

Abstract
The molecular mechanisms that control innate immune cell trafficking during chronic infection and inflammation, such as in tuberculosis (TB), are incompletely understood. During active TB, myeloid cells infiltrate the lung and sustain local inflammation. While the chemoattractants that orchestrate these processes are increasingly recognized, the posttranscriptional events that dictate their availability are unclear. We identified microRNA-223 (miR-223) as an upregulated small noncoding RNA in blood and lung parenchyma of TB patients and during murine TB. Deletion of miR-223 rendered TB-resistant mice highly susceptible to acute lung infection. The lethality of miR-223–/– mice was apparently not due to defects in antimycobacterial T cell responses. Exacerbated TB in miR-223–/– animals could be partially reversed by neutralization of CXCL2, CCL3, and IL-6, by mAb depletion of neutrophils, and by genetic deletion of Cxcr2. We found that miR-223 controlled lung recruitment of myeloid cells, and consequently, neutrophil-driven lethal inflammation. We conclude that miR-223 directly targets the chemoattractants CXCL2, CCL3, and IL-6 in myeloid cells. Our study not only reveals an essential role for a single miRNA in TB, it also identifies new targets for, and assigns biological functions to, miR-223. By regulating leukocyte chemotaxis via chemoattractants, miR-223 is critical for the control of TB and potentially other chronic inflammatory diseases.
Introduction
Tuberculosis (TB) is a chronic infectious disease that is characterized by ongoing inflammation. TB is caused by Mycobacterium tuberculosis (Mtb) and is responsible for excessive mortality and morbidity worldwide. The past decades have witnessed considerable efforts toward the elucidation of the immune mechanisms underlying protection and pathogenesis in TB (1, 2). The dual role of mononuclear phagocytes as effectors against, and habitats of, Mtb is well accepted (1). Less is known about polymorphonuclear neutrophils (PMNs), which have, however, recently received particular attention due to their functional duality. PMNs can either be beneficial or detrimental in TB (3). In both humans and mice, the recruitment of PMNs to the lung parenchyma is a feature of TB inflammation. While Mtb-infected PMNs have been detected in the sputum of TB patients (4), accelerated PMN influx is a hallmark of TB susceptibility in mice (5–7). The depletion of PMNs, in some models, extended the survival of TB-prone animals (8, 9). However, the precise molecular events that fine-regulate the abundance of chemoattractants and subsequently the PMN dynamics at the site of mycobacterial infection, await further clarification.
MicroRNAs (miRNAs) are short, conserved, noncoding RNA molecules that modulate mRNA translation and/or degradation (10). Recent investigations suggest that distinct miRNAs play a role in the maintenance of immune homeostasis and the induction of immune defense (11). Moreover, evidence has shown that certain miRNAs are involved in protection against bacterial infections (12). However, their propensity to tailor the trafficking of immune cells remains unknown. Chemotaxis is crucial for the initiation and sustenance of inflammation. Despite apparent functional redundancy, leukocyte migration is controlled by unique chemoattractants at a given stage of inflammation (13). Lipid mediators, complement cleavage products, and peptides initiate prompt cell migration, while cytokines, notably chemokines, tune leukocyte tissue recruitment at later stages (13). Information on how chemoattractants are regulated posttranscriptionally is sparse, although diseases associated with nonresolving inflammation are common (14). Several chemokine genes display adenylate uridylate–rich gene sequence elements in their 3′ untranslated region (3′-UTR) that influence the rate of mRNA degradation (15). Nonetheless, the role of miRNAs in controlling chemokines, particularly in relation to the chronic inflammation seen in TB, has not been investigated to date. Several studies suggest that key TB–protective cytokines, including TNF-α and IFN-γ, are regulated by miR-125b and miR-29, respectively (16, 17). Moreover, screening approaches have tentatively pointed to miRNA signatures characteristic of active TB (18–22). Yet, no functional role in TB for any of these miRNAs has been elucidated, including the effects on chemokines and cell migration.
To address this question, we focused on miR-223, which we found to be upregulated in the blood and lungs of TB patients and experimentally infected mice. The expression of miR-223 is principally found in cells of the myeloid lineage and is enriched in PMNs (23), where it controls their activation (24). In addition to its prominent role in PMN development (25), miR-223 regulates inflammatory responses of mononuclear phagocytes by modulating NF-κB kinase subunit α (IKKα) inhibitor expression during monocyte-to-macrophage lineage progression (26).
To identify the biological role of miR-223 during active TB and to further decipher the biological significance of miR-223 enrichment in distinct immune cells, we investigated experimental Mtb infection in the absence of miR-223 in mice. We observed that miR-223 gene KO mice (miR-223–/–) failed to control pulmonary TB, and that their increased susceptibility was a consequence of aberrant PMN migration and subsequent exacerbated lung inflammation, ultimately leading to death. We identified the chemokine C-X-C motif ligand 2 (CXCL2) and C-C motif ligand 3 (CCL3) as direct targets of miR-223, along with IL-6.
Here, we describe a unique biological role of a single miRNA in TB and uncover new targets of miR-223. Our findings suggest that PMNs negatively control leukocyte chemotaxis at late stages of inflammation by means of developmentally accumulated miR-223. Our results, therefore, not only single out a discrete regulatory function of miR-223, but also shed light on the hitherto controversial role of PMNs in TB (3).
Results
miR-223 is abundantly expressed during active TB in humans.
We evaluated blood miRNA profiles of patients with active pulmonary TB (TB+) and latently infected healthy individuals (tuberculin skin test–positive [TST+]) from an East-African cohort. Using a human miRNA microarray, we identified miR-629, miR-197, miR-625, miR-223, miR-22, and miR-335* as being significantly upregulated in diseased individuals. On the contrary, miR-451, miR-148b, miR-19b, miR-501-3p, miR-940, miR-296-5p, miR-33b*, miR-92b, miR-744, and miR-1238 were downregulated in active TB patients (Figure 1A and Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI67604DS1). Principal component analysis (PCA) was applied to the dataset of these differentially regulated miRNAs. TST+ and TB+ individuals segregated into two distinct clusters, suggesting that these miRNAs are robust disease discriminators (Figure 1B).
Figure 1. Differential miR-223 expression in active human TB.

(A) 2D cluster analysis across miRNA probe (left) and subjects (top) (n = 16). Heatmap shows downregulated (green) and upregulated (red) miRNA. (B) Identification of TB-active (n = 8) and TST+ (n = 8) subjects by PCA based on the miRNAs shown in A. (C) qRT-PCR assay of miR-223 in peripheral blood from TST– (n = 24), TB-active (n = 37), and TST+ subjects (n = 15). miR-103 was used as a reference gene. Data are presented as the mean ± SEM; ANOVA with Bonferroni’s post test. (D) qRT-PCR of miR-223 in paraffin-embedded lung tissue from pulmonary TB patients (n = 8) and healthy individuals (n = 8). miR-103 was used as a reference gene. One healthy sample was normalized to 1. Data are presented as the mean ± SEM; unpaired Student’s t test. *P < 0.05; **P < 0.01.
We focused on miR-223 because of its relevance to myeloid cell biology, given that this leukocyte lineage plays key roles in TB pathogenesis and protection (1). To validate the microarray data and to extend the analysis to another ethnic group, we used TaqMan miR real-time quantitative PCR (qPCR) to assay miR-223 expression in a mixed European–descent cohort. The analysis performed on blood samples from healthy individuals (TST–), active TB patients, and TST+ individuals (Figure 1C) revealed a similar differential signature in miR-223 expression in samples from active TB versus TST+ subjects. Thus, our data, together with observations of an Asian cohort, suggest a distinct regulation of miR-223 in TB (20). At the site of TB disease manifestation (i.e., lung parenchyma), we further examined whether miR-223 was regulated similarly. To this end, we analyzed lung biopsies from active TB patients and detected upregulated miR-223 expression compared with lung tissue collected at autopsy from healthy subjects (Figure 1D). Thus, Mtb induced an upregulation of miR-223 in pulmonary lesions and an abundant expression in peripheral blood cells from TB patients of different genetic backgrounds.
miR-223 controls susceptibility of mice to TB.
Since the mouse model represents a tractable in vivo tool for investigating TB pathogenesis in molecular terms, we examined the expression of miR-223 during experimental aerogenic TB in mice. We detected abundant miR-223 transcripts in lung tissue and blood during infection of C57BL/6 mice with virulent Mtb (Figure 2, A and B). Thus, similar to human TB, experimental infection of mice with Mtb induced the upregulation of miR-223 at the site of disease manifestation.
Figure 2. Expression of miR-223 during murine TB.

(A) qRT-PCR of miR-223 in the lungs of Mtb-infected C57BL/6 mice. Data are presented as the mean ± SEM and are pooled from two independent experiments (n = 8–10). (B) qRT-PCR of miR-223 in the blood of Mtb-infected C57BL/6 mice. Data are presented as the mean ± SEM and are pooled from two independent experiments (n = 8–10). Data in A and B were obtained using U6 small nuclear RNA (snRNA) as a reference gene and were normalized to uninfected mice.
Further, we used miR-223 KO mice to determine the role of miR-223 in TB. miR-223 KO animals succumbed (80%–100% mortality) to infection with either low (200 CFUs) or medium (400 CFUs, data not shown) inocula (Figure 3A). In contrast, their WT (C57BL/6) littermates survived (maximum 10% mortality). miR-223–deficient animals gradually lost weight and presented significantly higher bacterial burdens in the lungs, but not in the spleen (Figure 3B and Supplemental Figure 1). In the absence of miR-223, lung parenchyma of KO mice contained large infiltration foci packed with abundant bacteria (Figure 3C). Th1 responses are critical for TB control, thus we further examined whether an absence of miR-223 impacts the generation of Mtb-specific CD4+ T cells secreting IFN-γ and TNF-α. We detected similar frequencies of lung CD4+ lymphocytes, which secreted these cytokines after ex vivo stimulation with Mtb-derived proteins (purified protein derivative [PPD]) or polyclonal triggers (CD3/CD28) on day 21 postinfection (p.i.) (Figure 3E). Analogous investigations with CD8+ lymphocytes demonstrated that miR-223 KO T cells were intact in their propensity to produce IFN-γ and TNF-α during Mtb infection. The Th1 cytokine IFN-γ activates the enzyme-inducible NOS (iNOS) in phagocytes for the generation of NO, a potent mycobactericidal molecule in mice. We performed immunostaining for iNOS and observed numerous iNOS+ cells in the parenchyma of KO mice (Figure 3D), suggesting that susceptibility is not due to defective NO release. The source of NO in the lungs was mostly mononuclear phagocytes (Supplemental Figure 2). More abundant iNOS+ cells, paralleled by robust T cell responses, correlated with higher Mtb burdens, as observed in the KO mice. We conclude that miR-223 is critical for the control of murine TB, and this effect is apparently independent of adaptive immune responses.
Figure 3. Deletion of miR-223 renders resistant mice susceptible to TB.

(A) Survival of miR-223 KO (miR-223–/–) mice and C57BL/6 (WT) littermates after aerosol infection with Mtb (~200 CFUs). Data are representative of two independent experiments; Kaplan–Meier curves and log-rank test (n = 12). (B) Kinetics of bacterial load in lungs of infected mice. Data are presented as the median ± the interquartile range (iQR) and are representative of three independent experiments; Mann-Whitney U test (n = 5–6); **P < 0.01. (C) Large infiltration foci in lung (left panels, Giemsa staining) and numerous bacilli (right panels, acid-fast staining) in the absence of miR-223. Tissues were collected on days 21 and 25 p.i., respectively. Data are representative of two independent experiments. Scale bars: 1,000 μm (left panels) and 10 μm (right panels) (n = 5). (D) Immunohistochemistry for iNOS in lung tissue collected 21 days p.i. Data are representative of two independent experiments (n = 5). Scale bars: 100 μm. (E) Left panels: representative dot plots of CD4+ and CD8+ lymphocytes isolated on day 21 p.i. and stimulated with PPD or CD3/CD28 and then stained for intracellular TNF-α and IFN-γ. Right panels: frequency of cells double-positive for TNF-α and IFN-γ. Data are presented as the mean ± SEM and are pooled from two independent experiments (n = 8–9).
miR-223 modulates cytokine release in Mtb-infected myeloid cells.
Given the preponderant expression of miR-223 in myeloid cells (23) and the dual role of these cells in protection and pathogenesis in TB, we generated bone marrow–derived macrophages (BMDMs) and purified PMNs from bone marrow to assess their responses to Mtb infection. Following activation, IL-6 and TNF-α production were altered in KO BMDMs, whereas the secretion of antiinflammatory cytokines, notably IL-10, and the release of NO in response to IFN-γ activation remained unchanged (Figure 4A). Consistent with the latter finding, the miR-223 KO BMDMs, analogous to WT cells, restricted Mtb replication following classical activation. Resting macrophages of both genotypes similarly supported bacterial growth (Figure 4B). Strikingly, miR-223 KO PMNs secreted elevated amounts of TNF-α and IL-10 into cell-free supernatants at 6 and 20 hours p.i. (Figure 4C). The abundance of NO in supernatants of Mtb-infected PMNs was below the detection limit (data not shown). miR-223 is known to modulate NF-κB activation (26). In view of the effects of miR-223 on cytokine release and to better understand its impact on NF-κB activation during Mtb infection, we stably transfected a murine macrophage reporter cell line that overexpresses miR-223 to analyze NF-κB stimulation simultaneously with bacteria-triggered cell activation (Figure 4D). Overexpression of miR-223 reduced NF-κB activity p.i. and after TNF-α stimulation. We conclude that miR-223 directly regulates NF-κB activity in macrophages during TB and that miR-223 regulates cytokine release in myeloid cells. These observations suggest differential consequences of miR-223 regulation for cytokine release in macrophages and PMNs in TB.
Figure 4. miR-223 regulates the propensity of myeloid cells to release cytokines upon Mtb encounter.

(A) BMDMs were infected with Mtb at an MOI of 5, and cytokine concentrations in cell-free supernatants collected 24 hours p.i. were quantified using ELISA. Data are presented as the mean ± SEM and are representative of five experiments, with three replicates each; Student’s t test. N.D., not detected. (B) BMDMs were infected with Mtb at an MOI of 1, and bacterial growth was determined by CFUs. Data are presented as the mean ± SEM and are representative of five experiments, with three replicates each. (C) Neutrophils (PMNs) were infected with Mtb at an MOI of 10, and cytokine concentrations in cell-free supernatants collected at 6 and 20 hours p.i. were measured using ELISA. Data are presented as the mean ± SEM and are representative of three independent experiments with eight replicates each; Student’s t test. (D) NF-κB induction was measured in murine macrophage RAW 246.7 cells overexpressing miR-223 using an NF-κB reporter after stimulation with TNF-α or infection with Mtb. Data are presented as the mean ± SEM and are representative of three independent experiments with six replicates each; ANOVA with Bonferroni’s post test. *P < 0.05; **P < 0.01; ***P < 0.001.
miR-223 modulates neutrophil-driven inflammation in TB.
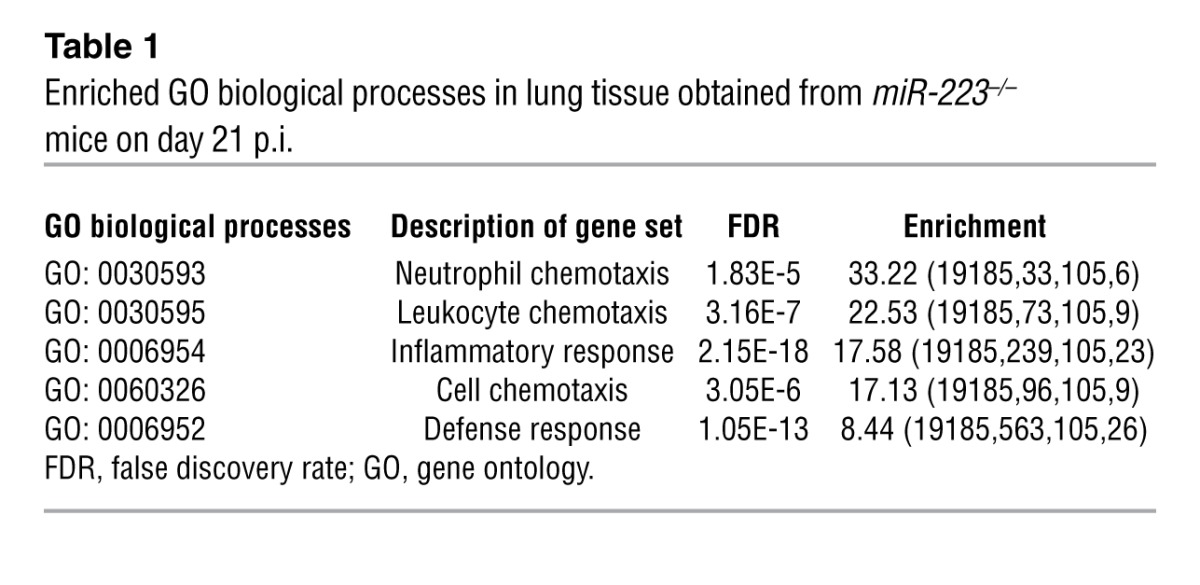
To identify target genes regulated by miR-223 during Mtb infection, we collected lung tissue on days 14 and 21 p.i. and performed microarray analysis of isolated RNA. We selected genes that were upregulated in miR223–/– mice and performed a pathway enrichment analysis to identify biological processes affected by miR-223. On day 14, no significant pathway enrichment specific for infectious processes was observed, and no immune-relevant genes exclusively regulated in miR-223 KO mice were identified (Supplemental Table 2, data not shown). On day 21, the KO mice presented more abundant transcripts for genes involved in inflammatory cell recruitment and function (Figure 5A and Supplemental Table 3). The gene showing the greatest upregulation in the absence of miR-223 was Cxcl2, a chemokine involved in PMN chemotaxis (27). Enrichment analysis using the web-based tool GORILLA (Gene Ontology enRIchment anaLysis and visuaLizAtion) pointed to chemotaxis as the biological process that dominated transcriptional responses in miR-223–deleted animals (Table 1). We validated the array data by RT-PCR for genes known to govern PMN and macrophage tissue migration (Cxcl2, Cxcl1, Il6, Ccl2, and Ccl3) (Figure 5B and Supplemental Figure 2). Subsequently, we determined the abundance of chemokines and cytokines involved in PMN and monocyte/macrophage recruitment. On day 21 p.i., protein concentrations of CXCL2, CCL3, and IL-6 were significantly increased in lung homogenates obtained from KO mice (Figure 5C). We did not observe differences in the total level of IKKα in lung homogenates from KO and WT mice (Supplemental Figure 3). This suggested that 21 days p.i., dissimilarities in chemokine abundance were not primarily due to miR-223 regulation of NF-κB. T cell chemoattractants, notably CXCL9 and CXCL10, were also elevated in KO lung tissue, and their concentrations correlated with IFN-γ levels, a T cell cytokine known to induce CXCL9 and CXCL10 synthesis (Supplemental Figure 2). In contrast, miR-223 did not modulate circulating levels of key cytokines and chemokines during TB (Supplemental Figure 2). Our data suggest an accelerated recruitment of PMNs and mononuclear phagocytes to lung parenchyma during TB progression in miR-223–deficient animals. Using Giemsa staining and immunohistochemistry, we confirmed that PMNs and inflammatory mononuclear phagocytes (myeloperoxidase, [MPO+] cells) represented the major cell types within inflammatory foci in miR223–/– mice (Figure 5D). Increased PMN recruitment was confirmed by phenotypic analysis of the cells purified from infected lungs (Figure 5E and see Supplemental Figure 4 for the gating strategy). The accumulation of PMNs was not due to reduced cell death events, as indicated by intensified TUNEL staining in lung parenchyma from miR223–/– mice (Supplemental Figure 5). We conclude that miR-223 controls neutrophil-driven inflammation during TB through the regulation of chemoattractants.
Figure 5. Lung influx of innate cells during TB is modulated by miR-223.

(A) Relative gene expression of selected genes in lung tissue of miR-223–/– compared with C57BL/6 (WT) mice on days 0 and 21 p.i., as identified by microarray gene chip assay. Data are presented as the mean ± SEM and are combined from two independent experiments (n = 9–11). (B) qRT-PCR of genes differentially expressed in respiratory parenchyma of miR-223–/– animals. Gapd was used as a reference gene, and data were normalized to uninfected mice. Data are presented as the mean ± SEM and are pooled from two independent experiments; Student’s t test (n = 9–11). (C) Concentrations of CXCL2, CCL3, and IL-6 were measured using a multiplex immunoassay in lung homogenates collected 21 days p.i. Data are presented as the mean ± SEM and are pooled from four independent experiments; ANOVA with Bonferroni’s post test (n = 18–24). (D) Giemsa staining and immunohistochemistry for neutrophils (PMNs) and inflammatory monocytes/macrophages (MPO) in lung tissue collected on days 25 and 21 p.i. Data are representative of two independent experiments (n = 5). Scale bars: 100 μm. (E) Frequency and number of alveolar macrophages (AM), inflammatory macrophages (iM), and neutrophils (PMN) isolated from the lungs of miR233–/– and WT infected animals on days 7, 14, and 21 p.i. Data are presented as the mean ± SEM and are pooled from two independent experiments; ANOVA with Bonferroni’s post test (n = 9–10). *P < 0.05; **P < 0.01; ***P < 0.001.
Table 1.
Enriched GO biological processes in lung tissue obtained from miR-223–/– mice on day 21 p.i.
CXCL2, CCL3, and IL-6 are direct targets of miR-223.
Matching lung gene expression results (Supplemental Tables 2 and 3) with miRecords, a software program for miRNA target analysis based on eleven different prediction databases (28), we proceeded with the identification of direct miR-223 targets. By focusing on genes involved in PMN chemotaxis and activation, we identified Cxcl2 as a potential target of miR-223. Consequently, we cloned the complete 3′-UTR of Cxcl2 into a dual luciferase reporter system (psi-CHECK2) downstream of the Renilla luciferase ORF. To avoid any interference with endogenous miR-223, we used HeLa cells for the target validation assays (29). The cotransfection experiment of HeLa cells with the reporter construct and the miR-223 mimic demonstrated a profound decrease in luciferase activity in the miR-223–transfected cells as compared with the control or scramble cotransfected cells (Figure 6A). To further validate the specificity of this assay, we mutated nucleotides in the 3′-UTR seed sequence of miR-223. In the presence of miR-223, luciferase activity was reestablished to levels similar to those of the controls, thus further verifying Cxcl2 as a direct target of miR-223. In a similar way, Ccl3 and Il6 were identified as direct targets of miR-223 (Figure 6, B and C). By contrast, genes encoding CXCL9 and CXCL10 were not validated as direct miR-223 targets, despite an increased protein abundance of respective chemokines in KO lung tissue upon infection (Supplemental Figure 6). Analogous results were observed for IFN-γ (data not shown). Our data reveal a previously unknown role of miR-223, namely, the control of tissue migration of inflammatory cells via the direct regulation of distinct chemokines and cytokines.
Figure 6. miR-223 directly targets the chemoattractants CXCL2, CCL3, and the cytokine IL-6.

(A) Luciferase activity of HeLa cells transfected with either WT or the seed sequence–mutated (mut) 3′-UTR of Cxcl2 plus scramble and miR-223 mimics. (B) Luciferase activity of HeLa cells transfected with either WT or mutated 3′-UTR of Il6 plus scramble and miR-223 mimics. (C) Luciferase activity of HeLa cells transfected with either WT or mutated 3′-UTR of Ccl3 plus scramble and miR-223 mimics. Renilla luciferase activity is normalized to that of the firefly luciferase and set to 1 in cells with no miRNA mimic or scramble. Mutated nucleotides are highlighted in red. *P < 0.05; **P < 0.01; ***P < 0.001. (A–C) Data are the mean ± SEM of three replicates of one representative experiment; Student’s t test.
miR-223 induces negative feedback control of PMN chemotaxis.
Both hematopoetic and tissue-resident cells produce chemoattractants upon bacterial infection (30). However, miR-223 is uniquely expressed in hematopoietic cells, where it regulates gene transcription via NF-κB directly, as well as through posttranscriptional modulation of mRNA stability. Thus, we determined whether myeloid cell–derived miR-223 affects CXCL2 and CCL3 released by PMNs and macrophages by assessing transcript abundance and chemokine concentrations in cultures of PMNs and BMDMs. In both cell populations, an absence of miR-223 resulted in augmented Cxcl2 and Ccl3 transcription (Figure 7, A and B) and in elevated CXCL2 and CCL3 protein secretion following Mtb infection (Figure 7, C and D). While IL-6 was not detected in PMNs (data not shown), the reduced levels observed in KO BMDMs (Figure 3A) primarily indicate differences in transcriptional regulation. Further, we evaluated the abundance of miR-223 in lung myeloid cell populations during TB. Alveolar macrophages (AMs), inflammatory macrophages (iMs), and PMNs were obtained for miRNA quantification by enzymatic digestion of the lungs, followed by FACS (see Supplemental Figure 4 for the gating strategy). Both on days 14 and 21 p.i., we observed the highest level of miR-223 transcripts in PMNs, suggesting that they were the main cell population expressing miR-223 (Figure 7E and data not shown). In addition, treatment with the mAb against Ly6G to deplete PMNs significantly reduced miR-223 abundance in the lung (Figure 7F). We applied the same FACS-sorting strategy to determine which cells presented in vivo differential regulation of Cxcl2 and Ccl3 on day 21 p.i. We identified significantly increased Cxcl2 and Ccl3 gene transcripts exclusively in PMNs deleted of miR-223 (Figure 7, G and H). We conclude that miR-223 affected PMN recruitment into the lungs via intrinsic chemokine regulation.
Figure 7. miR-223 targets Cxcl2 and Ccl3 in a cell-intrinsic manner during TB.

(A and B) Mouse PMNs and BMDMs were infected with Mtb at an MOI of 10 and 5, respectively. Gene expression of Cxcl2 and Ccl3 was measured at 6, 20 (A), and 24 (B) hours p.i. by qRT-PCR. Gapd was used as a reference gene, and data were normalized to uninfected cells. Data are presented as the mean ± SEM and are representative of two experiments with three to five replicates each; Student’s t test. (C and D) Mouse PMNs and BMDMs were infected with Mtb at an MOI of 10 and 5, respectively. Cytokine concentration in cell-free supernatants collected at 6, 20 (C), and 24 (D) hours p.i. was measured using ELISA. Data are presented as the mean ± SEM and are representative of two to five experiments with three to eight replicates; Student’s t test. (E) miR-223 expression in AM, PMN, and iM were FACS sorted from the lungs of C57BL/6 (WT) mice 21 days p.i. Data were obtained using U6 snRNA as a reference gene and were normalized to uninfected mice. Data are from one experiment and are presented as the mean ± SEM; ANOVA with Bonferroni’s post test (n = 4). (F) miR-223 expression was evaluated by qRT-PCR in total lung homogenates obtained on day 25 p.i. from C57BL/6 (WT) mice treated as indicated. Data were obtained using U6 snRNA as a reference gene and were normalized to uninfected mice. Data are presented as the mean ± SEM and are representative of two independent experiments; Student’s t test (n = 5). (G and H) Cxcl2 and Ccl3 expression in AM, PMN, and iM sorted from the lungs of Mtb-infected mice 21 days p.i. Gapd was used as a reference gene, and data were normalized to uninfected mice. Data are from one experiment and are presented as the mean ± SEM; Student’s t test (n = 4). *P < 0.05; **P < 0.01; ***P < 0.001.
CXCL2 and CCL3 orchestrate PMN chemotaxis, while IL-6 modulates granulopoesis (13). To assess whether chemokine release and, consequently, chemotaxis were affected by miR-223, we purified PMNs from WT and KO mice and defined their migratory capacities. The absence of miR-223 induced accelerated migration in the presence of CXCL2 or lung homogenates obtained from TB-diseased WT and KO animals (Figure 8A). We confirmed a higher abundance of CXCR2 in miR-223–/– PMNs, pointing to a role of this chemokine receptor in chemotaxis (Supplemental Figure 7) (24). Thus, PMNs from KO animals appeared particularly prone to migrate in response to chemotactic stimuli. Accordingly, this effect was exacerbated in miR-223–/– mice due to a higher abundance of relevant mediators. We next determined whether accelerated PMN dynamics at the site of infection caused heightened lethality of miR-223–/– mice with TB. The treatment of mice with mAbs against Ly6G to deplete PMNs upon infection resulted in prolonged survival of KO animals (Figure 8B), indicating that neutrophilic inflammation and subsequent tissue destruction were major causes of morbidity in the absence of miR-223. The specificity of the depleting antibody was verified by estimating blood and lung frequencies of PMNs and the presence of MPO+ cells in lung parenchyma (Supplemental Figures 8 and 9). Lung abundance of Cxcl2, Ccl3, and Il6 was reduced by Ly6G-neutralizing mAbs, further emphasizing the critical role of miR-223 as a regulator of chemokines, PMN chemotaxis, and local inflammation in TB (Supplemental Figure 8). To determine the role of direct cytokine targets of miR-223 in TB outcome, we performed antibody neutralization experiments. Simultaneous neutralization of CXCL2, CCL3, and IL-6 ameliorated TB susceptibility (Figure 8C). KO mice that received the cocktail of neutralizing antibodies had fewer MPO+ cells in their lungs 25 days p.i. (Figure 8D). This treatment did not affect Mtb burdens on days 18 and 25 p.i. (Figure 8E). In a complementary approach, we infected double-KO mice, deleted of miR-223 and CXCR2 (miR-223–/– Cxcr2–/–), with Mtb. The concurrent absence of miR-223 and CXCR2, which is a central receptor for chemokines orchestrating PMN chemotaxis including CXCL1, CXCL2, and CXCL5, completely reversed the severe TB susceptibility of the miR-223 KO animals (Figure 8F). Blood PMN frequencies were elevated in mir-223–/– Cxcr2–/– mice early during the disease (Supplemental Figure 10), but this pattern was reversed at the peak of inflammation. However, during steady state and early upon infection, PMN numbers did not differ between WT and mir-223–/– mice (Supplemental Figure 2). These observations rule out a direct effect of miR-223 on granulopoesis or PMN mobilization from the bone marrow. In summary, our findings suggest that the PMN-targeting chemokines CXCL2, CCL3, and the proinflammatory cytokine IL-6, induced lethal neutrophilic inflammation in the absence of miR-223, mostly through tissue destruction. We propose, first, that miR-223 modulates the propensity of myeloid cells to produce cytokines by direct interference with NF-κB activation. The cytokine IL-6 is particularly relevant for autocrine activation of the Mtb-infected phagocytes (31) and, in addition, modulates granulopoesis. Second, miR-223 orchestrates tissue migration of PMNs and inflammatory monocytes by posttranscriptional regulation of the leukocyte chemoattractants CXCL2 and CCL3 along with cell surface expression of CXCR2 (Supplemental Figure 11).
Figure 8. Autocrine regulation of PMN recruitment through miR-223–targeted cytokines.

(A) Percentage of neutrophils that migrated toward the indicated stimuli after 2 hours. LH, lung homogenate. Data are presented as the mean ± SEM and are representative of two independent experiments with three replicates each; Student’s t test. (B) miR-223–/– mice were treated with monoclonal antibodies against Ly6G (PMN depletion) or with control IgG or (C) against CXCL2/IL-6/CCL3 during TB, and survival was monitored. Data are representative of two independent experiments; Kaplan-Meier curves and log-rank test (n = 4–10). (D) Immunohistochemistry for neutrophil and inflammatory monocytes/macrophages (MPO) in lung tissue collected 25 days p.i. Data are representative of two experiments (n = 3–4). Scale bars: 100 μm. (E) Bacterial load in the lungs of infected mice treated with monoclonal antibodies against CXCL2/CCL3/IL-6. Data are the median ± iQR and are pooled from three experiments (n = 9–16). (F) Survival of miR-223–/–Cxcr2–/– (n = 7) and miR-223–/– (n = 4) mice after aerosol infection with Mtb; Kaplan-Meier curves and log-rank test. Data are from one experiment. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Our data demonstrate an essential role of miR-223 in TB and define the mechanisms by which it regulates the outcome of TB. In more general terms, our data reveal that a single miRNA can dictate the immune response in chronic infection and inflammation. miR-223 directly regulated CXCL2, CCL3, and IL-6, which in turn orchestrated inflammatory cell recruitment to lung parenchyma during TB. Thus, our data reveal what we believe to be a novel function of miR-223 in nonresolving inflammation, namely, the regulation of inflammatory leukocyte migration. We propose that PMNs not only incrementally accumulate miR-223 during their development (11), but also exploit the same miRNA to negatively regulate recruitment into infected tissue sites of PMNs generated by emergency granulopoiesis.
A fundamental feature of the immune response in infection and inflammation is leukocyte migration to the site of bacterial insult. While successful immune surveillance necessitates a prompt influx of innate cells into affected tissues, dysregulated chemotaxis can be detrimental. We examined whether, and how, a single miRNA can regulate inflammation in response to infection with Mtb. Distinct miRNAs have been correlated with specific groups of diseases and have been suggested as potential disease biomarkers (32). In stark contrast, only scant data exist regarding the specific biological functions of miRNAs on cellular and molecular levels during the infectious disease process (11). Our data demonstrate that miR-223, in addition to its known function in myeloid cell development under homeostasis (25), tunes the immune response during inflammation. Thus, our experiments assign an essential biological function of miR-223 enrichment in differentiated myeloid cells to the control of TB.
Consistent with previous findings (20), we identified a differential expression of miR-223 in the peripheral blood of active TB patients, healthy latently infected individuals (TST+), and uninfected individuals (TST–), with the highest expression levels found in TST– subjects. Ratios of myeloid cell subpopulations in TB can vary (33–35), and this observation could explain more the abundant expression of miR-223 in active TB patients over healthy latently infected (TST+) individuals, as well as the highest abundance of miR-223 in healthy uninfected subjects (TST–). In addition, the developmental stages at which PMNs are released into the blood stream can affect blood miR-223 levels. The highest expression in healthy subjects could indicate the presence of more mature PMNs, while active TB patients and TST+ individuals may have a substantial proportion of immature PMNs and/or monocytes in the periphery. Moreover, TB patients could present an upregulation of miR-223 in myeloid subsets as a consequence of infection/inflammation. miR-223 levels are influenced by nuclear factor I-A (NFI-A), CCAAT enhancer–binding proteins (C/EBPs), and the transcription factor purine-rich box 1 (PU.1) (25, 36). The effects of soluble mediators, i.e., cytokines, on these transcription factors await clarification. We did not detect changes in miR-223 levels in human or murine myeloid cells following ex vivo Mtb infection (data not shown). Based on these observations, we consider it likely that aberrant miR-223 regulation exacerbates TB disease. Intriguingly, miR-223 was enriched in granulomatous lung specimens from active TB patients, probably reflecting the accumulation of myeloid cells in lesions. Mononuclear infiltrations into TB granulomas have been repeatedly demonstrated (37), and abundant PMNs have been observed in the sputum of active TB patients (4). Our investigations of experimental murine TB strengthen our observations from human studies and emphasize that miR-223 levels were upregulated in both the lung and blood. Lung PMNs showed the highest miR-223 abundance among the myeloid populations analyzed, and treatment with an anti-Ly6G mAb to deplete PMNs substantially affected overall miR-223 lung levels. These results indicate that during TB, miR-223 influences disease outcome primarily through intracellular accumulation in PMNs.
To understand the role of miR-223 in TB in molecular terms, we monitored disease outcomes in miR-223–deficient mice and further dissected the biological role of miR-223. The miR-223–/– mice failed to control Mtb infection and succumbed to extensive pulmonary inflammation, dominated by massive PMN infiltration. Consequently, we evaluated the genes regulated by miR-223 during TB. miR-223 is a positive regulator of PMN differentiation and function (24), and lung infiltration with PMNs has been observed in aged mice due to incremental steady-state granulocytosis and hyperactivation of PMNs in the absence of miR-223 (24). These alterations are due to Mef2c activity, a gene targeted by miR-223. We did not observe any signs of neutrophilic pneumonia in miR-223–/– mice at 3 months of age (data not shown), when the TB experiments were initiated. In addition, Mef2c levels remained unchanged during infection (Supplemental Table 4), thus ruling out a major role for this homeostatic pathway in TB. In independent experiments, miR-223 has been shown to downregulate the activity of cyclic nucleotide response element–binding protein (CREB) by repressing upstream pathways (IGF-1, Ca2+ channel, and GPCR) (38). Since CREB is critical to the maintenance of cell survival and growth, the absence of miR-223 could sustain cell survival. A miR-223 regulatory pathway has been suggested that affects CEBP-β and LMO2 (CCAAT/enhancer-binding protein β and LIM domain only 2), in this way attenuating cell proliferation (39). We did not detect a differential regulation of the above-mentioned genes upon Mtb infection in the absence of miR-223 (Supplemental Tables 2–4), indicating that its absence did not activate cell proliferation. Moreover, the accumulation of PMNs in miR-223–/– lung tissue was accompanied by stronger TUNEL staining, suggesting that the absence of miR-223 did not result in sustained survival of PMNs in the infected lungs. Overall, we conclude that the accumulation of PMNs in infected lungs of miR-223–/– mice was independent of cell survival/proliferation and that other events triggered neutrophilic pneumonia.
Despite an exacerbated susceptibility to TB, T cell responses were apparently unaffected, and IFN-γ concentrations in lung tissue from miR-223–deficient mice were elevated. Although the critical role of IFN-γ in acquired immunity to Mtb is beyond doubt (1), it was insufficient for protection in the absence of miR-223. Along these lines, IFN-γ–dependent iNOS induction was not impaired in miR-223 KO mice. Altogether, these observations suggest that the induction and effector functions of adaptive immunity were virtually intact in the absence of miR-223. Evidence is accumulating that IFN-γ is essential, but not sufficient, for protection against TB (40, 41). Recent data suggest that IFN-γ impairs the accumulation of PMNs in the lung during TB and regulates their survival (42). In contrast, miR-223 KO mice showed accelerated lung PMN recruitment in the presence of abundant IFN-γ. Thus, these IFN-γ effects are probably redundant, and independent pathways likely control PMN accumulation in the lung during TB. Alternatively, a loss of IFN-γ responsiveness could explain the observed phenotype. However, we dismiss the latter hypothesis due to the heightened iNOS (IFN-γ–inducible) pattern observed in the lung parenchyma of miR-223–/– mice. Thus, the modulation of neutrophilic inflammation in the absence of miR-223 in murine TB was independent of adaptive immunity.
Gene expression profiling indicated that the genes involved in inflammatory cell recruitment and function were significantly upregulated in miR-223–/– mice. This was not apparent 14 days p.i., but became obvious later during infection due to the incremental expression of immune-relevant genes. In addition, gene network analysis identified a PMN-specific signature in the lung of miR-223–/– mutants 21 days p.i. that was characterized by an enrichment of genes involved in leukocyte chemotaxis. Cxcl2, Ccl3, and Il6 were among the top upregulated genes and emerged as potential targets of miR-223 in our in silico analysis. Elevated protein abundance of these cytokines further supports our finding that miR-223 modulates leukocyte migration to the lung. The Cxcl2, Cccl3, and Il6 genes were validated as direct targets of miR-223. PMNs were the main cell type regulating the expression of the above chemokines under the control of miR-223. Other chemokines, namely CXCL9 and CXCL10, were also increased in abundance in miR-223–/– mice, but this was likely due to the indirect effects including elevated IFN-γ production (43, 44). The expression of Cxcl2, Ccl3, and Il6 is dependent on NF-κB activation (45), and thus we assume that miR-223 plays a dual role in the control of NF-κB activity (26). However, besides NF-κB, other signaling pathways (MAPKs, phosphatidylinositide 3-kinases/protein kinase B [PI3K/AKT]) induce transcriptional responses, and their overall contribution to PMNs and monocytes/macrophages can differ. In addition, the contribution of these pathways to the transcriptional activation of a given gene is variable. For instance, the regulation of IL-6 depends on both NF-κB and MAPK. This could explain the opposite effects observed in myeloid populations upon an ex vivo Mtb encounter. In the absence of miR-223, Mtb-stimulated BMDMs expressed less IL-6, whereas the propensity of PMNs and BMDMs to release CCL3 and other mediators was heightened. Previous studies have identified activated PMNs as a source of CCL3 and IL-6 (46, 47). In Leishmania infection, PMNs can coordinate the recruitment of DCs via CCL3 and thus modulate protective T cell responses (48). A role of PMNs as facilitators of antigen presentation and induction of Th1 responses in TB was recently described (49, 50). PMN-derived CCL3, which impacts DC functions, could explain the excessive IFN-γ levels observed in miR-223 KO mice.
We found that the second chemokine under direct control of miR-223 is CXCL2. In response to surgical trauma and other insults, this chemokine is exclusively produced by inflammatory leukocytes (15, 51). Upon Mtb infection, both PMNs and BMDMs secreted copious amounts of CXCL2 in the absence of miR-223. Despite a redundancy of CXC chemokines (52), inhibition of PMN infiltration by blocking the CXCL2 receptors CXCR1/CXCR2 holds promise for cancer therapy (53) and hence could be targeted in inflammatory diseases as well. Granulopoiesis is regulated by the inflammatory cytokine IL-6 (54), and IL-6 has been shown to participate in protection against TB (31, 55). More recent studies demonstrate that IL-6 is indirectly controlled by miR-223 via STAT3 (56). In extension of this observation, we provide molecular evidence that Il6 is a direct target of miR-223. By controlling IL-6 abundance at different levels, miR-223 most likely modulates the generation of PMNs in bone marrow and thus determines general PMN availability. We observed that even though early upon infection, blood frequencies of PMNs did not differ between mouse strains, on day 21 p.i., miR-223–/– mice had higher percentages of PMNs in their circulation (Supplemental Figure 2). Although during steady state, miR-223 apparently did not affect PMN retention in marrow sinusoidal epithelium, further investigation is required to determine whether miR-223 plays a role in the mobilization of PMNs to the periphery via the CXCL12/CXCR4 axis during infection. It appears that miR-223 targets multiple genes involved in inflammation. Our data reveal that upon infection, miR-223 drives leukocyte chemotaxis by targeting Cxcl2, Ccl3, and Il6. Recent evidence has been provided by studies showing that miR-223 targets the Nlrp3 gene and thus regulates inflammasome function (57, 58). Altogether, miR-223 could represent a cellular regulatory hub in host inflammation. The Cxcl2, Ccl3, and Il6 genes were defined as novel miR-223 targets in the murine system. Due to the incomplete similarities at the 3′UTR miR-223 recognition sites within the target genes in murine and human homologs, we cannot exclude the possibility of different roles of miR-223 in human TB.
Previous experiments aimed at elucidating the murine response to TB have been confined to cells and mediators. To the best of our knowledge, our experiments describe for the first time defined functions of a single miRNA in TB control. At first sight, this might be surprising, since miRNAs are generally considered to fine-tune gene expression rather than induce complete on-off switches (59). Our data provide a conclusive explanation for the essential role of miR-223 in TB by: (a) identifying CXCL2, CCL3, and IL-6 as direct targets of miR-223 along with constitutively elevated CXCR2 expression; (b) demonstrating an amelioration of exacerbated PMN-dependent susceptibility of miR-223–/– mice by CXCL2/CCL3/IL-6 neutralization; and (c) revealing that concurrent gene deletion of CXCR2 and miR-223 reduced TB lethality. Principally, miR-223 restricts neutrophil inflammation by dampening the production of proinflammatory mediators, primarily chemokines, and subsequently PMN recruitment into the lungs. Hence, we define what we believe to be a novel effector role for miR-223 in immunity, infection, and inflammation. Understanding the tissue dynamics of leukocytes at the miRNA level can pave the way to therapeutic modulation of PMN-mediated unleashed inflammation.
Methods
Mice and Mtb infection.
C57BL/6 mice, 8–12 weeks of age at the beginning of the experiments, were kept under specific pathogen-free (SPF) conditions at the Max Planck Institute for Infection Biology in Berlin, Germany. miR223–/– mice (C57BL/6 background) (24) had been backcrossed at least eight times to C57BL/6 mice. The double KO miR-223–/–Cxcr2–/– mice were obtained by crossing Cxcr2–/– mice (The Jackson Laboratory) with miR-223–/– mice. A Glas-Col inhalation exposure system was used for Mtb infection. One day p.i., a group of mice was sacrificed to estimate the precise infection dose. Lung tissue was dispersed in water 1% w/v BSA, 1% v/v Tween 80 and plated on Middlebrook 7H10 agar plates supplemented with ampicillin (10 μg/ml). Mtb colonies were enumerated after 3 weeks of incubation at 37°C. For neutralization experiments on days 14, 18, and 21 p.i., mice received an i.p. injection of 100 μg of monoclonal antibodies against CXCL2, CCL3, and IL-6 (R&D Systems) or control Igs. Neutrophil depletion was performed as previously described (9).
Human blood and lung tissue specimens.
The 76 subjects of mixed European descent included in this study were recruited from the Monaldi Hospital in Naples, Italy. The diagnosis of pulmonary TB was based on a TST combined with a chest x-ray and sputum-smear microscopy. Further confirmation of a diagnosis was achieved by Mtb identification via PCR and bacteriological tests. The samples from active TB patients were obtained before chemotherapy. The subjects belonging to the TST– (n = 24) and TST+ (n = 15) groups were household contacts (genetically related or unrelated) of TB-diagnosed individuals (n = 37). These subjects (TST– and TST+) did not present any symptoms of TB. All subjects in this study were HIV– and immunocompetent.
Human blood samples (16 subjects) for the miRNA array analysis were collected at Makerere University in Kampala, Uganda.
Formalin-fixed, paraffin-embedded lung tissue blocks from patients with pulmonary TB were obtained from the L. Spallanzani National Institute for Infectious Diseases (INMI) in Rome, Italy. Specimens were from archived autopsies of patients who had succumbed to TB and were Mtb culture–positive or had positive stains for acid-fast bacilli. Eight specimens from TB+ HIV– patients were available for this investigation. Healthy lung tissue was commercially obtained from OriGene.
Total RNA extraction.
Total RNA extraction of both blood samples from human and mice was performed using TRIzol LS and TRIzol (Invitrogen), respectively, according to the manufacturer’s instructions. Mouse lung tissue and lung cells were processed in TRIzol. RNA purification from lung sections was achieved using a RecoverAll Total Nucleic Acid Isolation Kit for FFPE (Ambion) following the manufacturer’s manual. The RNA yield and A260/280 ratio were monitored with a NanoDrop ND 100 spectrometer (NanoDrop Technologies), and RNA integrity was verified using the 2100 Bioanalyzer (Agilent Technologies).
RNA quantification.
Total RNA (50 ng) was reverse transcribed to cDNA, and qRT-PCR was performed to quantify mature miR-223 expression using the TaqMan miRNA Assay (Applied Biosystems) according to the manufacturer’s instructions. Samples were measured in triplicate in three different experiments. Moreover, a no-template control and two interplate controls were carried out in each PCR. miR-103 was chosen as a reference for data analysis of human samples (60). For mRNA quantification, the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used to synthesize cDNA according to the manufacturer’s protocol. TaqMan qRT-PCR assays with specific probes for mouse Cxcl2, Il6, Ccl3, Cxcl1, Ccl2, Cxcl9, Cxcl10, Ifng, Lmo2, Mef2c, Ikka, and Cebpb (Applied Biosystems) were used. All probes were normalized to Gapd as an internal control (Applied Biosystems). All fold changes were calculated using the ΔΔCt method (61) and compared with uninfected mice. Amplifications were performed with ABI PRISM 7900HT (Applied Biosystems).
3′-UTR cloning and luciferase reporter assay.
Validation of miRNA targets (Cxcl2, Il6, Ccl3, Cxcl9, Cxcl10) was performed by cloning complete 3′-UTRs. PCR products were amplified using primers with restriction sites for XhoI and NotI and cloned into the psiCHECK-2 vector (Promega), and miRNA binding sites were mutated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene) (Supplemental Table 5). Sequencing was performed to verify the correct alignment of all constructs. For luciferase reporter assays, HeLa cells were cotransfected with psiCHECK-2 constructs and control miRNA (scramble, AM17110) or miR-223 (PM12301; Ambion) using Fugene HD (Promega) following the manufacturer’s instructions. Forty-eight hours after transfection, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. Total Renilla luciferase activity was calculated by normalizing to firefly luciferase in order to correct for differences in transfection efficiency. Control (scramble) and miR-233 were added at a 30-nM concentration.
Raw 264.7 cell lines overexpressing miR-223 and NF-κB reporter cell line.
Stable overexpression of miR-223 cells in RAW 264.7 macrophages was induced using shMIMIC lentiviral miR-223 particles (ABgene Ltd.), followed by selection with puromycin. miR-223 overexpression was validated by qRT-PCR. RAW 264.7 cells were infected with the Lenti NF-κB Reporter (SABiosciences), followed by selection with puromycin. NF-κB activation was validated upon stimulation of the reporter cell line with TNF-α. For Mtb infection, 5 × 104 cells were seeded in 96-well plates and infected with Mtb H37Rv at an MOI of 10. TNF-α was used as a positive control for NF-κB reporter activation.
miRNA and gene expression array analysis.
miRNA microarray experiments were carried out as one-color hybridizations on human catalog V2 8-plex 15K microarrays (AMADID 019118; Agilent Technologies). Data analysis was performed with the Rosetta Resolver Platform Build 7.2.2 SP1.31 (Rosetta Biosoftware). Gene expression microarray experiments were performed as dual-color hybridizations (Agilent Technologies) for mouse studies. Analysis was carried out in a manner similar to that described for miRNA microarrays. An agglomerative clustering algorithm on the reporter level of the normalized intensity profiles was used to structure the results. The hierarchical 2D structure of the miRNA reporters and intensity profiles was illustrated as a hierarchical tree, while the intensity data were displayed as a heatmap. A correlation coefficient cutoff (red lines) was used for intensity profiles and for reporters. The heuristic criteria “average link” and the similarity measure “cosine correlation” (correlation without mean subtraction) together with error weighting were used, while the data were transformed to a Z-score to remove the multiplicative error effect of the intensity measurement, such that the variation of high-intensity and low-intensity measurements made an equal contribution to the similarity measurement. Microarray data were deposited in the NCBI’s Gene Expression Omnibus (GEO accession number GSE39163).
Primary cell cultures and innate immunity assays.
BMDMs were prepared and activated as described (8). Classical activation of myeloid cells was achieved by treatment with recombinant mouse IFN-γ (100 U/ml) (Strathmann Biotech AG). PMNs were purified from bone marrow with Ly6G magnetic beads (Miltenyi Biotec). Cells were infected with Mtb H37Rv at various MOIs. Bacteria were grown in Middlebrook 7H9 broth (BD Biosciences) supplemented with 0.05% glycerol and Tween 80 and 10% ADC enrichment (BD Biosciences). Single bacterial cells resuspended in culture medium were obtained from early log-phase cultures. TNF-α, IL-6, IL-10, CXCL2, and CCL3 in cell culture supernatants were determined using commercial ELISA kits (R&D Systems). NO was determined by Griess reaction. Cell surface expression of CXCR2 in PMNs was assessed by FACS using the antibody clone TG11 (BioLegend).
Multiplex cytokine assays.
Lung tissue was disrupted in PBS 0.05% v/v Tween 20 supplemented with a protease inhibitor cocktail (Roche). Measurements were performed using multiplex bead–based immunoassay kits (Millipore) according to the manufacturer’s instructions. Samples were measured on a Bio-Rad instrument.
Lung cell isolation, flow cytometry, and fluorescence-activated cell sorting.
Single-cell suspensions from lungs of mice were prepared as described (8). To characterize and sort innate immune cells, isolated leukocytes were stained with antibodies against Ly6G (1A8; BD), CD11b (M1/70; eBioscience), F4/80 (BM8; eBioscience), CD11c (HL3), and Ly6C (AL-21; BD). For flow cytometric analysis, all stained cells were acquired on a Canto II flow cytometer (BD) and analyzed with FACSDiva (BD) and FACS Analyzer software. FACS was conducted on an Aria II cell sorter (BD). Purity of the sorted cells was verified immediately after sorting. Sorted leukocyte populations were resuspended in TRIzol. RNA was isolated and quantified as described above.
For T cell analysis, single-cell lung suspensions were incubated for 6 hours in the presence of brefeldin A, with mycobacterial antigens prepared in PPD (SSI, Denmark), polyclonal activators (anti-CD3 and anti-CD28), or media. Antibodies against the following markers, all from BD, were used for identification of the lymphocyte subsets and intracellular cytokines: CD4 (RM4-5), CD8 (53-6.7), IFN-γ (XMG1.2), and TNF-α (TN3-19.2). Cells were acquired on a Canto II flow cytometer (BD) and data were analyzed with FlowJo software (Tree Star Inc.).
Histology and immunohistochemistry of mouse specimens.
Lung lobes were fixed with 4% w/v PFA overnight and embedded in paraffin. Lung specimens cut in 2-μm-thick sections were evaluated following Giemsa staining. In situ Mtb was visualized by acid-fast Ellis staining. Commercially available antibodies were used to visualize MPO+ (DAKO) and iNOS+ cells (NeoMarkers).
Chemotaxis assay.
PMNs purified from bone marrow as described above were seeded on the upper compartment of a Transwell (Costar) at 106 cells per well. Cells were allowed to migrate for 2 hours against gradients of CXCL2 (10 ng/ml) or lung homogenates obtained 21 days p.i. from WT and miR-223–/– mice. The lung homogenates were pooled from five animals and diluted 1:20 in media. Cells migrating to the lower compartment were counted, and the percentage of migration was calculated as migrated cells × 100 / 106.
Western blot analysis.
Protein concentrations in lung homogenates were measured using a BCA Protein Assay kit (Thermo Scientific), and the same amount of protein was loaded for each sample. Primary antibodies against mouse IKKα and β-actin were purchased from Abcam, and secondary antibodies against mouse or rabbit were purchased from Cell Signaling Technology.
Blood PMN enumeration.
Blood was obtained from the tail vein or aorta abdominalis and classical hematological techniques were used to obtain smears, which were stained using the Diff-Quick kit (Fisher Scientific).
Statistics.
GraphPad Prism (GraphPad Software) was used for statistical analysis. Differences in RT-PCR data were assessed using ANOVA and a Student’s t test. Bacterial titers were analyzed using the Mann-Whitney U test. Concentrations of cytokines and chemokines were compared using a two-way ANOVA followed by a Bonferroni’s post test. P values ≤ 0.05 were considered statistically significant. Differentially expressed genes on days 14 and 21 p.i. were analyzed for biological functions using GORILLA software (62). Only genes that were upregulated more than 2-fold in KO mice after Mtb infection, and not deregulated in the naive KO mice, were analyzed.
Study approval.
Approval for study on human samples was obtained from the ethics committees of Monaldi Hospital in Naples, Italy, and Kampala University in Kampala, Uganda (GC6). Mouse experiments were performed in accordance with German Animal Protection Law (0221/12).
Supplementary Material
Acknowledgments
We thank Mary Louise Grossman and Diane Schad for their help in preparing the manuscript; Harriet Mayanja-Kizza and William H. Boom for providing whole blood through the Uganda-Case Western Reserve University Research Collaboration funded by NIH’s TB Research Unit and the GC6-74 Consortium; all donors for supplying samples; Shreemanta K. Parida for organizational support; and Fernando D. Camargo (Children’s Hospital, Boston, Massachusetts, USA) for providing the miR-223 KO mice. This work was supported by the European Union’s Seventh Framework Programme (EU FP7) projects “NEWTBVAC” (Health-F3-2009-241745) and “ADITEC” (HEALTH-F4-2011-280873).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(11):4836–4848. doi:10.1172/JCI67604.
References
- 1.Philips JA, Ernst JD. Tuberculosis pathogenesis and immunity. Annu Rev Pathol. 2012;7:353–384. doi: 10.1146/annurev-pathol-011811-132458. [DOI] [PubMed] [Google Scholar]
- 2.Kaufmann SHE. Tuberculosis: back on the immunologists’ agenda. Immunity. 2006;24(4):351–357. doi: 10.1016/j.immuni.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Lowe DM, Redford PS, Wilkinson RJ, O’Garra A, Martineau AR. Neutrophils in tuberculosis: friend or foe? Trends Immunol. 2012;33(1):14–25. doi: 10.1016/j.it.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Eum SY, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010;137(1):122–128. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eruslanov EB, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73(3):1744–1753. doi: 10.1128/IAI.73.3.1744-1753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. . Infect. Immun. 2006;74(7):4295–4309. doi: 10.1128/IAI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lyadova IV, et al. In mice, tuberculosis progression is associated with intensive inflammatory response and the accumulation of Gr-1 cells in the lungs. PLoS One. 2010;5(5):e10469. doi: 10.1371/journal.pone.0010469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorhoi A, et al. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med. 2010;207(4):777–792. doi: 10.1084/jem.20090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208(11):2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 11.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for micro-RNAs in the immune system. Nat Rev Immunol. 2010;10(2):111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 12.Eulalio A, Schulte L, Vogel J. The mammalian microRNA response to bacterial infections. RNA Biol. 2012;9(6):742–750. doi: 10.4161/rna.20018. [DOI] [PubMed] [Google Scholar]
- 13.Sadik CD, Luster AD. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol. 2012;91(2):207–215. doi: 10.1189/jlb.0811402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton T, et al. Diversity in post-transcriptional control of neutrophil chemoattractant cytokine gene expression. Cytokine. 2010;52(1–2):116–122. doi: 10.1016/j.cyto.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajaram MV, et al. Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. Proc Natl Acad Sci U S A. 2011;108(42):17408–17413. doi: 10.1073/pnas.1112660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma F, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-γ. Nat Immunol. 2011;12(9):861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Wang X, Jiang J, Cao Z, Yang B, Cheng X. Modulation of T cell cytokine production by miR-144* with elevated expression in patients with pulmonary tuberculosis. Mol Immunol. 2011;48(9–10):1084–1090. doi: 10.1016/j.molimm.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Fu Y, Yi Z, Wu X, Li J, Xu F. Circulating microRNAs in patients with active pulmonary tuberculosis. J Clin Microbiol. 2011;49(12):4246–4251. doi: 10.1128/JCM.05459-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, et al. Comparative miRNA expression profiles in individuals with latent and active tuberculosis. PLoS One. 2011;6(10):e25832. doi: 10.1371/journal.pone.0025832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maertzdorf J, et al. Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci U S A. 2012;109(20):7853–7858. doi: 10.1073/pnas.1121072109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh Y, et al. Mycobacterium tuberculosis controls microRNA-99b (miR-99b) expression in infected murine dendritic cells to modulate host immunity. J Biol Chem. 2013;288(7):5056–5061. doi: 10.1074/jbc.C112.439778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Connell RM, Zhao JL, Rao DS. MicroRNA function in myeloid biology. Blood. 2011;118(11):2960–2969. doi: 10.1182/blood-2011-03-291971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnnidis JB, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451(7182):1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 25.Fazi F, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005;123(5):819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 26.Li T, Morgan MJ, Choksi S, Zhang Y, Kim YS, Liu ZG. MicroRNAs modulate the noncanonical transcription factor NF-kappaB pathway by regulating expression of the kinase IKKalpha during macrophage differentiation. Nat Immunol. 2010;11(9):799–805. doi: 10.1038/ni.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, Cerami A. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci U S A. 1989;86(2):612–616. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for micro-RNA-target interactions. Nucleic Acids Res. 2009;37(Database issue):D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felli N, et al. MicroRNA 223-dependent expression of LMO2 regulates normal erythropoiesis. Haematologica. 2009;94(4):479–486. doi: 10.3324/haematol.2008.002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol. 2011;45(2):189–201. doi: 10.1165/rcmb.2011-0011RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ladel CH, Blum C, Dreher A, Reifenberg K, Kopf M, Kaufmann SH. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infec Immun. 1997;65(11):4843–4849. doi: 10.1128/iai.65.11.4843-4849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10(10):704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veenstra H, et al. Changes in leucocyte and lymphocyte subsets during tuberculosis treatment; prominence of CD3dimCD56+ natural killer T cells in fast treatment responders. Clin Exp Immunol. 2006;145(2):252–260. doi: 10.1111/j.1365-2249.2006.03144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berry MP, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466(7309):973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brahmbhatt S, et al. Immune markers measured before treatment predict outcome of intensive phase tuberculosis therapy. Clin Exp Immunol. 2006;146(2):243–52. doi: 10.1111/j.1365-2249.2006.03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukao T, et al. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell. 2007;129(3):617–631. doi: 10.1016/j.cell.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 37.Hunter RL, Jagannath C, Actor JK. Pathology of postprimary tuberculosis in humans and mice: contradiction of long-held beliefs. Tuberculosis. 2007;87(4):267–278. doi: 10.1016/j.tube.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, et al. MicroRNA expression and virulence in pandemic influenza virus-infected mice. J Virol. 2010;84(6):3023–3032. doi: 10.1128/JVI.02203-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun W, Shen W, Yang S, Hu F, Li H, Zhu TH. miR-223 and miR-142 attenuate hematopoietic cell proliferation, and miR-223 positively regulates miR-142 through LMO2 isoforms and CEBP-β. Cell Res. 2010;20(10):1158–1169. doi: 10.1038/cr.2010.134. [DOI] [PubMed] [Google Scholar]
- 40.Kagina BM, et al. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guérin vaccination of newborns. Am J Respir Crit Care Med. 2010;182(8):1073–1079. doi: 10.1164/rccm.201003-0334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mittrücker HW, et al. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc Natl Acad Sci U S A. 2007;104(30):12434–12439. doi: 10.1073/pnas.0703510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208(11):2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farber JM. A macrophage mRNA selectively induced by gamma-interferon encodes a member of the platelet factor 4 family of cytokines. Proc Natl Acad Sci U S A. 1990;87(14):5238–5242. doi: 10.1073/pnas.87.14.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luster AD, Jhanwar SC, Chaganti RS, Kersey JH, Ravetch JV. Interferon-inducible gene maps to a chromosomal band associated with a (4;11) translocation in acute leukemia cells. Proc Natl Acad Sci U S A. 1987;84(9):2868–2871. doi: 10.1073/pnas.84.9.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng SC, et al. Computational modeling with forward and reverse engineering links signaling network and genomic regulatory responses: NF-kappaB signaling-induced gene expression responses in inflammation. BMC Bioinformatics. 2010;11:308. doi: 10.1186/1471-2105-11-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hachicha M, Rathanaswami P, Naccache PH, McColl SR. Regulation of chemokine gene expression in human peripheral blood neutrophils phagocytosing microbial pathogens. J Immunol. 1998;160(1):449–454. [PubMed] [Google Scholar]
- 47.Cicco NA, et al. Inducible production of interleukin-6 by human polymorphonuclear neutrophils: role of granulocyte-macrophage colony-stimulating factor and tumor necrosis factor-alpha. Blood. 1990;75(10):2049–2052. [PubMed] [Google Scholar]
- 48.Charmoy M, et al. Neutrophil-derived CCL3 is essential for the rapid recruitment of dendritic cells to the site of Leishmania major inoculation in resistant mice. PLos Pathog. 2010;6(2):e1000755. doi: 10.1371/journal.ppat.1000755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J. Immunol. 2011;186(12):7110–7119. doi: 10.4049/jimmunol.1100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blomgran R, Desvignes L, Briken V, Ernst JD. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe. 2012;11(1):81–90. doi: 10.1016/j.chom.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Armstrong DA, Major JA, Chudyk A, Hamilton TA. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J Leukoc Biol. 2004;75(4):641–648. doi: 10.1189/jlb.0803370. [DOI] [PubMed] [Google Scholar]
- 52.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354(6):610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 53.Strieter RM, Belperio JA, Phillips RJ, Keane MP. CXC chemokines in angiogenesis of cancer. Semin Cancer Biol. 2004;14(3):195–200. doi: 10.1016/j.semcancer.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 54.Liu F, Poursine-Laurent J, Wu HY, Link DC. Interleukin-6 and the granulocyte colony-stimulating factor receptor are major independent regulators of granulopoiesis in vivo but are not required for lineage commitment or terminal differentiation. Blood. 1997;90(7):2583–2590. [PubMed] [Google Scholar]
- 55.Saunders BM, Frank AA, Orme IM, Cooper AM. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect Immun. 2000;68(6):3322–3326. doi: 10.1128/IAI.68.6.3322-3326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Q, et al. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β production in macrophages by targeting STAT3. PLoS One. 2012;7(8):e42971. doi: 10.1371/journal.pone.0042971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haneklaus M, et al. Cutting Edge: miR-223 and EBV miR-BART15 Regulate the NLRP3 Inflammasome and IL-1β Production. J Immunol. 2012;189(8):3795–8799. doi: 10.4049/jimmunol.1200312. [DOI] [PubMed] [Google Scholar]
- 58.Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. NLRP3 Inflammasome Activity Is Negatively Controlled by miR-223. J Immunol. 2012;189(8):4175–4181. doi: 10.4049/jimmunol.1201516. [DOI] [PubMed] [Google Scholar]
- 59.O’Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 60.Peltier HJ, Latham G.J. Normalization of microRNA expression levels in quantitative RT-PCR assays: Identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA. 2008;14(5):844–852. doi: 10.1261/rna.939908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Livack KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2 (Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 62.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.