

Abstract
A dynamic interaction occurs between the lymphoma cell and its microenvironment, with each profoundly influencing the behavior of the other. Here, using a clonogenic coculture growth system and a xenograft mouse model, we demonstrated that adhesion of mantle cell lymphoma (MCL) and other non-Hodgkin lymphoma cells to lymphoma stromal cells confers drug resistance, clonogenicity, and induction of histone deacetylase 6 (HDAC6). Furthermore, stroma triggered a c-Myc/miR-548m feed-forward loop, linking sustained c-Myc activation, miR-548m downregulation, and subsequent HDAC6 upregulation and stroma-mediated cell survival and lymphoma progression in lymphoma cell lines, primary MCL and other B cell lymphoma cell lines. Treatment with an HDAC6-selective inhibitor alone or in synergy with a c-Myc inhibitor enhanced cell death, abolished cell adhesion–mediated drug resistance, and suppressed clonogenicity and lymphoma growth ex vivo and in vivo. Together, these data suggest that the lymphoma-stroma interaction in the lymphoma microenvironment directly impacts the biology of lymphoma through genetic and epigenetic regulation, with HDAC6 and c-Myc as potential therapeutic targets.
Introduction
Despite intensive effort in the development of new therapies and improvement in overall survival in B cell lymphomas, significant proportions of patients relapse with incurable disease. Mantle cell lymphoma (MCL) is classically considered an aggressive lymphoma. However, some studies have described a subset of patients with an indolent clinical evolution (1). The emergence of clinical drug resistance continues to be an obstacle to the successful treatment of these lymphomas. Extensive evidence has shown that specific niches within lymphoma tumor microenvironment provide sanctuary for subpopulations of lymphoma cells through stromal cell–tumor cell interactions. These interactions notably dictate lymphoma cell growth, response to therapy, and resistance of residual lymphoma cells to chemotherapeutic agents. Depending on lymphoma type and location, cellular elements of stroma are composed of supportive fibroblast-like stromal cells, including mesenchymal stromal cells, dendritic cells, osteoclasts, osteoblasts, and endothelial cells, among others. B lymphocytes and lymphoma cells within the lymph node and bone marrow are likely to interact with their resident stromal cells, such as follicular dendritic cells (FDCs) and bone marrow stromal cells, and the interaction plays a critical role in lymphoma progression. Furthermore, this interaction plays a role in the resistance of residual lymphoma cells to chemotherapeutic agents, a problem that remains a major challenge in the treatment of MCL and other B cell lymphomas and consequently contributes to disease relapse. However, how the lymphoma microenvironment influences lymphoma cell survival and response to therapy, as well as the molecular mechanisms involved, remains unclear.
Several subsets of stromal cells, in particular FDCs and bone marrow stromal cells, are found within secondary lymphoid organs and bone marrow, in which they play a key role in the initiation and maintenance of efficient immune responses (2). FDCs are restricted to germinal centers and allow B cell migration, selection, and differentiation through a complex set of survival factors, including B cell receptor–mediated signaling, chemokines, cytokines, and adhesion molecules. Circulating resting B cells migrate through the FDC networks, whereas antigen-activated B cells undergo clonal expansion within the FDC network in a T cell–dependent fashion, thereby generating the germinal center (2). Gene expression profiling has revealed that lymphoma stroma networks might be associated with clinical outcome in follicular lymphoma and diffuse large B cell lymphomas (3–5). Furthermore, the diffuse distribution of FDCs in MCL may be associated with a worse clinical outcome (6). These observations suggest that interaction between stroma and B cell lymphoma cells contributes to drug resistance and supports the growth of MCL and other B lymphoma cell survival.
MicroRNAs (miRNAs) are non–protein coding genes that regulate the human transcriptome by pairing to the 3′-untranslated region (UTR) of target genes, inducing RNA cleavage and/or translational inhibition (7). miRNAs have been found to play key roles in a wide range of biological processes and to be aberrantly expressed in many types of cancer (8, 9). Given that physical interactions between B cells and stromal cells from the lymphoid tissue microenvironment are critical to the survival of normal and malignant B cells, we and others have recently demonstrated that miRNA expression is closely related to the stage of B cell maturation and identified a set of miRNAs regulated by interactions between stromal cells and B cells (10, 11). We illustrated that lymph node stroma induces expression of miRNA-181a, which in turn targets the proapoptotic protein BCL-2–interacting mediator of cell death (Bim) for silencing and contributes to cell adhesion–mediated drug resistance (CAM-DR) in lymphoma cells (12). Here, we demonstrate that adhesion of MCL and other B cell lymphoma cells to lymphoma stroma confers drug resistance, enhances lymphoma cell clonogenicity, and is associated with c-Myc/miR-548m feed-forward loop, leading to sustained c-Myc activation and miR-548m downregulation. Furthermore, c-Myc, through a corepressor complex with EZH2, downregulates miR-548m and contributes to stroma-mediated cell survival and colony formation through histone deacetylase 6 (HDAC6) upregulation. HDAC6 in turn is an upstream effector of the Bim signaling pathway in MCL and other B cell lymphomas. We also show that stroma-mediated drug resistance and growth are reversed by enforced expression of miR-548m as well as inhibition of c-Myc and HDAC6 ex vivo and in vivo. The HDAC6-selective inhibitor tubastatin A cooperates with bromodomain and extraterminal domain (BET) inhibitor, JQ1, which substantially enhanced cell death, abolished CAM-DR, and suppressed anchor-independent growth and lymphoma growth in vivo. These findings define a novel mechanism by which the tumor microenvironment alters specific miRNA and HDAC6 levels, leading to drug resistance and lymphoma survival, with HDAC6 and c-Myc as potential therapeutic targets to overcome drug resistance in MCL and other B cell lymphomas.
Results
Cell adhesion to stromal cells induces HDAC6 expression and downregulation of miR-548m in MCL and other B cell lymphoma cell lines and primary cells.
Recently, using global miRNA expression profiling, we reported a set of miRNAs that was dysregulated when lymphoma cells adhered to lymph node stromal cells (follicular dendritic-like cells, also called HK cells) in MCL and other B cell lymphomas, which revealed that expression of multiple miRNAs is altered in lymphoma cells upon adhesion to HK cells and associated with B cell differentiation and survival (12, 13). Among these miRNAs, we found that miR-548 family members miR-548f, miR-548h, and miR-548m were among the most downregulated miRNAs by stroma interaction, with miR-548m as the most strongly downregulated miRNA (11 fold), as shown in Figure 1A. Thus, in this study, we further explored miR-548m’s function in stroma-mediated survival and drug resistance in various types of B cell lymphoma. We used MCL cell lines and a germinal center cell–derived lymphoma cell line (SUDHL-4) in all mechanistic studies herein. To validate the miRNA microarray data, we examined the miR-548m level with and without HK cell adhesion by using quantitative RT-PCR (qRT-PCR), with results revealing that miR-548m was indeed downregulated upon cell adhesion to HK cells in MCL (Jeko-1 and HBL-2 cells) and SUDHL-4 cells (Figure 1B). Next, we searched the potential targets of miR-548m through TargetScan and found that the 3′-UTR of HDAC6 contains one sequence motif that perfectly matches with the “seed” sequence of miR-548m. This motif is well conserved among humans, mice, and rats, suggesting a potential regulation of HDAC6 by miR-548m. We therefore performed Western blotting to validate that cell adhesion indeed induces HDAC6 expression in MCL and other B cell lymphoma cells. As shown in Figure 1C, HDAC6 protein levels, when cultured in direct contact with HK cells, were markedly increased in Jeko-1, HBL-2, and SUDHL-4 lymphoma cells. In contrast, lymphoma cells exposed to soluble factors with no contact with HK cells (in Transwell inserts) revealed no significant changes in HDAC6 expression, supporting the role of lymphoma cell–HK cell contact in HDAC6 induction. The HDAC6 induction by HK cells was time dependent, observed as early as at 6 hours and sustained at 48 hours after coculture (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI64210DS1). Furthermore, we examined whether cell adhesion–mediated miR-548m downregulation and HDAC6 induction observed in lymphoma cell lines were operative in primary MCL and other B cell lymphoma specimens and examined the changes in miR-548m and HDAC6 following adhesion of the primary lymphoma cells to HK cells. As shown in Figure 1D, lymphoma samples (including 5 MCL samples, 3 diffuse large B cell lymphomas, and 5 follicular center cell lymphomas) responded to stromal cell adhesion with downregulation of miR-548m expression similar to that observed in lymphoma cell lines. We also determined the effect of cell adhesion on HDAC6 expression in primary lymphoma cells. As shown in Figure 1E, increased levels of HDAC6 were observed in these primary lymphoma cells after adhesion to HK cells. To this end, we used bone marrow stromal cells (HS-5 cells) in our coculture system to test whether bone marrow stroma induced similar HDAC6 changes. As shown in Figure 1F, HS-5 cells indeed triggered upregulation of HDAC6 expression and downregulation of miR-548m in HBL-2 and SUDHL-4 cells. These results imply that both lymph node and bone marrow stroma may control lymphoma cell response via a miR-548m/HDAC6 pathway in MCL and other B cell lymphomas.
Figure 1. Cell adhesion to stromal cells (HK cells) induces HDAC6 expression and downregulation of miR-548m in MCL and other B cell lymphoma cell lines and primary cells.

(A) Heat map showing downregulation of miR-548m/miR-548f/miR-548h expression after lymphoma cells adhered to HK cells for 24 hours, as measured by miRNA array. Sus, cells in suspension; HK-Ad, cells after adhesion to HK cells. (B and C) Cell adhesion induced changes of (B) miR-548m and (C) HDAC6 expression in SUDHL-4, HBL-2, and Jeko-1 lymphoma cells. Lymphoma cells in suspension adhered to the preestablished monolayer of HK cells or with a confluent HK monolayer but separated by Transwell inserts were treated for 24 hours (Transwell). The lymphoma cells were collected, and miR-548m and HDAC6 protein were analyzed by TaqMan microRNA qRT-PCR and Western blot. *P < 0.05; **P > 0.05. (D and E) Cell adhesion–induced changes of (D) miR-548m and (E) HDAC6 expression in primary lymphoma cells. DLBCL, diffuse large B cell lymphoma; FCL, follicular lymphoma. (F) Cell adhesion to bone marrow stromal cells (HS-5 cells) induced changes of miR-548m and HDAC6 expression in SUDHL-4 and HBL-2 cells. *P < 0.05. Results are representative of at least 4 experiments (mean ± SD). (C and E) Thin white lines were used to separate lanes run on different gels.
miR-548m directly targets HDAC6, and miR-548m downregulation contributes to cell adhesion–mediated HDAC6 induction.
We subsequently determined whether HDAC6 is directly regulated by miR-548m and whether the downregulation of miR-548m is necessary for adhesion-induced HDAC6 expression. To confirm this, the effect of ectopic expression of miR-548m on HDAC6 expression was investigated. As shown in Figure 2A, the increased expression of miR-548m by ectopic miR-548m expression downregulated HDAC6 mRNA and protein expression in Jeko-1 cells. In contrast, knockdown of miR-548m by transfection of anti–miR-548m increased HDAC6 expression (Figure 2B). We next established a stable miR-548m–inducible cell line (referred to herein as the HBL-2-i-miR-548m cell line) by using a inducible gene expression system (also called Cumate Switch Inducible Expression System, System Biosciences) for induction and overexpression of miR-548m. This system features a cumate-inducible pre–miR-548m gene, so that miR-548m expression can be turned on with addition of cumate to cell culture medium. HBL-2-i-miR-548m cells were treated with cumate for up to 72 hours, and the expression level of miR-548m was measured. As shown in Figure 2C, addition of cumate induced miR-548m expression in a time-dependent manner observed at 48 and 72 hours. Using this system, we examined the expression of the corresponding HDAC6 by Western blot, and reduction of HDAC6 was detected after addition of cumate, supporting the regulatory role of miR-548m on HDAC6 expression (Figure 2D). When we applied this stable, inducible cell line to the coculture experiment, induction of miR-548m by cumate abolished HK-induced HDAC6 expression (Supplemental Figure 2A). Further, we confirmed that HDAC6 is a direct target of miR-548m, since the relative luciferase activity of the wild-type construct of HDAC6 3′-UTR was reduced in the presence of pre–miR-548m (P < 0.05) in both Jeko-1 and SUDHL-4 cells, whereas such a suppressive effect of miR-548m on luciferase activity was not observed with the mutant construct of HDAC6 3′-UTR. The construction of the mutant and the effect of miR-548m on HDAC6 3′-UTR luciferase activity are shown in Figure 2, E and F. To further confirm that the 3′-UTR is responsible for the reduction of HDAC6 by miR-548m, we developed 2 stable cell lines using 2 pCMV6-AC-GFP constructs (OriGene): one containing HDAC6 ORF (without the 3′-UTR) and one control construct. After G418 (Neomycin) selection, these 2 stable cell lines were transfected with pre–miR-548m and pre-miR control, respectively, and HDAC6 levels were examined by Western blot. As anticipated, ORF construct blocked the miR-548m inhibitory effect on HDAC6 expression. As shown in Supplemental Figure 2B, transfection of pre–miR-548m resulted in a marked decrease of HDAC6 in control cells but not HDAC6 ORF cells, supporting that miR-548m regulates HDAC6 through targeting its 3′-UTR. These findings show a direct and specific interaction of miR-548m on HDAC6 expression, and HDAC6 is directly targeted and repressed by miR-548m. To explore the role of miR-548m in adhesion-induced HDAC6 expression, miR-548m was overexpressed or knocked down in lymphoma cells, and the expression of HDAC6 was assessed upon adhesion to HK cells. As shown in Figure 2, A and B, overexpression of miR-548m abolished adhesion-induced HDAC6 increase, and knockdown of miR-548m enhanced adhesion-induced HDAC6 increase, supporting the notion that cell adhesion–mediated HDAC6 induction is through miR-548m downregulation.
Figure 2. HDAC6 is a direct target of miR-548m, and cell adhesion–mediated HDAC6 induction is through miR-548m downregulation.

(A) Overexpression of miR-548m decreased HDAC6 expression and inhibited cell adhesion–mediated HDAC6 expression. The lanes separated by white lines were run on the same gel but were noncontiguous. (B) Knockdown of miR-548m upregulated HDAC6 mRNA and protein expression. Jeko-1 cells were transfected with (A) pre–miR-548m or pre-miR control and (B) anti-miR control or anti–miR-548m, and miR-548m expression was analyzed by qRT-PCR. (A and B) The relative level of HDAC6 protein was measured by quantitative densitometry and is indicated below each lane. (C) Time course of miR-548m induction in HBL-2-i-miR-548m cells after addition of cumate (30 μg/ml) to cell culture medium. qRT-PCR was used to measure miR-548m expression levels at the indicated times. (D) The effect of miR-548m induction on HDAC6 protein expression in HBL-2-i-miR-548m cells after addition of cumate at 48 and 72 hours. (E) Schematic diagram showing putative miR-548m binding site on HDAC6 3′-UTR and the sequence of mutant-type reporter constructs. The white rectangle indicates putative miR-548m binding site in HDAC6 3′-UTR. The long black rectangle indicates the human HDAC6 3′-UTR. The red letters indicate the mutation sites of HDAC6 3′-UTR (in HDAC6 3′-UTRm reporter). (F) Jeko-1 or SUDHL-4 cells were transfected with pmiR-Report control vector (Ctrl) or pmiR-Report.HDAC6 3′-UTR wild-type plasmids or pmiR-Report.HDAC6 3′-UTR mutant plasmids (MUT) harboring point mutations in the target sites for miR-548m. These reporter plasmids were cotransfected with pre–miR-548m (50 nM) or control. The firefly luciferase activities of these transfected cells were measured and normalized to Renilla luciferase activities. Data are shown with the mean ± SD of at least 4 experiments. *P < 0.05; **P > 0.05.
Cell adhesion–mediated downregulation of miR-548m and HDAC6 upregulation are required for cell adhesion–mediated drug resistance.
We next determined whether miR-548m downregulation is required for CAM-DR upon cell adhesion to the HK cells. Jeko-1 and SUDHL-4 cells were examined for ectopic overexpression of miR-548m and for changes in drug-induced apoptosis. As shown in Figure 3, A and B, ectopic miR-548m expression suppressed HK cell adhesion–mediated HDAC6 expression, induced cell apoptosis in the absence of adhesion, and enhanced mitoxantrone-induced apoptosis in the cells adhered to HK cells as well as overcame stroma-mediated drug resistance (CAM-DR) in Jeko-1, SUDHL-4, and HBL-2 cells (Supplemental Figure 3A). Similar results were observed after miR-548h ectopic expression (Supplemental Figure 3B) in HBL-2 cells. To determine whether miR-548m is involved in CAM-DR through cell adhesion–mediated HDAC6 upregulation, we next ablated HDAC6 expression using shRNA and examined the effects of HDAC6 knockdown on CAM-DR. As shown in Figure 3C and Supplemental Figure 3C, HDAC6 shRNAs were highly effective in depleting HDAC6 in both Jeko-1 and SUDHL-4 cells, respectively. Cell extracts from Jeko-1 and SUDHL-4 cells transfected with HDAC6 shRNAs or a control shRNA with or without cell adhesion to HK cells were prepared, followed by Western blot and apoptosis analysis. HDAC6 knockdown through shRNAs not only increased cell apoptosis and sensitized the response to the cytotoxic drug mitoxantrone but also abolished CAM-DR in Jeko-1 and SUDHL-4 cells, as measured by Annexin V (Figure 3D and Supplemental Figure 3D). We further tested the effects of overexpression of HDAC6 on CAM-DR. As revealed by Figure 3, E and F, overexpression of HDAC6 through ectopic transfection of HDAC6 plasmid increased HDAC6 protein expression and suppressed mitoxantrone-induced apoptosis as well as further promoted CAM-DR in Jeko-1 cells. Again, these results indicate that stromal (HK) cells use the HDAC6 pathway to promote lymphoma cell survival. Taken together, these results support the key role of miR-548m and its downstream protein, HDAC6, in CAM-DR in MCL as well as other B cell lymphomas. These results demonstrate for the first time that the level of HDAC6 and cell apoptosis is tightly controlled by the interaction with surrounding HK cells via downregulation of miR-548m and that HDAC6 can be a novel target for overcoming drug resistance in B cell lymphomas.
Figure 3. Cell adhesion–mediated downregulation of miR-548m and HDAC6 upregulation are required for CAM-DR.

(A and B) Overexpression of miR-548m induces HDAC6 downregulation and lymphoma cell apoptosis and enhances mitoxantrone-induced cell apoptosis. Jeko-1 and SUDHL-4 cells transfected with pre–miR-548m or pre-miR control were treated with vehicle control or mitoxantrone (0.2 μM) for 24 hours with or without HK cell adhesion. *P < 0.05; **P > 0.05. (A) The relative level of HDAC6 protein was measured by quantitative densitometry and is indicated below each lane. (C) HDAC6 knockdown after 48 hours of transfection with HDAC6 shRNA constructs 1 or 2 or nonsilencing control. (D) Knockdown of HDAC6 induces cell apoptosis and enhances mitoxantrone-induced cell apoptosis with and without HK cell adhesion. Jeko-1 or SUDHL-4 cells transfected with HDAC6 shRNA-2 (shHDAC6) or control shRNA (shCtrl) were incubated for 48 hours and then treated with vehicle control or mitoxantrone (0.2 μM) for 24 hours. Apoptosis was analyzed by Annexin V. (E and F) Overexpression of HDAC6 inhibits mitoxantrone-induced lymphoma cell apoptosis in Jeko-1 cells in the presence of HK cell adhesion. Jeko-1 cells were transfected with pCDNA3 control (pLCtrl) or pCDNA3-HDAC6 (pLHDAC6) for 24 hours. Cells, collected and washed in suspension versus in adhesion with HK, were treated with mitoxantrone (0.2 μM) for 12 hours. (E) HDAC6 protein levels were then analyzed by Western blot and (F) apoptosis was analyzed by Annexin V. Data are representative of at least 3 independent experiments (mean ± SD).
Stromal cells enhance lymphoma cell clonogenicity through the miR-548m/HDAC6 axis.
We further tested whether HK cells could alter the anchorage-independent clonogenic growth of lymphoma cells. We plated lymphoma cells (HBL-2 cells) alone or with HK and HS-5 cells in methylcellulose cultures. Plating lymphoma cells alone in this assay resulted in the growth of discrete tumor colonies, with an efficiency of 1% to 2% of cells plated. The addition of HK cells to these colony formation cultures constituted a novel clonogenic coculture growth system and led to a greater number of lymphoma colonies and increased the size of individual colonies when compared with addition of lymphoma cells alone (Figure 4A and Supplemental Figure 4A). Of note, most of these colonies are physically associated with HK cells, supporting the need of clonogenicity for cellular proximity (Figure 4A, inset). To determine that cell-cell contact between stroma (HK) and lymphoma cells is required for this clonogenicity, we used the Transwell insert assay by incubating methylcellulose with lymphoma cells in the bottom chamber, which was separated from HK cells in the top chamber by a Transwell membrane, and found no increased ability of HK cells to enhance lymphoma cell colony formation (data not shown). These findings suggest that cell-cell contact accounts for the majority of stroma-induced lymphoma cell growth. We further examined whether miR-548m and HDAC6 are involved in stroma-mediated lymphoma cell clonogenic growth. As shown in Figure 4B and Supplemental Figure 4B, transfection of pre–miR-548m or induction of miR-548m with cumate in HBL-2-i-miR-548m cells resulted in overexpression of miR-548m and significantly abolished stromal (HK) cell–induced clonogenic growth. Importantly, knockdown of HDAC6 with shRNA markedly abrogated stroma-mediated lymphoma colony formation (Figure 4C). The clonogenicity inhibition could be due to increased apoptosis and/or growth suppression. Therefore, stroma-enhanced lymphoma clonogenicity is mediated, at least in part, by the miR-548m/HDAC6 axis, and interaction between stromal cells and lymphoma cells activated HDAC6, leading to prolonged survival and growth of lymphoma cells.
Figure 4. Stromal cells enhance the clonogenicity of lymphoma cell lines through the miR-548m/HDAC6 pathway.

(A–C) Numbers of HBL-2 colonies after 2 weeks of culture. (A) HBL-2 cells were plated without HK cells (Sus) and with HK cells (HK-Ad) at ratio of 1:2 in methylcellulose medium. (B) HBL-2 cells were transfected with pre-miR control (pre-miR-Ctrl) or pre–miR-548m and plated without and with HK cells in methylcellulose medium. (C) HBL-2 cells were transfected with control shRNA (shCtrl) or HDAC6 shRNA (shHDAC6) and plated without and with HK cells in methylcellulose medium. (D and E) Targeting HDAC6 circumvents CAM-DR and stroma-induced clonogenicity. (D) Jeko-1 cells were treated with vehicle control or tubastatin A (0.5 μM) or mitoxantrone (0.2 μM) alone or tubastatin A plus mitoxantrone for 24 hours with or without HK cell adhesion. (E) HBL-2 cells were treated with DMSO or tubastatin A (0.5 μM) for 24 hours and plated without and with HK cells in methylcellulose medium. (F) Bim mRNA and (G) protein levels in HBL-2 cells after 48 hours of transfection with HDAC6 shRNA constructs or nonsilencing control shRNA as well as treatment with DMSO or tubastatin A (0.5 μM). EL, extra-long form of Bim; L, long form of Bim; S, short form of Bim. Data are representative of 3 to 4 independent experiments (mean ± SD). Original magnification, ×40 (A–C and E); ×100 (A, inset).
Targeting the miR-548m/HDAC6 axis circumvents CAM-DR, clonogenicity, and lymphoma formation in vivo.
The above results established a novel role of HDAC6 in stroma-mediated drug resistance and stroma-enhanced clonogenicity. We examined whether selective HDAC6 inhibitors induce lymphoma cell apoptosis and sensitize lymphoma cells to drug-induced apoptosis. As shown in Figure 4D, addition of the HDAC6-specific inhibitor tubastatin A resulted in increased apoptosis and sensitized lymphoma cells to mitoxantrone in the presence of stromal cells. Given that tubastatin A has been shown to be a new, potentially antitumor drug (14–18), we also tested whether inhibition of HDAC6 by tubastatin A suppresses lymphoma cell colony formation and blocks stroma-mediated clonogenicity. As revealed in Figure 4E, tubastatin A dramatically inhibited clonogenic growth of HBL-2 lymphoma cells in the absence and presence of stroma adhesion, further supporting the role of HDAC6 in cell adhesion–mediated clonogenicity.
Next, we determined the downstream molecular effector of HDAC6 in CAM-DR and clonogenicity. We have previously shown that HDAC pan-inhibitors, SAHA and LBH, induce Bim upregulation and revealed cell adhesion–mediated Bim downregulation in B cell lymphomas (13). Recently, inhibition of HDAC6 was shown to significantly increase protein levels of Bim in acute myeloid leukemia (19). We therefore speculated that HDAC6 is involved in stroma adhesion–mediated Bim downregulation. The effects of HDAC6 on Bim mRNA expression and protein levels were examined. HDAC6 shRNA was used to knockdown the HDAC6 expression, and then Bim expression levels in HBL-2 cells were examined with qRT-PCR in control and HDAC6 shRNA–treated cells. As shown in Figure 4, F and G, and Supplemental Figure 4, C and D, HDAC6 knockdown had no effect on Bim mRNA but increased Bim protein levels in HBL-2 as well as Jeko-1 and SUDHL-4 cells, supporting that HDAC6 downregulates Bim through a posttranslational mechanism. Similar effects were observed when HDAC6 was inhibited by tubastatin A (Figure 4, C and D, and Supplemental Figure 4, B and C). Given our previous findings that Bim played a role in HK CAM-DR (13), we believe that HDAC6 is involved in CAM-DR, at least partially, through Bim downregulation.
Having defined the functional role of stromal cells in regulation of stroma-mediated drug resistance and lymphoma growth in vitro, we next examined whether stromal cells similarly promote lymphoma cell growth in vivo. We used the NOD/SCID mouse model and injected HBL-2 cells and diffuse large B cell lymphoma cells, Toledo cells, with or without HK cells subcutaneously. One million lymphoma cells (HBL-2 and Toledo cells) were injected into the flanks of NOD/SCID mice (n = 4–6 mice per condition). As shown in Figure 5A and Supplemental Figure 5A, a more robust growth of tumors was observed in mice receiving HK and HBL-2 cells than in mice injected with HBL-2 cells alone. HK cells alone did not induce tumors in mice (data not shown). Further, immunohistochemical stains by using HK cell antibody (CD21) further confirmed the presence of dispersed single and clusters of HK cells in tumor coinjected with HBL-2 with HK cells and absence of HK in tumor injected with HBL-2 cell only (Supplemental Figure 5B). In order to assess whether downregulation of miR-548m is necessary for stroma-induced cellular transformation and lymphoma formation in vivo, HBL-2 cells were transfected with pre–miR-548m or control miRNA and injected into the flanks of NOD/SCID mice with and without HK cells (ratio of 1:2) to initiate lymphomagenesis. As revealed in Figure 5B, enforced expression of miR-548m in HBL-2 cells dramatically suppressed lymphoma formation and blocked stroma-induced lymphoma progression. Additionally, using HBL-2-i-miR-548m cells to induce miR-548m in vivo with cumate, we observed that induction of miR-548m with cumate attenuated lymphoma formation and stroma-enhanced tumor growth, supporting the role of HDAC6 in lymphoma growth (Supplemental Figure 5, C and D). Next, treatment with tubastatin A significantly inhibited tumor growth in NOD/SCID mice bearing HBL-2 xenografts (Figure 5C). In addition, tubastatin A also abolished stromal HK cell–mediated tumor growth of NOD/SCID mice bearing HBL-2 and HK xenografts (Figure 5C). Finally, when combined with mitoxantrone, tubastatin A enhanced mitoxantrone-induced lymphoma killing and overcame stroma-mediated drug resistance in vivo (Figure 5D). Collectively, stroma-mediated HDAC6 induction through unleashing of miR-548m repression is a novel therapeutic target for MCL and other B cell lymphomas.
Figure 5. Stromal (HK) cells enhance the lymphoma formation, and targeting HDAC6 inhibits lymphoma growth in vivo.

(A) Stromal (HK) cells support lymphoma formation in vivo. HBL-2 cells (1 × 106 cells) were subcutaneously injected with or without HK cells (5 × 105 cells) into the posterior flank of NOD/SCID mice. The graph shows tumor size measured at the indicated days after cell injection. Photographed tumors inoculated by HBL-2 alone or HBL-2 coinoculated with HK (HBL-2+HK) on day 26 are also shown. *P < 0.05. (B) Enforced expression of miR-548m suppressed lymphoma formation. HBL-2 cells were transfected with pre-miR control (pre-miR-Ctrl) or pre–miR-548m for 24 hours and injected without and with HK cells. *P < 0.05. (C) Treatment with tubastatin A significantly inhibited tumor growth in the absence and presence of HK cells. *P < 0.05. (D) Treatment with tubastatin A enhanced mitoxantrone-induced lymphoma killing in vivo in NOD/SCID mice bearing HBL-2 xenografts. (B–D) NOD/SCID mice were inoculated subcutaneously in the right flank with 1 × 106 HBL-2 cells with and without HK cells (5 × 105 cells). Two weeks later, when palpable tumors (≥5 mm in diameter) developed, mice were treated with intraperitoneal injections of (C) tubastatin A alone (25 mg/kg/d) for 14 consecutive days or (D) mitoxantrone alone (1.5 mg/kg/d) on days 1, 2, 3, 4, 8, 9 10, and 11 or the tubastatin A–mitoxantrone combination for 14 consecutive days. Each group consisted of 4 to 6 mice. *P < 0.05, significantly delayed tumor growth. The inset shows a typical representative of each treatment group. Data are shown with mean ± SD.
c-Myc/miR-548m feed-forward circuit contributes to sustained stroma-mediated c-Myc activation and miR-548m downregulation in lymphoma microenvironment.
We next investigated the underlying molecular mechanism of stroma-mediated miR-548m downregulation. Our recent studies revealed that c-Myc recruited HDAC3 and EZH2, a histone methyltransferase and a member of the polycomb group of proteins, to the promoters of miR-29 and miR-15a/16 as corepressors to downregulate miRNA expression (20, 21). We, thus, tested whether miR-548m expression is c-Myc dependent and its repression is mediated through histone modification. Using P493-6 cells, a transformed human B cell line featuring a tetracycline-repressible c-MYC gene that allows c-Myc expression to be turned on or off without altering the survival of these cells (22), the miR-548m expression and corresponding c-Myc expression levels were examined in c-Myc–overexpressed and knocked down P493-6 cells. We found induction of miR-548m in Myc-off P496-3 cells and miR-548m downregulation in Myc-on P496-3 cells, supporting the role of c-Myc in miR-548m expression (Figure 6A). Recently, a small molecule termed JQ1, a substituted 6H-thieno[3,2-f][1,2,4]triazolo[4,3-a]azepine that functions as an inhibitor of BET, was identified to specifically prevent bromodomain from binding to acetylated histone and silencing MYC transcription, thus preventing transactivation of MYC target genes (23). By using JQ1 and the EZH2 inhibitor, DZNep, respectively, to block the activities of c-Myc and EZH2, we observed that inhibition of c-Myc with JQ1 and of EZH2 with DZNep each increased miR-548m expression in HBL-2 and SUDHL-4 cells, as shown in Figure 6B and Supplemental Figure 6A. Moreover, to confirm that the above EZH2 and c-Myc are indeed involved in miR-548m expression, a more specific and independent genetic approach was used; siRNAs against c-Myc and EZH2 were used and shown to increase miRNA-548m expression (Figure 6B and Supplemental Figure 6B). These data are in line with the notion that c-Myc represses miR-548m expression through EZH2. To determine whether the repression effect can be attributed to direct binding of c-Myc and EZH2 to miR-548m gene promoter, close examination of the highly conserved miR-548m promoter using the University of California at Santa Cruz Genome Browser conserved transcription factor–binding site track revealed the presence of at least 2 E-box Myc binding sites, S1 and S2 (Figure 6C). ChIP assay was performed to explore whether EZH2 could be recruited to the miR-548m promoters by c-Myc and whether EZH2 binding is c-Myc dependent. As revealed by Figure 6C, antibodies against both c-Myc and EZH2 efficiently immunoprecipitated the miR-548m promoter regions. EZH2 binding is c-Myc dependent, since EZH2 binding is abolished in Myc-off P493-6 cells, further supporting the recruitment role of c-Myc, and c-Myc cooperates with EZH2 to regulate miR-548m expression. thus, these results provided further support of the underlying mechanistic of Myc-induced miR-548m repression.
Figure 6. c-Myc/miR-548m feed-forward circuit contributes to stroma-mediated c-Myc activation and miR-548m downregulation in lymphoma microenvironment.

(A) miR-548m expression inversely correlated with c-Myc expression in tetracycline-treated (Myc off) and untreated (Myc on) P493-6 cells after addition and withdraw of tetracycline. (B) Inhibition of c-Myc and EZH2 induced miR-548m expression in HBL-2 and SUDHL-4 cells. (C) Schematic diagram of the location of Myc-binding sites of the miR-548m regulatory region. S1 and S2 represent Myc-binding sites with E-box sequence. The black bar represents the miR-548m locus. NC, negative control. Both S1 and S2 are highly conserved in their putative promoter region. ChIP assay shows c-Myc and EZH2 enrichment on miR-548m promoter in c-Myc on and c-Myc off P493-6 cells and Jeko-1 cells. (D) Ectopic expression of miR-548m downregulated c-Myc expression (left) and directly targeted 3′-UTR of c-Myc (right). Jeko-1 or SUDHL-4 cells were transfected with pmiR-Report control vector (Ctrl) or pmiR-Report.Myc 3′-UTR wild-type or pmiR-Report.Myc 3′-UTR mutant (Mut) plasmids harboring point mutations in the target sites for miR-548m. The luciferase activities were normalized against firefly luciferase activities. Data are representative of at least 3 independent experiments (mean ± SD). (E) Cell adhesion to HK cells induced upregulation of c-Myc and EZH2. (F) Silencing of c-Myc with JQ1 (1 μM, 48 hours) inhibited c-Myc and stroma-induced c-Myc upregulation and increased miR-548m expression. (G) Overexpression of miR-548m with pre–miR-548m decreased stroma-induced c-Myc expression, and knockdown of miR-548m by anti–miR-548m increased c-Myc expression. Results are mean ± SD or representative from at least 3 biological replicates.
We next tested that miR-548m directly targets c-Myc and therefore forms a forward-feedback circuit to sustain c-Myc activation and miR-548m downregulation. Bioinformatic analysis revealed that miR-548m is predicted to target c-Myc. We examined the effects of miR-548m on c-Myc expression. Overexpression of miR-548m through transfection of pre–miR-548m reduced c-Myc mRNA as well as protein abundance in Jeko-1, HBL-2, and SUDHL-4 cells (Figure 6D and Supplemental Figure 6C), and, further, this ectopic expression of miR-548m inhibited wild-type but not mutant c-Myc 3′-UTR luciferase reporter activity (Figure 6D). Overall, these data confirmed that miR-548m regulates c-Myc. To test that c-Myc activation is operative in lymphoma microenvironment, we cocultured lymphoma cells with stromal cells, HK cells, and observed that c-Myc as well as EZH2 were upregulated upon cell adhesion to HK cells in HBL-2, Jeko-1, and SUDHL-4 cells (Figure 6E). To determine whether c-Myc activation is required for stroma-induced miR-548m downregulation, we used JQ1 to silence c-Myc expression in lymphoma cells in the absence and presence of HK cells. As shown in Figure 6F, JQ1 reduced c-Myc expression, increased miR-548m expression, and blocked stroma-induced miR-548m downregulation. In contrast, as shown in Figure 6G, overexpression of miR-548m by transfection of pre–miR-548m decreased c-Myc expression and suppressed stroma-induced c-Myc expression. Moreover, knockdown of miR-548m with anti–miR-548m transfection upregulated c-Myc expression and enhanced HK-induced c-Myc expression (Figure 6G and Supplemental Figure 6D), supporting the notion that c-Myc/miR-548m feed-forward circuit contributes to stroma-mediated c-Myc activation and miR-548m downregulation in lymphoma microenvironment.
Targeting c-Myc overcomes stroma-mediated drug resistance, which cooperates with HDAC6 inhibition to synergistically suppress lymphoma survival and growth.
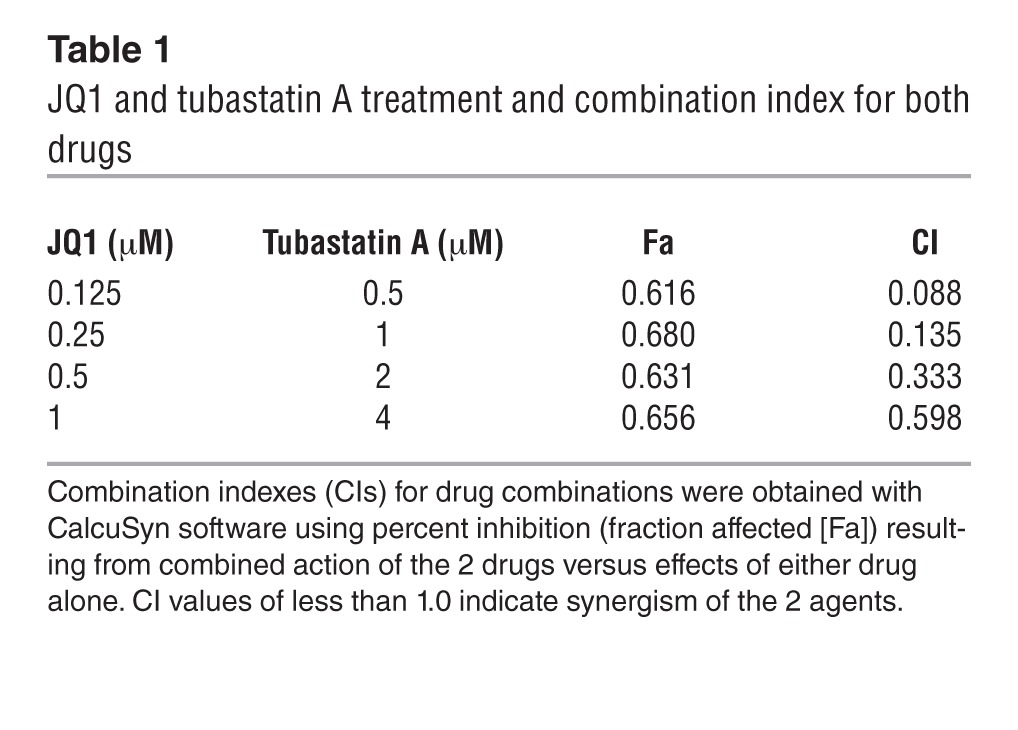
Given cell adhesion–induced c-Myc expression, c-Myc function in tumor genesis, and the ability of inhibition of c-Myc to enhance apoptosis (23, 24), the combination of c-Myc inhibitor and cytotoxic drug may be a more optimal treatment for B cell malignancies. We next examined whether inhibition of c-Myc sensitizes lymphoma cells to drug-induced apoptosis. The following experiments were performed to assess whether silencing of c-Myc by JQ1 enhances lymphoma cell response to chemotherapy. As shown in Figure 7A, addition of JQ1 sensitized lymphoma cells to mitoxantrone in the absence and presence of HK cells as well as overcame CAM-DR. In addition, as shown in Figure 7B, JQ1 also enhanced tubastatin A–induced cell apoptosis in the absence and presence of HK cells, indicting a combinatorial therapy against lymphoma cells. Therefore, we further evaluated these results using median effect analysis (CalcuSyn, Biosoft) to determine whether the greater growth inhibitory activity of combined treatment reflected an additive or synergistic effect in Jeko-1 cells. We found all combinations of c-Myc and HDAC6 inhibitors to be synergistic in comparison with single-agent treatment (Figure 7C and Table 1). Taken together, these data suggest that the lymphoma-stroma interaction in lymphoma microenvironment directly impacts the biology of lymphoma through genetic and epigenetic regulation, with HDAC6 and c-Myc as potential therapeutic targets.
Figure 7. Targeting c-Myc overcomes CAM-DR and cooperates with HDAC6 to regulate stroma-mediated drug resistance.

(A) Lymphoma cells were treated with JQ1 (1 μM) and/or mitoxantrone (0.2 μM) for 48 hours with or without HK cell adhesion as indicated, and apoptosis was analyzed by flow cytometry using Annexin V. (B) Lymphoma cells were treated with JQ1 (1 μM) and/or tubastatin A (1 μM) for 48 hours with or without HK cell adhesion as indicated, and apoptosis was analyzed by flow cytometry using Annexin V. (C) Cell proliferation assay (CCK8) shows that JQ1 and tubastatin A cotreatment synergistically inhibits lymphoma cell growth. Cells were treated with tubastatin A and/or JQ1 as indicated for 48 hours, and CCK8 assay was performed. (B and C) Data are representative of 4 independent experiments (mean ± SD). (D) Stroma-mediated c-Myc/miR-548m/HDAC6 amplification loop drives drug resistance, clonogenic growth, and tumor progression in B cell lymphomas. The stroma-lymphoma interaction induces c-Myc expression and miR-548m downregulation, leading to HDAC6 overexpression, and contributes to lymphoma cell survival, drug resistance, growth, and tumor progression. c-Myc and miR-548m generate a forward-feedback loop to ensure persistently high protein levels of c-Myc and low levels of miR-548m in B cell lymphoma microenvironment.
Table 1.
JQ1 and tubastatin A treatment and combination index for both drugs
Discussion
It has become increasingly clear that proliferation and survival of B cell lymphomas is not only driven by genetic changes but also by the close interaction with the immune microenvironment and stromal cells. Cellular interactions between the neoplastic lymphocytes and the numerous surrounding cells are critical determinants for lymphoma initiation, progression, response to therapy, and drug resistance (25). Lymphoma stromal cells, such as FDCs and bone marrow stromal cells, are intermingled with B lymphocytes and lymphoma cells in lymphoid tissue of lymph nodes and bone marrow. With their cytoplasmic ramifications, they form a dense network that contains the B lymphocytes. Presence of DC gene expression signature in lymphoma indicates prognosis (26). Our and other studies have shown that cell adhesion to stroma cells (HK and HS-5 cells) confers a multidrug-resistant phenotype and that disruption of cell adhesion–mediated signaling may increase the efficacy of cytotoxic agents (2, 27–30). To this end, our laboratory has focused on identifying targets that contribute to stroma-mediated drug resistance and tumor progression. In this study, we provide the in vitro and in vivo evidence that lymphoma stromal cells directly impact the clonogenic growth of lymphoma cells and lymphoma progression. To our knowledge, this is the first report to characterize c-Myc, miR-548m, and HDAC6 in the interaction of the lymphoma cell with its stroma. We further explored the rationale for specifically targeting this interaction as a novel approach for the therapy of lymphoma. As summarized in Figure 7D, we show that (a) adhesion of MCL and other non-Hodgkin lymphoma cells to lymph node and bone marrow stroma promotes lymphoma cell clonogenic growth and survival by using a clonogenic growth coculture system and is associated with induction of HDAC6 and Myc; (b) stromal cell adhesion–downregulated miR-548m contributes to HDAC6 upregulation; (c) stroma triggered c-Myc/miR-548m feed-forward loop, linking sustained c-Myc activation, miR-548m downregulation, and subsequent HDAC6 upregulation, and is a key determinant for stroma-mediated cell survival and colony formation in cell lines and primary lymphoma samples of MCL and other B cell lymphomas. Further, HDAC6-selective inhibitor tubastatin A, in combination with JQ1, markedly enhances cell death, abolishes CAM-DR, and suppresses colony formation and tumor growth in vivo. Thus, c-Myc and HDAC6 are potential therapeutic targets to overcome stroma-mediated drug resistance and growth in MCL and other B cell lymphomas.
Among the 11 human zinc-dependent histone deacetylases, HDAC6 is a structurally and functionally unique class IIb HDAC. Class IIb HDACs contains 2 catalytic domains, are primarily cytoplasmic proteins, and have nonhistone proteins as primary targets (31). HDAC6 has been associated with many cell functions, including tubulin stabilization, cell motility, and regulation of the binding between Hsp90 and its cochaperone (32–34). A high level of HDAC6 has been associated with ovarian cancer (16), breast cancer (35), and acute myeloid leukemia (19), but it is unclear whether HDAC6 plays a role in lymphoma development. In the present study, we investigated the role of HDAC6 in cell adhesion–mediated lymphoma cell survival and growth in vitro and in vivo. To this end, we demonstrate that lymphoma stromal cells directly enhance anchorage-independent growth (colony formation), and lymphoma progression and inhibition of HDAC6 lead to the suppression of stroma-mediated enhancement of clonogenic growth in lymphoma cells and lymphoma growth. Our study therefore reveals a novel molecular signaling mechanism for interaction between lymphoma cells and the lymphoma microenvironment. Our findings that HDAC6 inhibition by shRNAs or tubastatin A impaired malignant growth in soft agar are consistent with a recent report showing that HDAC6 is required for efficient oncogenic transformation and anchorage-independent proliferation in solid tumors (36). Furthermore, HDAC6 likely modulates tumor formation by several different mechanisms. We found that HDAC6 downregulated Bim, at least in part, by facilitating the degradation of Bim through a posttranscriptional regulation and likely contributes to tumor genesis. It has been well documented that Bim is critical for HDAC inhibitor-induced apoptosis of solid tumor and leukemia cells (19, 31, 37–41). Our previous study demonstrated that cell adhesion–mediated decrease in Bim is involved in CAM-DR in lymphoma cell lines and primary lymphoma cells, further implying that HDAC6 could be a therapeutic target and may have clinical utility in lymphoma therapy (13).
Advances in HDAC basic biology have provided a better understanding of how HDAC6 functions; however, how HDAC6 expression and activity are regulated has not been fully characterized. In this study, we demonstrate that HDAC6 is a direct target of miR-548m via translational inhibition, providing a novel mechanism for HDAC6 regulation. Our findings indicate that miR-548m downregulation is a key factor inducing protection from drug-primed apoptosis in tumor microenvironment. Indeed, reexpression of miR-548m is sufficient to promote lymphoma cell apoptosis and overcome CAM-DR. This occurs through downmodulation of the HDAC6 through direct binding of miR-548m to the HDAC6 mRNA 3′-UTR. The effect of miR-548m is in agreement with a recent finding that overexpression of miR-548d resulted in increased apoptosis in pancreatic cancer (42). Here, we describe an oncogenic pathway underlying lymphoma development, whereby cell adhesion downregulates miR-548m, which, in turn, unleashes HDAC6 and further downmodulates the tumor suppressor, Bim. Our study offers a molecular mechanism for the potential role of miR-548m in microenvironment-dependent tumor growth and response to therapy. Aberrant expression of the miR-548m/HDAC6 axis in the lymphoma results in downregulation of Bim and enhances the survival of B lymphoma cells. It is logical to predict that HDAC6 inhibitor alone, or in conjunction with other anticancer agents, could function as an effective alternate therapeutic regimen against MCL and other types of B cell malignancies.
We further elucidate the underlying mechanism associated with miR-548m downregulation in lymphoma microenvironment. Downregulation of a subset of miRNAs is a commonly observed feature of cancers, suggesting that these miRNAs, such as miR-548m, may act as tumor suppressors. Recent studies revealed that c-Myc activation resulted in widespread direct repression of miRNA expression and c-Myc–induced miRNA repression contributes to lymphoma aggressive progression (43–45). The stroma-induced miR-548m repression can be the result of c-Myc overexpression. In this study, we demonstrate that, upon lymphoma cell adhesion to FDC cells, c-Myc is indeed activated, and c-Myc further recruits EZH2 as a corepressor complex to the miR-548m promoter to suppress miR-548m expression. These data are further supported by our loss-of-function experiments showing that knockdown of c-Myc and EZH2, using their specific inhibitors and siRNAs, respectively, induced miR-548m expression and CHIP assay revealing the direct bindings of c-Myc and EZH2 and c-Myc dependency of EZH2 binding. Conversely, we demonstrate that miR-548m regulated c-Myc expression through direct targeting of c-Myc 3′-UTR and downregulation of miR-548m, in turn, contributes to c-Myc overexpression, thus generating a positive c-Myc/miR-548m feedback loop to ensure persistent high protein levels of c-Myc and further repression of miR-548m expression in lymphoma microenvironment. Moreover, the finding of c-Myc–induced EZH2-mediated miR-548m repression is in line with findings of our previous study that miR-29 expression is c-Myc and EZH2 dependent, implicating a genetic silencing mechanism for c-Myc–induced widespread miRNA repression (20, 21, 43). JQ1 functions as a specific inhibitor of BET proteins, including bromodomain-containing protein 4 (BRD4) and BRD2 through interference with the acetyl-lysine recognition domains (bromodomains). However, BRD4 not only regulates c-Myc but may also affect other oncogenic proteins, and the observed effect of JQ1 may not be exclusively attributed to the effect on c-Myc. Our results provide the first evidence of a critical role of c-Myc and associated genes in tumor microenvironment: c-Myc activation contributes to stroma-induced clonogenic growth and lymphoma progression. More importantly, we show a novel, promising therapeutic approach through targeting c-Myc, using newly developed JQ1 and other c-Myc inhibitors to enhanced mitoxantrone-induced apoptosis and overcome microenvironment-promoted drug resistance and lymphoma growth. These novel insights indicate that targeting HDAC6 and c-Myc may not only support their future use as single agents or in combination with other cytotoxic agents, exploiting microenvironment-mediated c-Myc dependence, but also indicate that when combined, they might synergistically suppress lymphoma survival, overcome drug resistance, and eradicate lymphoma progression.
Methods
Cell lines, cell cultures, and patient lymphoma specimens.
The human lymphoma cell lines Jeko-1, HBL-2, SUDHL-4, and SUDHL-10 were provided and maintained as previously described (28). SUDHL-4 and SUDHL-10 are germinal center–type large B cell lymphoma cell lines, and Jeko-1 and HBL-2 are MCL cell lines. The FDC line, HK, was obtained from Y.S. Choi (Ochsner Clinic Foundation, New Orleans, Louisiana, USA) (2). The cells were grown in RPMI 1640 medium (Cellgro, Fischer Scientific), supplemented with 5% fetal bovine serum (Omega Scientific) and 1% (v/v) penicillin (100 U/ml), streptomycin (100 U/ml), and 1% (v/v) l-glutamine (GIBCO-BRL). Diffuse large B cell lymphoma and MCL cells were obtained from fresh biopsy-derived lymphoma tissues (lymph nodes) after informed consent from patients. The primary lymphoma cells and CD19-positive cells were isolated by using CD19 microbeads with the AutoMACS Magnetic Cell Sorter according to the manufacturer’s instructions (Miltenyi Biotec), as described previously (28). Either lymphoma cell lines or CD19-sorted lymphoma cells (from diffuse large cell lymphomas, 1.5 × 106 cells/ml) were adhered to a preestablished monolayer of HK cells or kept in suspension for 12 to 24 hours. MCL cells were used for experiments without CD19 sorting since all of our MCL samples had >90% lymphoma cells in purity. Lymphoma cells were then carefully removed, with the monolayer of HK cells kept intact. The purity of the lymphoma cell population was greater than 95%, as shown by flow cytometry. Cells were then analyzed as described below. In Transwell experiments, lymphoma cells were separated from the HK cells by Transwell inserts.
Stable cell lines.
Inducible stable clones expressing miR-548m in HBL-2 cells were generated by using CymR repressor virus along with the SparQ Lentiviral Cumate Switch Inducible System and were analyzed according to the manufacturer’s protocol (System Biosciences). In this system, the bacterial operons cmt and cym have been engineered to regulate gene expression in mammalian cells. In the repressor configuration, regulation is mediated by strong binding of the CymR repressor to the cumate operator site (CuO), which is downstream of the CMV5 promoter. Addition of cumate, a small nontoxic molecule, relieves the repression. HBL-2 cells were initially transfected with pCDH-EF1-CymR-T2A-Puro plasmid and selected by 0.5 μg/ml puromycin for 2 weeks. Then, the selected cells were transfected with pCDH-CuO-MCS-EF1-GFP+Puro lentivector, containing dual promotor and dual marker with hsa-pre–miR-548m, invented by System Biosciences, using Amaxa Nucleofector (program T-001) in T solution. The constructs were verified by DNA sequencing before the transfection. Forty-eight hours later, transfected cells were selected by adding 1 μg/ml puromycin (Santa Cruz Biotechnology Inc.) to growth medium. At 14 days after transfection, stably transfected lymphoma cells were analyzed by flow cytometry to identify those GFP-positive cells. The GFP-positive cells were enriched for by subsequent GFP cell sorting using a FACSVantage SE (Becton Dickinson), resulting in a homogeneous population of 90% GFP-positive cells.
To establish HBL-2-Ctrl.ORF and HBL-2-HDAC6.ORF stable cell lines, HBL-2 cells were transfected with 1 μg of a pCMV6-AC-GFP–tagged ORF clone of Homo sapiens HDAC6 or a pCMV6-AC-GFP–tagged ORF clone of control (OriGene) using Amaxa Nucleofector (programme T-001) in T solution. Forty-eight hours after the transfection, 500 μg/ml G418 (Gibco/Life Technology) was added to the growth medium. After 2 weeks, these transfected cells were analyzed by flow cytometry to identify and select GFP-positive cells. The selected GFP-positive cells were enriched for by subsequent GFP sorting using a FACSVantage SE (Becton Dickinson). The final percentage of GFP-positive cells in the pool was 90%.
Antibodies, reagents, Western blotting, apoptosis detection assay, and qRT-PCR analysis.
The following monoclonal antibodies were purchased: HDAC6 and c-Myc (Santa Cruz Biotechnology Inc.), EZH2 and Bim (Cell Signaling Technology), and Actin (Sigma-Aldrich). Tubastatin A was purchased from BioVision Incorporated, and mitoxantrone was purchased from Sigma-Aldrich. At the indicated times, cells were collected and washed, and Western blot and apoptosis analyses were then performed as previously described (28, 46). JQ1 was provided by James E. Bradner (Dana-Farber Cancer Institute, Boston, Massachusetts, USA), and DZNep was provided by Victor E. Marquez (National Cancer Institute, NIH, Frederick, Maryland, USA).
RNA was isolated from samples in TRIzol as recommended by the manufacturer (Invitrogen). The samples were further purified into total RNA fractions using RNeasy columns as described by the manufacturer (Qiagen). For miRNA quantitation, miR-548m levels in cell lines and patient lymphoma cells were determined using TaqMan miRNA assays (Applied Biosystems) following manufacturer-recommended protocols and performed as described previously (13). For quantitative detection of HDAC6 mRNAs, cDNAs were synthesized from 1 μg of total RNA using the SuperScript First-Strand Kit (Invitrogen). Real-time PCR was conducted by using the ABI PRISM 7000 Sequence Detection System (Applied Biosystems), as described previously (46). Primers for real-time detection of HDAC6 (assay ID: Hs00195869), BIM (assay ID: Hs00708019), and endogenous control gene GAPDH mRNA were purchased from Applied Biosystems. Real-time PCR was performed by using human TaqMan predeveloped assay reagents (Applied Biosystems).
Transfection of pre-miRNAs and anti-miRNAs, HDAC6 overexpression by transient transfection, and HDAC6 knockdown by small hairpin RNA.
Optimized nucleofection protocols generated by Amaxa (Cologne) were followed for the transfection of Jeko-1 cells, HBL-2 cells (Nucleofector Kit T, Program T-001), and SUDHL-4 cells (Nucleofector Kit V, Program X-001) with miRNA precursors (Ambion) or anti-miRNA inhibitors (Ambion), respectively. For transient overexpression, the Jeko-1 cell line was transfected by electroporation using Nucleofector (Amaxa) according to the manufacturer’s instructions and a Nucleofector TM II (Amaxa). Briefly, 5 × 106 cells from each lymphoma cell line were suspended in 100 μl Nucleofector T solution with 1 μg of either the control empty vector (pCDNA3) or a pCDNA3-HDAC6 expression plasmid (provided by E. Seto, H. Lee Moffitt Cancer Center) and then electroporated using program T-001. Cells were collected at 24 to 48 hours after electroporation and subjected to Western blot analysis for determination of HDAC6 expression. HDAC6 small hairpin RNAs (pSM2 shRNA vector) and control shRNAs (pSM2 nonsilencing) were obtained from Cold Spring Harbor Laboratory. For transient expression, cell lines were transfected by electroporation using Nucleofector according to the manufacturer’s instructions and as described previously (28). For additional shRNA knockdown experiments, Jeko-1 or SUDHL-4 cells were transfected with 0.5 μg psiHIV-U6 of OmicsLink shHDAC6 RNA Expression Clones: shHDAC6.GC1, shHDAC6.GC2, shHDAC6.GC3, shHDAC6.GC4, and sh-scrambled control (GeneCopoeia). Forty-eight hours later, cells were harvested and analyzed by Western blot to examine the efficiency of HDAC6 knockdown.
HDAC6 gene 3′-UTR luciferase reporter assay.
Putative target sites for miRNA in the 3′-UTR of human, mouse, and rat Bim mRNA were identified using the miRanda Human miRNA Targets website ( http://cbio.mskcc.org/cgi-bin/mirnaviewer/mirnaviewer.pl), RNA22 miRNA target detection ( http://cbcsrv.watson.ibm.com/rna22.html), and the TargetScan (version 5.4) website ( http://genes.mit.edu/targetscan) based on the target prediction algorithms.
To create HDAC6 3′-UTR luciferase reporter constructs, 60-bp sequences containing the putative miR-548m binding sites were synthesized and ligated into the pmiR-Report vector (Ambion) at SpeI and HindIII sites. To create a mutant 3′-UTR, point mutations were introduced at the first 2 miR-548m–matching nucleotides within selected putative seeding sequence regions with the following rules: A changed to T and vice versa and G changed to C and vice versa. Jeko-1, HBL-2, and SUDHL-4 cells in 24-well plates were transfected with 0.10 μg of the pmiR-Report-3′-UTR/HDAC6 luciferase reporter, 0.05 μg of the normalization plasmid Renilla, and 0.6 μg of miR-548m or non-GFP–expressing control vector. Luciferase assays were performed using a luciferase assay system (Promega), and activities were normalized to β-galactosidase activity.
Clonogenic assays and in vivo xenograft model.
Clonogenic growth of tumor cell lines was evaluated by plating 500 HBL-2 cells alone or coculturing with 1,000 HK cells in duplicate wells in a 24-well plate containing 1.0 ml Methocult, the methylcellulose medium (StemCell Technologies), per well. The plates were placed in an incubator at 37°C with 5% CO2 for 10 to 14 days. After this incubation, colonies consisting of >40 cells in each well were counted using an inverted microscope, and percentage of colony growth inhibition compared with that of the untreated control cells was calculated.
Six- to eight-week-old male NOD/SCID mice were purchased from The Jackson Laboratory. The NOD/SCID mice were inoculated subcutaneously in the right flanks with 1 × 106 HBL-2 cells suspended in 100 μl PBS with and without HK cells with ratio of 1:2. Each treatment group and control group consisted of 4 to 6 mice. Treatment was initiated when mean tumor volume was approximately 200 mm3. Mice were treated intraperitoneally with PBS (vehicle control), tubastatin A alone (25 mg/kg/d for 14 consecutive days), or mitoxantrone alone (1.5 mg/kg/d) or with a combination of tubastatin A and mitoxantrone. Tumor size was assessed 3 times per week using a digital caliper. Tumor volumes were determined by measuring the length (l) and the width (w) of the tumor and calculating the volume (V = l [w]2/2).
For the HBL-2-i-miR-548m xenograft model, HBL-2-i-miR-548m cells were treated with 30 μg/ml cumate (System Biosciences) for 24 hours. Then, the NOD/SCID mice were inoculated subcutaneously in the right flanks with 1 × 106 HBL-2-i-miR-548m cells suspended in 100 μl PBS with or without HK cells with ratio of 1:2.
Accession number.
The GEO database accession number for the microarray data is GSE49253.
Statistics.
One-tailed Student’s t test for homoscedastic variances was used to evaluate RT-PCR and apoptosis assay data. Statistical significance between the control and treated mice was evaluated using a 1-tailed Student’s t test. P values of less than 0.05 were considered significant. Data are shown with the mean ± SD of at least 4 experiments.
Study approval.
The human specimen study was approval by the Institutional Review Board at the University of South Florida, and patients provided signed informed consent forms. All experiments involving the use of mice were performed in accordance with protocols approved by the Animal Care and Use Committees of the University of South Florida College of Medicine.
Supplementary Material
Acknowledgments
We are grateful to the Tissue Procurement, Molecular Core Laboratory, and Flow Cytometry Core Facilities at H. Lee Moffitt Cancer Center and Research Institute for providing specimens, molecular analysis, and cell apoptosis analysis. We thank Rasa Hamilton for editorial assistance. This work was supported by grants from the National Cancer Institutes (R01CA137123, to J. Tao), Lymphoma Research Foundation (to J. Tao), and Maher Fund (to J. Tao).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(11):4612–4626. doi:10.1172/JCI64210.
References
- 1.Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest. 2012;122(10):3416–3423. doi: 10.1172/JCI61272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park CS, Choi YS. How do follicular dendritic cells interact intimately with B cells in the germinal centre? Immunology. 2005;114(1):2–10. doi: 10.1111/j.1365-2567.2004.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dave SS, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351(21):2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 4.Lenz G, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359(22):2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dave SS. Host factors for risk and survival in lymphoma. Hematology American Soc Hemato Educ Program. 2010;2010:255–258. doi: 10.1182/asheducation-2010.1.255. [DOI] [PubMed] [Google Scholar]
- 6.Schrader C, et al. Growth pattern and distribution of follicular dendritic cells in mantle cell lymphoma: a clinicopathological study of 96 patients. Virchows Arch. 2006;448(2):151–159. doi: 10.1007/s00428-005-0049-5. [DOI] [PubMed] [Google Scholar]
- 7.Croce CM. MicroRNAs and lymphomas. Ann Oncol. 2008;19(suppl 4):iv39–iv40. doi: 10.1093/annonc/mdn192. [DOI] [PubMed] [Google Scholar]
- 8.Fabbri M, Croce CM, Calin GA. MicroRNAs in the ontogeny of leukemias and lymphomas. Leuk Lymphoma. 2009;50(2):160–170. doi: 10.1080/10428190802535114. [DOI] [PubMed] [Google Scholar]
- 9.Klein U, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17(1):28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 10.Calin GA, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99(24):15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, et al. Patterns of microRNA expression characterize stages of human B-cell differentiation. Blood. 2009;113(19):4586–4594. doi: 10.1182/blood-2008-09-178186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin J, et al. Follicular dendritic cell-induced microRNA-mediated upregulation of PRDM1 and downregulation of BCL-6 in non-Hodgkin’s B-cell lymphomas. Leukemia. 2011;25(1):145–152. doi: 10.1038/leu.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lwin T, et al. Follicular dendritic cell-dependent drug resistance of non-Hodgkin lymphoma involves cell adhesion-mediated Bim down-regulation through induction of microRNA-181a. Blood. 2010;116(24):5228–5236. doi: 10.1182/blood-2010-03-275925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22(5):1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tran AD, et al. HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J Cell Sci. 2007;120(pt 8):1469–1479. doi: 10.1242/jcs.03431. [DOI] [PubMed] [Google Scholar]
- 16.Bazzaro M, et al. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin Cancer Res. 2008;14(22):7340–7347. doi: 10.1158/1078-0432.CCR-08-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100(8):4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132(31):10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, Xie C, Edwards H, Zhou H, Buck SA, Ge Y. Inhibition of histone deacetylases 1 and 6 enhances cytarabine-induced apoptosis in pediatric acute myeloid leukemia cells. PLoS One. 2011;6(2):e17138. doi: 10.1371/journal.pone.0017138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene. 2012;31(24):3002–3008. doi: 10.1038/onc.2011.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-cell lymphomas. Cancer Cell. 2012;22(4):506–523. doi: 10.1016/j.ccr.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Pajic A, et al. Cell cycle activation by c-myc in a burkitt lymphoma model cell line. Int J Cancer. 2000;87(6):787–793. doi: 10.1002/1097-0215(20000915)87:6<787::AID-IJC4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 23.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dews M, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalton WS, Hazlehurst L, Shain K, Landowski T, Alsina M. Targeting the bone marrow microenvironment in hematologic malignancies. Semin Hematol. 2004;41(2 suppl 4):1–5. doi: 10.1053/j.seminhematol.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 26.Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. 2005;87:163–208. doi: 10.1016/S0065-2776(05)87005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lwin T, et al. Bone marrow stromal cells prevent apoptosis of lymphoma cells by upregulation of anti-apoptotic proteins associated with activation of NF-kappaB (RelB/p52) in non-Hodgkin’s lymphoma cells. Leukemia. 2007;21(7):1521–1531. doi: 10.1038/sj.leu.2404723. [DOI] [PubMed] [Google Scholar]
- 28.Lwin T, et al. Lymphoma cell adhesion-induced expression of B cell-activating factor of the TNF family in bone marrow stromal cells protects non-Hodgkin’s B lymphoma cells from apoptosis. Leukemia. 2009;23(1):170–177. doi: 10.1038/leu.2008.266. [DOI] [PubMed] [Google Scholar]
- 29.Xargay-Torrent S, et al. Sorafenib inhibits cell migration and stroma-mediated bortezomib resistance by interfering B-cell receptor signaling and protein translation in mantle cell lymphoma. Clin Cancer Res. 2013;19(3):586–597. doi: 10.1158/1078-0432.CCR-12-1935. [DOI] [PubMed] [Google Scholar]
- 30.Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9(9):665–674. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, et al. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11(4):313–324. doi: 10.1593/neo.81358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao R, et al. HDAC6 inhibition enhances 17-AAG–mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood. 2008;112(5):1886–1893. doi: 10.1182/blood-2008-03-143644. [DOI] [PubMed] [Google Scholar]
- 33.Bali P, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;90(29):26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 34.Kovacs JJ, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18(5):601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 35.Park SY, et al. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol Rep. 2011;25(6):1677–1681. doi: 10.3892/or.2011.1236. [DOI] [PubMed] [Google Scholar]
- 36.Lee YS, et al. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008;68(18):7561–7569. doi: 10.1158/0008-5472.CAN-08-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deacetylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol Cell Biol. 2009;29(23):6149–6169. doi: 10.1128/MCB.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gillespie S, Borrow J, Zhang XD, Hersey P. Bim plays a crucial role in synergistic induction of apoptosis by the histone deacetylase inhibitor SBHA and TRAIL in melanoma cells. Apoptosis. 2006;11(12):2251–2265. doi: 10.1007/s10495-006-0283-6. [DOI] [PubMed] [Google Scholar]
- 39.Mandl-Weber S, Meinel FG, Jankowsky R, Oduncu F, Schmidmaier R, Baumann P. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br J Haematol. 2010;149(4):518–528. doi: 10.1111/j.1365-2141.2010.08124.x. [DOI] [PubMed] [Google Scholar]
- 40.Emanuele S, et al. SAHA induces apoptosis in hepatoma cells and synergistically interacts with the proteasome inhibitor Bortezomib. Apoptosis. 2007;12(7):1327–1338. doi: 10.1007/s10495-007-0063-y. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Adachi M, Zhao X, Kawamura R, Imai K. Histone deacetylase inhibitors FK228, N-(2-aminophenyl)-4-[N-(pyridin-3-yl-methoxycarbonyl)amino- methyl]benzamide and m-carboxycinnamic acid bis-hydroxamide augment radiation-induced cell death in gastrointestinal adenocarcinoma cells. Int J Cancer. 2004;110(2):301–308. doi: 10.1002/ijc.20117. [DOI] [PubMed] [Google Scholar]
- 42.Heyn H, Schreek S, Buurman R, Focken T, Schlegelberger B, Beger C. MicroRNA miR-548d is a superior regulator in pancreatic cancer. Pancreas. 2012;41(2):218–221. doi: 10.1097/MPA.0b013e318224b701. [DOI] [PubMed] [Google Scholar]
- 43.Chang TC, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40(1):43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao P, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Lisio L, Martinez N, Montes-Moreno S, Piris-Villaespesa M, Sanchez-Beato M, Piris MA. The role of miRNAs in the pathogenesis and diagnosis of B-cell lymphomas. Blood. 2012;120(9):1782–1790. doi: 10.1182/blood-2012-05-402784. [DOI] [PubMed] [Google Scholar]
- 46.Lwin T, et al. Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas. Blood. 2007;110(5):1631–1638. doi: 10.1182/blood-2006-11-060350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.