

Abstract
Exposure to ionizing radiation during childhood markedly increases the risk of developing papillary thyroid cancer. We examined tissues from 26 Ukrainian patients with thyroid cancer who were younger than 10 years of age and living in contaminated areas during the time of the Chernobyl nuclear reactor accident. We identified nonoverlapping somatic driver mutations in all 26 cases through candidate gene assays and next-generation RNA sequencing. We found that 22 tumors harbored fusion oncogenes that arose primarily through intrachromosomal rearrangements. Altogether, 23 of the oncogenic drivers identified in this cohort aberrantly activate MAPK signaling, including the 2 somatic rearrangements resulting in fusion of transcription factor ETS variant 6 (ETV6) with neurotrophic tyrosine kinase receptor, type 3 (NTRK3) and fusion of acylglycerol kinase (AGK) with BRAF. Two other tumors harbored distinct fusions leading to overexpression of the nuclear receptor PPARγ. Fusion oncogenes were less prevalent in tumors from a cohort of children with pediatric thyroid cancers that had not been exposed to radiation but were from the same geographical regions. Radiation-induced thyroid cancers provide a paradigm of tumorigenesis driven by fusion oncogenes that activate MAPK signaling or, less frequently, a PPARγ-driven transcriptional program.
Introduction
There is strong epidemiological evidence that ionizing radiation is the triggering event leading to development of papillary thyroid cancers (PTCs) in children who were exposed to fallout after the Chernobyl nuclear reactor accident (1, 2). Thyroid cancers arising in these exposed children have a high prevalence of RET/PTC rearrangements, which lead to constitutive expression and activation of the kinase domain of the RET tyrosine kinase receptor (3–6). The association between radiation exposure and generation of recombination events is likely direct and causal. Thus, RET/PTC fusion transcripts are generated by ionizing radiation in human thyroid cell lines and human fetal thyroid tissue xenografts (7, 8). The gene loci involved in these fusion events lie in close spatial proximity to one another within the human thyroid nucleus during interphase and are likely predisposed to recombination of adjacent chromosomal regions after radiation-induced DNA damage (9, 10).
The role for radiation in generating fusion oncogenes in thyroid cells prompted us to screen a large number of post-Chernobyl cancer specimens to identify yet undiscovered driver translocations in this disease. We succeeded in identifying driver events in every case, including novel fusions that predict for alternative mechanisms of thyroid cell transformation.
Results
Screening for known genetic events in radiation-exposed pediatric thyroid cancer reveals a high prevalence of fusion oncogenes.
We obtained tissue samples from 26 Ukrainian patients who were less than 10 years old and living in contaminated areas at the time of the Chernobyl accident to identify likely somatic oncogenic drivers of the disease (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI69766DS1). We first screened them for known oncogenic events (4, 11–13) and found driver mutations in tumors of 21 out of 26 patients: 18 out of 26 (69%) patients had rearrangements (15 RET/PTC [including 6 RET/PTC3, 4 RET/PTC1, 2 RET/PTC6, 1 RET/PTCΔ-3, 1 RET/PTC9, and 1 RET/PTC with unknown partner], 1 TPR-NTRK1 [TRK-T2], 1 PAX8-PPARG, and 1 AKAP9-BRAF) and 3 out of 26 (12%) patients had point mutations/indels (2 BRAFV600E and 1 BRAFV600_K601>E) (Figure 1). Five radiation-exposed thyroid tumors had no known driver alterations identified by this candidate gene approach and were therefore selected for paired-end RNA sequencing (RNA-seq).
Figure 1. Screen for genetic alterations in radiation-exposed pediatric thyroid cancer.

(A) Analysis of RET unbalanced expression (exons 10 and 11/exons 12 and 13) to detect samples with RET fusion oncogenes. The figure shows representative CT plots from quantitative RT-PCR of exons 10 and 11 (encoding extracellular domain) and 12 and 13 (encoding cytoplasmic domain) of a sample without RET fusion oncogene (ratio ~1:1) (left) and a sample with RET fusion (right). (B) RT-PCR and Sanger sequencing for the identification and validation of TPR-NTRK1 and AKAP9-BRAF fusion oncogenes in 2 radiation-exposed cancers. (C) Summary of genetic alterations found in 26 radiation-exposed pediatric thyroid cancers. All but 5 harbored one of the previously described oncogenic events. The 5 tumors with no identifiable defect on the candidate gene screen underwent RNA-seq.
Detection of novel fusion oncogenes in radiation-exposed pediatric thyroid cancer by RNA-seq.
We performed RNA-seq in the 5 tumors without known alterations, as well as in 1 case with a RET/PTC3 fusion as a positive control, using 75 × 75 paired-end reads on the Illumina HiSeq2000. We generated an average of 150 million reads for each sample and aligned them to the human genome (hg19) using TopHat, which showed consistently high base quality on mapped reads and gene coverage for all samples (Supplemental Figure 1). We used the Snowshoes program to search for gene fusions (14), with which we successfully identified RET/PTC3 in the positive control (Supplemental Figure 2). After stringent filtering for read depth and unique matching, we detected 16 somatic fusions that were tumor specific (Supplemental Table 2). For each putative fusion we looked for reads supporting the inverse fusion (e.g., AGK→BRAF to BRAF→AGK). We identified 3 somatic fusions in 4 out of 5 samples with likely oncogenic properties: 1 with CREB3L2-PPARG, 1 with acylglycerol kinase–BRAF (AGK-BRAF), and 2 with ETS variant 6–neurotrophic tyrosine kinase receptor, type 3 (ETV6-NTRK3).
The CREB3L2-PPARG fusion juxtaposes exons 1 and 2 of CREB3L2 to the entire PPARG coding sequence (exons 1–6). CREB3L2-PPARG t(3;7)(p25;q34) was previously identified in 1 case of follicular thyroid cancer (15). The tumor harboring this fusion in our cohort showed a 3.6-fold increase in PPARG mRNA levels when compared with its matched normal. By contrast, all other tumors submitted to RNA-seq showed reduced levels of PPARG mRNA when compared with their corresponding normal tissue (Supplemental Figure 3). All driver fusions identified by RNA-seq were validated by RT-PCR and Sanger sequencing (Figures 2 and 3 and Supplemental Figure 3).
Figure 2. Characterization of ETV6-NTRK3 fusion oncogene in radiation-exposed pediatric thyroid cancer.

(A) Genomic location, mRNA and protein structure, and validation by Sanger sequencing of NTRK3 fusion oncogene. Exons 1–4 of ETV6 are shown in blue and exons 12–18 of NTRK3 are shown in red. PTK, protein tyrosine kinase; SAM, sterile α motif. (B) Western blot of NIH3T3 cells transiently transfected with empty vector or pMSCV-ETV6-NTRK3. Cells were depleted of serum for 24 hours prior to collection. ETV6-NTRK3–expressing cells had higher levels of pAKT, pMEK, pERK, and cyclin D1. tAKT, total AKT; tMEK, total MEK; tERK, total ERK. (C) Increased growth (*P < 3 × 10–4 vs. vector transfected) and agar colony formation of NIH-3T3 cells expressing ETV6-NTRK3. Data represent mean ± SEM.
Figure 3. Characterization of AGK-BRAF fusion oncogene in radiation-exposed pediatric thyroid cancer.

(A) Genomic location, mRNA and protein structure, and validation by Sanger sequencing of BRAF fusion oncogenes. Exons 1 and 2 of AGK are shown in brown and exons 8–18 of BRAF are shown in green. (B and C) Western blot of serum-depleted (B) NIH3T3 or (C) COS7 cells transiently transfected with pLVX-AcGFP-N1 or pLVX-AGK-BRAF-GFP. AGK-BRAF expressing cells had robust induction of pMEK and pERK. (D) Increased growth (*P < 3 × 10–3 vs. vector transfected) and agar colony formation of NIH-3T3 cells expressing AGK-BRAF. Data represent mean ± SEM. (E) Confocal fluorescence imaging of COS7 cells transfected with pLVX-AGK-BRAF-GFP and stained with MitoTracker. The distribution of green fluorescence overlaps but does not colocalize with that of MitoTracker (red). Merged images (bottom row) show mitochondria surrounded by a green fluorescent rim (original magnification, ×252 [top and bottom left]; ×840 [bottom right]). Scale bars: 10 μm. (F) The BRAF kinase domain faces the cytosol in cells transfected with AGK-BRAF. Western blotting of lysates of mitochondrial fractions of NIH3T3 cells stably transfected with pLVX-AcGFP-N1 (GFP) or pLVX-AGK-BRAF-GFP (AGK-BRAF) treated with or without proteinase K. Membranes were incubated with antibodies to BRAF, hexokinase 1, or ATP synthase β. All 3 proteins, including the AGK-BRAF-GFP fusion protein, are seen in intact mitochondrial lysates. AGK-BRAF is depleted by proteinase K, as is the outer mitochondrial-bound protein hexokinase 1, whereas the inner mitochondrial protein ATP synthase β is not.
ETV6-NTRK3 activates MAPK and PI3K pathways and transforms NIH3T3 cells.
ETV6-NTRK3 results from an interchromosomal translocation t(12;15)(p13;q25) that juxtaposes exons 1–4 of ETV6 to exons 12–18 of NTRK3, a kinase not previously implicated in thyroid cancer. The breakpoint of congenital fibrosarcomas and secretory breast cancers, which include exons 1–5 of ETV6 and 13–18 of NTRK3, differs from that of ETV6-NTRK3 (16, 17). The thyroid-specific chimeric transcript encodes a protein with the sterile α motif dimerization domain of ETV6 fused to the protein tyrosine kinase domain of NTRK3 (Figure 2A). Expression of the novel fusion of exons 1–4 of ETV6 to exons 12–18 of NTRK3 in NIH-3T3 cells activated PI3K and MAPK signaling (Figure 2B). Expression of the oncoprotein promoted growth of NIH-3T3 cells in low serum and colony formation in soft agar (Figure 2C).
The AGK-BRAF fusion localizes to the outer mitochondrial membrane, activates MAPK signaling, and induces NIH3T3 cell growth and colony formation.
We identified a novel fusion of AGK (encoding the mitochondrial protein acylglycerol kinase) and BRAF. These 2 genes are 626 kb apart in the opposite orientation at 7q34. AGK-BRAF juxtaposes exons 1 and 2 of AGK to exons 8–18 of BRAF (Figure 3A). The oncoprotein contains the kinase domain but lacks the autoinhibitory N-terminal RAS-binding domain of BRAF. Expression of AGK-BRAF in NIH3T3 and COS-7 cells resulted in constitutive activation of MEK and ERK phosphorylation, overexpression of cyclin D1 (Figure 3, B and C), and transformation in NIH3T3 cells (Figure 3D). Exons 1 and 2 of AGK encode for the mitochondrial localization domain of the protein. Consistent with this, the fusion protein localized to the periphery of mitochondria (Figure 3E). To establish the topography of the aberrantly expressed kinase, we isolated mitochondrial preparations of NIH3T3 cells expressing AGK-BRAF. Incubation with proteinase K degraded the fusion protein as well as hexokinase 1, a protein that is known to bind to the outer mitochondrial membrane, whereas the inner mitochondrial protein ATP synthase β was unaffected. This indicates that the BRAF kinase domain faces the cytosol (Figure 3F), which explains its ability to recruit MEK and activate MAPK signaling.
Analysis of somatic SNPs and indels in RNA-seq samples of radiation-associated thyroid cancers identifies an activating TSHR mutation.
We used Varscan to identify somatic SNPs and indels in the RNA-seq samples (18). This identified a somatic missense mutation, c.1274T>G, in exon 10 of the TSHR gene, which encodes for the thyrotropin receptor, in the remaining case with no known oncogenic driver. This mutation substitutes a serine for an isoleucine in codon 425 (p.S425I) within transmembrane domain 1 of this G protein–coupled receptor (Figure 4). Activating mutations of TSHR stimulate thyrocyte proliferation and expression of thyroid differentiation genes. Accordingly, the tumor with the TSHR p.S425I allele had markedly increased expression of thyroid-specific genes, including SLC5A5 (also known as NIS), whereas all other tumors showed predominant downregulation of this genetic program (Figure 4D). We also examined the RNA-seq data of the tumor samples from the radiation-exposed patients for all other somatic mutations and indels (Supplemental Table 3) but did not identify other potential driver genetic abnormalities that may have contributed to radiation-induced thyroid tumorigenesis, indicating that a fairly small set of genetic aberrations likely account for tumor induction in most thyroid cancers.
Figure 4. Identification of a TSHR gain-of-function mutation (TSHRS425I) in a radiation-exposed PTC by RNA-seq.

(A) Identification of TSHRS425I mutation in a radiation-exposed patient by RNA-Seq as seen by Integrative Genomics Viewer plot. (B) Validation of the somatic mutation by Sanger sequencing using genomic DNA (gDNA) from tumor and matched normal sample tissue. (C) Mutation localizes to the first transmembrane domain of TSHR. (D) Increased expression of genes required for thyroid differentiated function and thyroid hormone biosynthesis is restricted to the tumor with the activating TSHR.
Gene expression analysis of RNA-seq data shows transcriptome signatures associated with the underlying oncogenic driver mutations.
We next used a previously validated MAPK expression signature to determine which of the tumors analyzed by RNA-seq had induction of this pathway (19). The cases with RET/PTC3, AGK-BRAF, and ETV6-NTRK3 fusions had increased expression of MAPK-regulated genes, unlike the tumors with the CREB3L2-PPARG fusion and TSHR mutation (Supplemental Figure 4). The tumor with the PPARG rearrangement showed expression changes consistent with a PPARG-driven transcriptional program (20), underscoring the likely role of these genetic alterations as driver events in these tumors.
Sporadic pediatric thyroid cancers have lower prevalence of fusion oncogenes.
We next analyzed 27 sporadic pediatric thyroid cancers from the same geographical region that were carefully matched for age and tumor size (Supplemental Table 1). We then screened these tumors for all known genetic alterations, including the novel fusions identified in the radiation-exposed cohort. Only 9 out of 27 (33%) tumors had fusions (5 RET/PTC1, 2 ETV6-NTRK3, 1 RET/PTC3, 1 RET/PTCΔ-3), and 9 out of 27 (33%) tumors had somatic oncogenic point mutations (7 BRAFV600E and 2 NRASQ61R). There were no cases of AGK-BRAF or CREB3L2-PPARG. The prevalence of driver fusions in tumors from this patient cohort was significantly different from that seen in the radiation-exposed group (sporadic [9 out of 27] vs. exposed [22 out of 26] tumors; P = 6.5E-0.5); Figure 5).
Figure 5. Genomic landscape of PTCs from children exposed to radiation.

(A) Summary of genetic alterations found in radiation-exposed (left; T1–T26) and sporadic (right; T27–T53) pediatric thyroid tumors. Fusion oncogenes were significantly more prevalent in radiation-exposed cases (P = 6.5E-0.5, Fisher’s exact test). (B) Circos plot of rearrangements detected in radiation-exposed cancers. Breakpoints were assumed to lie in the middle of the introns immediately upstream and downstream of the fused exons, respectively. Rearrangements were color coded as follows: red, drivers; gray, unvalidated fusions. The thickness of the lines indicates the frequency of the fusion. Intrachromosomal rearrangements were the most prevalent events.
Pathological characteristics of pediatric thyroid tumors correlate with driver alterations.
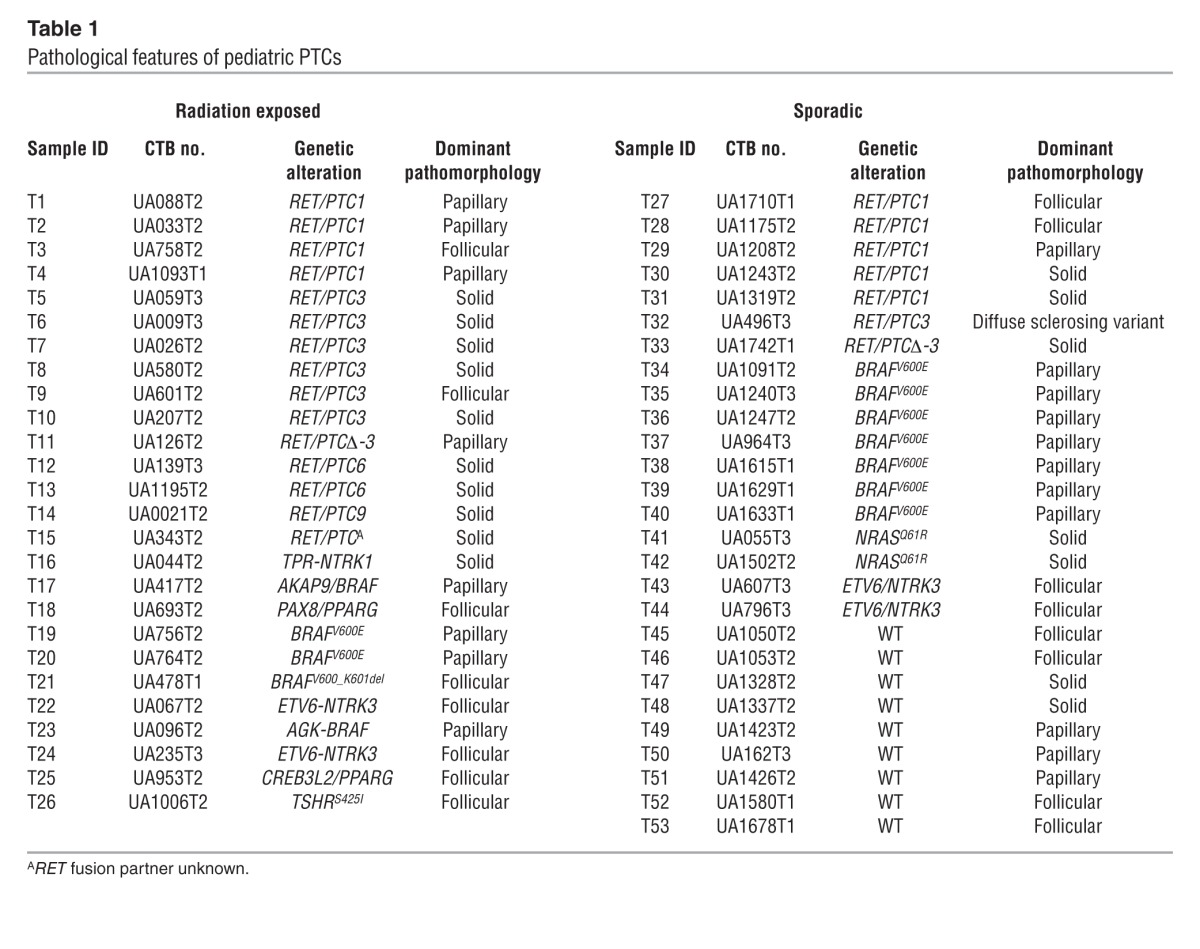
The pathological characteristics of the radiation-induced and sporadic cases appeared to correlate primarily with the nature of the underlying driver mutation, as shown in Table 1. All 4 cases harboring ETV6-NTRK3 rearrangements were follicular-variant PTCs, as were the cases with PPARG rearrangements. By contrast, all 9 tumors with BRAFV600E mutations were classical PTCs, as were the 2 cases with BRAF rearrangements. The sole exception was a radiation-associated case with BRAFV600_K601del, which was a follicular-patterned lesion.
Table 1.
Pathological features of pediatric PTCs
Low-pass whole-genome sequencing shows no difference in the number of somatic SNPs, indels, or chromosomal rearrangements between sporadic and radiation-exposed tumors.
To obtain a fuller picture of the genomic landscape of the radiation-exposed and sporadic pediatric thyroid cancers, we performed low-pass whole-genome sequencing (WGS) of 10 tumors and matched normal pairs (5 radiation exposed and 5 sporadic), with an average coverage of 7 to 8 times (each base in the genome was sequenced 7 to 8 times on average), and aligned the reads to the human genome (hg19). Eight out of these ten patients had previously identified driver mutations that were found during screening for the known oncogenic events, and all were confirmed in the low-pass WGS data. We detected known drivers for tumor samples T1, T2 (RET/PTC1), T8 (RET/PTC3), and T16 (TPR/NTRK1) using BreakDancer (21). We also identified WGS reads supporting T5 (RET/PTC3) and T27 (RET/PTC1). The full list of somatic chromosomal rearrangements is shown in Supplemental Table 4. There were 19 ± 3.1 rearrangements in the sporadic group and 15.8 ± 4 rearrangements in the radiation-exposed group (Supplemental Figures 5 and 6). There was no significant difference in the total number of rearrangements, or of intrachromosomal rearrangements, between the radiation-exposed and sporadic groups (P = 0.288, Mann-Whitney-Wilcoxon test). Thus, despite the higher prevalence of fusion oncogenes in the radiation-exposed cohort, the overall frequency of somatic rearrangements is not different between the 2 groups.
Then, we applied Varscan again to detect somatic single nucleotide variants (SNVs) and indels in the low-pass WGS data files (18). We found pair-end reads supporting the known BRAF mutations in T35 and T38. Altogether, we found no difference in the average count of nonsynonymous somatic SNVs in the sporadic and radiation-exposed groups (11.6 ± 3.9 and 12 ± 4.2, respectively; P = 1, Mann-Whitney-Wilcoxon test) (Supplemental Table 5). No additional candidate tumor drivers emerged from the analysis of the SNV/indels in the wild-type sporadic samples (Supplemental Tables 6 and 7).
Discussion
Exposure of children to ionizing radiation after the Chernobyl reactor accident in 1986 increased the incidence of clinically significant thyroid cancers dramatically compared with that seen in the same regions of Belarus and Ukraine prior to the accident. By 1995, the incidence of childhood thyroid cancer had increased to 4 cases per 100,000 children per year compared with 0.03 to 0.05 cases per 100,000 children per year prior to the accident (2). The risk was greatest in those exposed at a very young age. The incidence of thyroid cancer in children born after 1986 returned to baseline levels, highlighting the likely causal role of radiation in the pathogenesis of the disease. The tumor samples chosen for genetic analysis in this study were carefully selected to maximize the likely association with radiation exposure by selecting patients from the contaminated regions who were exposed at a very young age (most were less than 5 years old at the time of the accident) and who had clinically significant disease.
Previous studies identified RET/PTC fusions as the most prevalent genetic abnormalities in radiation-associated thyroid cancers (4–6). Although RET/PTC1 and RET/PTC3 are the most common rearrangements, RET can also recombine with a number of other upstream partner genes to generate fusion proteins with constitutively activated RET kinase activity (12). The prevalence of RET rearrangements in this series (15 out of 26 tumor samples; 58%), and the relative distribution of the various RET/PTC fusions, is largely consistent with previous reports. Studies of both RET/PTC and NTRK rearrangements have shown that the gene loci participating in the fusions lie in close spatial proximity to one another during interphase in thyroid cells, likely predisposing to recombination of adjacent chromosomal regions after radiation-induced DNA damage (9, 10). The spatial organization of the genome is now thought to be a key contributing factor to the generation of chromosomal translocations in cancer (22). The rationale to select RNA-seq rather than whole-exome sequencing as the primary modality to screen for new oncogenic drivers was based on these factors and the consequent prediction that fusion oncogenes were likely to predominate in this particular patient population.
Two of the radiation-exposed and two of the sporadic cases had ETV6-NTRK3 rearrangements. This fusion kinase is found in congenital fibrosarcomas and in secretory breast cancers (23) but has not been previously reported in thyroid cancer. ETV6 is a member of the ETS family of transcription factors, and, for unclear reasons, its gene is frequently involved in chromosomal translocations in cancer. The resulting fusion proteins usually contain the sterile α motif of ETV6, a helix-loop-helix domain with oligomerization properties, either fused to DNA-binding transcription factors in myeloid malignancies (24) or to the protein tyrosine kinase domains of genes such as PDGFRB, ABL, JAK2, ARG, or FGFR3 (reviewed in ref. 23). The chimeric ETV6-NTRK3 gene found in thyroid cancers differs in potentially significant ways from the fusion found in breast cancers and fibrosarcomas. This is because the thyroid ETV6-NTRK3 chimeric transcript includes exon 12 of NTRK3, which is excluded in the canonical fusion oncogene. Exon 12 of NTRK3 encodes tyrosine 516, which when phosphorylated binds the adaptor proteins SHC, SH2B, or the PI3K p85 subunit, which are required to activate MAPK and PI3K signaling, respectively. The absence of this domain in the breast and fibrosarcoma fusion protein dictates a requirement for concomitant activation of insulin receptor substrate 1 (IRS1) by insulin-like growth factor 1 for downstream signaling and transformation. IRS1 is believed to associate with the C terminus of the fusion protein (25, 26). Although we have not systematically compared the 2 constructs, the fact that the thyroid fusion activates MEK-ERK and AKT in serum-free conditions is consistent with the prediction that the NTRK3 domains encoded by exons 12–18 are sufficient for signaling and transformation following constitutive oligomerization mediated by the ETV6 sterile α motif domain.
BRAF is by far the most prevalent thyroid oncogene, particularly in PTCs, and is usually activated by the V600E substitution.
We characterized previously the functional properties of AKAP9-BRAF, the first reported fusion of BRAF in post-Chernobyl thyroid cancers (13). One of the radiation-exposed patient samples in this cohort harbored this defect, and another was found to express a BRAF fusion with AGK. The AGK domain involved in the chimeric protein targeted the BRAF kinase to the outer mitochondrial membrane. Despite this aberrant subcellular localization, the BRAF kinase faced the cytosol and was competent for signaling and transformation. The histological appearance of the tumors with BRAF fusions did not differ from that of those harboring the BRAFV600E mutation, as they were all classical PTCs.
Two of the twenty-six radiation-exposed cases had PPARG fusions, adding these to the spectrum of alterations present in radiation-exposed patients. Consistent with reports in the literature, these were associated with follicular-patterned histological variants of PTC (27).
Activating mutations of TSHR and GNAS are common in autonomously functioning thyroid adenomas and far less frequently in carcinomas but have not been previously identified in radiation-induced cancers. The TSHR 1274T>G mutation identified here was previously found in 4 cases of autonomously functioning thyroid adenomas and is known to be a gain-of-function defect, as expression of the mutant protein increased cAMP levels by approximately 8 fold in functional assays (28). Consistent with this finding, the follicular-variant PTC harboring the TSHR mutation had higher expression of thyroid differentiation markers regulated by TSH signaling via adenylyl cyclase. Although the criteria used to select cases in this study were designed to obtain a cohort enriched for radiation-associated cancers, we accept that one or more sporadic cases may have been intermingled with this set. The only TSHR mutant case we found is insufficient to claim an association with radiation exposure.
The prevalence of driver fusion oncogenes in tumors of the cohort with sporadic cases was much lower than that in the radiation-exposed group and consistent with that seen in children and adolescents from other geographical regions (29). These data challenge the notion that fusion oncogenes are about equally prevalent in pediatric thyroid cancers, regardless of whether or not they are radiation induced. This led us to test the hypothesis that the overall genomic landscape of cancers from these two classes of tumors would also differ. Interestingly, the low-pass WGS of representative samples from the 2 groups failed to identify differences in the average number of somatic SNPs, indels, or rearrangements. The underlying predisposing factor(s) leading to development of sporadic pediatric thyroid cancers is not known. The relatively high frequency of passenger recombination events that they harbor suggests that there may be some deficit in DNA repair. Although radiation also leads to a high frequency of recombination events, the mechanisms accounting for these are likely to differ and may explain the higher likelihood of generation of fusion oncogenes.
In conclusion, we identified nonoverlapping driver alterations for each one of the 26 radiation-exposed cases studied, highlighting the power of RNA-seq to reveal such alterations. Twenty-two out of twenty-six (84%) cases had fusion events. At least 14 of these resulted from intrachromosomal rearrangements, lending further support to the evidence that spatial proximity favors generation of recombination events after radiation exposure and DNA damage (9, 10). Moreover, 23 out of 26 tumors express oncoproteins that activate MAPK signaling, confirming the critical role of constitutive activation of this pathway in thyroid tumor initiation, including the kinase fusions ETV6-NTRK3 and AGK-BRAF, which encode activated kinases that can be targeted pharmacologically. Although the signaling pathways activated by the fusion oncogenes are similar to those engaged by the oncogenic drivers of thyroid cancers from adult patients, the spectrum of lesions is quite different, which may account for the differences in their biological and clinical behavior. The driver events found in radiation-exposed children arise within a mutational landscape induced by radiation damage, which underscores the need for rapid responses to mitigate the consequences of radiation exposure in children to avoid generation of oncogenic fusions that lead to increased risk of thyroid cancer.
Methods
Tumor samples and nucleic acid isolation
DNA and RNA from 53 PTCs and the corresponding normal thyroid tissue were obtained from the Chernobyl Tissue Bank (Imperial College, London): 26 samples were from patients exposed to radiation and 27 samples had no known radiation exposure. All radiation-exposed patients were from regions of Ukraine close to the Chernobyl nuclear reactor (Supplemental Table 1) and were less than 10 years old in April 1986 (24 out of 26 were less than 5 years old). Patients were considered not to have been exposed if they were born after 1986 and were from the same geographical regions. All primary tumors were at least 1 cm in size. DNA and RNA were isolated using the QIAamp DNA Kit (Qiagen) and the Rneasy Fibrous Tissue Kit (Qiagen), respectively.
Detection of known fusion oncogenes and point mutations
The cDNA of the 53 tumors was screened for recombination events previously described in PTC (30). For RET/PTC rearrangements, we screened for unbalanced expression of exons 12 and 13 relative to exons 10 and 11 of RET, which flank the common breakpoint in intron 11, by quantitative RT-PCR (Supplemental Figure 1). Samples in which expression of exons 12 and 13 was greater than that of exons 10 and 11 were amplified with primer pairs spanning the specific breakpoints in all known RET/PTC variants. AKAP9-BRAF, PAX8-PPARG, and NTRK1 fusions were screened by RT-PCR. Genomic DNA was also analyzed by Sequenom mass spectrometry to interrogate hot spot mutations in thyroid cancer genes, including RET, NRAS, KRAS, HRAS, BRAF, PIK3CA, and AKT1 (31).
Paired-end RNA-seq
cDNA libraries were prepared using the mRNA-Seq Sample Prep Kit v2 (Illumina). Briefly, poly-A(+) RNA purified from 2 μg total RNA was fragmented by incubation at 94°C for 5 minutes in fragmentation buffer and used for double-stranded cDNA synthesis. After ligation of the paired-end adapter, the 250- to 300-bp fraction was gel purified and amplified with 15 cycles of PCR. The resulting libraries were subjected to paired-end sequencing of 75-bp reads on HiSeq 2000 (Illumina).
Bioinformatic analyses
We used Snowshoes v2.0 (14) to detect fusion events and Varscan 2.3.2 (18) to detect somatic SNVs and calculated gene expression using r-make ( http://physiology.med.cornell.edu/faculty/mason/lab/r-make/) for read count value. A more detailed description is provided in the Supplemental Methods. The RNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (32) and are accessible through GEO Series accession number GSE48850 ( http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48850).
RT-PCR, genomic PCR, and Sanger sequencing for validation of genetic alterations
Total RNA (500 ng) was reverse transcribed to cDNA using SuperScript III Reverse Transcriptase (Invitrogen). cDNA (corresponding to 10 ng total RNA) or 10 ng genomic DNA was subjected to PCR amplification using Platinum Taq DNA Polymerase (Invitrogen). The reactions were carried out in a thermal cycler under the following conditions: 35 cycles at 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, with a 5-minute final extension at 72°C. The gene encoding GAPDH was amplified to estimate the efficiency of cDNA synthesis. PCR products were directly sequenced in both directions using the BigDye Terminator Kit and an ABI 3130xl DNA Sequencer (Applied Biosystems). The PCR primers for detection of ETV6-NTRK3, AGK-BRAF, CREB3L2-PPARG, and TSHR mutations are listed in Supplemental Table 8.
Signaling effects of ETV6-NTRK3 and AGK-BRAF
The cDNAs for ETV6-NTRK3 and AGK-BRAF were cloned into pMSCVpuro and pLVX-AcGFP-N1 (Clontech), respectively. NIH3T3 cells were transfected with the empty vector, pMSCV-ETV6-NTRK3, pLVX-AcGFP-N1, or pLVX-AGK-BRAF-GFP and treated with 1.5 μg/ml puromycin for 2 weeks. COS7 cells were transiently transfected with pLVX-AcGFP-N1 or pLVX-AGK-BRAF-GFP.
Cells were incubated in the indicated conditions for 48 hours and then lysed in a buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Triton, 5 mM EGTA, 1.5 mM MgCl2, and 20 mM Na4O7P2 supplemented with protease inhibitor cocktail (Roche). Equal amounts of total protein were resolved by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with the following primary antibodies: p-ERK1/2 Thr202/Tyr204, 1:1,000 (Cell Signaling); ERK1 (137F5), 1:1,000 (Cell Signaling); p-MEK1/2 S217/221 (no. 9121S), 1:1,000 (Cell Signaling); MEK (no. 9122S), 1:1,000 (Cell Signaling); p-AKT S473 (D9E), 1:1,000 (Cell Signaling); p-AKT Thr308 (244F9), 1:1,000 (Cell Signaling); AKT (C2667), 1:1,000 (Cell Signaling); anti-PI3Kinase-p85 N-SH2 domain, 1:1,000 (Millipore); Cyclin D1 (92G2), 1:1,000 (Cell Signaling); HA-Tag (C29F4), 1:1,000 (Cell Signaling); Hexokinase I (C35C4), 1:1,000 (Cell Signaling); anti-ATP Synthase β (612518), 1:1,000 (BD Bioscience); BRAF (ab59357), 1:1,000 (Abcam); and β-actin (A2228), 1:1,000 (Sigma-Aldrich). Bands were visualized with enhanced chemiluminescence (GE Healthcare Biosciences) as directed by the manufacturer.
Studies in mitochondrial lysates.
A confluent 15-cm dish was washed with PBS, cells were spun down, and the pellet was resuspended in 1 ml of ice-cold HIM buffer (200 mM mannitol, 70 mM sucrose, 1 mM EGTA, and 10 mM HEPES) supplemented with protease inhibitor cocktail (Roche), homogenized using a Polytron and then passed through a syringe with 20-gauge and 30-gauge needles (5 times each). Homogenates were then centrifuged at 700 g at 4°C for 10 minutes (pellet, nuclei and unbroken cells), and the supernatant was centrifuged at 10,000 g at 4°C for 10 minutes to collect the heavy membrane pellet (supernatant, cytosol and light membranes). The pellet was resuspended in 1 ml HIM buffer followed by another cycle of 700 g and 10,000 g centrifugation. The pellet was resuspended in HIM buffer to obtain the mitochondrial fraction. Fresh isolated mitochondria were incubated with or without proteinase K (10 μg/ml) at 4°C for 10 minutes. Digestions were stopped by the addition of 2 mM PMSF and SDS-PAGE–loading buffer added prior to electrophoresis.
Confocal microscopy.
COS7 cells were plated in chamber slides and transfected with pLVX-AcGFP or pLVX-AGK-BRAF-AcGFP the following day. Two days later they were stained with MitoTracker (Life Technologies), fixed in 4% PFA, washed with PBS, and mounted in Vectorshield (Vector Laboratories). Confocal imaging was performed with a Leica TCS SP5-II using a 63×/1.4NA oil immersion objective. Laser lines used for each fluorophore were 488 nm (GFP) and 594 nm (MitoTracker). Serial z-stack images were taken at 0.2-micron steps, and individual slices were chosen for figures.
Paired-end WGS
High-molecular-weight DNA was extracted from freshly frozen normal and tumor tissues from 5 radiation-exposed and 5 sporadic papillary thyroid carcinomas. DNA of the appropriate size was gel purified from the short fragmented DNA library to exclude any inappropriate DNA fusions during library construction. The short fragment DNA libraries were generated using the TruSeq Nano DNA Sample Preparation Kit (catalog no. FC-121-4001, Illumina) following the manufacturer’s protocols. Clustering of the libraries was performed on the cBot clustering generation instrument (Illumina), following manufacturer’s specifications (catalog no. CT-402-4001). Paired-end sequencing (76-bp read length) was performed using the Hiseq 2500 sequencer (Illumina) using the latest chemistry (v3) for the 200 cycle reagent kits (catalog no. FC-402-4001).
Bioinformatics of WGS
Paired-end reads were aligned to the hg19 human genome using Burrows-Wheeler Aligner (version 0.7.5). The mean ± standard deviation of the genome coverage was 7.22 ± 0.60. Somatic mutations were detected using Varscan (version 2.3.2), and somatic chromosomal rearrangements were identified using BreakDancer (version 1.1.2) (21). Only rearrangements spanning partners that were at least 50 kb apart were taken for further analysis. Structural variations were annotated using Refseq ( http://www.ncbi.nlm.nih.gov/refseq/). Mann-Whitney-Wilcoxon tests were used to compare the number of SNVs or rearrangements between the 2 groups. The paired-end sequencing data have been deposited in the NCBI sequence read archive under accession number SRA903315.
Statistics
A 2-tailed Student’s t test was used to compare effects of transfected oncoproteins on growth and colony formation assays. The Mann-Whitney-Wilcoxon test was used to compare the total number of rearrangements between the radiation-exposed and sporadic PTCs. The same test was applied to compare the frequency of somatic nonsynonymous SNVs between the 2 groups. Fisher’s exact test was used to compare the prevalence of fusion oncogenes in radiation-exposed and sporadic cases. A more detailed description of the bioinformatic analyses used can be found in the Supplemental Methods. P values of less than 0.05 were considered statistically significant.
Study approval
The study was conducted with the approval of the NIH IRB, Bethesda, Maryland, USA (protocol no. 0H00-C-N024) and the Imperial College Research Ethics Committee (protocol no. ICREC-8-2-4).
Supplementary Material
Acknowledgments
This work was supported by NIH grants CA50706, CA72597, R01HG006798, R01NS076465, and R44HG005297; the Margot Rosenberg Pulitzer Foundation; the Byrne fund; and the Lefkofsky Family Foundation. We thank Doron Betel from Weill Cornell Medical College for his extraordinary help in generating the WGS data. We are grateful to the Chernobyl Tissue Bank for providing patient specimens (CTB project number 002/2009) and to the members of the International Pathology Panel of the Chernobyl Tissue Bank for confirmation of the diagnoses: A. Abrosimov, T.I. Bogdanova, N. Dvinskikh, G. Fadda, J. Hunt, M. Ito, V. LiVolsi, J. Rosai, and E.D. Williams.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(11):4935–4944. doi:10.1172/JCI69766.
See the related Commentary beginning on page 4566.
References
- 1.Williams D. Cancer after nuclear fallout: lessons from the Chernobyl accident. Nat Rev Cancer. 2002;2(7):543–549. doi: 10.1038/nrc845. [DOI] [PubMed] [Google Scholar]
- 2.Cardis E, et al. Risk of thyroid cancer after exposure to 131I in childhood. J Natl Cancer Inst. 2005;97(10):724–732. doi: 10.1093/jnci/dji129. [DOI] [PubMed] [Google Scholar]
- 3.Bounacer A, et al. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene. 1997;15(11):1263–1273. doi: 10.1038/sj.onc.1200206. [DOI] [PubMed] [Google Scholar]
- 4.Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997;57(9):1690–1694. [PubMed] [Google Scholar]
- 5.Klugbauer S, Lengfelder E, Demidchik EP, Rabes HM. High prevalence of RET rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene. 1995;11(12):2459–2467. [PubMed] [Google Scholar]
- 6.Fugazzola L, et al. Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Res. 1995;55(23):5617–5620. [PubMed] [Google Scholar]
- 7.Mizuno T, et al. Preferential induction of RET/PTC1 rearrangement by X-ray irradiation. Oncogene. 2000;19(3):438–443. doi: 10.1038/sj.onc.1203343. [DOI] [PubMed] [Google Scholar]
- 8.Caudill CM, Zhu Z, Ciampi R, Stringer JR, Nikiforov YE. Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to gamma-radiation: a model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J Clin Endocrinol Metab. 2005;90(4):2364–2369. doi: 10.1210/jc.2004-1811. [DOI] [PubMed] [Google Scholar]
- 9.Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290(5489):138–141. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- 10.Roccato E, et al. Proximity of TPR and NTRK1 rearranging loci in human thyrocytes. Cancer Res. 2005;65(7):2572–2576. doi: 10.1158/0008-5472.CAN-04-4294. [DOI] [PubMed] [Google Scholar]
- 11.Santoro M, et al. Gene rearrangement and Chernobyl related thyroid cancers. Br J Cancer. 2000;82(2):315–322. doi: 10.1054/bjoc.1999.0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabes HM, et al. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: biological, phenotypic, and clinical implications. Clin Cancer Res. 2000;6(3):1093–1103. [PubMed] [Google Scholar]
- 13.Ciampi R, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest. 2005;115(1):94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asmann YW, et al. A novel bioinformatics pipeline for identification and characterization of fusion transcripts in breast cancer and normal cell lines. Nucleic Acids Res. 2011;39(15):e100. doi: 10.1093/nar/gkr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lui WO, et al. CREB3L2-PPARgamma fusion mutation identifies a thyroid signaling pathway regulated by intramembrane proteolysis. Cancer Res. 2008;68(17):7156–7164. doi: 10.1158/0008-5472.CAN-08-1085. [DOI] [PubMed] [Google Scholar]
- 16.Tognon C, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2(5):367–376. doi: 10.1016/S1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 17.Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18(2):184–187. doi: 10.1038/ng0298-184. [DOI] [PubMed] [Google Scholar]
- 18.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pratilas CA, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106(11):4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giordano TJ, et al. Delineation, functional validation, and bioinformatic evaluation of gene expression in thyroid follicular carcinomas with the PAX8-PPARG translocation. Clin Cancer Res. 2006;12(7 pt 1):1983–1993. doi: 10.1158/1078-0432.CCR-05-2039. [DOI] [PubMed] [Google Scholar]
- 21.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6(9):677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roukos V, Burman B, Misteli T. The cellular etiology of chromosome translocations. Curr Opin Cell Biol. 2013;25(3):357–364. doi: 10.1016/j.ceb.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lannon CL, Sorensen PH. ETV6-NTRK3: a chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin Cancer Biol. 2005;15(3):215–223. doi: 10.1016/j.semcancer.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Haferlach C, et al. ETV6 rearrangements are recurrent in myeloid malignancies and are frequently associated with other genetic events. Genes Chromosomes Cancer. 2012;51(4):328–337. doi: 10.1002/gcc.21918. [DOI] [PubMed] [Google Scholar]
- 25.Martin MJ, et al. The insulin-like growth factor I receptor is required for Akt activation and suppression of anoikis in cells transformed by the ETV6-NTRK3 chimeric tyrosine kinase. Mol Cell Biol. 2006;26(5):1754–1769. doi: 10.1128/MCB.26.5.1754-1769.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison KB, Tognon CE, Garnett MJ, Deal C, Sorensen PH. ETV6-NTRK3 transformation requires insulin-like growth factor 1 receptor signaling and is associated with constitutive IRS-1 tyrosine phosphorylation. Oncogene. 2002;21(37):5684–5695. doi: 10.1038/sj.onc.1205669. [DOI] [PubMed] [Google Scholar]
- 27.Castro P, et al. PAX8-PPARgamma rearrangement is frequently detected in the follicular variant of papillary thyroid carcinoma. J Clin Endocrinol Metab. 2006;91(1):213–220. doi: 10.1210/jc.2005-1336. [DOI] [PubMed] [Google Scholar]
- 28.Palos-Paz F, et al. Prevalence of mutations in TSHR, GNAS, PRKAR1A and RAS genes in a large series of toxic thyroid adenomas from Galicia, an iodine-deficient area in NW Spain. Eur J Endocrinol. 2008;159(5):623–631. doi: 10.1530/EJE-08-0313. [DOI] [PubMed] [Google Scholar]
- 29.Sassolas G, et al. Oncogenic alterations in papillary thyroid cancers of young patients. Thyroid. 2012;22(1):17–26. doi: 10.1089/thy.2011.0215. [DOI] [PubMed] [Google Scholar]
- 30.Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol. 2011;7(10):569–580. doi: 10.1038/nrendo.2011.142. [DOI] [PubMed] [Google Scholar]
- 31.Ricarte-Filho JC, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69(11):4885–4893. doi: 10.1158/0008-5472.CAN-09-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett T, et al. NCBI GEO: mining millions of expression profiles — database and tools. Nucleic Acids Res. 2005;33(Database issue):D562–D566. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.