

Abstract
Calnexin and ERp57 act cooperatively to ensure a proper folding of proteins in the endoplasmic reticulum (ER). Calnexin contains two domains: a lectin domain and an extended arm termed the P-domain. ERp57 is a protein disulfide isomerase composed of four thioredoxin-like repeats and a short basic C-terminal tail. Here we show direct interactions between the tip of the calnexin P-domain and the ERp57 basic C-terminus by using NMR and a novel membrane yeast two-hybrid system (MYTHS) for mapping protein interactions of ER proteins. Our results prove that a small peptide derived from the P-domain is active in binding ERp57, and we determine the structure of the bound conformation of the P-domain peptide. The experimental strategy of using the MYTHS two-hybrid system to map interaction sites between ER proteins, together with NMR, provides a powerful new strategy for establishing the function of ER complexes.
Keywords: calnexin, ERp57, ER yeast two-hybrid, NMR
Introduction
Secretory proteins are translocated into the lumen of the endoplasmic reticulum (ER), where they are N-glycosylated, disulfide bonds are formed, and they become folded by interaction with a mechanism termed the calnexin cycle (Ellgaard et al, 1999; Schrag et al, 2001). The central component of this process is the membrane protein calnexin (Wada et al, 1991), which selectively recognizes monoglucosylated glycoproteins. Disulfide bond formation and exchange are catalyzed by the members of the family of disulfide isomerases. While the general mechanism of disulfide bond formation and the specialized functions of the different members of this family are not known, the oxidoreductase ERp57 has been shown to act specifically on glycoproteins bound to calnexin (Zapun et al, 1998; Oliver et al, 1999; Daniels et al, 2003).
Calnexin and its ER lumenal homolog calreticulin specifically bind Glc1Man9GlcNAc2 glycoproteins irrespective of their conformation, and therefore function as lectins as well as molecular chaperones (Ou et al, 1993; Zapun et al, 1997). This lectin function is suggested by some in vivo studies and confirmed by the structure of calnexin determined to 2.9 Å by X-ray crystallography (Ou et al, 1993; Schrag et al, 2001). The structure revealed a characteristic lectin fold, and the identification of a glucose residue in a location similar to that found for sugar binding in a structurally related class of lectins (Schrag et al, 2001, 2003). The protein is characterized by two main structural components, a globular lectin domain and an extended region or loop termed the P-domain (Schrag et al, 2001). The X-ray structure also provides information on how this remarkable structure is organized with the lectin domain. The P-domain of calnexin leaves the lectin domain at residue P270, with four copies of a repeat motif (termed type 1), and then returns to the lectin domain at residue F415, with four copies of another repeat motif (type 2) in a ‘11112222' configuration, thus forming a hook-like arm consisting of four similar P-modules. The structure of the extended P-domain of calreticulin has been determined by NMR by Ellgaard et al (2001, 2002).
In vivo studies of glycoprotein binding to calnexin have shown that different glycoproteins bind with different half-lives. Proteins with many disulfide bonds, such as transferrin, have prolonged association times in vivo with calnexin, implying that there is a correlation between disulfide bond formation and association with calnexin (Ou et al, 1993). The functional relationship between calnexin, calreticulin, and ERp57 was shown in vitro by experiments using purified proteins to study disulfide exchange in Glc1Man9GlcNAc2 RNaseB glycoproteins that were bound to calnexin or calreticulin (Zapun et al, 1998). It was shown that ERp57 catalyzes disulfide bond exchange of monoglucosylated RNaseB, bound to calnexin or calreticulin, in contrast with protein disulfide isomerase (PDI), which interacts with glycoproteins and nonglycoproteins (Zapun et al, 1998). Furthermore, it is interesting that this calnexin- or calreticulin-mediated interaction greatly increases the disulfide-isomerase activity of ERp57 on the monoglucosylated protein, whereas the same type of interaction has been indicated to reduce PDI activity and peptide binding (Zapun et al, 1997, 1998). Further experiments demonstrated a physical association between ERp57 and calnexin, and this was confirmed by crosslinking (Oliver et al, 1999). Thus, there is a functional, and probably physical, interaction between ERp57, calnexin, and calreticulin, which is not found with PDI.
ERp57 and PDI are members of the protein disulfide isomerase family and have similar overall organization of their a b b′ a′ thioredoxin domains with CGHC catalytic site motifs in the a and a′ domains. They also show significant sequence similarity in regions corresponding to the b and b′ domains, with an overall identity of 33% (Ferrari and Söling, 1999; Pirneskoski et al, 2000). Although ERp57 has the same modular structure of active and inactive domains as PDI, it lacks the C-terminal acidic region, and instead has a basic C-terminal region that is found in ERp57 orthologs.
In previous studies using NMR spectroscopy, it was found that ERp57 binds to the P-domain of calreticulin, and by chemical shift mapping it was shown that interactions with ERp57 occur exclusively through amino-acid residues that form the tip of the P-domain (Frickel et al, 2002). Leach et al (2002) used glutathione S-transferase (GST) pull-down assays with fragments of calnexin and calreticulin, and the data support the NMR findings that for both calnexin and calreticulin the site of ERp57 binding is located within their P-domains.
Here, we have used genetic and NMR methods to determine the residues on ERp57 that are responsible for its interaction with calnexin. To do this, we have developed a novel membrane yeast two-hybrid system (termed MYTHS), which detects interactions between ER proteins, to initially map and then determine specific residues responsible for the interaction. We confirmed these interactions by NMR as well as in vitro binding assays. These results show that the C-terminal region that is found in ERp57 is responsible for the specific interaction with the tip of the P-domain of calnexin. Confirmation of this interaction was obtained by construction of hybrid PDI/ERp57 molecules, and by mutagenesis, which also identifies residues in the b′ and a′ domains of ERp57 that influence these interactions. The conformation of the bound form of the calnexin P-domain to ERp57 was determined by transferred NOE spectroscopy, and it was shown that the peptide that we have identified within ERp57 binds to the tip of the P-domain in the same manner.
Results
Calnexin P-domain modular organization
All genetic constructs and peptides for NMR analysis used in this study are based on the structural information of calnexin that has recently been solved at a 2.9 Å resolution (Schrag et al, 2001). We have also analyzed NMR spectra of the 15N and 15N/13C-enriched calnexin P-domain fragments comprising two and four modules and homonuclear data for the one-module fragment. The 1H–15N HSQC spectra of the two-module fragment and four-module fragment display good signal dispersion characteristics of a folded protein.
The structure of the two P-module fragment consists of two similar modules connected through a short β-sheet. The hydrophobic core of each is formed by the two perpendicularly positioned tryptophan side chains and a lysine side chain in contact with both rings (Figure 1A), and the tip of the calnexin P-domain has a helix-like conformation. The overall structure of the calnexin P-domain is similar to the calreticulin P-domain structure also established by NMR, but is one repeat longer, and is better defined than in the calnexin crystal structure (Schrag et al, 2001, 2003). Also, the addition of residual dipolar couplings here more accurately defines the relative orientation of the modules to each other.
Figure 1.
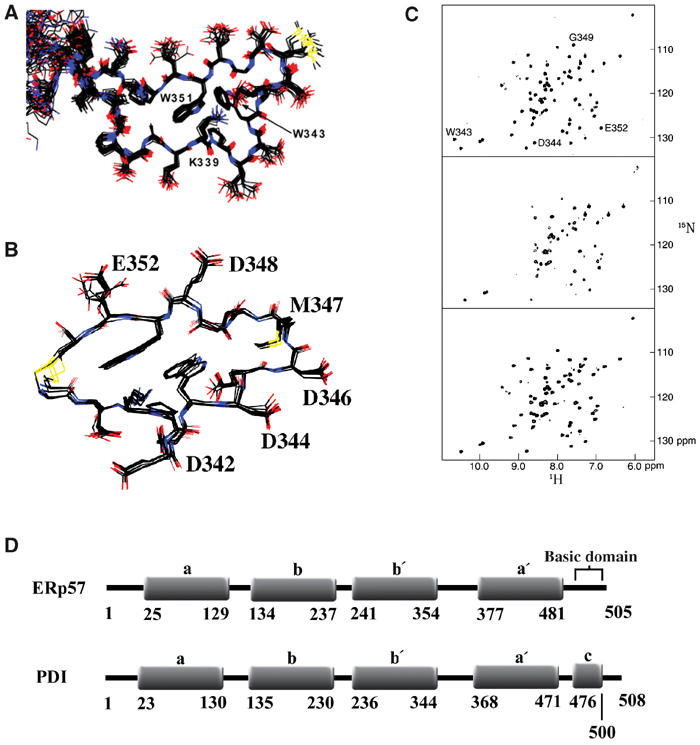
NMR structure and spectra of the calnexin tip module fragment upon binding to ERp57 and ERp57 domain organization. (A) Overlay of the tip module from the 20 lowest-energy two-module calnexin structures. Two perpendicularly positioned tryptophan rings (W343 and W351) and the side chain of K339 form the hydrophobic core of the module. (B) Overlay of the 20 lowest-energy structures of the cyclic peptide derived from the tip module sequence, when bound to ERp57. The residues that are involved in ERp57 interactions are labeled. (C) Effect of binding to ERp57 as detected by amide signals of the calnexin two-module fragment (residues 310–381). 1H–15N HSQC spectra of 15N-labeled calnexin in its free form (top), with a ratio of 2:1 of ERp57 to calnexin (middle), and a ratio of 1:10 (bottom) of ERp57 to calnexin. The signals most affected are annotated. (D) Organization of the a b b′ a′ domains of ERp57 compared with PDI. The residue numbers of the individual thioredoxin-like domains are given. The a b b′ a′ domains of ERp57 and PDI are similar, except that ERp57 has a basic C-terminus and PDI has an acidic c domain.
Chemical shift mapping of P-domain/ERp57 interactions
Two and four P-module fragments were tested for their ability to bind ERp57. Initial experiments showed that ERp57 binding is similar for both fragments. Therefore, further binding studies were performed predominantly using the shorter, two-module fragment. The interactions between calnexin and ERp57 were followed using 15N-enriched P-domain and unlabeled ERp57. As shown in Figure 1C, a small subset of amide signals is influenced by complex formation. The extent of broadening depends on the molar ratio of calnexin to ERp57, which is typical for an intermediate exchange rate. Titration in the range of 2:1 to 1:10 of the ERp57 to calnexin P-module allowed the recovery of practically all signals at the interface. This titration also allowed us to determine that most of the broadened signals do not experience extensive chemical shift, with differences ranging between 0.2 ppm in the 1H dimension and 0.4 ppm in the 15N dimension. These small changes are indicative of a weak binding affinity. Mapping the changes to the structure of calnexin showed that the tip module of its P-domain was most affected by ERp57 binding. Therefore, our NMR studies are focused on the tip module of the calnexin P-domain.
Structure of the single-module fragment bound to ERp57
To further investigate the unique modular nature of the calnexin P-domain, we analyzed the spectra of a single P-domain module. Two fragments of 25 and 17 amino acids from the tip of the P-domain were analyzed. Linear peptides were only partially folded, but peptides cyclized by the addition of two cysteine residues and a disulfide bond gave high-quality spectra indicative of a folded structure. The cyclized peptide was designed by replacing alanines that are close together in two-module structure with cysteines. The cyclized structure is assumed to be representative of the structure of the P-domain tip in the intact calnexin molecule; however, there may be small differences. NMR spectra were sequentially assigned using two-dimensional NOESY and TOCSY experiments and used to compare the structure of the P-tip in the context of the longer calnexin fragments and when bound to ERp57. Structural statistics for the single-module calnexin peptide are given as Supplementary data (Appendix I).
Interactions between the cyclized 17-residue peptide from the tip module of calnexin P-domain and ERp57 were monitored by recording homonuclear NOESY spectra of the peptide at different ERp57/peptide ratios. This transferred NOEs approach was used because of intermediate/weak binding affinity and the large size of ERp57. The transferred NOEs allowed us to determine the structure of the cyclized peptide bound to ERp57 (Figure 1B). The most affected amides were W343, D344, G349, and E352, which are all located on the same side of the tip module, defining the binding surface. More specifically, the results show that side chains of D342, D344, D346, M347, D348, and E352 are involved in calnexin P-domain/ERp57 interactions.
The ER membrane yeast two-hybrid system
Our two-hybrid system to detect interactions in the ER uses the unfolded protein response (UPR) in Saccharomyces cerevisiae to signal the interaction of proteins in the ER. In wild-type cells, this response is initiated by the type I membrane protein Ire1p, which has its N-terminus in the lumen of the ER, and senses and oligomerizes in response to the accumulation of misfolded proteins within the ER (Shamu and Walter, 1996). Oligomerization induces autophosphorylation of the cytosolic kinase domains, and activates its endoribonuclease domain (Shamu and Walter, 1996) that specifically cleaves the mRNA of HAC1. Hac1p is a bZIP transcriptional activator that binds the UPR element (UPRE) upstream of many genes that encode ER proteins required for proper protein folding, and induces their expression. The intron within HAC1 mRNA has been shown to attenuate the translation of this mRNA (Chapman and Walter, 1997), and its removal leads to the expression of a full-length and more active version of Hac1p, resulting in the upregulation of the expression of many molecular chaperones and folding enzymes, as well as many components of the ER-associated degradation pathway (ERAD) (Travers et al, 2000). In this study, we have used these elements of the UPR to construct a reporter system that senses protein interactions in the ER. As shown in Figure 2A, the system uses Ire1p fusion proteins, where interactions between the proteins in the ER cause the Ire1p cytosolic domains to auto- and or trans-phosphorylate and activate the UPR. A LacZ reporter gene is located downstream of the UPRE in order to measure these interactions.
Figure 2.
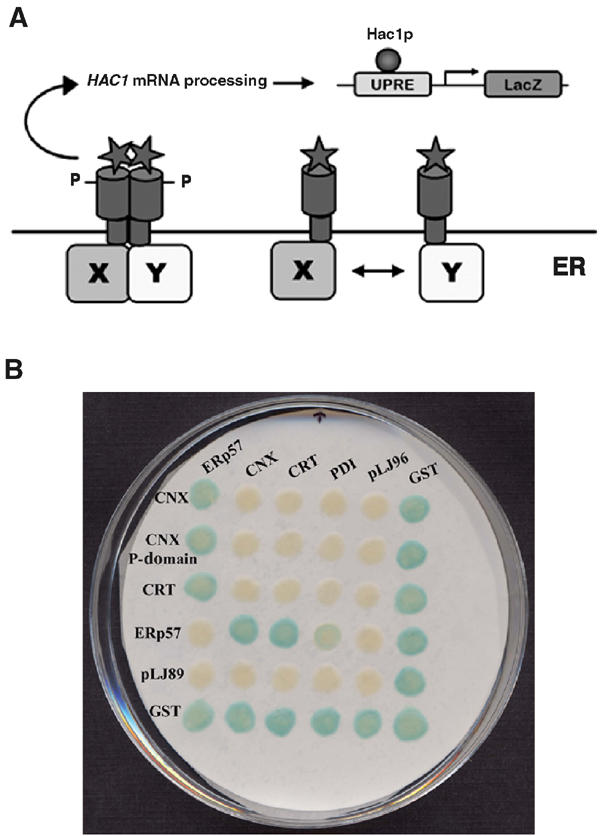
Model of the ER MYTHS and validation using known ER interactors. (A) Fusions of proteins ‘X' and ‘Y' to the transmembrane kinase/endoribonuclease Ire1p that result in oligomerization/dimerization induce autophosphorylation of the Ire1p kinase domains. This leads to activation of the Ire1p endoribonuclease domain, which then leads to processing of the mRNA of the UPR transcriptional activator, HAC1. With the intron removed (HAC1*), expression of the active Hac1p is induced, which binds to the minimal yeast UPRE upstream of LacZ, initiating the expression of the reporter, β-galactosidase. (B) Sample filter lift of diploid yeast created by crossing MATa yeast containing calnexin, calnexin P-domain, calreticulin, ERp57, pLJ89, and GST (located along the vertical axis) with MATα yeast containing ERp57, calnexin, calreticulin, PDI, pLJ96, and GST (located along the horizontal axis). Results of the β-galactosidase filter assays are shown where a blue color demonstrates a positive interaction between and/or among the two Ire1p fusion proteins.
Interactions in vivo between calnexin, calreticulin, ERp57, and PDI
To test our experiment, MATα strains expressing a set of Ire1p fusion proteins were mated with MATα strains expressing a second set of fusion proteins, and diploid strains were selected for on -Leu-His selective media. The MATa set consisted of fusions containing the lumenal domain of calnexin (CNXH21-P482), the calnexin P-domain (CNXP270-F415), calreticulin (CRTE18-L417), ERp57 (ERp57A24-L505), the parental vector pLJ89 (negative control), and glutathione S-transferase (GSTM1-G225) from Escherichia coli as a positive control. GST is used as a positive control in these experiments as it is known to form homodimers that are seen in our system as LacZ positives in diploid cells as well as in haploids. The MATα set of fusions consisted of ERp57 (ERp57A24-L505), the lumenal domain of calnexin (CNXH21-P482), the ER protein disulfide isomerase (PDID18-L508), the parental vector pLJ96, and GST. Diploids expressing all combinations of fusion proteins were isolated and assayed for β-galactosidase activity. The results presented in Figure 2B demonstrated that we can detect the specific interactions of calnexin, the calnexin P-domain, and calreticulin with ERp57, but there are no detectable interactions with PDI. From these results, we also conclude that calnexin does not interact with calreticulin, and also that ERp57 interacts slightly with PDI. In addition, we did not find self-interaction for any of these fusion proteins other than GST. The domain organization of ERp57 compared to PDI is shown in Figure 1D.
Next, we mapped the protein interactions between ERp57 and the P-domain of calnexin and of calreticulin. The calreticulin loop is shorter than that of calnexin, as it is missing the first type 1 repeat, as well as the last type 2 repeat. Thus, it resembles a truncated calnexin P-domain with a 111222 configuration.
When tested in our two-hybrid system, the calnexin lectin domain failed to mediate a specific interaction with ERp57 (data not shown), while its P-domain (CNXP270-F415) conferred this specificity. We designed further deletion constructs that defined the region of interaction between calnexin and calreticulin to the last two modules of the P-domain (Table I).
Table 1.
Mapping interactions between calnexin, calreticulin, ERp57, and PDI using MYTHS
| CNX H21P482 | ERp57 A24-L505 | PDI D18-L508 | ERp57 K362R | ERp57 F386S | ERp57 W405R | ERp57 F476S | pLJ96 | GST | |
|---|---|---|---|---|---|---|---|---|---|
| ERp57 A24-L505 | +++ | − | + | − | − | − | − | − | +++ |
| CNX H21−P482 | − | +++ | − | +++ | +++ | +++ | +++ | − | +++ |
| CNX P270−F415 (11112222) | − | +++ | − | +++ | +++ | +++ | +++ | − | +++ |
| CNX D289−N393 (111222) | − | +++ | − | +++ | +++ | +++ | +++ | − | +++ |
| CNX P310−P378 (1122) | − | +++ | − | +++ | +++ | +++ | +++ | − | +++ |
| CRT E18-L417 | − | +++ | − | +++ | +++ | +++ | +++ | − | +++ |
| CRT D201-A307 (111222) | − | +++ | − | +++ | +++ | +++ | +++ | − | +++ |
| CRT A223-P283 (1122) | − | +++ | − | +++ | +++ | +++ | +++ | − | +++ |
| PDI D18-L508 | − | + | − | n/a | n/a | n/a | n/a | − | +++ |
| CNX2pmA | − | + | − | +++ | +++ | +++ | +++ | − | +++ |
| CNX6pmA | − | − | − | − | − | − | − | − | +++ |
| pLJ89 | − | − | − | − | − | − | − | − | +++ |
| GST |
+++ |
+++ |
+++ |
+++ |
+++ |
+++ |
+++ |
+++ |
+++ |
| Results of using several calnexin and calreticulin P-domain constructs to map their interactions with ERp57. CNX and CRT deletion mutants are indicated, with the number of P-domain repeat motifs indicated in parenthesis. Specific point mutations in calnexin confirm acidic residues necessary for interaction. Error-prone PCR identifies point mutations in ERp57 that complement the mutated calnexin P-domain. The nature of each mutant fusion protein is described in the text. Plus symbols indicate the presence and strength of an interaction (‘+++' strongest to ‘+' weakest), and ‘−' indicates no interaction detected. ‘n/a' indicates data not available. | |||||||||
Specific residues involved in calnexin/ERp57 interaction
Using the results obtained from NMR for specific calnexin residues interacting with ERp57, we further defined the calnexin interaction surface. Mutation of individual residues on the P-domain tip did not show a significant reduction in the interaction with ERp57 (data not shown). The double mutant of D344A/E352A (CNX2pmA) showed a measurable reduction in signal (Table I, Figure 3A). We thus mutated the acidic residues D342, D344, D346, D348, E350, and E352 to alanines. The six-point mutant of calnexin (CNX6pmA) was used along with a double-point mutant (CNX2pmA) to test for interaction with ERp57. The results are shown in Table I and Figure 3A, where compared with wild-type levels, the interaction with ERp57 is lost for the CNX6pmA mutant, and significantly reduced for CNX2pmA. These results support the NMR findings that the acidic surface found on the tip of the P-module of calnexin is indeed the area responsible for its interaction with ERp57. Based on these results, we conclude that calnexin residues D342, D344, D346, D348, E350, and E352 are all involved in ERp57 binding, and D344 and E352 are likely the two most important residues contributing to this interaction.
Figure 3.
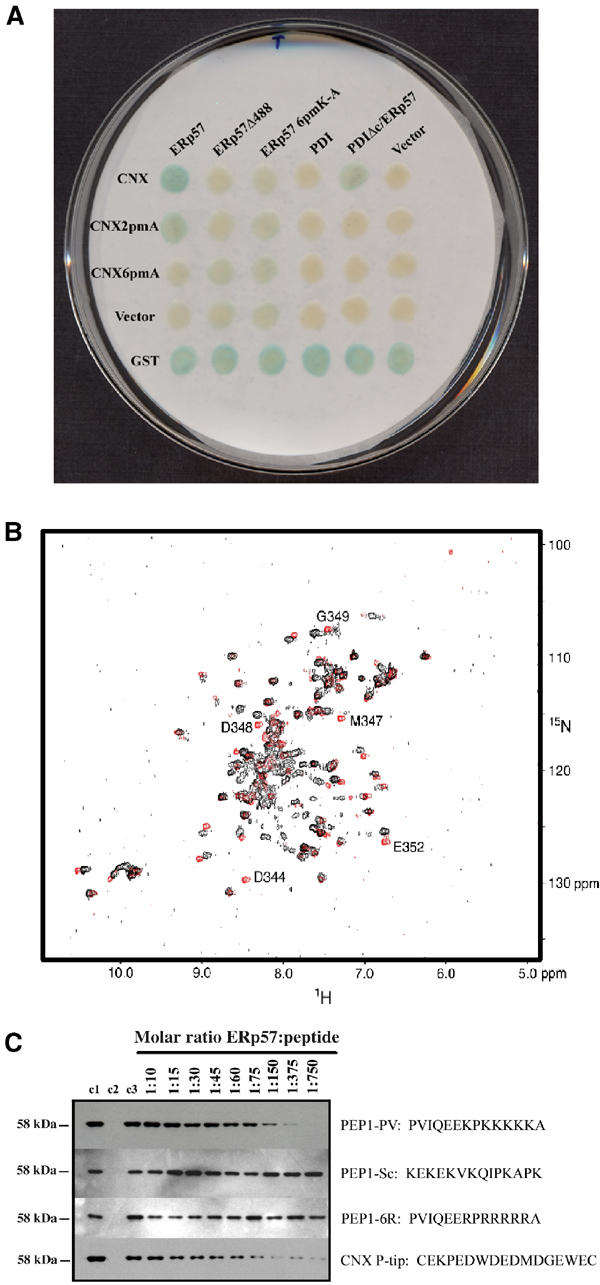
The C-terminal domain of ERp57 is directly involved in an interaction with the calnexin P-domain. (A) β-Galactosidase filter assay of diploid yeast patched onto selective media after mating of two sets of fusion proteins. One set expressed in MATa yeast listed on the vertical axis is mated with another set expressed in MATα yeast listed on the horizontal axis. The nature of each fusion construct is described in the text. With the removal of the ERp57 C-terminus (deletion at residue P488), its interaction with calnexin is lost. The same observation is made when the six-point mutation that removes all lysines within this region (ERp57-6pmA) is introduced into the same system. The fusion of PDI/C-terminal ERp57 also gives a signal stronger than PDI alone when tested against calnexin, however not as strong as the wild-type calnexin/ERp57 interaction. This suggests that residues P488-A501 of ERp57 are necessary, and responsible, for the interaction with the calnexin P-domain. (B) Superimposed 1H–15N HSQC spectra of the 15N-labeled calnexin three-module fragment in its free form (in red), and at a ratio of 1:5 of calnexin to the PEP1-PV peptide from ERp57 (in black). The most affected signals are annotated. Residues affected upon addition of ERp57 peptide correspond to those affected upon addition of the full 58 kDa protein. (C) Western blot analysis of GST pull-down assays using an anti-ERp57 probe. Purified ERp57 was run as a positive control in lane 1 (c1). Lanes 2 (c2) and 3 (c3) are pull-down experiments performed using GST and GST–CNX, respectively, and purified ERp57 as described in the Methods section. Lanes 4–12 contain GST–CNX samples that were incubated with the same amount of ERp57 as in lanes 2 and 3, however with the addition of increasing amounts of each peptide listed. Molar ratios of ERp57 to peptide are noted on each lane. Results demonstrate that with increasing amounts of the C-terminal ERp57 peptide, less full-length ERp57 is able to be isolated. With a molar ratio of 1:60, 50% of the full-length ERp57 interaction is inhibited, and with a ratio of 1:750 the interaction is fully inhibited by the peptide. Although the concentrations necessary for inhibition are quite high, the competition for binding is specific enough so that a scrambled version of the same peptide does not interfere. In addition, a peptide that replaces all lysine residues with arginines also fails to compete, demonstrating the necessity of the poly-lysine repeat. The calnexin P-tip cyclized peptide was included as a positive control. This peptide (representing the tip of the calnexin P-domain) competes with the full P-domain for binding to ERp57 in the same concentrations observed for the PEP1-PV peptide.
We attempted to define the site of interaction on ERp57 by screening for compensating mutations that restore interaction with the compromised calnexin P-domain, CNX2pmA. ERp57 was mutated using random mutagenesis by error-prone PCR, and the resulting library of mutant ERp57 amplification products was used for in vivo recombination into the pLJ96 plasmid. In all, 1500 individual colonies were picked and mated with CNX2pmA (in MATa strain) in an attempt to identify ERp57 mutants that would complement the mutations in the calnexin P-domain, and restore the signal observed between the wild-type proteins. The ERp57 mutations were tested for self-activation, interaction with wild-type calnexin, CNX2pmA, CNX6pmA, and with vector alone. Six ERp57 mutants were isolated, which restored the interaction with CNX2pmA. This group includes four single-point mutants (ERp57_K362R, ERp57_F386S, ERp57_W405R, and ERp57_F476S) as listed in Table I, and two double-point mutants (ERp57_N298Y_R351G and ERp57_D358G_L462P). Looking at the distribution of mutations along the ERp57 protein sequence (Figure 1D), all seem to cluster in the b′ a′ domains of ERp57, identifying these domains as possible sites of interaction with calnexin. Based on these results, we focused on the C-terminal end of ERp57, and made random 3′ truncations of the cDNA in an effort to identify deletion mutants that do not interact with wild-type calnexin P-domain.
ERp57 cDNA fragments ranging from 100 bases to the full-length protein were generated by a nested deletion method (see Methods), introduced into pLJ96, and screened for an interaction with wild-type calnexin. A total of 1500 individual ERp57 clones were screened in total, and 34 positive interactors were isolated. Upon DNA sequencing, 20 turned out to be full-length ERp57, 10 were truncated at A501, and four were truncated at K499. We focused on the C-terminus of ERp57 and particularly on a stretch of six lysine residues beginning at K494 that is conserved in many ERp57 orthologs (see Supplementary data, Appendix II). Based on these results and further observations, we made a specific truncation of ERp57 (ERp57Δ488) that removes the six lysine sequence, along with other surrounding conserved residues, and tested this for an interaction with calnexin. As seen in Figure 3A, we observe no interaction between calnexin and ERp57Δ488, which implies that the last 18 amino acids (most likely K494–K500) of ERp57 are necessary for the interaction with calnexin. A six-point mutant of ERp57 (ERp57-6pmA) was also created where all lysine residues in this region were mutated to alanine, changing the sequence from KPKKKKK to APAAAAA. The interaction of this ERp57-6pmA with calnexin was measured and, again, a complete loss of interaction was observed (Figure 3A). Western blot analysis shows that all mutant proteins are indeed expressed (data not shown). Next, two ERp57 fragments were introduced into ER MYTHS: one containing the C-terminus of the protein alone (ERp57A482-L505) and another containing the C-terminus along with the complete b′ a′ domains (ERp57L241-L505). Whereas ERp57A482-L505 does not interact with calnexin, ERp57L241-L505 gave a signal comparable to the wild-type interaction (data not shown). Whether the C-terminal fragment is too small (24 amino acids) to form an interaction with calnexin in our system, or whether it requires extra interactions supplied by the structured b′ a′ domains, or whether the b′ a′ domains are also somehow involved in the interaction, are all possibilities.
Comparison of ERp57 and PDI
ERp57 is a PDI family member that specifically catalyzes disulfide bond rearrangement of glycoproteins bound to calnexin and calreticulin (Zapun et al, 1998). These two disulfide isomerases differ in the sequence of their C-terminal regions (see Figure 1D). PDI contains a final c domain that is extremely acidic (Pirneskoski et al, 2000), and thus we made deletion and fusion constructs with PDI in order to remove the c domain, and then to replace it with the C-terminus of ERp57.
First, a deletion of PDI was synthesized, which removed the c domain, PDI D18-E471. Then a fusion was made where the c domain of PDI was replaced with the C-terminal end of ERp57, PDIΔc/ERp57A482-L505. Both constructs were introduced into the pLJ96 vector. The c domain truncation of PDI (PDID18-E471) failed to interact with calnexin as did the full-length PDI protein (PDID18-L508). PDIΔc/ERp57A482-L505 demonstrated an interaction stronger than the wild-type calnexin/PDI interaction; however, the signal was slightly weaker than for the wild-type calnexin/ERp57 interaction (Figure 3A). This result also suggests that the C-terminal tail of ERp57 is involved in the interaction with calnexin, but since the signal strength was slightly weaker with the PDI/ERp57 fusion (compared to the calnexin/ERp57 signal strength) it suggests that there may be other domains of ERp57 that are also involved in this interaction. This idea supports our theory of the b′ a′ domains of ERp57 also being somehow involved in the interaction with calnexin.
Confirmation of the specific interaction of the C-terminus of ERp57 with calnexin
In order to confirm our results obtained from ER MYTHS, we tested binding between the calnexin P-domain and a C-terminal peptide of ERp57 by NMR and pull-down assays. The peptide PEP1-PV contains the 14-amino-acid residues in the ERp57 C-terminal sequence P488-A501 (PVIQEEKPKKKKKA).
The addition of the PEP1-PV peptide of ERp57 to the 15N-labeled three-module fragment of the P-domain resulted in shifts and the disappearance of some amide signals, indicating binding (Figure 3B). The largest changes involved the amides of D344, M347, D348, G349, and E352. These changes are grouped in the tip module of the calnexin P-domain and are in excellent agreement with the binding results obtained with complete ERp57. The binding between the negatively charged tip of the calnexin P-domain and the conserved poly-K stretch from ERp57 is likely to be based on electrostatic interactions, although the interactions between tryptophan and lysine side chains may contribute to the binding and make it stronger and more specific. Interestingly, moderate shifts were also observed for amides of E362, G366, and G368 that are in close proximity to the C361–C367 disulfide bond. These shifts may reflect slight conformational changes in the disulfide region that are caused by the binding of the peptide, although this effect is not likely to be relevant for P-domain/ERp57 interactions since this disulfide bond is not present in the P-domain of calreticulin.
GST pull-down assays along with competition assays were performed in order to confirm the full-length and peptide interactions from ER MYTHS and NMR studies, respectively. Fusions of the full lumenal domain of CNX (GST–CNXK46-M417), and GST as a control, were used to test for full ERp57 and PEP1-PV binding. As shown in Figure 3C, ERp57 binds specifically to the GST–CNX fusion; however, with increasing amounts of PEP1-PV added in the incubation, a decreasing amount of full-length ERp57 is able to bind due to the peptide competing for the calnexin P-domain binding surface. The results show a 50% reduction in ERp57 binding with the addition of PEP1-PV to a molar ratio of 1:60 (ERp57:PEP1-PV), and a 100% decrease at a molar ratio of 1:750.
Control pull-down experiments were also performed using a scrambled version (PEP1-Sc) of the PEP1-PV peptide along with another peptide that replaces all the lysine residues with arginines (PEP1-6R). Using the same molar ratios as those presented above for PEP1-PV, both PEP1-Sc and PEP1-6R fail to compete with full-length ERp57 for binding to the P-domain of calnexin (Figure 3C). This result demonstrates the specificity of the interaction between calnexin and the ERp57 peptide, meaning the binding is not a result of nonspecific electrostatic interactions, and demonstrates the requirement of the lysine repeats to mediate this interaction.
A final competition pull-down assay was performed using the same P-tip peptide of calnexin (cyclized 17-residue peptide) used in the NMR experiments. As shown in Figure 3C, the calnexin P-tip peptide successfully competes with the P-domain of calnexin for binding to ERp57 at roughly the same concentrations required for PEP1-PV (Figure 3C).
Discussion
We have determined the specific interaction sites of calnexin and ERp57 by a combination of structural and genetic methods. The data presented here define a small region in the calnexin P-domain that determines its interaction with ERp57. The region comprises amino acids 337–353 and is highly conserved in the calnexin/calreticulin family of proteins (see Supplementary data, Appendix II). The interactions with ERp57 are centered close to the tip of the P-domain, as the largest effect is seen on the amides of residues W343, D344, G349, and E352.
The novel ER yeast two-hybrid system, MYTHS, implemented in this study provides a tool for mapping protein interactions for both membrane and soluble ER proteins. The yeast ER provides similar redox potential, calcium and ionic concentrations to that found in mammalian ER, enabling specific interactions to be detected and studied. Using this genetic system and the structural information as a guide, we detected specific interactions between calnexin/ERp57 and calreticulin/ERp57. The specific interaction of ERp57 with the P-domains of both calnexin and calreticulin was also confirmed by mutagenesis, with only the tip of the loop being necessary for this interaction.
The region at the tip of the calnexin and calreticulin P-domains forms acidic surfaces that interact with ERp57, and when this region in calnexin was mutated (CNX6pmA) the interaction was lost. The calnexin double-point mutant (CNX2pmA) also causes a significant decrease in interaction strength when compared to the full-length calnexin, and since NMR data identified these two residues as the two side chains demonstrating broadened signals upon the addition of ERp57, we conclude that they are most responsible for the interaction. From these results, there is no influence on residues in the remaining part of calnexin, and since binding results in broadening rather than shifting of NMR signals, we also conclude that interactions with ERp57 do not cause major structural changes in the calnexin P-domain, despite its elongated, nonglobular nature and intrinsic flexibility.
Using a method designed to create random 3′ deletions of the ERp57 cDNA, we mapped the major site of interaction of ERp57 with calnexin to its C-terminus. This region includes residues 494–500 of ERp57 (with the sequence KPKKKKK), and forms a basic surface that, when removed (ERp57Δ488), causes the interaction with calnexin to be lost. From an analysis of protein sequences of several ERp57 orthologs, it seems that this lysine repeat and the surrounding residues are highly conserved, and always located at the C-terminal end (see Supplementary data, Appendix II). In addition, PDI does not contain such a sequence, but instead has an extremely acidic c domain (see Figure 1D).
Our genetic evidence from point mutations of ERp57 that specifically restore interaction with the CNX2pmA mutant detected mutations that were localized to the b′ a′ domains. In addition, we were unable to make fusions of the C-terminus of ERp57 directly with PDI that could fully recreate the interaction. Thus, there may be a contribution of other domains in the interaction. This idea is supported by the results which show that the C-terminus alone could not interact with calnexin; however, with the addition of only the b′ a′ domains, the interaction was fully restored.
Although other domains of ERp57 are possibly indirectly involved in the interaction with calnexin, the C-terminus has been identified and confirmed as the region directly responsible for this interaction, and a new model based on these findings is presented in Figure 4. This was proven using NMR analysis, which showed that addition of PEP1-PV to the calnexin P-module resulted in the same shifting and disappearance of resonances on the tip of the module as with the full-length ERp57 protein. GST pull-down assays confirmed these results by demonstrating inhibition of ERp57 binding to calnexin with increased concentrations of PEP1-PV due to their competition for the P-domain binding surface. Another novel and interesting finding is that of a newly defined ERp57/PDI interaction. Although this finding is still at a preliminary stage, we suggest the possibility of this interaction occurring through the formation of intermolecular disulfide bonds between the two proteins.
Figure 4.
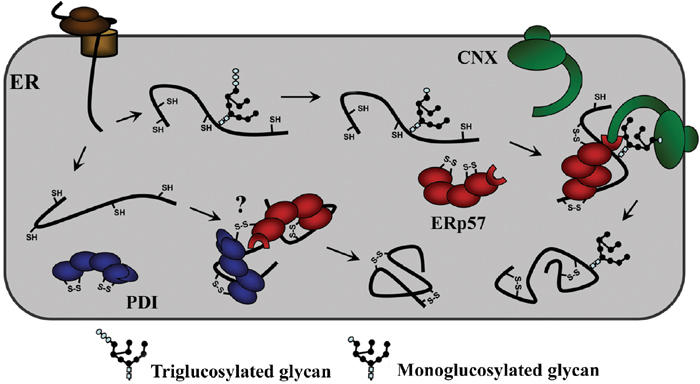
Model of the interaction of monoglucosylated folding intermediates with calnexin and ERp57. The monoglucosylated substrate binds to the chaperone through both lectin–oligosaccharide interactions as well as polypeptide-based associations. The terminal glucose residue of the substrate binds to the lectin site of calnexin within its globular domain, while the arm-like P-domain recruits ERp57, by binding to its C-terminal domain, and brings it into proximity with the polypeptide where it will catalyze disulfide bond formation and isomerization. The model also raises the possibility of ERp57 interacting with PDI through the formation of intermolecular disulfide linkages.
At this stage, the organization and interaction of ER proteins are relatively unknown, and the well-characterized interaction motifs of nuclear and cytoplasmic proteins are not found in ER proteins. There is evidence that proteins in the ER interact functionally, and have specific physical interactions; thus, this general strategy of the ER-specific MYTHS combined with structural studies of the interacting domains will aid in their elucidation.
Experimental procedures
Manipulations, plasmids, and yeast strains
For NMR experiments, fragments of canine calnexin cDNA (Wada et al, 1991; Schrag et al, 2001) corresponding to two modules of the P-domain were expressed as affinity tag fusions in E. coli. The full-length calnexin P-domain (residues 267–411) was cloned into plasmid pGEX-4T1 (Amersham Biosciences, Piscataway, NJ). The two-module fragment (residues 310–381) was cloned in plasmid pET15b as a His6 fusion. All constructs were expressed in E. coli BL21.
The S. cerevisiae reporter strain used in the ER two-hybrid system was constructed by integrating UPR-Y::CYC1::LacZ::URA3, derived from pLG-Δ17834 kindly provided by Dr Claude A Jacob (Zurich, Switzerland). It was integrated into W303a (MATa; ura3-52; trp1D2; leu2-3-112; his3-11; ade2-1; can1-100), which was then crossed to BY4742 MATα Δire1::KanMX (ATCC # 4011907). The diploids were sporulated, and haploids of both mating types were isolated (yLJ29 MATa; ura3; trp1; leu2; his3; ΔIRE1::KanMX; UPR-Y::CYC1::LacZ::URA3 and yLJ31 MATα ura3; trp1; leu2;his3; ade2; Δire1::KanMX; UPR-Y::CYC1::LacZ::URA3). Unless otherwise mentioned, all plasmids were made by recombinational cloning directly in yeast as described elsewhere (Jansen et al, in preparation). The parent plasmid pLJ89 was made in three steps: (1) A 2 μm yeast plasmid with a selectable LEU2 marker (pGreg505) was linearized and recombined with a 477 bp PCR product (amplified using the Expand High Fidelity™ system (Roche, Laval, Canada) following the manufacturer's protocol) that spanned the −411 to +66 bp coding region of IRE1 (i.e. IRE1 promoter and ER signal sequence). (2) A 66 bp linker that has both NotI and SalI sites was added 3′ to the sequence encoding the ER signal sequence, without disrupting the reading frame. (3) The region encoding the IRE1 transmembrane domain, kinase, and endoribonuclease was amplified and was similarly introduced. To produce pLJ96, the IRE1 cassette was removed from pLJ89 by PmeI and XbaI (New England Biolabs, Mississauga, Canada) and subcloned into pGreg503, which contains a selectable HIS3 marker. In the final products, plasmids pLJ89 and pLJ96, with LEU2 and HIS3 markers, respectively, both encode Ire1p with the N-terminal lumenal domain deleted up to the transmembrane domain (TM). The Ire1p signal sequence is intact and followed by a 23-amino-acid linker to ensure that the signal peptidases do not cleave the nascent protein subcloned in place of the Ire1 lumenal domain. Expression vectors used in the two-hybrid assays are under the control of the native yeast IRE1 promoter so as to avoid high concentrations of proteins. This low level of expression is to avoid nonspecific interactions, which might occur if two fusion proteins are brought into proximity, although there has been no evidence of this in the proteins studied so far. All constructs used for testing protein–protein interactions were derived from plasmids pLJ89 and pLJ96. cDNA templates for expression were amplified by PCR using forward and reverse primers containing 5′ sequences that are homologous to those upstream and downstream of the SalI site of each expression vector, respectively. PCR products and linear vectors were transformed into competent yeast (either the MATa or MATα strain) using standard methods, allowing the PCR fragment to in vivo recombine into the vector at its SalI site, always in the proper orientation and frame.
Plasmid encoding GST–CNXK46-M417 was constructed using plasmid pGEX6P-1 (Amersham Biosciences, Piscataway, NJ), and cloning CNXK46-M417 in between BamH1 and Not1 sites.
Protein expression, preparation, and purification
The recombinant fragments were labeled by the growth of E. coli BL21 in M9 minimal medium, with 15N ammonium sulfate or 15N ammonium sulfate/13C glucose as the sole sources of nitrogen and carbon. Cells were harvested and broken in PBS, pH 7.8. The GST fusion protein was purified by affinity chromatography on glutathione-Sepharose resin (Amersham Biosciences, Piscataway, NJ) and tags were removed by cleavage with thrombin, leaving Gly-Ser N-terminal extension. The cleaved protein was exchanged into NMR buffer using Centricon 3000 concentrators. The His6 fusion P-domain two-module fragment was purified by affinity on NiNTA resin (Qiagen, Mississauga, Canada), and then by ion exchange chromatography using Q-Sepharose (Amersham Biosciences, Piscataway, NJ). ERp57 was expressed in E. coli BL21 and grown in rich (LB) medium. Purification was performed following two-step chromatography including heparin and ion exchange (Q-Sepharose, Amersham Biosciences). In both steps, ERp57 was eluted by a linear increase of salt concentration as described elsewhere.
Peptide synthesis and purification
Peptides corresponding to the single-module fragment of 17-mer residues 337–353 and 25-mer residues 332–356 were synthesized using standard Fmoc solid-phase peptide synthesis and purified by reverse-phase chromatography on a C18 Vydac (Hesperia, CA) column using 0.1% TFA in water/0.1% TFA in acetonitrile gradient. The 17-residue peptide was synthesized with N- and C-terminal cysteine residues replacing the alanines in the normal sequence, and the intramolecular disulfide bond was formed by air oxidation at pH 8.4 in 1% ammonium bicarbonate solution. The single-module fragments correspond to the sequence repeats at the tip of the calnexin P-domain. All ERp57 peptides were synthesized by the Sheldon Biotechnology Institute (McGill University, Canada).
NMR spectroscopy
NMR resonance assignments of the calnexin P-domain two-module fragment were carried out by using standard triple-resonance techniques on 13C, 15N-labeled samples. Protein signal assignments were performed using standard techniques, including three-dimensional experiments HNCACB, CBCA(CO)NH, HNCA, 15N-noesyHMQC, and 15N-tocsyHMQC. NMR samples contained 1.5 mM calnexin in 50 mM phosphate, 150 mM NaCl at pH 6.5 or 40 mM MES, 150 mM NaCl, pH 6.5. 15N–1HN dipolar couplings were measured from IPAP-HSQC experiments (Ottiger et al, 1998) using a protein sample with 15 mg/ml of Pf1 phage (Hansen et al, 1998). NMR experiments were performed at 25°C and 30°C using Bruker DRX500 and Varian 750 MHz spectrometers. NMR spectra were processed using GIFA (Pons et al, 1996) and NMRPIPE (Delaglio et al, 1995) and analyzed with XEASY (Bartels et al, 1995).
Structure determination
For determination of the 17-mer peptide structure, a set of 261 transferred NOEs was collected from a homonuclear NOESY spectrum of the peptide/ERp57 mixture in a 10:1 ratio. 3J couplings for non-proline residues were measured using the DFQ-COSY experiment. The structure was calculated using standard protocols in CNS v. 0.9 (Brunger et al, 1998). In the final 20 structures, all residues were well defined with an average pairwise root mean squared deviation (RMSD) of 0.40±0.16 Å for backbone heavy atoms. PROCHECK-NMR (Laskowski et al, 1996) showed over 98% residues in the most favored and allowed regions of the Ramachandran plot.
Yeast mating and β-galactosidase assays
Yeast strains MATa and MATα were grown for 2 days and mated on YPD for 12 h. Diploids were isolated by replica plating onto synthetic dropout medium (-Leu -His), and allowed to grow for 24 h. Cells were then patched out on the same synthetic media, where they were grown for another 24 h, and used in filter β-galactosidase assays. Cell patches were transferred to nylon-enforced nitrocellulose membranes (Stratagene, La Jolla, CA), dipped in liquid nitrogen for 10 s, and placed on Whatman filter paper saturated in Z buffer with 4 mg/ml X-Gal. Filters were incubated for 45 min to 1 h at 37°C.
Preparation of calnexin point mutants
Site-directed mutagenesis was performed on calnexin cDNA using mismatched primers and PCR with TaqPlus Precision (Stratagene, La Jolla, CA), following the manufacturer's protocol. Automated DNA sequencing was performed to verify all sequence modifications. CNX6pmA was created by introducing the following point mutations: D342A, D344A, D346A, D348A, E350A, and E352A, where the standard alanine codon introduced was GCT. The double-point mutant CNX2pmA contained both the D344A and E352A mutations.
Preparation of ERp57 fragments and point mutants
Random mutagenesis of human ERp57 was performed by amplifying the cDNA template using previously described PCR methods, and replacing the dNTP mixture with a new mixture containing dITP (20 mM dATP, 20 mM dTTP, 20 mM dCTP, 20 mM dGTP, and 20 mM dITP) so as to introduce random mismatches within the amplification products. In all, 1500 ERp57 mutants were tested for self-activation and screened against CNX2pmA for restoration of the full interaction strength. Those that did not self-activate were sequenced in order to determine the nature/location of the mutation(s) within the cDNA. A total of 48 ERp57 clones were also chosen at random for sequencing to ensure that mutations were introduced evenly throughout the cDNA, and not biased towards a given area. A library of ERp57 truncations was created by amplifying ERp57 cDNA with a biotinylated primer containing the upstream SalI sequence for recombination into pLJ96. ddNTPs were added to the standard PCR reaction and made up 1% of all nucleotides in the mix. Separation of the single-stranded PCR products from the full-length ERp57 template was achieved by purification on a HiTrap Streptavidin Affinity Column (Amersham Biosciences, Piscataway, NJ). Products were made double stranded by PCR with a mixture of primers designed to have three random nucleotides preceded by the downstream SalI sequence in pLJ96, and cycled 25 times. All other ERp57 constructs were created by PCR as previously described. ERp57-6pmA was created by introducing the following mutations—K494A, K496A, K497A, K498A, K499A, and K500A—where the standard alanine codon introduced was GCT. PDIΔc/ERp57A482-L505 was created by replacing the C-terminal c domain of PDI (residues 472–508) with the C-terminal sequence of ERp57 (residues 482–505).
Production of GST fusion proteins and GST pull-down assays
The expression and purification of GST fusions were performed as previously described (Zheng and Guan, 1993). In all, 300 ng of GST–CNXK46-M417, or GST was incubated along with 20 μl of 50% glutathione Sepharose 4B resin (Amersham Biosciences, Piscataway, NJ), pre-equilibrated with 1 × PBS pH 7.0, in 1.0 ml 1 × PBS total volume, at room temperature with rotation for 1 h. The resin was washed three times with 250 μl of 1 × PBS, and 250 ng of ERp57 was loaded onto the resin along with varying concentrations of peptide, again in a 1.0 ml total volume of 1 × PBS. Tubes were allowed to incubate with rotation at room temperature for 1 h, and the resin was washed three times.
Western blot analyses
All Western blots were performed using WesternBreeze (Invitrogen, Carlsbad, CA), following the manufacturer's protocol. For ERp57 blots, a rabbit polyclonal anti-ERp57 antiserum was provided by Daniel Tessier (BRI/NRC, Montreal, Canada).
Supplementary Material
Supplementary data Appendix I
Supplementary data Appendix II
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research to K Gehring and DY Thomas, and from Genome Quebec/Canada. NRC publication number 48181.
References
- Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K (1995) The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR 6: 1–10 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu WS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 1: 905–921 [DOI] [PubMed] [Google Scholar]
- Chapman RE, Walter P (1997) Translational attenuation mediated by a mRNA intron. Curr Biol 7: 850–859 [DOI] [PubMed] [Google Scholar]
- Daniels R, Kurowski B, Johnson AE, Hebert DN (2003) N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell 11: 79–90 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Bettendorff P, Braun D, Herrmann T, Fiorito F, Jelesarov I, Güntert P, Helenius A, Wüthrich K (2002) NMR structures of 36 and 73-residue fragments of the calreticulin P-domain. J Mol Biol 322: 773–784 [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Herrmann T, Günter P, Braun D, Helenius A, Wüthrich K (2001) NMR structure of the calreticulin P-domain. Proc Natl Acad Sci USA 98: 3133–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Molinari M, Helenius A (1999) Setting the standard: quality control in the secretory pathway. Science 286: 1882–1888 [DOI] [PubMed] [Google Scholar]
- Ferrari DM, Söling H-D (1999) The protein disulfide-isomerase family: unravelling a string of folds. Biochem J 339: 1–10 [PMC free article] [PubMed] [Google Scholar]
- Frickel E-M, Riek R, Jelesarov I, Helenius A, Wüthrich K, Ellgaard L (2002) TROSY-NMR reveals interaction between ERp57 and the tip of the calreticulin P-domain. Proc Natl Acad Sci USA 99: 1954–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MR, Mueller L, Pardi A (1998) Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat Struct Biol 5: 1065–1074 [DOI] [PubMed] [Google Scholar]
- Jansen G, Wu C, Schade B, Thomas DY, Whiteway M (2004) Drag&Drop Cloning in Yeast. In Preparation
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477–486 [DOI] [PubMed] [Google Scholar]
- Leach MD, Cohen-Doyle MF, Thomas DY, Williams DB (2002) Localization of the lectin, ERp57 binding, and polypeptide binding sites of calnexin and calreticulin. J Biol Chem 277: 29686–29697 [DOI] [PubMed] [Google Scholar]
- Oliver JD, Roderick HL, Llewellyn DH, High S (1999) ERp57 Functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell 10: 2573–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottiger M, Delaglio F, Marquardt JL, Tjandra N, Bax A (1998) Measurement of dipolar couplings for methylene and methyl sites in weakly oriented macromolecules and their use in structure determination. J Magn Reson 134: 365–369 [DOI] [PubMed] [Google Scholar]
- Ou WJ, Cameron PH, Thomas DY, Bergeron JJM (1993) Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature 364: 771–776 [DOI] [PubMed] [Google Scholar]
- Pirneskoski A, Ruddock LW, Klappa P, Freedman RB, Kivirikko KL, Koivunen P (2000) Domains b′ and a′ of protein disulfide isomerase fulfill the minimum requirement for function as a subunit of prolyl 4-hydroxylase. J Biol Chem 276: 11287–11293 [DOI] [PubMed] [Google Scholar]
- Pons JL, Malliavin TE, Delsuc MA (1996) GIFA v.4: a complete package for NMR data set processing. J Biomol NMR 8: 445–452 [DOI] [PubMed] [Google Scholar]
- Schrag JD, Bergeron JJM, Li Y, Borisova S, Hahn M, Thomas DY, Cygler M (2001) The structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol Cell 8: 633–644 [DOI] [PubMed] [Google Scholar]
- Schrag JD, Procopio DO, Cygler M, Thomas DY, Bergeron JJM (2003) Lectin control of protein folding and sorting in the secretory pathway. TIBS 28: 49–57 [DOI] [PubMed] [Google Scholar]
- Shamu CE, Walter P (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J 15: 3028–3039 [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101: 249–258 [DOI] [PubMed] [Google Scholar]
- Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ 2nd, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJM (1991) SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem 266: 19599–19610 [PubMed] [Google Scholar]
- Zapun A, Darby NJ, Tessier DC, Michalak M, Bergeron JJM, Thomas DY (1998) Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J Biol Chem 273: 6009–6012 [DOI] [PubMed] [Google Scholar]
- Zapun A, Petrescu SM, Rudd PM, Dwek RA, Thomas DY, Bergeron JJM (1997) Conformation-independent binding of monoglucosylated ribonuclease B to calnexin. Cell 88: 29–38 [DOI] [PubMed] [Google Scholar]
- Zheng CF, Guan KL (1993) Properties of MEKs, the kinases that phosphorylate and activate the extracellular signal-regulated kinases. J Biol Chem 268: 23933–23939 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data Appendix I
Supplementary data Appendix II