grown in YPD at 25°C, heat-treated for 20 min at 37°C, and cross-linked with HCHO for 15 min (A) or 1 h (B, C). (A) WCEs were separated and analyzed as in Figure 1A. (B, C) WCEs were separated and treated as in Figure 1B, except that each fraction was divided and analyzed by Western and Northern blotting.
Comparison of equivalent A260 units of WCEs from HCHO-treated WT and prt1-1 cells revealed the expected reduction in polysomes and accumulation of 80S monosomes in the mutant cells incubated at 37°C (Figure 2A). Interestingly, the 40S fraction from the prt1-1 cells had similar levels of eIF3, eIF5 and eIF1, and relatively greater amounts of eIF2, eIF4G, eIF1A, tRNAiMet and mRNA compared to that seen in WT (Figure 2B and C). Quantification of the results from several experiments revealed that the amounts of 40S-bound eIF2γ, tRNAiMet and mRNA were increased in the prt1-1 cells by 166±58, 180±10 and 250±60%, respectively. The amount of free 40S subunits was generally greater in the mutant cells due to polysome run-off. If we normalize the amounts of 40S-bound eIF2, tRNAiMet and mRNA for the RPS22 signals, then the levels of these 40S-bound factors are nearly identical between prt1-1 and WT cells. However, because it is unknown whether 40S subunits are limiting for 43S/48S formation, we chose to compare the absolute levels of 40S-bound factors determined in replicate experiments. Regardless of how the data are quantified, the results indicate that 40S binding of TC, mRNA and eIF4G is not diminished in prt1-1 cells at 37°C. We also found that similar amounts of eIF2 and eIF4G were present in the 80S and polysome fractions of WT and prt1-1 mutant cells at 37°C, representing ∼50% of the total pools of these factors, despite the low polysome content of the mutant at 37°C (data not shown). Thus, high levels of eIF2 and eIF4G are associated with both free and polysome-associated 48S complexes in the mutant. We conclude that the rate-limiting defect occurs at a step following assembly of 48S complexes in prt1-1 cells at 37°C.
To validate the cross-linking technique, we wished to show that mutations in eIF2 would reduce the 40S binding of tRNAiMet in cross-linked cells. Hence, we constructed a strain harboring a temperature-sensitive degron allele, sui3-td, encoding a fusion protein containing ubiquitin and a thermolabile dihydrofolate reductase moiety attached to the N-terminus of eIF2β, and expressed from a copper-dependent promoter. The strain also expresses the ubiquitin ligase UBR1 from a galactose-inducible promoter. Shifting the sui3-td cells from medium containing copper and raffinose at 25°C to medium containing galactose and lacking copper at 36°C represses new synthesis of the degron-tagged protein (eIF2βtd), and the pre-existing eIF2βtd is rapidly eliminated by proteosomal degradation (Dohmen et al, 1994; Labib et al, 2000). After incubating under the nonpermissive condition for 16 h to deplete eIF2βtd, the sui3-td cells were treated with HCHO to cross-link the 43–48S complexes.
As expected, we observed polysome run-off in the cross-linked sui3-td cells under the nonpermissive condition (Figure 3A). Furthermore, eIF2βtd was undetectable and little or no eIF2γ and eIF2α cosedimented with free 40S subunits (Figure 3B, right panel). Importantly, the amount of tRNAiMet in the 40S fraction (Figure 3C, right panel) declined to 14±4% of that seen under permissive conditions. There was little reduction in 40S binding of eIF3 subunits, and a small increase in 40S-bound eIF1A under the nonpermissive condition (Figure 3B), indicating a specific loss of TC from preinitiation complexes. We also examined the effects of a nonconditional mutation in eIF2γ, gcd11-506, which reduces the affinity of eIF2 for Met-tRNAiMet in vitro (Erickson and Hannig, 1996). Consistently, we observed a threefold reduction in the amount of eIF2 on 40S subunits in cross-linked gcd11-506 cells compared to WT (data not shown).
Figure 3.
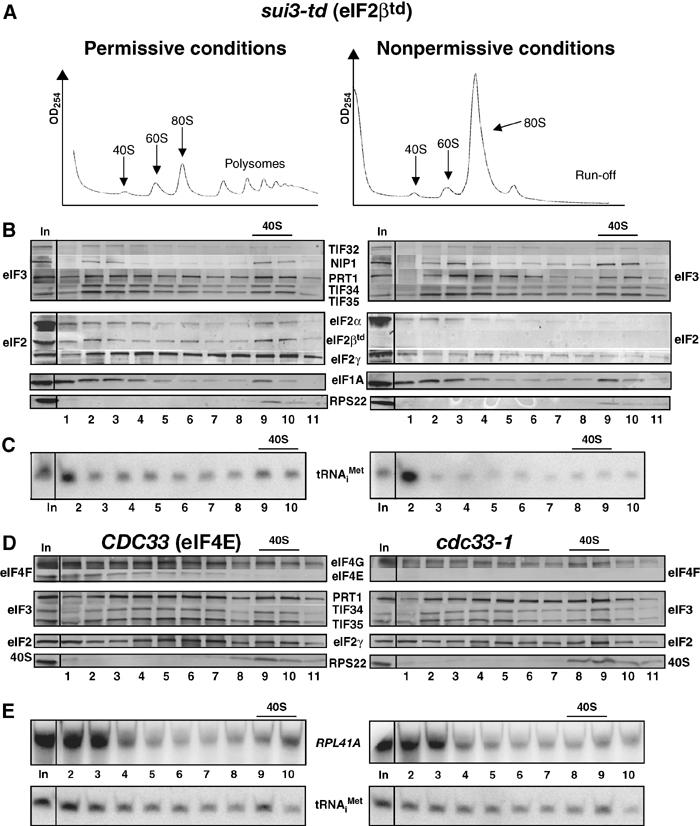
sui3-td and cdc33-1 mutations reduce the binding of TC and mRNA to 40S subunits in vivo. (A–C) sui3-td (YAJ18-3) cells were grown in SC-raffinose in the presence of 0.1 M copper sulfate at 25°C and shifted to SC-raffinose+galactose in the absence of copper and grown overnight at 37°C. Cells were cross-linked with HCHO and analyzed as described in Figure 2A–C, except that eIF2βtd was detected in (B) using HA antibody. (D) Strain F324 was transformed with YEplac195-CDC33-URA3 (CDC33) or empty vector (cdc33-1) and the resulting transformants were grown in SC-Ura medium at 25°C, heat treated for 2 h at 37°C, and cross-linked with HCHO. WCEs were prepared and analyzed as (B–C).
We next examined the cdc33-1 mutant to examine the consequences of depleting the cap-binding protein, eIF4E, on mRNA recruitment. Incubating these cells at 37°C for 2 h leads to the disappearance of eIF4E, presumably due to proteolytic degradation (Figure 3D). The amount of 40S-bound mRNA also declined in the cdc33-1 cells at 37°C to 47±8% of the level observed in WT cells, while the level of tRNAiMet was reduced to only 86±10% of WT (Figure 3E). The ability of eIF4G to interact with both poly(A)-binding protein and eIF4E (Sachs, 2000) likely accounts for the residual mRNA binding to 40S ribosomes in cdc33-1 cells at 37°C. As expected, we saw little reduction in the amounts of eIF3 or eIF2 subunits bound to free 40S subunits in the cdc33-1 mutant (Figures 3D). Thus, it appears that HCHO cross-linking provides a faithful depiction of the composition of preinitiation complexes in living cells.
If the rate-limiting defect in prt1-1 cells is downstream from 48S complex assembly, we would expect to observe a halfmer shoulder on the 80S monosome formed by mRNAs containing an elongating 80S ribosome plus a 48S complex in the mRNA leader. As most monosomes in this mutant are 80S couples lacking mRNA (data not shown), the concentration of such 1½-mers should be small. Nevertheless, we consistently detected a halfmer on the 80S peak in cross-linked prt1-1 cells incubated for 20 min or 1 h at 37°C (Figure 4B and data not shown). In some experiments, the 1½-mer was less prominent (Figure 2A, right panel), possibly due to the shorter HCHO treatment in those cases; however, we never detected a 1½-mer peak in cdc33-1 (Figure 4D) or sui3-td cells (Figure 3A, right panel). (We attribute the absence of the halfmer peak in Figure 1A to the presence of heparin, which could compete with mRNA for 40S binding.)
Figure 4.
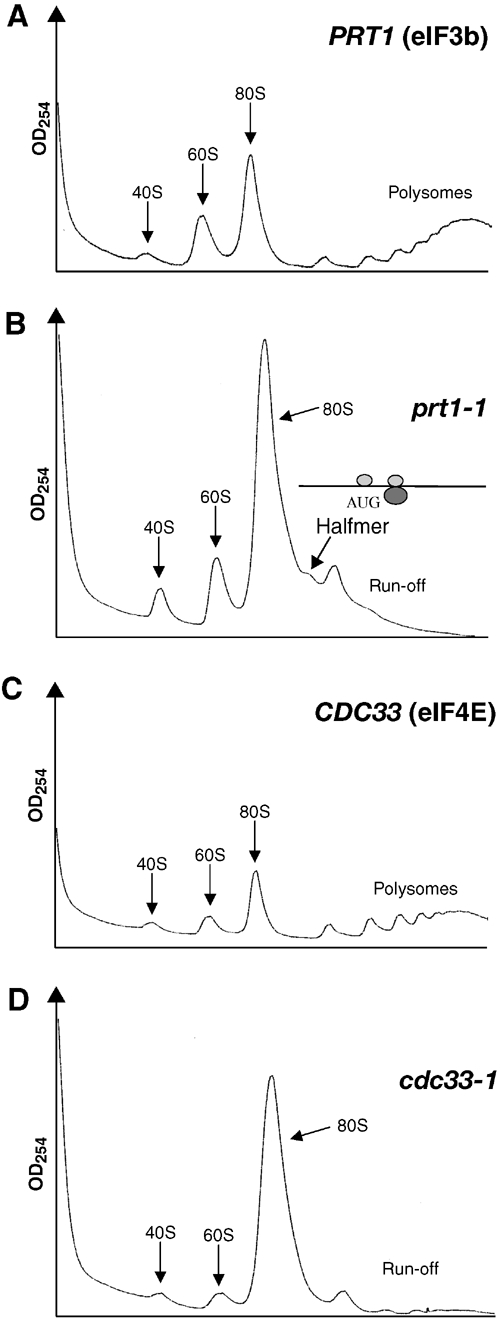
The prt1-1 mutant displays a halfmer phenotype at the nonpermissive temperature. Polysome profiles of (A) PRT1 (H2879) and (B) prt1-1 (H1676) cells after 20 min at 37°C. The halfmer shoulder on the 80S peak is indicated along with an explanatory schematic of a 1½-mer (see text). Polysome profiles of the (C) CDC33 and (D) cdc33-1 transformants described in Figure 3D after 2 h at 37°C.
Genetic evidence that TC binding to 40S subunits is not rate-limiting in prt1-1 cells
If binding of TC to 40S ribosomes was the principal deficiency in the prt1-1 mutant, then its growth defect should be reduced by overexpressing TC from an hc plasmid. As expected, hc TC suppressed the growth defect of a gcd1-502 mutant containing a defective subunit of eIF2B (Figure 5A, right panels) (Dever et al, 1995). By contrast, the growth defect of prt1-1 cells at a semipermissive temperature of 33°C was not suppressed by hc TC (Figure 5A, left panels).
Figure 5.
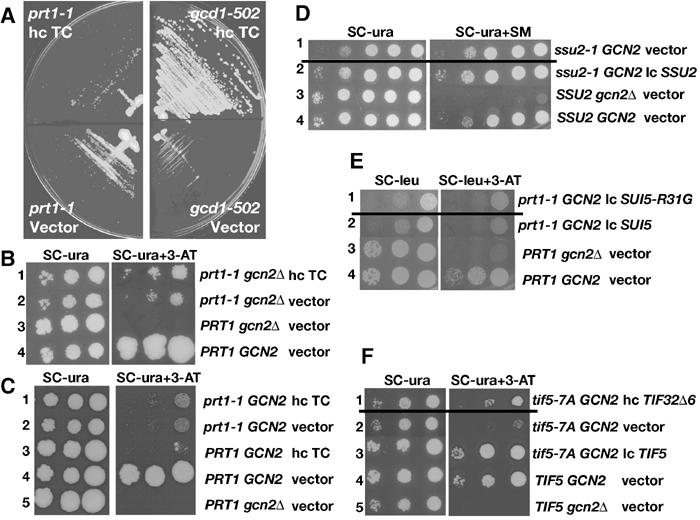
The prt1-1 strain has a strong Gcn− phenotype and its Ts− phenotype is not suppressed by TC overexpression. (A) prt1-1 (H1676) and gcd1-502 (H70) cells were transformed with hc plasmid p3000 encoding hc TC, or empty vector, streaked on SC-Ura plates and incubated for 4 (left panels) or 2 days (right panels) at 33°C. (B) prt1-1 (YKHN60) cells were transformed with p3000 (hc TC) or empty vector, grown overnight in SC-Ura, and 10-fold serial dilutions were spotted in rows 1 and 2 on SC-Ura plates or SC-Ura-His plates containing 40 mM leucine with 30 mM 3-AT and incubated for 7 and 10 days, respectively, at 33°C. PRT1 GCN2 (H2879) and PRT1 gcn2Δ (H2881) cells transformed with empty vector were analyzed in parallel in rows 3 and 4. (C) PRT1 (H2879) and prt1-1 (H1676) cells were transformed with p3000 (hc TC) or empty vector and analyzed essentially as in (B). (D) ssu2-1 (F708) cells were transformed with empty vector or p3342 encoding SSU2 (TIF5), and analyzed in rows 1 and 2 as in (B), except using SC-Ura and SC-Ura-Ile-Val+1 μg/μl SM plates and incubating for 2 or 3 days (row 1) and 1 or 2 days (rows 2–4) at 30°C. SSU2 gcn2Δ (H2881) and SSU2 GCN2 (H2879) cells, transformed with empty vector, were analyzed in parallel in rows 3 and 4. (E) prt1-1 (H1676) cells were transformed with p3993 encoding SUI5-R31G or p3992 encoding SUI5 (TIF5) and analyzed essentially as in (B), except using SC-Leu plates or SC-Leu-His plates containing 10 mM 3-AT and incubating for 5 (row 1) or 3 days (row 2) at 33°C. PRT1 gcn2Δ (H2881) and PRT1 GCN2 (H2879) cells, transformed with empty vector, were analyzed in parallel in rows 3 and 4, incubating for 2 days. (F) tif5-7A GCN2 (YKHN206) cells were transformed with p3927 encoding TIF32Δ6–His or empty vector or p3342 encoding TIF5 and analyzed essentially as in (B), except that plates were incubated at 30°C for 2 or 3 days (rows 2–5) and 3 or 4 days (row 1) due to the synthetic growth defect of the tif5-7A TIF32Δ6–His strain. TIF5 GCN2 (YKHN205) and TIF5 gcn2Δ (H2898), transformed with empty vector, were analyzed in parallel in rows 4 and 5 and incubated for 2 or 3 days.
If prt1-1 lowers the rate of TC binding to 40S subunits, then it should confer a Gcd− phenotype. Gcd− mutations suppress the sensitivity of gcn2Δ cells to 3-aminotriazole (3-AT), an inhibitor of the HIS3 product, because they derepress GCN4 translation, with attendant derepression of HIS3, independently of eIF2α phosphorylation by GCN2. We found that prt1-1 gcn2Δ cells displayed a weak Gcd− phenotype, allowing slightly better growth on 3-AT plates at 33°C compared to PRT1 gcn2Δ cells (Figure 5B, rows 2 and 3). This weak Gcd− phenotype was not suppressed by hc TC, however (Figure 5B, rows 1 and 2), suggesting that it does not result from impaired TC recruitment.
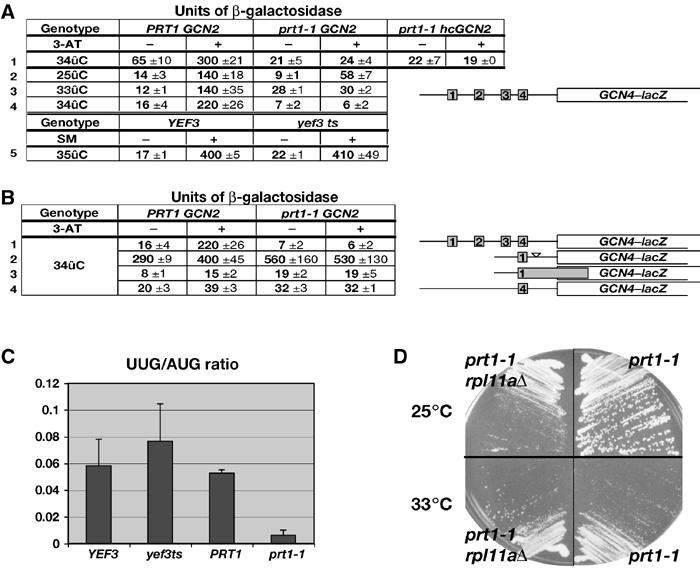
Unexpectedly, prt1-1 cells containing GCN2 are sensitive to 3-AT (3-ATs) at 33°C (Figure 5C, rows 2 and 4), the phenotype characteristic of Gcn− mutants. Consistently, derepression of a GCN4–lacZ reporter containing all four uORFs was conditionally defective in the prt1-1 GCN2 cells, showing nearly WT induction by 3-AT at 25°C, but no induction at 33°C (Figure 6A, rows 2–4, 28 versus 30 U). Compared to the PRT1 gcn2Δ strain, GCN4–lacZ was partially derepressed under noninducing conditions at 33°C in the prt1-1 GCN2 mutant (weak Gcd− phenotype) but did not increase further upon 3-AT induction. At 34°C, we observed only the strong Gcn− phenotype in the prt1-1 GCN2 strain (Figure 6A, row 4, 7 versus 6 U), suggesting that it reflects the most severe defect produced by prt1-1.
Figure 6.
The prt1-1 mutation impairs GCN4 translational control, leads to hyperaccurate start codon selection, and does not show a synthetic growth defect with depletion of 60S subunit protein RPL11A. (A) prt1-1 (H1676), PRT1 (H2879), YEF3 (F1006) or yef3 (F650S) (F1007) cells were transformed with p180 containing the GCN4–lacZ fusion with all four uORFs and grown in SC-Ura in the presence or absence of 10 mM 3-AT, or 0.06 μg/μl SM, as shown, at the indicated temperatures. In row 1, the prt1-1 strain was also transformed with hc LEU2 plasmid (p832) containing GCN2 (hcGCN2) or empty vector, and the PRT1 strain also harbored an empty vector. β-Galactosidase activities were measured in WCEs and expressed in units of nmol of o-nitrophenyl-β-D-galactopyranoside hydrolyzed per min per mg of protein. The mean activities and standard errors obtained from independent transformants are indicated. (B) The prt1-1 (H1676) and PRT1 (H2879) strains were transformed with plasmids p180, pM199, pM226 or p226 (rows 1–4), respectively, and contained the GCN4–lacZ constructs shown schematically to the right and analyzed as in (A). Row 1 contains the same data as in row 4 of (A) shown for comparison. (C) prt1-1 (H1676), PRT1 (H2879), YEF3 (F1006) or yef3 ts (F1007) cells were transformed with p367 or p391 containing a HIS4–lacZ reporter harboring AUG or UUG start codons, respectively. The PRT1 and prt1-1 transformants were grown at 34°C, while the YEF3 and yef3 ts transformants were grown at 35°C, in SC-Ura medium, and β-galactosidase was assayed in WCEs. The graph shows the mean ratios of expression from the UUG to the AUG reporter measured for four independent transformants of each strain (for each reporter), with standard errors indicated as error bars. (D) Growth of prt1-1 (H1676) and prt1-1 rpl11aΔ (H2925) strains in YPD medium for 3 days at 25°C (upper panel) or 4 days at 33°C (lower panel).
The Gcn− phenotype of prt1-1 cells could arise from a defect in eIF2α phosphorylation by GCN2. Western analysis using antibodies specific for eIF2α phosphorylated on Ser-51 (eIF2α-P) revealed an ∼40% reduction in the level of eIF2α-P in the prt1-1 mutant grown at 34°C with 10 mM 3-AT. Introducing GCN2 on an hc plasmid into the prt1-1 strain restored eIF2α-P to levels greater than or equal to those seen in the PRT1 strain (data not shown), but derepression of GCN4–lacZ expression remained impaired (Figure 6A, row 1). Hence, the partial reduction in eIF2α phosphorylation in the prt1-1 strain cannot account for its strong Gcn− phenotype.
It was possible that the reduced rate of translation initiation in prt1-1 cells at 34°C would diminish the consumption of TC and restore TC to high levels even when eIF2α is phosphorylated. To test this, we asked whether a Ts− mutation in elongation factor eEF3 (encoded by YEF3) would produce a Gcn− phenotype at a semipermissive temperature (35°C) where the doubling time of the yef3 mutant (∼20 h) is similar to that of prt1-1 cells at 34°C. The yef3 mutant showed no Gcn− phenotype at 35°C (Figure 6A, row 5), making it improbable that the Gcn− phenotype of prt1-1 cells results from a reduced rate of translation.
A third possibility to explain the Gcn− phenotype of prt1-1 cells would be if 40S subunits cannot resume scanning following translation of uORF1, as only rescanning subunits can bypass uORFs 2–4 and reinitiate at GCN4 (Mueller and Hinnebusch, 1986). For example, 40S subunits might dissociate from the mRNA at the uORF1 stop codon or while scanning downstream from uORF1. To address this possibility, we examined a GCN4–lacZ construct containing only uORF1 and lacking the segment containing uORFs 2–4, (pM199). Expression of this construct is constitutively high in WT cells because a large proportion of ribosomes that translate uORF1 can resume scanning and reinitiate at GCN4 regardless of TC levels (Grant et al, 1994). As shown in Figure 6B (row 2), there was no significant difference in the expression of this construct between prt1-1 and PRT1 cells in the presence of 3-AT. Hence, the proportion of ribosomes able to resume scanning and reach GCN4 following uORF1 translation was unaltered by prt1-1. The same result was obtained for a construct containing solitary uORF1 at its natural distance from GCN4 (data not shown).
The fourth mechanism we addressed was the possibility that ribosomes leaky scan uORF1 in prt1-1 cells, making it impossible for them to bypass the remaining uORFs and reinitiate at GCN4. To test this possibility, we analyzed a GCN4–lacZ construct in which uORF1 is elongated and overlaps the beginning of GCN4. This elongated version of uORF1 blocks 98–99% of all scanning ribosomes from reaching the GCN4 start site, indicating that only 1–2% of ribosomes leaky scan uORF1 in WT cells (Grant et al, 1994). We observed little or no increase in GCN4–lacZ expression from this construct under 3-AT-inducing conditions in prt1-1 cells (Figure 6B, row 3). We also observed no increase in leaky scanning past uORF4 by assaying a construct containing solitary uORF4 at its normal location (Figure 6B, row 4). The results in rows 2–4 additionally eliminate the possibility that induction of GCN4–lacZ is reduced in prt1-1 cells by a reduction in reporter mRNA level, as this effect should apply equally to the mutant constructs containing solitary uORFs 1 or 4, yet expression of these constructs was unaffected by prt1-1.
The fact that eliminating uORFs 2–4 completely suppresses the inability of prt1-1 cells to induce GCN4 expression (Figure 6B) provides strong genetic evidence that the Gcn− phenotype results from the inability of rescanning ribosomes to bypass uORFs 2–4 under starvation conditions when eIF2 is highly phosphorylated (Mueller and Hinnebusch, 1986). We envision two ways in which this could occur: (i) a reduction in the rate of scanning or (ii) a delay in GTP hydrolysis by 48S complexes at the start codons of uORFs 2–4, creating a barrier to movement of all 40S subunits through the leader to GCN4. Either defect would increase the time required for 40S subunits lacking TC to scan past uORFs 2–4 and thereby compensate for the reduced rate of TC binding that results from eIF2α phosphorylation.
If a delay in GTP hydrolysis was responsible for the Gcn− defect in prt1-1 cells, then the 3-ATs phenotype should be reduced by expressing eIF5–G31R (encoded by SUI5-R31G), as eIF5–G31R has greater than WT activity in stimulating GTP hydrolysis by 40S-bound TC in vitro (Huang et al, 1997). However, SUI5-R31G did not alter the 3-ATs phenotype of prt1-1 even though it reduced the growth rate in nonstarvation medium when introduced into the prt1-1 strain on a low-copy (lc) plasmid (Figure 5E, rows 1 and 2). (To compensate for its Slg− phenotype, the prt1-1 lcSUI5-R31G strain was incubated 2 days longer than the prt1-1 lcSUI5 control strain.) We also asked whether the recessive Ts− ssu2-1 mutation in eIF5, shown to impair GAP activity in vitro (Asano et al, 2001), would mimic prt1-1 and confer a Gcn− phenotype. As shown in Figure 5D (rows 1 and 2), this was not the case, even though this mutation confers an Slg− phenotype. Thus, alterations in the GAP function of eIF5 do not impair the regulation of GCN4 translation, pointing to a delay in scanning as the more likely explanation for the Gcn− phenotype of prt1-1 cells.
The tif5-7A mutation in eIF5, which disrupts the connection between eIF2 and eIF3 in the MFC (Asano et al, 2000), does not affect GAP function in vitro, and we argued previously that it impairs the scanning process as the rate-limiting defect in vivo (Asano et al, 2001). Interestingly, tif5-7A shows a moderate Gcn− phenotype (Figure 5F, rows 2 and 3), consistent with the idea that optimal scanning is dependent on MFC integrity. Furthermore, the Gcn− phenotype of tif5-7A is suppressed by overexpressing the truncated eIF3a subunit TIF32-Δ6–His (Figure 5F, rows 1 and 2). As shown below, overexpressing TIF32-Δ6–His impairs TC binding to 40S subunits in tif5-7A cells. Hence, we propose that a scanning delay conferred by tif5-7A is compensated by the reduced TC binding produced by hc-TIF32-Δ6–His, restoring efficient GCN4 induction in the double mutant.
The stringency of AUG recognition is altered in prt1-1 cells
To investigate the possibility that prt1-1 alters the stringency of AUG selection during scanning, we measured the expression of HIS4–lacZ reporters containing either UUG or AUG as a start codon. Interestingly, the selection of UUG was dramatically reduced in the mutant at 34°C, as the ratio of expression from the UUG versus AUG reporters (UUG/AUG ratio) was lower by a factor of 8–9 in prt1-1 versus WT cells (Figure 6C). By contrast, the yef3 Ts− mutation had little or no effect on the UUG/AUG ratio at a semipermissive temperature of 35°C. Northern analysis confirmed that the HIS4–lacZ mRNAs containing UUG or AUG were present at similar levels in prt1-1 cells at 34°C (data not shown).
Evidence that subunit joining is not the rate-limiting defect in prt1-1 cells
Following hydrolysis of GTP by the TC, joining of the 60S subunit produces the 80S initiation complex in a reaction stimulated by eIF5B, encoded by FUN12. We wished to determine whether prt1-1 impairs this final step in the pathway. Deletion of FUN12 causes a strong Slg− phenotype (doubling time (Td) of 5 versus 2 h for WT). The rpl11aΔ mutant, lacking one of two genes encoding 60S ribosomal protein RPL11, exhibits reduced subunit joining and an Slg− phenotype (Td=3 h) resulting from a decreased amount of 60S subunits (Rotenberg et al, 1988). Combining fun12Δ and rpl11AΔ leads to a synthetic growth defect (Td=8 h), consistent with an additive impairment of subunit joining in the double mutant. By contrast, the prt1-1 rpl11aΔ double mutant grows faster than the single prt1-1 mutant at semipermissive temperature (Figure 6D), with doubling times of 15 and 22 h for the prt1-1 rpl11aΔ and prt1-1 mutants, respectively, at 34°C. These results are inconsistent with the idea that the prt1-1 mutant has a rate-limiting defect in 40S–60S subunit joining.
Biochemical evidence from in vivo cross-linking that eIF2–eIF3 contacts in the MFC enhance TC recruitment in vivo
We showed previously that overexpressing TIF32-Δ6–His leads to the formation of a defective MFC lacking eIF2, due to loss of the TIF32–CTD/eIF2β interaction (Valášek et al, 2002). The tif5-7A mutation in eIF5 disrupts the indirect contact between eIF2β and eIF3c/NIP1 (see the schematic in Figure 7B). Combining tif5-7A and hc-TIF32-Δ6–His produces a synthetic reduction in translation initiation and growth rate (Asano et al, 1999; Valášek et al, 2002). Although we expected to observe a reduction in TC binding to 40S subunits in the tif5-7A hc-TIF32-Δ6–His double mutant, this was not observed in WCEs prepared with heparin (Valášek et al, 2002). Hence, we used in vivo cross-linking to re-examine this prediction here.
Figure 7.
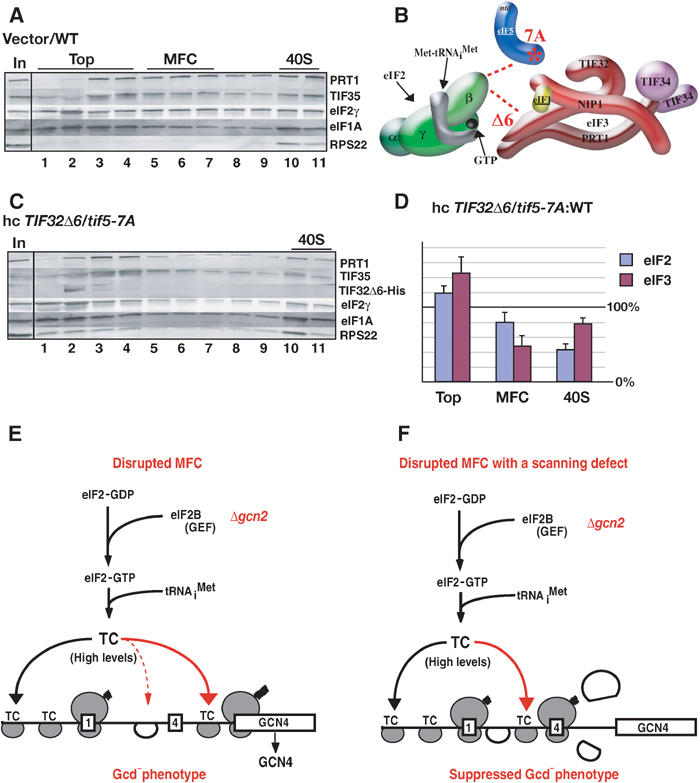
Redistribution of eIF2 from the 40S-bound to the 40S-free state in the hc-TIF32Δ6 tif5-7A mutant (A–D) and schematic model to explain the differing effects of mutations in MFC components on GCN4 translational control (E). (A) WT strain H2898 transformed with empty vector and (C) tif5-7A strain H2899 transformed with hc plasmid p3927 containing TIF32Δ6–His were grown in SC-Ura medium and cross-linked with HCHO. WCEs were separated and analyzed as in Figure 2B. (B) Schematic representation of the defective MFC in hc-TIF32Δ6 tif5-7A cells lacking the contact between eIF2β and TIF32–CTD and that between eIF2β and NIP1–NTD bridged by eIF5 (see text). (D) The amounts of eIF2γ (eIF2) or PRT1 (eIF3) in fractions 1–4 (top), 5–7 (MFC), and 10–11 (40S) were quantified with a PhosphorImager or by videodensitometry using NIH image 1.63 software, and the resulting values for the hc-TIF32Δ6 tif5-7A strain were normalized to the corresponding WT values. The results from four independent experiments were averaged and the mean normalized values and standard errors were plotted, with 100% corresponding to the WT values. The assignment of the MFC to fractions 5–7 was based on our previous analysis of mutant MFC complexes (Valášek et al, 2003). (E) Mutations in MFC components (prt1-1, TIF32Δ6 or tif5-7A) that decrease the rate of TC binding to 40S subunits scanning downstream from uORF1 should allow a fraction of 40S subunits to bypass uORF4 and reinitiate at GCN4, even in the absence of eIF2α phosphorylation in gcn2Δ cells where TC levels are high (Gcd− phenotype). (F) The rate-limiting defects conferred by mutations in MFC components reduce the rate of scanning by 40S subunits following uORF1 translation. This compensates for the reduced rate of TC recruitment caused by these mutations and thereby suppresses the Gcd− phenotype predicted from the recruitment defects.
As expected (Valášek et al, 2003), TIF32-Δ6–His did not bind to 40S ribosomes in tif5-7A cells and was located at the top of the gradient (Figure 7C). The eIF2γ and eIF3 subunits were present in the 40S fraction of the hc-TIF32-Δ6–His tif5-7A mutant (Figure 7A and C); however, quantification of four replicate experiments showed a reduction to 43±8% of WT in the proportion of eIF2 in the 40S fraction, plus an increased proportion of unbound eIF2 at the top of the gradient (Figure 7D). The eIF3 subunits also showed a redistribution from the 40S-bound to unbound fractions and a decreased proportion in the MFC in the mutant cells (Figure 7D). These results indicate that destabilizing the MFC reduces TC recruitment in vivo. Our ability to detect a reduced level of 40S-bound eIF2 in the slow-growing tif5-7A hc-TIF32-Δ6–His cells, but not in the growth-arrested prt1-1 cells at 37°C, underscores our conclusion that prt1-1 impairs a critical postassembly function of eIF3. The fact that the reduction in TC binding is not greater than ∼60% in tif5-7A hc-TIF32-Δ6–His cells, despite the marked reduction of polysomes (Valášek et al, 2002), is likely due to the fact that impaired TC binding is not the only defect in this double mutant.
Discussion
Yeast eIF3 has a critical function downstream of 48S assembly
The prt1-1 (eIF3b) mutant shows a severe defect in 40S binding by Met-tRNAiMet in heat-treated extracts (Danaie et al, 1995; Phan et al, 1998). However, we observed greater than WT amounts of eIF2 and tRNAiMet associated with 40S subunits in prt1-1 cells at the nonpermissive temperature, despite nearly complete inhibition of initiation (Figure 2A–C). Consistently, overexpression of TC did not reduce the Ts− phenotype of prt1-1 cells at a semipermissive growth temperature (Figure 5A). We also observed an accumulation of eIF4G and RPL41A mRNA bound to 40S subunits (Figure 2B and C) and the appearance of a 48S halfmer on the 80S peak in prt1-1 cells at 37°C. Thus, we conclude that the rate-limiting defect in the prt1-1 mutant lies downstream of 48S assembly. We presented genetic evidence that 40S–60S subunit joining is not the rate-limiting defect in prt1-1 cells by showing that prt1-1 did not exacerbate the subunit joining defect in rpl11aΔ cells produced by a low level of 60S subunits. Together, our findings place the rate-limiting defect in prt1-1 cells at one or more steps between 48S assembly and subunit joining, which include scanning, AUG recognition or GTP hydrolysis by the TC.
Our data do not necessarily indicate that binding of TC and mRNA to 40S subunits occurs at WT rates in prt1-1 cells at 37°C. If prt1-1 has a greater effect on conversion of 48S to 80S complexes than on 48S formation, there will be a net accumulation of 48S complexes at 37°C, as we observed. It is worth noting that endogenous eIF3, eIF2, eIF1 and eIF5 were found dissociated from 40S ribosomes when prt1-1 extracts were heat treated at 37°C (Phan et al, 2001). Thus, the prt1-1 product may be fully inactivated in vitro, but only partially impaired in vivo, producing a strong defect in 48S assembly only in extracts.
Our finding that prt1-1 cells are more accurate than WT in selecting AUG versus UUG as the start codon, a hyperaccurate initiation (Hai−) phenotype (Figure 6C), implicates eIF3 in AUG selection. A higher rate of eIF5-stimulated GTP hydrolysis by the TC increases the utilization of a near-cognate start codon (Sui− phenotype) (Huang et al, 1997). Thus, the Hai− phenotype of prt1-1 mutant could result from a delay in GTP hydrolysis, although we discounted this defect as an explanation for its Gcn− phenotype. A reduced rate of scanning might indirectly affect AUG selection, but it is difficult to predict whether it would favor or disfavor the use of near-cognate codons. A third possibility is that prt1-1 could destabilize Met-tRNAiMet base-paired to near-cognate start codons in the P-site.
Mutations that impair TC recruitment confer a Gcd− phenotype by allowing ribosomes to scan past uORFs 2–4 and reinitiate at GCN4 in the absence of eIF2α phosphorylation. A C-terminal truncation of eIF1A has a Gcd− phenotype that can be suppressed by hc TC, providing evidence that eIF1A stimulates 43S complex formation in vivo (Olsen et al, 2003). By contrast, the prt1-1 mutant does not show a Gcd− phenotype that is suppressible by hc TC (Figure 5B); moreover, it fails to induce GCN4 translation when eIF2α is phosphorylated under starvation conditions (Figures 5C and 6A)—a strong Gcn− phenotype. As removal of uORFs 2–4 overcomes the effect of prt1-1 on GCN4 translation, we can attribute the Gcn− phenotype to the failure of any 40S ribosomes rescanning after uORF1 translation to bypass uORFs 2–4 when TC levels are reduced by eIF2α phosphorylation (Mueller and Hinnebusch, 1986). Interestingly, deletion of eIF5B leads to a strong Gcn− phenotype that arises partly from an increase in leaky scanning past uORF1 (Choi et al, 1998; Lee et al, 2002; Shin et al, 2002). We ruled out this possibility for prt1-1, indicating distinct mechanisms for the Gcn− phenotypes of fun12Δ and prt1-1 mutants.
The simplest way to explain the Gcn− phenotype of prt1-1 cells is to propose a decrease in the rate of scanning. This would compensate for the reduced rate of TC binding produced by eIF2α phosphorylation and ensure that all 40S subunits will rebind TC before reaching uORF4 (Figure 7E and F). This would be equivalent to increasing the distance between uORFs 1 and 4, which also produces a Gcn− phenotype (Abastado et al, 1991). An alternative possibility would be that the fraction of rescanning ribosomes which rebind TC in the uORF1–uORF4 interval become stalled at AUG codons 2, 3 or 4 by a defect in GTP hydrolysis. This would produce a roadblock at these uORFs and increase the time required for all free 40S ribosomes to reach uORF4. This latter mechanism is disfavored by our finding that expressing eIF5–R31G, shown to have hyperactive GAP function (Huang et al, 1997), did not diminish the Gcn− phenotype of prt1-1 cells (Figure 5E). Moreover, a mutant containing the GAP-defective eIF5–G62S protein (Asano et al, 2001) does not exhibit a Gcn− phenotype (Figure 5D). On the other hand, the tif5-7A allele, whose product shows WT GAP function (Asano et al, 2001), does confer a moderate Gcn− phenotype (Figure 5F) and we presented evidence consistent with defective scanning in this strain (Asano et al, 2001). Thus, we favor the model that the Gcn− phenotype of prt1-1 results from a defect in scanning rather than impaired GTP hydrolysis by eIF2. There is evidence that eIF1 is required for efficient scanning by a 48S complex in vitro (Pestova et al, 1998); hence, prt1-1 could impair scanning indirectly by impeding eIF1 function. Indeed, prt1-1 weakens the association between the eIF3/eIF5 subcomplex and eIF1 (Phan et al, 2001).
Integrity of the MFC is required for optimal TC recruitment in vivo
Our previous genetic data led us to conclude that the direct eIF2β–TIF32 contact in the MFC disrupted by hc-TIF32-Δ6 and the indirect eIF2β–NIP1 contact eliminated by tif5-7A have additive stimulatory effects on TC recruitment (Valášek et al, 2002). However, we were unable to detect a reduction in eIF2 binding to 40S subunits in heparin-stabilized extracts. Here, using HCHO cross-linking, we consistently observed an ∼60% reduction in eIF2 binding to 40S subunits in the hc-TIF32-Δ6 tif5-7A double mutant (Figure 7C and D). We have also confirmed by in vivo cross-linking that TC binding to 40S subunits is not significantly impaired in the tif5-7A single mutant (data not shown). These data provide the first biochemical evidence that disrupting multiple contacts between eIF2 and eIF3 in the MFC reduces TC recruitment in vivo.
Despite the reduced TC recruitment in the hc-TIF32-Δ6 tif5-7A mutant, this strain does not show a Gcd− phenotype (Valášek et al, 2002). The solution to this paradox may be provided by our finding that the tif5-7A single mutant has a Gcn− phenotype, which is suppressed by overexpressing TIF32-Δ6–His (Figure 5F). Thus, the reduced rate of scanning conferred by tif5-7A could compensate for the diminished TC binding that results from TIF32-Δ6–His overexpression. This would restore efficient reinitiation at uORFs 2–4 and suppress the Gcd− phenotype expected from defective TC recruitment (Valášek et al, 2002) (Figure 7E and F). Presumably, prt1-1 confers a Gcn− phenotype because it has a relatively stronger effect on scanning and a minimal impact on TC binding.
Concluding remarks
Our combined genetic and biochemical analysis of the prt1-1 mutant has allowed us to demonstrate that eIF3 has critical functions downstream of the 48S complex assembly and prior to 60S subunit joining, which impact ribosomal scanning on GCN4 mRNA and AUG selection. Similarly, the results described here and elsewhere (Asano et al, 2001; Valášek et al, 2002) indicate that disrupting contacts between eIF3 and eIF2 in the MFC reduces the rate of TC recruitment in vivo and also impairs postassembly functions of the 48S complex involved in GCN4 control. Several key results supporting these conclusions were obtained using HCHO cross-linking of live cells to fix native initiation complexes. This new technique obviates the use of stabilizing compounds that alter the composition or abundance of 48S complexes, and it was shown previously that HCHO cross-linking also prevents artifactual 43S formation in cell extracts (Kumar et al, 1989). The experiments we conducted to validate this technique gave interesting results in their own right, which deserve further exploration. These include the fact that eIF1A and eIF3 bind to 40S subunits independently of eIF2, that the affinity of eIF2 for the 40S subunit is promoted by interaction with Met-tRNAiMet, and that high-level binding of eIF4G to 40S subunits can occur in the absence of eIF4E in vivo.
The Gcn− phenotypes of prt1-1 and tif5-7A mutants provide the first evidence that both eIF3 activity and MFC integrity are required for the specialized reinitiation mechanism underlying GCN4 translational control. It now seems likely that eIF3 and eIF5 must rebind to the 40S subunits following uORF1 translation to insure the proper rate of scanning from uORF1 to uORFs 2–4. This in turn dictates the fraction of 40S subunits that do not rebind TC before reaching uORF4 and thus go on to reinitiate at GCN4 instead. It is possible that translation of GCN4, or other mRNAs similarly controlled by uORFs, will be modulated by signaling pathways that target eIF3 function or assembly of the MFC.
Materials and methods
Yeast strains and plasmids
Strains and plasmids used in this study are listed in Tables 1 and 2, respectively, and details of their construction are provided in the Supplementary Materials.
Table 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| H1676a | MATa prt1-1 leu2-3,112 ura3-52 | Phan et al (1998) |
| H2879a | MATa PRT1 leu2-3,112 ura3-52 | This study |
| H2880a | MATa trp1Δ leu2-3,112 ura3-52 | This study |
| H2881a | MATa trp1Δ leu2-3,112 ura3-52 gcn2∷hisG | This study |
| YAJ18-3a | H2881 sui3∷cup1p-ubi-DHFRts-HA-sui3td-URA3 ubr1∷Gal1-10p-mycUBR1-TRP1 | This study |
| YKHN60a | MATa trp1Δ leu2-3,112 ura3-52 gcn2∷hisG prt1∷hisG pKHN7[prt1-1 LEU2] | This study |
| H2925a | MATa prt1-1 leu2-3,112 ura3-52 rpl11aΔ | This study |
| H2926a | MATa PRT1 leu2-3,112 ura3-52 rpl11aΔ | This study |
| F324 | MATα cdc33 leu1 ura3 trp1 ade8 | K Matsumoto |
| H2898b | MATa ura3-52 leu2-3 leu2-112 trp1-Δ63 gcn2Δ tif5Δ∷hisG tif34Δ∷hisG p[TIF5-FL TRP1] p[TIF34-HA LEU2] | Asano et al (2000) |
| H2899 b | MATa ura3-52 leu2-3 leu2-112 trp1- Δ63 gcn2Δ tif5Δ∷hisG tif34Δ∷hisG p[tif5-7A-FL TRP1] p[TIF34-HA LEU2] | Asano et al (2000) |
| YKHN205b | MATa ura3-52 leu2-3 leu2-112 trp1-Δ63 GCN2+ tif5Δ∷hisG tif34Δ∷hisG p[TIF5-FL TRP1] p[TIF34-HA LEU2] | This study |
| YKHN206b | MATa ura3-52 leu2-3 leu2-112 trp1- Δ63 GCN2+ tif5Δ∷hisG tif34Δ∷hisG p[tif5-7A-FL TRP1] p[TIF34-HA LEU2] | This study |
| H70 | MATα his1-29 gcn2-101 gcn3-101 ura3-52 gcd1-502 (HIS4-lacZ, ura3-52) | Harashima and Hinnebusch (1986) |
| F1006c | MATa ura3-52 leu2-3, 112 trp1-7 lys2 met2-1 his4-713 yef3∷LEU2 p[YEF3 TRP1] | Anand et al (2003) |
| F1007c | MATa ura3-52 leu2-3, 112 trp1-7 lys2 met2-1 his4-713 yef3∷LEU2 p[yef3-F650S TRP1] | Anand et al (2003) |
| J115d | MATα ura3-52 leu2-3,112 fun12Δ p[FUN12 URA3](pC479) | Lee et al (2002) |
| J116Fd | MATα ura3-52 leu2-3,112 fun12Δ | Lee et al (2002) |
| J113d | MATα ura3-52 leu2-3,112 fun12Δ rpl11aΔ p[FUN12 URA3](pC479) | Lee et al (2002) |
| J113Fd | MATα ura3-52 leu2-3,112 fun12Δ rpl11aΔ | Lee et al (2002) |
| F708 |
MATa ura3-52 his4- tif5-G62S |
T Donahue |
| aIsogenic strains. | ||
| bIsogenic strains. | ||
| cIsogenic strains. | ||
| dIsogenic strains. | ||
Table 2.
Plasmids used in this study
| Name | Description | Source or reference |
|---|---|---|
| YEplac195–CDC33 (B3351) | High copy vector expressing CDC33 | Cruz et al (1997) |
| YEp24–hc TC (B3000) | High copy URA3 vector expressing SUI2, SUI3, GCD11 and IMT4 | Asano et al (1999) |
| Yep13–GCN2 (B832) | High copy LEU2 vector expressing GCN2 | Ramirez et al (1991) |
| YCp50–GCN4–lacZ (B180) | Low copy URA3 vector containing wild-type GCN4 leader | Mueller and Hinnebusch (1986) |
| pM199 | Low copy URA3 vector containing only uORF1 at uORF4s position | Grant et al (1994) |
| pM226 | Derived from pM199; ORF of uORF1 extends into GCN4 sequence | Grant et al (1994) |
| Ycp50–GCN4–lacZ (B226) | Low copy URA3 vector containing GCN4 leader with only uORF4 | Mueller and Hinnebusch (1986) |
| YCp50–HIS4–AUG–lacZ | Low copy URA3 vector containing HIS4–AUG–lacZ fusion (p367) | Donahue and Cigan (1988) |
| YCp50–HIS4–AUU–lacZ | Low copy URA3 vector containing HIS4–AUU–lacZ fusion (p391) | Donahue and Cigan (1988) |
| YEplac–TIF32-Δ6–His (B3927) | High copy URA3 vector expressing TIF32-Δ6–His | Valášek et al (2002) |
| YCplac33–TIF5 (B3342) | Low copy URA3 vector expressing TIF5 | Asano et al (1999) |
| pRS415 (B3992) | Low copy LEU2 vector expressing SUI5 (TIF5) | Huang et al (1997) |
| pRS415 (B3993) | Low copy LEU2 vector expressing SUI5-R31G | Huang et al (1997) |
HCHO cross-linking, WCE preparation and fractionation of extracts for analysis of preinitiation complexes
WCE extracts were made from 200 ml of cells grown to an OD600 of ∼1.5 in YPD or SC medium. Cells were transferred to a 500 ml centrifuge tube containing 50 g of shaved ice and the tube was inverted five times. HCHO was added to 1% and the tube was inverted 10 times and left on ice for 1 h. Glycine was added to 0.1 M and the cells were collected by centrifugation for 5 min at 7000 r.p.m. in a Sorvall RC5B rotor. The pellet was resuspended in 10 ml of buffer B (20 mM Tris (pH 7.5), 50 mM KCl, 10 mM MgCl2) supplemented with EDTA-free protease inhibitor tablet (Roche), 5 mM NaF, 1 mM dithiothreitol, 1 mM phenylmethylsulfonylfluoride (PMSF) and 1 μg/ml of the following protease inhibitors–pepstatin A, aprotenin and leupeptin. For Northern analysis, 0.2 μg/ml of diethyl pyrocarbonate was also added. The cell suspension was transferred to a 15 ml conical tube and centrifugated for 5 min at 4200 r.p.m. in a Beckman J-6B centrifuge and the supernatant was decanted. One vol of cells was resuspended in ∼1.3 vol of buffer B and 1.3 vol of glass beads, and cells were lysed by vortexing eight times for 30 s with 30 s intervals on ice. The lysate was centrifugated for 5 min at 4200 r.p.m. and the supernatant was transferred to an Eppendorf tube. The extract was cleared by two consecutive centrifugations at 13 000 r.p.m. for 5 and 10 min in an Eppendorf 5415D centrifuge, collecting the supernatant while avoiding the lipid layer at the top and the pellet. The WCEs were separated by sedimentation through sucrose gradients as described previously (Asano et al, 2000). For Western analysis, the fractions were precipitated with 1 ml of 100% ethanol at −20°C overnight. Boiling of the samples in SDS-loading buffer for 10 min was sufficient to reverse the cross-linking. RNA was extracted from the fractions essentially as described (Cigan et al, 1991), except that the RNA was extracted twice with hot (70°C) phenol for 15 min, which was sufficient to reverse the cross-linking.
Other biochemical methods
Please refer to Supplementary Materials.
Supplementary Material
Supplementary Materials
Acknowledgments
We thank Patrick Linder, Terry Kinzy and Tom Donahue for providing plasmids and yeast strains, Ernest Hannig for GCD11 antiserum, Tom Dever for suggestions and a critical reading of the manuscript, and members of the Hinnebusch and Dever laboratories for helpful discussions. BS received a PhD fellowship grant from the Bay Zoltan Foundation for Applied Research in Hungary.
References
- Abastado JP, Miller PF, Jackson BM, Hinnebusch AG (1991) Suppression of ribosomal reinitiation at upstream open reading frames in amino acid-starved cells forms the basis for GCN4 translational control. Mol Cell Biol 11: 486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algire MA, Maag D, Savio P, Acker MG, Tarun SZ Jr, Sachs AB, Asano K, Nielsen KH, Olsen DS, Phan L, Hinnebusch AG, Lorsch JR (2002) Development and characterization of a reconstituted yeast translation initiation system. RNA 8: 382–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand M, Chakraburtty K, Marton MJ, Hinnebusch AG, Kinzy TG (2003) Functional interactions between yeast translation eukaryotic elongation factor (eEF) 1A and eEF3. J Biol Chem 278: 6985–6991 [DOI] [PubMed] [Google Scholar]
- Asano K, Clayton J, Shalev A, Hinnebusch AG (2000) A multifactor complex of eukaryotic initiation factors eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev 14: 2534–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano K, Krishnamoorthy T, Phan L, Pavitt GD, Hinnebusch AG (1999) Conserved bipartite motifs in yeast eIF5 and eIF2Bɛ, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J 18: 1673–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano K, Shalev A, Phan L, Nielsen K, Clayton J, Valášek L, Donahue TF, Hinnebusch AG (2001) Multiple roles for the carboxyl terminal domain of eIF5 in translation initiation complex assembly and GTPase activation. EMBO J 20: 2326–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SK, Lee JH, Zoll WL, Merrick WC, Dever TE (1998) Promotion of Met-tRNAiMet binding to ribosomes by yIF2, a bacterial IF2 homolog in yeast. Science 280: 1757–1760 [DOI] [PubMed] [Google Scholar]
- Cigan AM, Foiani M, Hannig EM, Hinnebusch AG (1991) Complex formation by positive and negative translational regulators of GCN4. Mol Cell Biol 11: 3217–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaie P, Wittmer B, Altmann M, Trachsel H (1995) Isolation of a protein complex containing translation initiation factor Prt1 from Saccharomyces cerevisiae. J Biol Chem 270: 4288–4292 [DOI] [PubMed] [Google Scholar]
- Dever TE, Yang W, ström S, Byström AS, Hinnebusch AG (1995) Modulation of tRNAiMet, eIF-2 and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2.GTP.Met-tRNAiMet ternary complexes. Mol Cell Biol 15: 6351–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohmen RJ, Wu P, Varshavsky A (1994) Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science 263: 1273–1276 [DOI] [PubMed] [Google Scholar]
- Donahue TF, Cigan AM (1988) Genetic selection for mutations that reduce or abolish ribosomal recognition of the HIS4 translational initiator region. Mol Cell Biol 8: 2955–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson FL, Hannig EM (1996) Ligand interactions with eukaryotic translation initiation factor 2: role of the γ-subunit. EMBO J 15: 6311–6320 [PMC free article] [PubMed] [Google Scholar]
- Grant CM, Miller PF, Hinnebusch AG (1994) Requirements for intercistronic distance and level of eIF-2 activity in reinitiation on GCN4 mRNA varies with the downstream cistron. Mol Cell Biol 14: 2616–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harashima S, Hinnebusch AG (1986) Multiple GCD genes required for repression of GCN4, a transcriptional activator of amino acid biosynthetic genes in Saccharomyces cerevisiae. Mol Cell Biol 6: 3990–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, McLaughlin CS (1968) Temperature-sensitive mutants of yeast exhibiting a rapid inhibition of protein synthesis. J Bacteriol 96: 1664–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey JWB, Merrick WC (2000) Pathway and mechanism of initiation of protein synthesis. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds), pp 33–88. Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Hinnebusch AG (1996) Translational control of GCN4: gene-specific regulation by phosphorylation of eIF2. In Translational Control, Hershey JWB, Mathews MB, Sonenberg N (eds), pp 199–244. Cold Spring Harbor, NewYork: Cold Spring Harbor Laboratory Press [Google Scholar]
- Hinnebusch AG (2000) Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds), pp 185–243. Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Huang H, Yoon H, Hannig EM, Donahue TF (1997) GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev 11: 2396–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RV, Wolfman A, Panniers R, Henshaw EC (1989) Mechanism of inhibition of polypeptide chain initiation in calcium-depleted Ehrlich ascites tumor cells. J Cell Biol 108: 2107–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K, Tercero JA, Diffley JF (2000) Uninterrupted MCM2-7 function required for DNA replication fork progression. Science 288: 1643–1647 [DOI] [PubMed] [Google Scholar]
- Lee JH, Pestova TV, Shin BS, Cao C, Choi SK, Dever TE (2002) Initiation factor eIF5B catalyzes second GTP-dependent step in eukaryotic translation initiation. Proc Natl Acad Sci USA 99: 16689–16694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar R, Bandyopadhyay A, Maitra U (2003) Mammalian translation initiation factor eIF1 functions with eIF1A and eIF3 in the formation of a stable 40 S preinitiation complex. J Biol Chem 278: 6580–6587 [DOI] [PubMed] [Google Scholar]
- Mueller PP, Hinnebusch AG (1986) Multiple upstream AUG codons mediate translational control of GCN4. Cell 45: 201–207 [DOI] [PubMed] [Google Scholar]
- Olsen DS, Savner EM, Mathew A, Zhang F, Krishnamoorthy T, Phan L, Hinnebusch AG (2003) Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J 22: 193–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Borukhov SI, Hellen CUT (1998) Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394: 854–859 [DOI] [PubMed] [Google Scholar]
- Phan L, Schoenfeld LW, Valášek L, Nielsen KH, Hinnebusch AG (2001) A subcomplex of three eIF3 subunits binds eIF1 and eIF5 and stimulates ribosome binding of mRNA and tRNAiMet. EMBO J 20: 2954–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan L, Zhang X, Asano K, Anderson J, Vornlocher HP, Greenberg JR, Qin J, Hinnebusch AG (1998) Identification of a translation initiation factor 3 (eIF3) core complex, conserved in yeast and mammals, that interacts with eIF5. Mol Cell Biol 18: 4935–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez M, Wek RC, Hinnebusch AG (1991) Ribosome-association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisae. Mol Cell Biol 11: 3027–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotenberg MO, Moritz M, Woolford JL Jr (1988) Depletion of Saccharomyces cerevisiae ribosomal protein L16 causes a decrease in 60S ribosomal subunits and formation of half-mer polyribosomes. Genes Dev 2: 160–172 [DOI] [PubMed] [Google Scholar]
- Sachs A (2000) Physical and functional interactions between the mRNA cap structure and the poly(A) tail. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds), pp 447–465. Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Shin BS, Maag D, Roll-Mecak A, Arefin MS, Burley SK, Lorsch JR, Dever TE (2002) Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 111: 1015–1025 [DOI] [PubMed] [Google Scholar]
- Valášek L, Mathew A, Shin BS, Nielsen KH, Szamecz B, Hinnebusch AG (2003) The yeast eIF3 subunits TIF32/a and NIP1/c and eIF5 make critical connections with the 40S ribosome in vivo. Genes Dev 17: 786–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valášek L, Nielsen KH, Hinnebusch AG (2002) Direct eIF2–eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J 21: 5886–5898 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials