

Abstract
Site-specific activation of the Rho-type GTPase Cdc42p by its guanine-nucleotide exchange factor (GEF) Cdc24p is critical for the establishment of cell polarity. Here we show that binding of Cdc24p to the small GTPase Rsr1p/Bud1p is required for its recruitment to the incipient bud site. Rsr1p/Bud1p binds to the CH-domain of Cdc24p, which is essential for its function in vivo. We have identified a cdc24-mutant allele, which is specifically defective for bud-site selection. Our results suggest that Cdc24p is auto-inhibited by an intramolecular interaction with its carboxy-terminal PB1-domain. Rsr1p/Bud1p appears to activate the GEF activity of Cdc24p in vivo, possibly by triggering a conformational change that dissociates the PB1-domain from its intramolecular binding site. Genetic experiments suggest that Bem1p functions as a positive regulator of Cdc24p by binding to the PB1-domain of Cdc24p, thereby preventing its re-binding to the intramolecular inhibitory site. Taken together, our results support a two-step molecular mechanism for the site-specific activation of Cdc24p, which involves Rsr1p/Bud1p and the adaptor protein Bem1p.
Keywords: auto-inhibition, Cdc24p, Cdc42p, cell polarity, GEF regulation, Rsr1p/Bud1p
Introduction
The establishment of asymmetry or cell polarity is critical for many different biological processes in both single- and multicellular organisms, including cell migration, differentiation, proliferation and morphogenesis (Drubin, 2000). The understanding of how cellular components can generate asymmetry represents one of the key steps toward understanding the molecular basis of cellular organization. The polarized assembly of the actin and microtubule cytoskeletons is regulated by site-specific activation of Rho-type GTPases, and many critical downstream targets have been identified (Aspenstrom, 1999; Johnson, 1999). However, much less is known about the spatial and temporal regulation of the Rho-type GTPases themselves.
The yeast Saccharomyces cerevisiae has proven to be an excellent and genetically tractable model organism to study the biochemical pathways leading to the establishment of cell polarity (Pruyne and Bretscher, 2000). During vegetative growth, polarization of the actin cytoskeleton toward a single position on the cell cortex (incipient bud site) leads to bud formation, while during mating a pointed projection (shmoo) develops to allow contact and fusion with a cell of the opposite mating type. Central to the initiation of actin polarization during mating and budding is the local activation of the GTPase Cdc42p (Johnson, 1999). Regulation of Cdc42p involves its sole GEF Cdc24p (Bi et al, 2000) and several putative GAPs (Bem2p, Bem3p, Rga1p and Rga2p). In addition, Cdc24p is positively regulated by binding to the adaptor protein Bem1p, which is required to keep Cdc24p at sites of polarized growth (Butty et al, 2002). Finally, Cdc24p is sequestered in the nucleus during the G1-phase of the haploid cell cycle by binding to the adaptor Far1p (Toenjes et al, 1999; Nern and Arkowitz, 2000b; Shimada et al, 2000). Like most Rho-GEFs (Hoffman and Cerione, 2002), Cdc24p contains a domain with strong similarity to the Dbl-family of exchange factors (residues 284–452, Dbl-homology domain or DH) and a nearby pleckstrin-homology (PH) domain (residues 477–667; Toenjes et al, 1999; Zheng, 2001). Cdc24p also contains a calponin-homology (CH) domain in its amino-terminus (residues 154–226), which in some proteins has been implicated in binding to actin (Stradal et al, 1998). Finally, Cdc24p has a PB1-domain at its carboxy-terminus (residues 781–854; Ito et al, 2001; Butty et al, 2002). While the PH-domain is thought to serve as a membrane-targeting signal (Irvine, 1998), the functions of the CH- and PB1-motifs for the regulation of Cdc24p are not clear.
We are interested in the spatial and temporal regulation of Cdc24p. During mating, cell polarity is determined by a gradient of mating pheromones secreted by the mating partner, which is interpreted with the help of the adaptor protein Far1p (Arkowitz, 1999; Gulli and Peter, 2001). Far1p interacts with Cdc24p and Gβγ and is thought to recruit Cdc24p to the site of receptor activation. Likewise, during vegetative growth, the position on the cell cortex where Cdc24p is activated is not random, but is regulated by several gene products in a cell-type-dependent manner (Chant, 1999). The transmembrane protein Bud10p/Axl2p is required to guide the budding machinery to the correct position in haploid cells, while Bud8p and Bud9p mark the site of polarization in diploid cells. The GTPase Rsr1p/Bud1p is regulated by its GEF Bud5p and its GAP Bud2p, both of which localize to the incipient bud site (Park et al, 1999). Bud5p co-localizes with the transmembrane protein Axl2p/Bud10p, a component of the budding landmark, and directly interacts with its cytoplasmic tail (Kang et al, 2001), suggesting that Bud5p may be recruited to the incipient bud site by binding to Axl2p/Bud10p. Several lines of evidence suggest a crucial role for the small GTPase Rsr1p/Bud1p in the local activation of Cdc24p at bud emergence. Rsr1p/Bud1p was originally isolated as a multicopy suppressor of cdc24-4-mutant cells (Bender and Pringle, 1989), and subsequently shown to bind directly to Cdc24p in a GTP-dependent manner (Park et al, 1997). Moreover, the recruitment of Cdc24p to the proper bud site depends on Rsr1p/Bud1p (Park et al, 2002). These results suggest that Rsr1p/Bud1p is an important regulator of Cdc24p, which may recruit Cdc24p to the incipient bud site.
In this study, we investigated the molecular mechanisms of Cdc24p activation. Our results suggest that Rsr1p/Bud1p is not only responsible for the localization of Cdc24p but may also be involved in its activation in vivo. In addition, we propose that Cdc24p is auto-inhibited by binding of its carboxy-terminal PB1-motif to an intramolecular site. As Bem1p also interacts with this PB1-motif, we favor a model for Cdc24p activation where Bem1p may specifically stabilize the active conformation of Cdc24p by preventing intramolecular inhibition by its PB1-domain.
Results
The CH-domain of Cdc24p is required for its site-specific localization of Cdc24p
Besides the DH- and PH-domains typically found in Rho-GEFs (Hoffman and Cerione, 2002), Cdc24p contains a CH-domain in its amino-terminal regulatory domain, and a PB1-domain at its carboxy-terminus (Figure 1A). Both Cdc24p-ΔCH and Cdc24p-ΔDH were unable to complement the growth defect of cdc24Δ cells (Figure 1B and C), implying that both domains are essential for their function in vivo. For control, we also analyzed a Cdc24p mutant harboring a 10 amino-acid deletion within its catalytic DH-domain (amino acids 406–415). As expected, Cdc24p-ΔCH-GFP did not accumulate in the cell nucleus of G1 cells (Toenjes et al, 1999; Nern and Arkowitz, 2000b; Shimada et al, 2000), but surprisingly was also unable to localize to the incipient bud site or tips of mating projections (Figure 1, panel D). In contrast, Cdc24p-ΔDH-GFP was found at cortical sites and the cell nucleus in a cell cycle-dependent manner (Figure 1, panel D), implying that GEF activity is not necessary for proper localization of Cdc24p. Taken together, these results suggest that the CH-domain is required for the recruitment of Cdc24p to the cell cortex.
Figure 1.
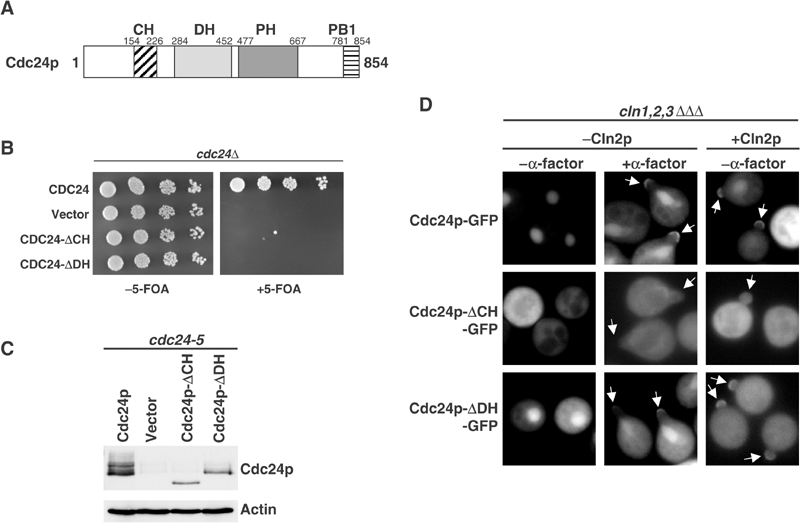
An intact CH- but not DH-domain is required for membrane localization of Cdc24p. (A) Schematic representation of the domain structure of Cdc24p. The numbers indicate amino acids starting from the start methionine. CH: calponin-homology domain; DH: Dbl-homology domain; PH: pleckstrin-homology domain, PB1: Phox-Bem1-homology domain. (B) cdc24Δ cells transformed with a URA3-marked plasmid carrying wild-type Cdc24p (YMG315) and, as indicated, either an empty plasmid (vector), wild-type Cdc24p, Cdc24p-ΔCH or Cdc24p-ΔDH. The cells were grown at 25°C until the mid-log phase, and five-fold serial dilutions were spotted on plates with (right panel) or without 5-FOA (left panel), a drug that selects against the URA-plasmid. (C) The expression of wild-type and the indicated Cdc24p mutants was analyzed in cdc24-5 cells, which express very low levels of endogenous Cdc24p (Butty et al, 2002). Actin was used as a loading control. (D) The localization of wild-type Cdc24p-GFP, Cdc24p-ΔCH-GFP or Cdc24p-ΔDH-GFP was analyzed in YMG258 (cln1Δ cln2Δ cln3Δ MET-CLN2) cells arrested in G1 by depletion of the G1-cyclins (−Cln2p) or 20 min after release from the arrest by induction of Cln2p (+Cln2p). Where indicated, the cells were treated with α-factor for 2 h. Note that, in contrast to Cdc24p-ΔDH-GFP, Cdc24p-ΔCH-GFP fails to localize to sites of polarized growth (arrows) during budding or mating.
Binding of Cdc24p to Rsr1p/Bud1p is required for its recruitment to the axial bud site
Cdc24p was previously shown to function as an effector of Rsr1p/Bud1p (Zheng et al, 1995; Park et al, 1997). To determine the functional significance of this interaction, we searched for cdc24 mutants, which are unable to interact with Rsr1p/Bud1p. Interestingly, cdc24-4 cells exhibited a random budding pattern when grown at the permissive temperature (Figure 2A), and their growth defect at restrictive temperature (36°C) was suppressed by increased levels of Rsr1p/Bud1p (Figure 3A). In contrast, cdc24-4 cells did not exhibit a mating defect even when assayed toward orientation-defective far1-c cells (Figure 2B; Chenevert et al, 1994), implying that they are able to efficiently interpret a natural pheromone gradient (Valtz et al, 1995). Immunoblotting with Cdc24p antibodies revealed that the Cdc24p-4-mutant protein was not grossly truncated and present in normal amounts (Figure 3B). However, the phosphorylation pattern of Cdc24p (upper blot) and the specific accumulation of the Cdc42p-effector Gic2p (middle blot) indicated that the Cdc24p-4 protein is inactive when the cells are shifted to the restrictive temperature. We recovered the cdc24-4-mutant allele by PCR, and identified a single point mutation at position 503, which changed glycine 168 to a glutamic acid residue (G168D; Figure 2C). Site-directed mutagenesis confirmed that this mutation is solely responsible for the observed defects (Figure 2D).
Figure 2.
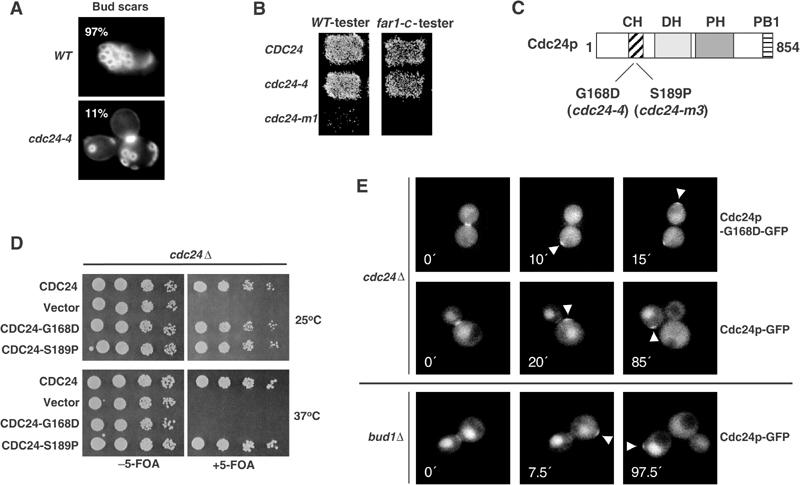
Cells expressing Cdc24p-G168D are specifically defective for bud-site selection. (A) The budding pattern of wild-type (YEF241) and cdc24-4 (YEF313) cells grown at 25°C until the mid-log phase was examined by staining the bud scars with calcofluor white. The defect was quantified and presented as the percentage (%) of cells with an axial budding pattern. (B) Wild-type (YEF241), cdc24-4 (YEF313) and cdc24-m1 (YPW81) cells were tested at 25°C for their ability to mate with orientation-defective far1-c (YMP325) mating testers. (C, D) The positions of the identified mutation in cdc24-4 (G168D) and cdc24-m3 (S189P, Nern and Arkowitz, 1998) are marked on the schematic domain structure of Cdc24p (C). Site-directed mutagenesis was used to confirm that this single amino-acid change is sufficient to confer the temperature-sensitive phenotype (D). The assay was performed as described in the legend of Figure 1B. (E) The localization of Cdc24p-GFP or Cdc24p-G168D-GFP expressed from the CYC1-promoter was analyzed by time-lapse microscopy in cdc24Δ (YMP315) or bud1Δ cells (YACB191). The arrowheads mark the position of Cdc24p-GFP or Cdc24p-G168D-GFP. The numbers indicate the time (min) after the first image was taken (time 0). Note that Cdc24p-G168D-GFP is recruited to random positions, analogous to wild-type Cdc24p-GFP in bud1Δ cells.
Figure 3.
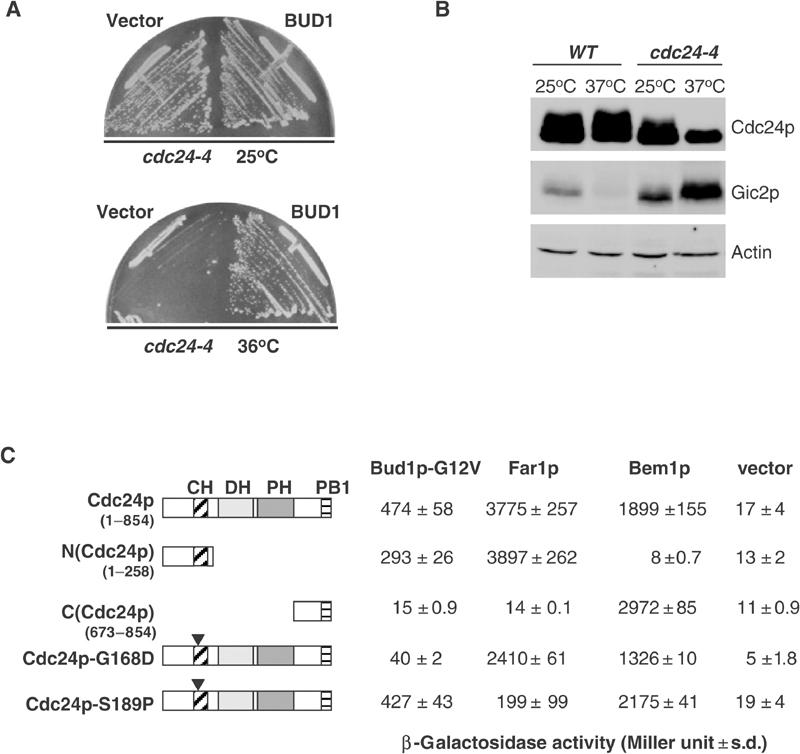
Interaction of Rsr1p/Bud1p with the CH-domain of Cdc24p is required for correct bud-site selection. (A) Wild-type (YEF241) and cdc24-4 (YEF313) cells were transformed with an empty control plasmid (vector) or a multi-copy plasmid expressing Rsr1p/Bud1p, and tested for their ability to grow at 36°C (A). (B) Extracts prepared from wild-type (YEF241) or cdc24-4 (YEF313) cells grown to the mid-log phase at 25°C and, where indicated, shifted to 37°C for 2 h were immunoblotted with antibodies against Cdc24p (upper panel), Gic2p (middle panel) or for control actin (bottom panel). (C) The interaction of Bud1p-G12V, Far1p and Bem1p with full-length or the indicated mutants of Cdc24p was analyzed by two-hybrid analysis using EGY48 cells grown at 25°C. The numbers indicate Miller units with standard deviations. An empty vector was included as control. A schematic representation of the various constructs is shown on the left.
We next used two-hybrid assays and time-lapse microscopy to test whether Cdc24p-G168D is defective for its interaction with activated Rsr1p/Bud1p (Bud1p-G12V; Figures 2E and 3C). Deletion analysis of Cdc24p revealed that Rsr1p/Bud1p and Far1p interact with the amino-terminal domain, while Bem1p binds to its C-terminal PB1-domain (Ito et al, 2001; Butty et al, 2002). Importantly, the interaction between Cdc24p-G168D and Bud1p-G12V was strongly reduced, while its interaction with Far1p and Bem1p remained largely intact. Conversely, Cdc24p-S189P was defective for its binding to Far1p (Figure 3C; Nern and Arkowitz, 1998), while it was still interacting with Bud1p-G12V. Finally, time-lapse experiments confirmed that Cdc24p-G168D-GFP expressed in cdc24Δcells was recruited to random positions on the cell cortex with respect to the position of the previous bud (Figure 2E). In contrast, wild-type Cdc24p-GFP accumulated at the expected axial position in wild-type cells, and at random positions when expressed in bud1Δ, bud2Δ and bud5Δ cells (Figure 2E and data not shown; Park et al, 2002). Taken together, these results demonstrate that binding of Rsr1p/Bud1p to Cdc24p is specifically required for its correct recruitment to the axial position on the cell cortex.
Rsr1p/Bud1p is sufficient to recruit Cdc24p to the cell cortex and may also be involved in its activation in vivo
We observed that strong overexpression of an activated mutant form of Rsr1p/Bud1p (Bud1p-G12V) was toxic for cells (Figure 4A), and the cells arrest predominantly with a large unbudded morphology and an unpolarized cytoskeleton (data not shown). This toxicity required an intact effector domain of Rsr1p/Bud1p, as overexpression of Bud1p-G12V/T35A had no effect. We speculated that Rsr1p/Bud1p may recruit Cdc24p to the membrane leading to uniform activation of Cdc42p, which in turn may interfere with polarized growth toward a single bud site. To test whether Bud1p-G12V was able to recruit Cdc24p to the plasma membrane (Figure 4B), we arrested YMG258 cells expressing Cdc24p-GFP (upper panels) or for control Cdc24p-G168D-GFP (lower panels) in G1 by depletion of the G1-cyclin Cln2p (Gulli et al, 2000). The expression of Bud1p-GTP (Bud1p-G12V) or for control no protein (vector) was then induced by the addition of 2% galactose, and the localization of Cdc24p-GFP was examined by GFP microscopy after 2 h. As expected, Cdc24p and Cdc24p-G168D-GFP were predominantly nuclear with some cytoplasmic staining under these conditions (Figure 4B). However, Cdc24p-GFP, but not Cdc24p-G168D-GFP, was uniformly recruited to the plasma membrane after expression of Bud1p-G12V from the inducible GAL1-10-promoter (Mumberg et al, 1995). These results suggest that binding of Rsr1p/Bud1p to Cdc24p is required for the recruitment of Cdc24p to the cell cortex.
Figure 4.
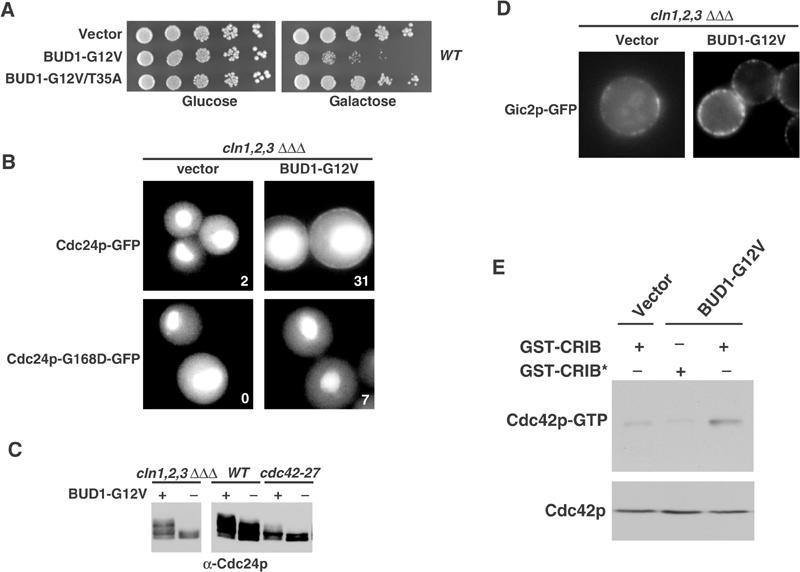
Overexpression of an activated mutant form of Rsr1p/Bud1p is toxic and may recruit and uniformly activate Cdc24p at the plasma membrane in vivo. (A) Wild-type (K699) cells were transformed with an empty control plasmid (vector) or plasmids expressing Bud1p-G12V or Bud1p-G12V/T35A from the inducible GAL1-10-promoter. The cells were grown at 30°C until mid-log phase, and five-fold serial dilutions were spotted on plates containing 2% glucose (left panel) or 2% galactose (right panel). Plates were photographed after 3 days at 30°C. (B) YMG258 (cln1Δ cln2Δ cln3Δ MET-CLN2) cells expressing from the GAL1-10-promoter either no protein (vector, left panels) or activated Bud1p-G12V (BUD1-G12V, right panels) were grown in media containing 2% raffinose (GAL-promoter off) and arrested in G1 by depletion of the G1-cyclins. The localization of Cdc24p-GFP (upper panels) or Cdc24p-G168D-GFP (lower panels) was analyzed by GFP microscopy 2 h after the addition of 2% galactose. The percentage (%) of cells with Cdc24p-GFP or Cdc24p-G168D-GFP, which were uniformly located at the plasma membrane, is indicated. (C) The phosphorylation state of Cdc24p was analyzed by immunoblotting in YMG258 (cln1Δ cln2Δ cln3Δ MET-CLN2) cells arrested in G1 by depletion of the G1-cyclins (left panel), or wild-type (DLY1) and cdc42-27 (MOSY0124) cells 2 h after expression of either no protein (‘−') or activated Bud1p-G12V (‘+') from the GAL1-10-promoter. (D) YMG258 (cln1Δ cln2Δ cln3Δ MET-CLN2) cells expressing from the GAL1-10-promoter either no protein (vector) or activated Bud1p-G12V (BUD1-G12V) were grown in media containing 2% raffinose (GAL-promoter off) and arrested in G1 by depletion of the G1-cyclins. The recruitment of the Cdc42p-target Gic2p-GFP to the plasma membrane was analyzed 2 h after addition of 2% galactose. (E) Extracts prepared from wild-type (K699) cells transformed with an empty control plasmid (vector) or a plasmid expressing Bud1p-G12V from the inducible GAL1-10-promoter were incubated with glutathione–sepharose beads coated, as indicated, with GST-CRIB, or for control a nonfunctional GST-CRIB-mutant protein (GST-CRIB*) purified from E. coli. Bound Cdc42p (upper panel) and a fraction of the total Cdc42p present in the extract (lower panel) were detected by immunoblotting.
We used three assays to determine whether activated Rsr1p/Bud1p is involved in the activation of Cdc24p at the cell cortex. First, Cdc24p accumulated in its hyperphosphorylated form in YMG258 cells expressing Bud1p-G12V (Figure 4C), and this hyperphosphorylation was largely dependent on functional Cdc42p. Second, the Cdc42p-target Gic2p-GFP was uniformly localized at the plasma membrane when Bud1p-GTP was overexpressed in G1-arrested YMG258 cells (Figure 4D; Jaquenoud and Peter, 2000). Finally, pull-down assays with GST-CRIB beads revealed that Cdc42p-GTP levels were increased in YMG258 cells expressing Bud1p-G12V (Figure 4E). Taken together, these data imply that membrane recruitment of Cdc24p by Rsr1p/Bud1p is sufficient for uniform activation of Cdc42p at the plasma membrane.
Two possibilities could account for the observed increase in Cdc42p-GTP in vivo. First, membrane localization of Cdc24p may be sufficient for its activation. Alternatively, binding of Rsr1p/Bud1p to Cdc24p may activate Cdc24p. To distinguish between these possibilities, we fused the amino-terminus of Cdc24p to the myristoylation signal of c-Src (Figure 5A; myr-Cdc24p), and overexpressed the fusion protein from the weak, but inducible GALL-promoter (Mumberg et al, 1995). For control, we used a mutated myristoylation signal, where the myristoylated glycine residue was replaced with a non-myristoylatable alanine residue (myrG2A-Cdc24p). As expected, myr-Cdc24p-GFP, but not myrG2A-Cdc24p-GFP, was uniformly recruited to the plasma membrane and some internal membranes (Figure 5B). Interestingly, cells overexpressing myr-Cdc24p were viable (Figure 5C). In contrast, co-expression of myr-Cdc24p with low levels of Bud1p-G12V was toxic (Figure 5, panel C), and these cells accumulated with a large unbudded morphology and an unpolarized actin cytoskeleton (Figure 5, panel D). This lethality required an intact DH-domain of Cdc24p (Figure 5, panel E), and was specific for activated Rsr1p/Bud1p, as neither wild-type Rsr1p/Bud1p nor the GDP-locked Bud1p-K16N or the effector-mutant Bud1p-G12V/T35A was able to interfere with polarization in the presence of myr-Cdc24p (Figure 5, panels C and D). Finally, co-expression of Bud1p-G12V was able to induce lethality with myr-Cdc24p and myr-Cdc24p-S189P, but not with myr-Cdc24p-G168D, which is unable to interact with Rsr1p/Bud1p (see Figure 3). Together, these results support the notion that cells co-expressing myr-Cdc24p and Bud1p-G12V are unable to polarize, because Cdc42p may be activated uniformly over the cell cortex and thus interfere with polarized growth toward a single bud site. Indeed, GST-CRIB pull-down assays confirmed that these cells express increased levels of Cdc42p-GTP (Figure 5F). Based on these results, we conclude that membrane localization is not sufficient to activate Cdc24p in vivo, and that the interaction between Rsr1p/Bud1p and Cdc24p may be required for this process.
Figure 5.
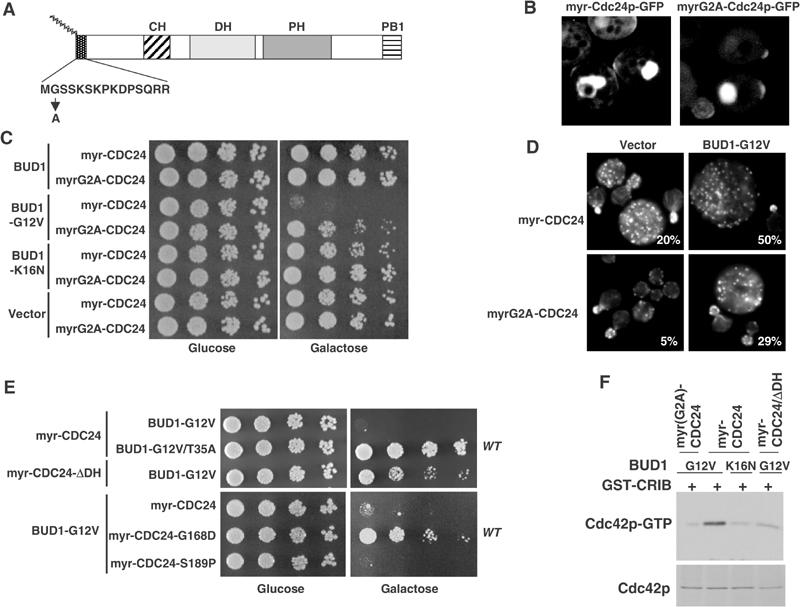
Binding of Rsr1p/Bud1p to Cdc24p may be required to activate Cdc24p in vivo. (A) Schematic representation of Cdc24p fused at its amino-terminus to the wild-type (myr-Cdc24p) or mutant (myrG2A-Cdc24p) myristoylation signal of c-Src. (B) Fusion of a wild-type (myr) but not a mutant (myrG2A) myristoylation signal is able to uniformly target Cdc24p-GFP to the plasma membrane. The proteins were expressed in wild-type (K699) cells from the weak, but inducible GALL-promoter, and analyzed after 3 h of induction by confocal microscopy. (C) Wild-type (K699) cells expressing either myr-Cdc24p or myrG2A-Cdc24p from the GALL-promoter were transformed with an empty control plasmid (vector) or multicopy plasmids carrying the indicated wild-type or mutant BUD1 alleles expressed from their endogenous promoter. Five-fold serial dilutions of cultures in mid-log phase were spotted on media containing glucose or galactose. (D) The morphology and actin polarization of the cells shown in (C) were examined after staining with rhodamine-phalloidin. The percentage (%) of large unbudded, unpolarized cells was quantified. (E) Wild-type (K699) cells co-expressing wild-type or the indicated myr-Cdc24p mutants from the inducible GALL-promoter, together with multicopy plasmids carrying wild-type or the indicated mutant forms of BUD1, were analyzed as described in (C). (F) Extracts prepared from wild-type (K699) cells co-expressing wild-type or the indicated myr-Cdc24p mutants from the inducible GALL-promoter, together with multicopy plasmids carrying BUD1-G12V or BUD1-K16N, were analyzed for the presence of Cdc42p-GTP by GST-CRIB pull-down assays. Bound Cdc42p (upper panel) and a fraction of the total Cdc42p present in the extract (lower panel) were detected by immunoblotting.
Isolation of constitutively activated alleles of Cdc24p
As membrane-targeted Cdc24p required binding of Rsr1p/Bud1p for its activation, we reasoned that Cdc24p may be present in an auto-inhibited, inactive conformation. If this model were correct, we should be able to isolate activated alleles of myr-Cdc24p. We thus randomly mutagenized myr-Cdc24p expressed from the inducible GALL-promoter by error-prone PCR, and screened for mutants that failed to grow in the presence of galactose (Figure 6A). Interestingly, these cells arrested with an unbudded morphology and an unpolarized actin cytoskeleton (Figure 6D and data not shown). This defect was not caused by increased levels of myr-Cdc24p, as the mutant alleles were expressed at levels comparable to myr-Cdc24p (Figure 6C). Sequencing revealed that all mutations mapped to the carboxy-terminal part of Cdc24p after the DH-motif (Figure 6B), and we identified several point mutations within the PH- and the carboxy-terminal domain. Surprisingly, one mutation introduced a stop codon already at position 457, which results in a truncated Cdc24 protein lacking its PH-domain. We wanted to exclude the possibility that some of the recovered alleles may cause a dominant-negative phenotype by interfering with the function of wild-type Cdc24p. First, expression of myr-Cdc24-G644C, but not myr-Cdc24-G644C-ΔDH, was toxic, implying that either Cdc42p binding or its ability to produce Cdc42p-GTP was required (Figure 6E). Second, the growth defect of cells overexpressing myr-Cdc24-G644C was suppressed by simultaneous overexpression of the Cdc42p GAP Bem2p (Figure 6F). Third, GST-CRIB pull-down assays revealed that cells overexpressing myr-Cdc24-G644C, but not myr-Cdc24-G644C-ΔDH, possess increased levels of Cdc42p-GTP (Figure 6G). Finally, we tested the functionality of the Cdc24p mutants by expressing them without the myristoylation signal from the endogenous promoter in cdc24Δ cells. Indeed, all tested Cdc24p mutants were able to support growth in this context (Figure 6H and data not shown), demonstrating that the mutant alleles are functional. Based on these results, we conclude that the identified mutations are constitutively activating Cdc24p in vivo.
Figure 6.
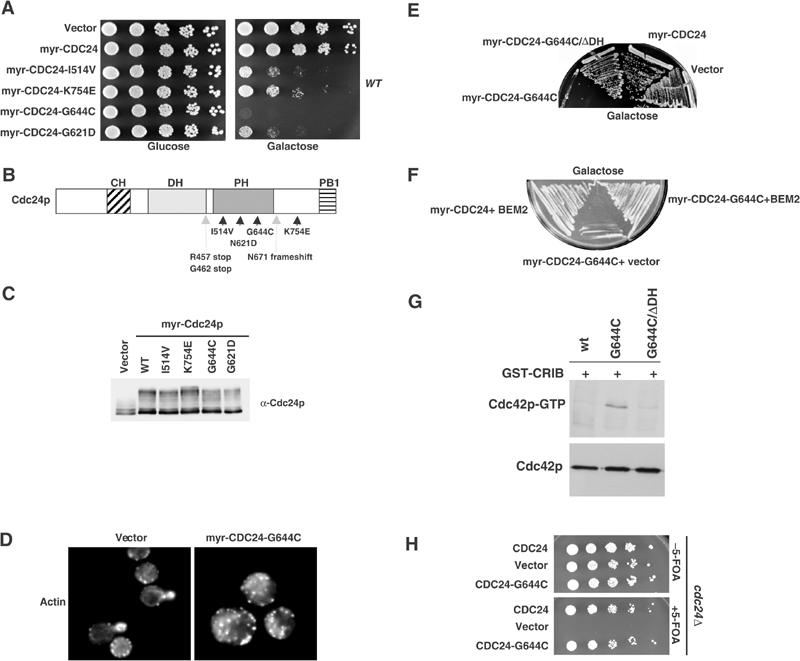
Identification and characterization of constitutively active alleles of Cdc24p. (A–C) Specific mutations within myr-Cdc24p that confer toxicity when expressed in wild-type cells (K699) from the GALL-promoter were screened as described in Material and methods. Five-fold serial dilutions were spotted on plates containing glucose or galactose, and grown for 3 days at 30°C (A). The responsible amino-acid changes were determined and are shown in (B). Arrowheads show single amino-acid changes, while stifled arrows point to mutations that result in a truncated protein by introducing a stop codon. The expression level of myr-Cdc24p and the indicated myr-Cdc24p mutants in wild-type cells (K699) was determined by immunoblotting with Cdc24p-antibodies (C). (D) The morphology and actin polarization of wild-type (K699) cells expressing for 3 h either no protein (vector) or myr-Cdc24p-G644C from the GALL-promoter were examined after staining with rhodamine-phalloidin. (E, F) Wild-type (K699) cells expressing from the GALL-promoter, as indicated, either no protein (vector), myr-Cdc24p, myr-Cdc24p-G644C or myr-Cdc24p-G644C/ΔDH were grown for 3 days at 30°C on plates containing galactose. The cells shown in (F) were transformed with a plasmid expressing the GAP BEM2 from the Gal1-10 promotor. (G) The levels of Cdc42p-GTP were compared by GST-CRIB pull-down assays in extracts prepared from wild-type (K699) cells expressing myr-Cdc24p, myr-Cdc24-G644C or myr-Cdc24p-G644C/ΔDH from the inducible GALL-promoter. Bound Cdc42p (upper panel) and a fraction of the total Cdc42p present in the extract (lower panel) were detected by immunoblotting. (H) cdc24Δ cells transformed with a URA3-marked plasmid carrying wild-type Cdc24p (YMG315) and, as indicated, either an empty plasmid (vector), wild-type Cdc24p or Cdc24p-G644C were grown at 25°C until mid-log phase, and serial dilutions were spotted on plates with (lower panel) or without 5-FOA (upper panel), as described in the legend to Figure 1B.
The C-terminal PB1-domain of Cdc24p is required to prevent activation of myr-Cdc24p
We next tested the importance of different Cdc24p-domains (schematically shown in Figure 7A) to prevent unregulated activation when fused to a myristoylation signal. Although all amino-terminal truncation mutants are produced (Figure 7, panel B), they did not induce lethality when overexpressed in wild-type cells (Figure 7, panel C). Likewise, expression of myr-Cdc24p-ΔCH had no effect, suggesting that an intact CH-domain is required even when Cdc24p is artificially targeted to the plasma membrane. In contrast, deletion of the carboxy-terminal PB1-domain of myr-Cdc24p was toxic (Figure 7, panel C), and the cells arrested with a phenotype characteristic for uniformly activated Cdc42p (data not shown). Taken together, these results suggest that an intact PB1Cdc24-domain is required to keep Cdc24p inactive, possibly by an auto-inhibitory mechanism.
Figure 7.
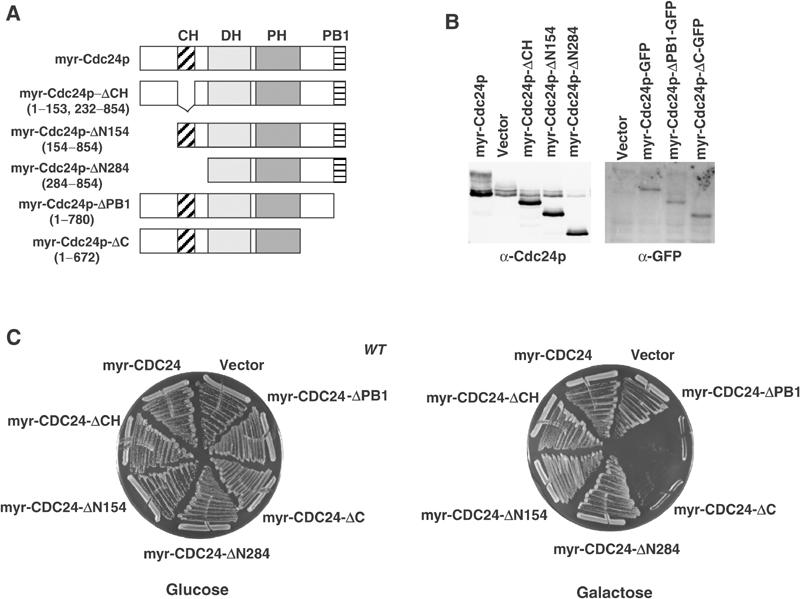
Expression of myr-Cdc24p lacking its carboxy-terminal PB1Cdc24-domain is toxic. (A, B) Schematic representation of the Cdc24p-deletion constructs that were fused to the myristoylation signal (A). The expression levels of the indicated constructs were examined in wild-type cells by immunoblotting with Cdc24p- (B, left blot) or GFP-antibodies (B, right blot). (C) Wild-type (K699) cells overexpressing either no protein (vector), myr-Cdc24p or the indicated myr-Cdc24-mutant proteins from the GALL-promoter were photographed after 3 days at 30°C on plates containing glucose (left plate) or galactose (right plate).
If the PB1Cdc24-domain is required to inhibit Cdc24p activity in vivo, we predict that overexpression of this domain may interfere in trans with the activation of endogenous Cdc24p. Indeed, expression of the PB1Cdc24-domain was toxic in cdc24-4 cells (Figure 8A), although we were unable to detect a clear effect when the PB1Cdc24-domain was expressed in wild-type cells. Consistent with these results, overexpression of the PB1Cdc24-domain suppressed the lethality caused by hyperactive myr-Cdc24p-ΔPB1 (Figure 8B), demonstrating that the PB1Cdc24-domain functions as an inhibitor of Cdc24p. This inhibitory effect required a domain in the carboxy-terminal region of Cdc24p, as expression of the PB1Cdc24-domain was unable to suppress the lethality induced by myr-Cdc24p-ΔC. We used two-hybrid analysis to examine whether the PB1Cdc24-domain indeed interacts with a motif in the carboxy-terminus of Cdc24p. As shown in Figure 8C, the PB1Cdc24-domain strongly interacted with an internal domain of Cdc24p. No binding between the PB1Cdc24-domain and Cdc24p-ΔC was detected, suggesting that the PB1Cdc24-domain binds to a motif between amino acids 673 and 780 in the carboxy-terminus of Cdc24p. This fragment contains a motif with significant sequence homology to the PB1-domain of Bem1p (A Petit and M Peter, unpublished results), indicating that the intramolecular binding site may resemble a PB1-fold. Strikingly, while the PB1Cdc24-domain strongly interacted with the internal motif when the carboxy-terminal PB1-domain of Cdc24p was deleted, PB1Cdc24 was unable to interact with full-length Cdc24p (Figure 8C). We interpret this result to indicate that the PB1Cdc24-domain in full-length Cdc24p is already engaged in an intramolecular interaction, such that the PB1Cdc24-domain expressed in trans may be unable to bind in this context.
Figure 8.
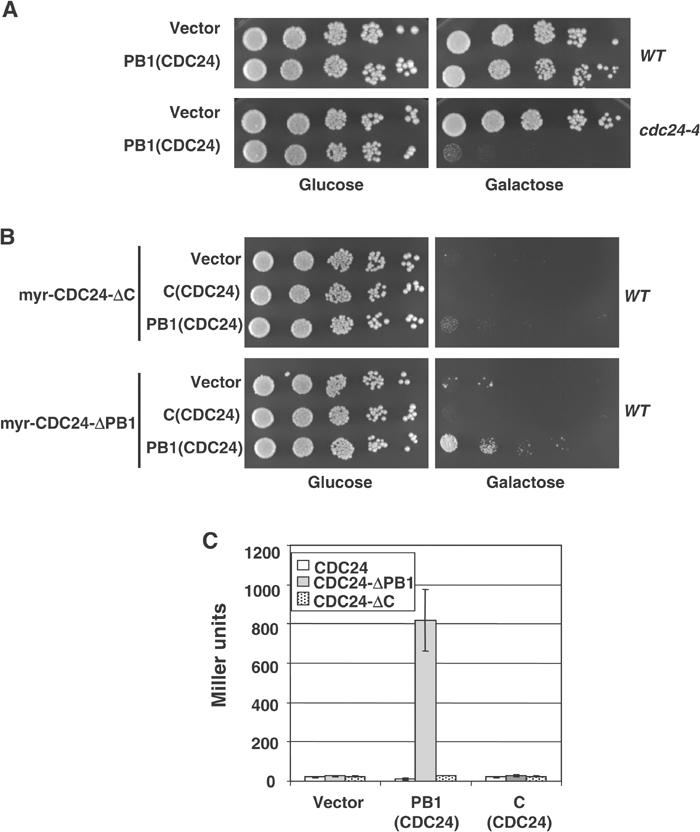
The PB1Cdc24-domain interacts with an intramolecular site and plays an inhibitory role in vivo. (A) Wild-type (YEF241) or cdc24-4 (YEF313) cells were transformed with an empty control plasmid (vector) or a multicopy plasmid expressing the PB1-domain of Cdc24p (amino acids 781–854) from the inducible GAL1-10-promoter. Five-fold serial dilutions were spotted on plates containing glucose (left panel) or galactose (right panel) and photographed after 3 days at 25°C. (B) Wild-type (K699) cells expressing constitutively active myr-Cdc24-ΔC or myr-Cdc24p-ΔPB1 were transformed with a plasmid containing the PB1-domain of Cdc24p (amino acids 781–854) from the inducible GAL1-10-promoter, and analyzed as described in (A). Note that the PB1Cdc24-domain was able to suppress the toxicity induced by overexpression of myr-Cdc24p-ΔPB1. (C) The PB1Cdc24-domain was tested by two-hybrid assay for its ability to interact with full-length Cdc24p, Cdc24p-ΔC and Cdc24p-ΔPB1. The numbers indicate Miller units with standard deviations.
The PB1-domain of Bem1p may function as an activator of Cdc24p
To examine whether the PB1-domain of Bem1p may contribute to the activation of Cdc24p in vivo, we co-expressed full-length and the PB1-domain of Bem1p with myr-Cdc24p. For control, we analyzed the effect of a mutant PB1Bem1-domain (PB1Bem1-m1), which is unable to interact with Cdc24p (Butty et al, 2002). Interestingly, expression of the wild-type PB1Bem1-domain, but not PB1Bem1-m1, was toxic in this assay (Figure 9A), although immunoblotting confirmed that both proteins were expressed at comparable levels (data not shown). As this toxicity required a functional DH-domain of myr-Cdc24p, we conclude that the PB1Bem1-domain contributes to the activation of Cdc24p in vivo, most likely by preventing the intramolecular interaction of its inhibitory PB1-motif.
Figure 9.
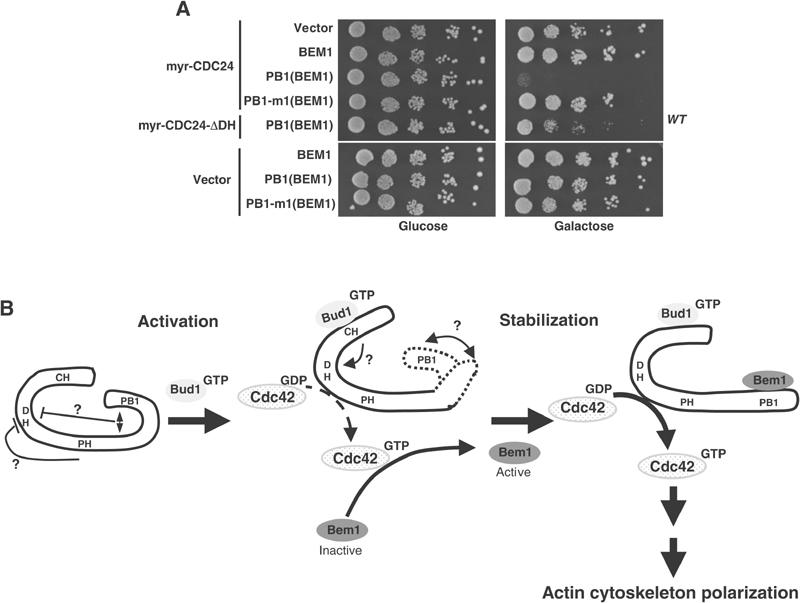
The PB1Bem1-domain enhances Cdc24p-function in vivo. (A) Wild-type (K699) cells expressing no protein (vector), myr-Cdc24p or catalytically-inactive myr-Cdc24p-ΔDH were transformed with an empty control plasmid (vector) or plasmids expressing either the PB1-domain of Bem1p (amino acids 463–551, PB1(Bem1)) or the PB1-domain harboring the K480E mutation (PB1-m1(Bem1); Butty et al, 2002) from the inducible GAL1-10-promoter. Five-fold serial dilutions were spotted on plates containing glucose (left panel) or galactose (right panel). (B) A speculative model for the site-specific activation of Cdc24p by Rsr1p/Bud1p and Bem1p in vivo. Our results suggest that Cdc24p is auto-inactivated by an intramolecular interaction of its carboxy-terminal PB1-domain. Binding of Rsr1/Bud1p-GTP to the CH-domain of Cdc24p may trigger a conformation change, which causes dissociation of the PB1-domain, thereby increasing the GEF activity of Cdc24p. Locally activated Cdc42p then recruits Bem1p, which interacts with the PB1-domain of Cdc24p, thereby preventing its re-binding to the intramolecular site. As a result, active Cdc24p is stabilized at the site of polarization.
Discussion
An intact CH-domain is required for the interaction of Rsr1p/Bud1p and Cdc24p in vivo
Our results strongly suggest that an intact CH-domain is required for Cdc24p function and its specific subcellular localization. Surprisingly, an amino-terminal fragment encompassing the CH-domain of Cdc24p was necessary and sufficient for its interaction with Rsr1p/Bud1p. A previous study suggested that Rsr1p/Bud1p may also interact with the carboxy-terminus of Cdc24p (Park et al, 1997). Although we were unable to confirm this interaction by two-hybrid assay, it is possible that Rsr1p/Bud1p may have multiple binding sites. We identified a point mutant in a residue conserved in a large number of CH-domains (G168D), which specifically reduces the binding of Rsr1p/Bud1p. Cells expressing cdc24-G168D exhibit a random budding pattern, but are able to polarize correctly toward their mating partner. Conversely, cells expressing cdc24-S189P, which specifically affects binding to Far1p (Butty et al, 1998; Nern and Arkowitz, 1999), exhibit a normal budding pattern, but show defects during mating (Nern and Arkowitz, 1998). Both mutations are located within the CH-domain of Cdc24p, and are only separated by 21 amino acids. Our results thus suggest that the CH-domain of Cdc24p interacts with proteins, which are involved in the recruitment of Cdc24p to specific sites on the cell cortex. As bud1Δ far1Δ double mutants are viable (Nern and Arkowitz, 2000a), we speculate that additional proteins may exist, which bind to the CH domain and thereby recruit Cdc24p to the plasma membrane. Moreover, additional proteins with CH-domains are present in yeast, and it is possible that they may also function as effectors of Rsr1p/Bud1p.
Rsr1p/Bud1p may be a site-specific activator of Cdc24p
Our results suggest that Rsr1p/Bud1p may play a dual role in the site-specific activation of Cdc24p. First, activated Rsr1p/Bud1p recruits Cdc24p to the incipient bud site (Park et al, 2002). Recent experiments suggest that the GEF Bud5p may directly interact with the polarity cues Axl2/Bud10p, and Bud8p and Bud9p (Kang et al, 2001), respectively, thereby ensuring specific activation of Rsr1p/Bud1p at the incipient bud site. Second, binding of Rsr1p/Bud1p to Cdc24p at the cell cortex may be involved in the activation of Cdc24p. In support of this activation function, we found that membrane recruitment of Cdc24p via a nonspecific targeting signal is not sufficient for its activation. Although we cannot exclude that the myristoylation signal may interfere with the function of Cdc24p in vivo, co-expression of myr-Cdc24p with low levels of activated Rsr1p/Bud1p was toxic. Importantly, these cells arrested with increased levels of activated Cdc42p, provided that (1) Cdc24p had an intact catalytic DH-domain and (2) that Cdc24p was able to interact with Rsr1p/Bud1p. The simplest explanation for our findings is that Rsr1p/Bud1p not only recruits Cdc24p to the correct site but may also be required for its activation in vivo. In the accompanying paper (Wiget et al, 2004), we similarly found that Far1p is able to recruit and activate Cdc24p during mating, suggesting that both Rsr1p/Bud1p and Far1p may function as site-specific activators of Cdc24p in response to distinct signals.
Cdc24p may be auto-inhibited by its carboxy-terminal PB1Cdc24-domain
Although purified Cdc24p was previously shown to be active in vitro (Zheng et al, 1995), our results suggest that Cdc24p is regulated in vivo by auto-inhibition by its carboxy-terminal PB1Cdc24-domain. First, the PB1Cdc24-domain interacts with an internal site located close to the PH-domain, which itself shares significant homology to PB1-domains (A Petit and M Peter, unpublished results). However, full-length Cdc24p is unable to interact with the PB1Cdc24-domain expressed in trans, suggesting that the intramolecular binding domain is already quantitatively bound to its own PB1Cdc24-domain. Second, overexpression of Cdc24p proteins lacking the PB1Cdc24-domain is constitutively active in vivo when artificially targeted to the plasma membrane. Finally, overexpression of the PB1Cdc24-domain interferes with Cdc24p activity and is able to suppress the toxicity induced by a constitutively active Cdc24p-mutant protein. Thus, our results imply that activation of Cdc24p in vivo requires the dissociation of its carboxy-terminal PB1Cdc24-domain from the intramolecular binding site. We speculate that binding of the site-specific activators Rsr1p/Bud1p and Far1p may trigger this process. At present, it is not clear how the PB1Cdc24-domain interferes with Cdc24p activity. It is possible that intramolecular binding of the PB1Cdc24-domain may prevent the association of Cdc24p and Cdc42p. Alternatively, the bound PB1Cdc24-domain may directly interfere with the catalytic activity of Cdc24p. The mammalian exchange factors Vav and Dbl have previously been shown to be regulated by intramolecular events (Aghazadeh et al, 2000; Bi et al, 2001; Bustelo, 2002), suggesting that this mechanism may be generally important to control the activity of GEFs.
Bem1p may stabilize the active form of Cdc24p by interacting with the PB1Cdc24-domain
As Bem1p interacts with the PB1-domain of Cdc24p (Ito et al, 2001; Butty et al, 2002) that we identified here as the inhibitory domain, Bem1p may function as an activator of Cdc24p by stabilizing its active conformation. Supporting this model, overexpression of the PB1-domain of Bem1p is toxic in combination with membrane-targeted Cdc24p. Interestingly, full-length Bem1p is unable to function as an activator in the same assay, suggesting that Bem1p itself needs to be activated, possibly by binding to Cdc42p. Taken together, our results suggest the following pathway to ensure specific activation of Cdc24p at the incipient bud site (Figure 9B). Cdc24p is kept inactive by an auto-inhibition mechanism, which involves its carboxy-terminal PB1Cdc24-domain. In the G1-phase of the cell cycle, activated Rsr1p/Bud1p recruits Cdc24p to sites of polarized growth by binding to its CH-domain. This interaction triggers a conformational change, which leads to the dissociation of its PB1Cdc24-domain from its internal binding site. This initial activation of Cdc24p is predicted to initiate local production of Cdc42p-GTP, which in turn recruits Bem1p into the complex (Butty et al, 2002). Binding of Cdc42p-GTP to Bem1p may allow Bem1p to interact with the PB1-domain of Cdc24p, thereby stabilizing active Cdc24p by preventing its PB1Cdc24-domain from looping back and inhibiting its GEF activity. As Bem1p also contains a PX-domain, which is likely to interact with membranes (Itoh and Takenawa, 2002), the Cdc24p–Bem1p complex may be stably anchored at sites of polarized growth and produce a strong local Cdc42p-GTP signal, which in turn organizes the actin cytoskeleton. While such a two-step mode of Cdc24p activation is strongly supported by the available in vivo results, direct GEF assays are now needed to demonstrate that Rsr1p/Bud1p and Bem1p are able to directly activate Cdc24p in vitro (Zheng et al, 1995). Nevertheless, such a two-step mode of activation ensures that Cdc24p is activated predominantly at the incipient bud site, and may explain how active Cdc24p is maintained during polarized growth. It is possible that two signals are generally necessary to fully activate molecules that are regulated by auto-inhibition (Pufall and Graves, 2002). First, the auto-inhibitory domain needs to be dissociated, and in the second step this inhibitory domain may be bound by a molecule that prevents it from re-binding to the intramolecular site.
Materials and methods
Strains constructions and genetic manipulations
Yeast strains are described in Supplementary Table I. The genotypes of the yeast strains are: W303 (ade2-1, trp1-1, can1-100, leu2-3,112, his3-11,15, ura3, ssd1-d2), EG123 (trp1-Δ99, leu2-Δ1, his4-519, ura3-52, ade2-101) and S288C (ade2-101, ura3-52, lys2-801, trp1-Δ1, his3Δ 200, leu2-Δ), unless noted otherwise. Standard yeast growth conditions and genetic manipulations were used as described (Guthrie and Fink, 1991). CDC24 constructs were integrated as described previously (Shimada et al, 2000).
PCR mutagenesis
Random mutations in CDC24 were introduced by error-prone PCR (Muhlrad et al, 1992) using the template pYS150 and the primers oTP1003 (GCACTGCTCCGAACAATAAAG) and oTP1004 (AGTAGTGACAAGTGTTGGC). A library of mutagenized PCR products was transformed into wild-type cells (K699) with pYS150 digested with XbaI and XhoI to obtain plasmids that contain mutagenized CDC24 by GAP repair (Guthrie and Fink, 1991). Approximately 3000 transformants were picked and tested for their abilities to grow on media containing 2% galactose or 2% glycerol. The plasmids from strains that were unable to form colonies on galactose plates were recovered and characterized further.
DNA manipulations and two-hybrid assays
Plasmids are described in Supplementary Table II. Standard procedures were used for recombinant DNA manipulations (Ausubel et al, 1991). The details of plasmid constructions and oligo sequences are available upon request. Site-directed mutagenesis was performed by PCR, and confirmed by sequencing. Two-hybrid assays were performed in EGY48 cells containing the LacZ-reporter plasmid pSH18.34 (Gyuris et al, 1993). Miller units are the average of at least three independent experiments with standard deviations.
Microscopy
Proteins tagged with GFP were visualized on a Zeiss Axiovert 100 fluorescence microscope using a Chroma GFPII filter. Proteins expressed from the GAL1-10-promoter were induced by the addition of 2% galactose for 2 h. For quantitation, at least 200 cells were analyzed. For time-lapse microscopy, agarose pads and cell mounting on microscope slides was carried out as described previously (Hoepfner et al, 2000). Cells were imaged by acquiring three GFP focal planes 1 μm apart from each other every 150 s for up to 10 h (Gulli et al, 2000).
Arrest/release experiments with the cln1,2,3Δ METCLN2 (EY569) cells
Exponentially growing cln1,2,3Δ METCLN2 (YMG258) cells were arrested in G1 by repressing CLN2 for 3 h in a selective medium containing 2 mM methionine. Induction of proteins under the control of the GAL1-10-promoter was achieved by addition of 2% galactose (Gulli et al, 2000). Where indicated, the mating pathway was induced by addition of 30 μg/ml α-factor (LIPAL-Biochemicals, Zurich). For release experiments, the arrested YMG2589 cells were washed, released in medium lacking methionine (time 0), and aliquots were analyzed by GFP microscopy or immunoblotting after the times indicated.
GST-CRIB pull-down experiments
Wild-type or mutant CRIB domains of Gic2p (amino acids 189–213; Brown et al, 1997) fused to GST were purified from E. coli on gluthathione–sepharose beads, and incubated for each pull-down reaction with 500 μg of precleared yeast extracts for 30 min at 4°C (Gulli et al, 2000). The beads were washed three times, and bound Cdc42p was eluted with gel-loading buffer and visualized by immunoblotting with Cdc42p antibodies. An aliquot of the extract before the binding assay was immunoblotted to determine the total Cdc42p levels.
Antibodies and immunoblotting experiments
Western blotting was performed using α-11HA (Babco) and α-MYC(9E10) (ISREC) monoclonal antibodies. Antibodies against actin were used as recommended by the manufacturer (Roche). Polyclonal antibodies against Cdc24p, Cdc42p and Gic2p have been described previously (Brown et al, 1997; Butty et al, 1998). Standard conditions were used for yeast cell extracts and immunoblotting (Harlow and Lane, 1988).
Supplementary Material
Supplementary data
Acknowledgments
We thank H-O Park, D Kellogg, R Arkowitz, C Boone, M Jaquenoud and D Drubin for providing plasmids, strains and antibodies. We are grateful to N Perrinjaquet and M Gersbach for expert technical assistance. We thank members of the group for stimulating discussion, and Y Barral and M Sohrmann for critical reading of the manuscript. EB is supported by an NIH-grant (GM59216), and work in the laboratory of MP is supported by the Swiss National Science Foundation, the ETHZ and the Swiss Cancer League.
References
- Aghazadeh B, Lowry WE, Huang XY, Rosen MK (2000) Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell 102: 625–633 [DOI] [PubMed] [Google Scholar]
- Arkowitz RA (1999) Responding to attraction: chemotaxis and chemotropism in Dictyostelium and yeast. Trends Cell Biol 9: 20–27 [DOI] [PubMed] [Google Scholar]
- Aspenstrom P (1999) Effectors for the Rho GTPases. Curr Opin Cell Biol 1: 95–102 [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1991) Current Protocols in Molecular Biology. New York: Greene Publishing Associates and Wiley-Interscience [Google Scholar]
- Bender A, Pringle JR (1989) Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes including the ras-related gene RSR1. Proc Natl Acad Sci USA 86: 9976–9980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E, Chiavetta JB, Chen H, Chen GC, Chan CS, Pringle JR (2000) Identification of novel, evolutionarily conserved Cdc42p-interacting proteins and of redundant pathways linking Cdc24p and Cdc42p to actin polarization in yeast. Mol Biol Cell 11: 773–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi F, Debreceni B, Zhu K, Salani B, Eva A, Zheng Y (2001) Autoinhibition mechanism of proto-Dbl. Mol Cell Biol 21: 1463–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Jaquenoud M, Gulli MP, Chant J, Peter M (1997) Novel Cdc42-binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes Dev 11: 2972–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR (2002) Regulation of Vav proteins by intramolecular events. Front Biosci 7: d24–d30 [DOI] [PubMed] [Google Scholar]
- Butty AC, Perrinjaquet N, Petit A, Jaquenoud M, Segall JE, Hofmann K, Zwahlen C, Peter M (2002) A positive feedback loop stabilizes the guanine-nucleotide exchange factor Cdc24 at sites of polarization. EMBO J 21: 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butty AC, Pryciak PM, Huang LS, Herskowitz I, Peter M (1998) The role of Far1p in linking the heterotrimeric G protein to polarity establishment proteins during yeast mating. Science 282: 1511–1516 [DOI] [PubMed] [Google Scholar]
- Chant J (1999) Cell polarity in yeast. Annu Rev Cell Dev Biol 15: 365–391 [DOI] [PubMed] [Google Scholar]
- Chenevert J, Valtz N, Herskowitz I (1994) Identification of genes required for normal pheromone-induced cell polarization in Saccharomyces cerevisiae. Genetics 136: 1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG (2000) Cell Polarity. Oxford: Oxford University Press [Google Scholar]
- Gulli MP, Jaquenoud M, Shimada Y, Niederhauser G, Wiget P, Peter M (2000) Phosphorylation of the Cdc42 exchange factor Cdc24 by the PAK-like kinase Cla4 may regulate polarized growth in yeast. Mol Cell 6: 1155–1167 [DOI] [PubMed] [Google Scholar]
- Gulli MP, Peter M (2001) Temporal and spatial regulation of Rho-type guanine-nucleotide exchange factors: the yeast perspective. Genes Dev 15: 365–379 [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR (1991) Guide to Yeast Genetics and Molecular Biology. San Diego, CA, USA: Academic Press Inc [Google Scholar]
- Gyuris J, Golemis E, Chertkov H, Brent R (1993) Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell 75: 791–803 [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Hoepfner D, Brachat A, Philippsen P (2000) Time-lapse video microscopy analysis reveals astral microtubule detachment in the yeast spindle pole mutant cnm67. Mol Biol Cell 11: 1197–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GR, Cerione RA (2002) Signaling to the Rho GTPases: networking with the DH domain. FEBS Lett 513: 85–91 [DOI] [PubMed] [Google Scholar]
- Irvine R (1998) Inositol phospholipids: translocation, translocation, translocation. Curr Biol 8: R557–R559 [DOI] [PubMed] [Google Scholar]
- Ito T, Matsui Y, Ago T, Ota K, Sumimoto H (2001) Novel modular domain PB1 recognizes PC motif to mediate functional protein–protein interactions. EMBO J 20: 3938–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Takenawa T (2002) Phosphoinositide-binding domains: functional units for temporal and spatial regulation of intracellular signalling. Cell Signal 14: 733–743 [DOI] [PubMed] [Google Scholar]
- Jaquenoud M, Peter M (2000) Gic2p may link activated Cdc42p to components involved in actin polarization, including Bni1p and Bud6p (Aip3p). Mol Cell Biol 20: 6244–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DI (1999) Cdc42: an essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol Mol Biol Rev 63: 54–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang PJ, Sanson A, Lee B, Park HO (2001) A GDP/GTP exchange factor involved in linking a spatial landmark to cell polarity. Science 292: 1376–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Hunter R, Parker R (1992) A rapid method for localized mutagenesis of yeast genes. Yeast 8: 79–82 [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122 [DOI] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA (1998) A GTP-exchange factor required for cell orientation. Nature 391: 195–198 [DOI] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA (1999) A Cdc24p-Far1p-G beta gamma protein complex required for yeast orientation during mating. J Cell Biol 144: 1187–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA (2000a) G proteins mediate changes in cell shape by stabilizing the axis of polarity. Mol Cell 5: 853–864 [DOI] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA (2000b) Nucleocytoplasmic shuttling of the Cdc42p exchange factor Cdc24p. J Cell Biol 148: 1115–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HO, Bi E, Pringle JR, Herskowitz I (1997) Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc Natl Acad Sci USA 94: 4463–4468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HO, Kang PJ, Rachfal AW (2002) Localization of the Rsr1/Bud1 GTPase involved in selection of a proper growth site in yeast. J Biol Chem 277: 26721–26724 [DOI] [PubMed] [Google Scholar]
- Park HO, Sanson A, Herskowitz I (1999) Localization of Bud2p, a GTPase-activating protein necessary for programming cell polarity in yeast to the presumptive bud site. Genes Dev 13: 1912–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne D, Bretscher A (2000) Polarization of cell growth in yeast I. Establishment and maintenance of polarity states. J Cell Sci 113: 365–375 [DOI] [PubMed] [Google Scholar]
- Pufall MA, Graves BJ (2002) Autoinhibitory domains: modular effectors of cellular regulation. Annu Rev Cell Dev Biol 18: 421–462 [DOI] [PubMed] [Google Scholar]
- Shimada Y, Gulli MP, Peter M (2000) Nuclear sequestration of the exchange factor Cdc24 by Far1 regulates cell polarity during yeast mating. Nat Cell Biol 2: 117–124 [DOI] [PubMed] [Google Scholar]
- Stradal T, Kranewitter W, Winder SJ, Gimona M (1998) CH domains revisited. FEBS Lett 431: 134–137 [DOI] [PubMed] [Google Scholar]
- Toenjes KA, Sawyer MM, Johnson DI (1999) The guanine-nucleotide-exchange factor cdc24p is targeted to the nucleus and polarized growth sites. Curr Biol 9: 1183–1186 [DOI] [PubMed] [Google Scholar]
- Valtz N, Peter M, Herskowitz I (1995) FAR1 is required for oriented polarization of yeast cells in response to mating pheromones. J Cell Biol 131: 863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiget P, Shimada Y, Butty A-C, Bi E, Peter M (2004) Site-specific activation of the nucleotide exchange factor Cdc24p by the scaffold protein Far1p during yeast mating. EMBO J, accompanying paper [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y (2001) Dbl family guanine nucleotide exchange factors. Trends Biochem Sci 26: 724–732 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Bender A, Cerione RA (1995) Interactions among proteins involved in bud-site selection and bud-site assembly in Saccharomyces cerevisiae. J Biol Chem 270: 626–630 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data