

Abstract
Global, sustained production of ROS has deleterious effects on tissue structure and function and gives rise to biochemical and physiological changes associated with organ senescence. Specific, localized ROS metabolites generated by mitochondria and NADPH oxidases also transduce homeostatic information in response to metabolic, mechanical, and inflammatory cues. In this issue of the JCI, Dugan and colleagues demonstrate that mitochondrial-derived ROS, which is maintained by a feed-forward AMP kinase activation cascade, is reduced in diabetes and plays an adaptive role in preserving renal glomerular function during hyperglycemia. This enlightened view of mitochondrial ROS biology forces us to reconsider therapeutic approaches to metabolic disease complications such as diabetic nephropathy.
Diabetes management: a complicated issue
With an estimated 366 million individuals afflicted worldwide, the importance of diabetes’ effects on human health and healthcare cannot be overstated (1). In the United States alone, the direct and indirect costs are staggering. Every year in the US, 250 billion dollars, approximately 1.5% of the country’s entire gross domestic product, is spent on diabetes and its complications (2). The intrinsic cellular responses and vascular injury that arise from hyperglycemia result in a variety of costly complications such as neuropathies, retinopathies, and cardiovascular disease, which includes heart failure, stroke, myocardial infarction, and arteriosclerosis. Improvements in glycemic control alone are insufficient to fully mitigate diabetes-associated complications; therefore, clinical differences in the pathobiology of diabetic end organ complications should drive the search for adjunctive therapeutic approaches above and beyond glycemic control. For example, intensive control of both glucose and lipid intake has emerged as an effective strategy for reducing retinopathy (3), but other diabetes-associated complications such as nephropathy (4) and macrovascular disease (5) remain refractory to this focused metabolic strategy. Moreover, interactions between the metabolites produced in association with diabetes and chronic kidney disease synergize and have devastating effects on cardiovascular health (6). Innovative therapeutic approaches are necessary for diabetes treatment, and the pathways involved in mitochondrial oxidative stress have become attractive targets (7).
Oxidative stress in diabetes
It is widely appreciated that extracellular oxidative stress globally increases in both type 1 diabetes (T1D) and type 2 diabetes (T2D) (8, 9). Current opinion holds that diabetes-associated mitochondriopathy (7) directly contributes to ROS generation, as has been observed in isolated endothelial cells (10). ROS and oxylipid metabolites are critical for the microbiocidal activity of phagocytes, wound healing, and the pathobiology of several inflammatory diseases including atherosclerosis. Since ROS can induce DNA damage and both are increased in diabetes (11, 12), the accumulation of both diabetes-associated mitochondrial and genomic DNA alterations are considered to be the consequence of abnormal mitochondrial ROS production. Multiple cellular enzymes generate ROS signatures in distinct subcellular venues (13), primarily superoxide (e.g., NOX1, NOX2, mitochondrial complexes), with rapid dismutation to H2O2 or direct H2O2 elaboration (e.g., NOX4, acyl-CoA oxidase, xanthine oxidase). These local intracellular ROS signatures, which are dynamic and elicited in response to intracellular and extracellular cues, are highly regulated and critical second messengers in metabolism and signal transduction (14). The interrelationships between distinct intracellular ROS metabolism and extracellular oxidative stress are poorly understood but are of emerging importance in the pathogenesis of organ dysfunction associated with aging and chronic diseases including diabetes (14).
In this issue of the JCI, Dugan and colleagues examine the role of mitochondrial-derived superoxide in diabetic kidney disease (15). Implementing multiple validated and unconventional methods of superoxide assessment, they demonstrate that renal mitochondria in two models of T1D (streptozotocin-treated [STZ-treated] mice and Ins2Akita mice) produced reduced levels of superoxide even though total urinary ROS were increased. Diabetes-associated reductions in mitochondrial ROS resulted in the concomitant downregulation of the redox sensor AMPK along with reduced pyruvate dehydrogenase (PDH) activity and proliferator-activated receptor γ coactivator 1 α (Pgc1α) expression (Figure 1). Deficiencies in PGC1α expression and activity presumably contribute to diabetes-associated reductions in total mitochondrial density (16, 17). Even though mitochondrial ROS was reduced in diabetes, mitochondrial DNA (mtDNA) damage was increased and fully uncoupled from mitochondrial oxidative stress (15). Importantly, mtDNA damage was directly related to the diabetic state in both Ins2Akita and STZ-treated mice and unrelated to possible chemical alkylation from STZ treatment (Figure 1). Administration of the AMPK activator 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) (18) reversed AMPK inhibition, which restored renal mitochondrial numbers, ROS elaboration (15), and PGC1α induction, and reduced tissue mtDNA fragmentation (Figure 1). It is unclear how AMPK activation reduces mtDNA damage, but it likely involves enhanced mitochondrial biogenesis (via PCG1α) as well as mitochondrial turnover/mitophagy (19, 20). Albuminuria, which is a clinically relevant index of diabetic nephropathy and renal podocyte dysfunction (21), developed in both STZ and Ins2Akita T1D models and was reversed by AICAR administration (15). Finally, the authors found that renal albumin responses to AICAR were lost in mice lacking AMPK, confirming that the relief of diabetic complications by AICAR is mediated via AMPK activation. Of note, AICAR treatment globally reduced renal ROS formation while simultaneously promoting mitochondrial superoxide generation (15). These data indicate that renal mitochondrial-derived ROS are not the major source of renal oxidative stress in diabetes. This leads to a somewhat iconoclastic concept: mitochondrial-derived renal ROS, which are stimulated by AICAR and amplified by a feed-forward AMPK cascade (Figure 1), are protective in a hyperglycemic setting, and failure of mitochondrial ROS generation contributes to diabetic kidney disease (15).
Figure 1. A feed-forward cycle of AMPK-activated mitochondrial metabolism and ROS generation by the kidney reduces diabetes-induced albuminuria.
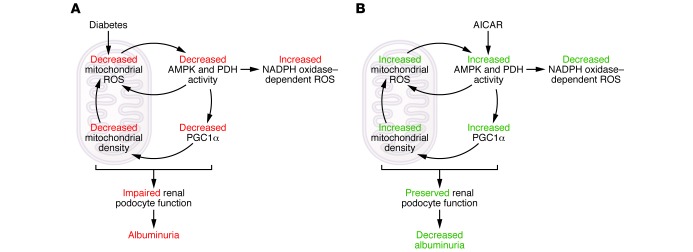
(A) Diabetes results in decreased renal mitochondrial superoxide production, which is associated with decreased AMPK and PDH activity. Decreases in AMPK and PDH activity further reduce mitochondrial ROS production directly and through decreased PGC1α, which promotes decreased mitochondrial density, ultimately resulting in impaired renal podocyte function and albuminuria. Decreased AMPK also results in increased NADPH oxidase–dependent ROS production. (B) Restoration of renal mitochondrial ROS production by treatment with the AMPK activator AICAR reduces albuminuria and total renal oxidative stress. Mitochondrially derived ROS, which is stimulated by AICAR and amplified by a feedforward AMPK cascade, is protective in the setting of hyperglycemia. The failure of mitochondrial ROS generation contributes to diabetic kidney disease. Furthermore, AMPK activation reduces NADPH oxidase–dependent ROS formation.
Reexamining the role of ROS and other diabetes-associated metabolites
Why is this study important? It forces us to reexamine our understanding of diabetic nephropathy and pathobiology as well as several other diabetes- and aging-related complications. A more detailed understanding of the mechanisms underlying diabetes-induced mtDNA damage is needed, since this antecedent genomic alteration likely contributes to impaired mitochondrial function (Figure 1). The role of mitochondria as sophisticated signaling organelles must be carefully considered when crafting diabetes-related treatment strategies (14, 22). In addition to ROS, several non-ROS metabolites that are regulated by hyperglycemia, PDH activation, and mitochondrial carbon flux, such as succinate and α-ketoglutarate, are ligands for G protein–coupled receptors found in the kidney (23, 24). Certainly, the indiscriminate “scavenging” of cellular ROS as a therapeutic approach to chronic disease may not be logical given the important role of cellular H2O2 as a second messenger in both adaptive and maladaptive responses (14). The concepts and pathways identified in ischemic preconditioning, which is a process that activates AMPK as a component of myocardial protection to subsequent hypoperfusion (25, 26), may be applicable to early diabetic kidney disease. Since mitochondrial ROS signaling couples NADPH oxidase/NOX activation with prosclerotic responses in renal mesangial cells, vascular smooth muscle cells, arterial myofibroblasts, and cardiomyocytes (27–30), the details of cell-type specificity will be important as we unravel how temporospatial alterations in ROS generation affect diabetic complications (8, 14). Additionally, the net effect of enhanced mitochondrial ROS production in one organ may dramatically differ depending on the phase of disease. For example, ROS may differentially affect events associated with diabetes initiation, such as early epithelial podocyte injury, compared with progressive complications such as advancing glomerulosclerosis (31–34).
Some challenges exist as we seek to optimally integrate these important insights into the development of therapeutic approaches to patient care. For example, mitochondrial-derived ROS participate in the vascular calcification characteristic of diabetes, uremia, and hyperphosphatemia (35). The onset of vascular calcification is related in part to the recruitment of prosclerotic signaling cascades downstream of NADPH oxidase, NOX, and NF-κB activation (36). Thus, indiscriminate activation of mitochondrial ROS signals is likely an important pitfall to avoid. Importantly, AMPK activity suppresses NOX activation (37) while sustaining mitochondrial ROS tone (15), thereby, uncoupling these two cellular ROS pathways (Figure 1). A strategy that mimics this bipartite action of AMPK activation is most likely to convey renal benefit and concomitantly mitigate arteriosclerotic vascular stiffening. An inconvenient truth that was elucidated by Zou and colleagues is that AMPK α2 activation downstream of nicotine exposure engenders aneurysm formation (38). Thus, the Aristotelian call for moderation in all things will likely apply to therapeutics targeting AMPK isoforms or upstream activators (20). Since AMPK isoforms α1 and α2 differ in cell specificity, sensitivity to ROS activation, and impact on aneurysmal remodeling, strategies that selectively target these two catalytic isoforms or their β/γ regulatory subunits may afford unique therapeutic windows (20). This potentially promising strategy has yet to be pharmacologically explored (39, 40), but has yet to be pharmacologically exploited. Moreover, in the setting of chronic kidney disease, metabolomic strategies that interrogate and integrate renal epithelial secretory and glomerular filtration functions (41) with mitochondrial metabolic markers including oxylipids (42) should be informative and guide stage-specific treatment decisions for any AMPK modulator (41). All in all, the insights afforded by the elegant study of Dugan and colleagues (15) are truly exciting and will no doubt prove extremely useful as we craft new therapeutic strategies to mitigate or prevent the end-organ complications arising in our patients with diabetes (1, 2).
Acknowledgments
D.A. Towler is supported by grants HL069229, HL081138, and HL114806 from the NIH, and the Sanford-Burnham Medical Research Institute.
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(11):4573–4576. doi:10.1172/JCI72326.
See the related article beginning on page 4888.
References
- 1.Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract. 2011;94(3):311–321. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 2.American Diabetes Association Economic costs of diabetes in the US in 2012. Diabetes Care. 2013;36(6):1033–1046. doi: 10.2337/dc12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chew EY, et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med. 2010;363(3):233–244. doi: 10.1056/NEJMoa1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coca SG, Ismail-Beigi F, Haq N, Krumholz HM, Parikh CR. Role of intensive glucose control in development of renal end points in type 2 diabetes mellitus: systematic review and meta-analysis intensive glucose control in type 2 diabetes. Arch Intern Med. 2012;172(10):761–769. doi: 10.1001/archinternmed.2011.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giorgino F, Leonardini A, Laviola L. Cardiovascular disease and glycemic control in type 2 diabetes: now that the dust is settling from large clinical trials. Ann N Y Acad Sci. 2013;1281:36–50. doi: 10.1111/nyas.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Debella YT, Giduma HD, Light RP, Agarwal R. Chronic kidney disease as a coronary disease equivalent — a comparison with diabetes over a decade. Clin J Am Soc Nephrol. 2011;6(6):1385–1392. doi: 10.2215/CJN.10271110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12(4):537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VanderJagt DJ, Harrison JM, Ratliff DM, Hunsaker LA, Vander Jagt DL. Oxidative stress indices in IDDM subjects with and without long-term diabetic complications. Clin Biochem. 2001;34(4):265–270. doi: 10.1016/S0009-9120(01)00204-1. [DOI] [PubMed] [Google Scholar]
- 9.Keaney JF, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol. 2003;23(3):434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 10.Sangle GV, Chowdhury SK, Xie X, Stelmack GL, Halayko AJ, Shen GX. Impairment of mitochondrial respiratory chain activity in aortic endothelial cells induced by glycated low-density lipoprotein. Free Radic Biol Med. 2010;48(6):781–790. doi: 10.1016/j.freeradbiomed.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Wu LL, Chiou CC, Chang PY, Wu JT. Urinary 8-OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin Chim Acta. 2004;339(1–2):1–9. doi: 10.1016/j.cccn.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Kang D, Hamasaki N. Mitochondrial oxidative stress and mitochondrial DNA. Clin Chem Lab Med. 2003;41(10):1281–1288. doi: 10.1515/CCLM.2003.195. [DOI] [PubMed] [Google Scholar]
- 13.Oakley FD, Abbott D, Li Q, Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal. 2009;11(6):1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al Ghouleh I, et al. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med. 2011;51(7):1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dugan L, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123(11):4888–4899. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung N, et al. Training-induced alterations of skeletal muscle mitochondrial biogenesis proteins in non-insulin-dependent type 2 diabetic men. Can J Physiol Pharmacol. 2012;90(12):1634–1641. doi: 10.1139/y2012-144. [DOI] [PubMed] [Google Scholar]
- 17.Choi CS, et al. Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc Natl Acad Sci U S A. 2008;105(50):19926–19931. doi: 10.1073/pnas.0810339105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sriwijitkamol A, Musi N. Advances in the development of AMPK-activating compounds. Expert Opin Drug Discov. 2008;3(10):1167–1176. doi: 10.1517/17460441.3.10.1167. [DOI] [PubMed] [Google Scholar]
- 19.Zhu J, Wang KZ, Chu CT. After the banquet: mitochondrial biogenesis, mitophagy and cell survival. Autophagy. 2013;9(11) doi: 10.4161/auto.24135. [published online ahead of print May 3, 2013]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111(6):800–814. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fogo AB. The targeted podocyte. J Clin Invest. 2011;121(6):2142–2145. doi: 10.1172/JCI57935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy MP. Modulating mitochondrial intracellular location as a redox signal. Sci Signal. 2012;5(242):pe39. doi: 10.1126/scisignal.2003386. [DOI] [PubMed] [Google Scholar]
- 23.Deen PM, Robben JH. Succinate receptors in the kidney. J Am Soc Nephrol. 2011;22(8):1416–1422. doi: 10.1681/ASN.2010050481. [DOI] [PubMed] [Google Scholar]
- 24.Toma I, et al. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest. 2008;118(7):2526–2534. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamberts RR, et al. Reactive oxygen species-induced stimulation of 5′AMP-activated protein kinase mediates sevoflurane-induced cardioprotection. Circulation. 2009;120(11 suppl):S10–S15. doi: 10.1161/CIRCULATIONAHA.108.828426. [DOI] [PubMed] [Google Scholar]
- 26.Kim AS, et al. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51(1):24–32. doi: 10.1016/j.yjmcc.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee DY, et al. Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric oxide synthase and fibronectin expression in response to angiotensin II. Role of mitochondrial reactive oxygen species. J Biol Chem. doi: 10.1074/jbc.M113.470971. [published online ahead of print August 12, 2013]. doi: 10.1074/jbc.M113.470971 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain M, et al. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem. 2013;288(2):770–777. doi: 10.1074/jbc.M112.431973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai DF, et al. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58(1):73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai CF, Shao JS, Behrmann A, Krchma K, Cheng SL, Towler DA. TNFR1-activated reactive oxidative species signals up-regulate osteogenic Msx2 programs in aortic myofibroblasts. Endocrinology. 2012;153(8):3897–3910. doi: 10.1210/en.2012-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lebleu VS, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19(8):1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Najafian B, Alpers CE, Fogo AB. Pathology of human diabetic nephropathy. Contrib Nephrol. 2011;170:36–47. doi: 10.1159/000324942. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Bertram JF. Review: Endothelial-myofibroblast transition, a new player in diabetic renal fibrosis. Nephrology (Carlton). 2010;15(5):507–512. doi: 10.1111/j.1440-1797.2010.01319.x. [DOI] [PubMed] [Google Scholar]
- 34.Steinke JM, Mauer M. Lessons learned from studies of the natural history of diabetic nephropathy in young type 1 diabetic patients. Pediatr Endocrinol Rev. 2008;5(suppl 4):958–963. [PubMed] [Google Scholar]
- 35.Shao JS, Cheng SL, Sadhu J, Towler DA. Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension. 2010;55(3):579–592. doi: 10.1161/HYPERTENSIONAHA.109.134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao MM, et al. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011;79(10):1071–1079. doi: 10.1038/ki.2011.18. [DOI] [PubMed] [Google Scholar]
- 37.Wang S, et al. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res. 2010;106(6):1117–1128. doi: 10.1161/CIRCRESAHA.109.212530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, et al. Activation of AMP-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012;18(6):902–910. doi: 10.1038/nm.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dasgupta B, et al. The AMPK β2 subunit is required for energy homeostasis during metabolic stress. Mol Cell Biol. 2012;32(14):2837–2848. doi: 10.1128/MCB.05853-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bogachus LD, Turcotte LP. Genetic downregulation of AMPK-alpha-isoforms uncovers the mechanism by which metformin decreases FA uptake and oxidation in skeletal muscle cells. Am J Physiol Cell Physiol. 2010;299(6):C1549–C1561. doi: 10.1152/ajpcell.00279.2010. [DOI] [PubMed] [Google Scholar]
- 41.Rhee EP, et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J Am Soc Nephrol. 2013;24(8):1330–1338. doi: 10.1681/ASN.2012101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiebish MA, et al. Dynamic simulation of cardiolipin remodeling: greasing the wheels for an interpretative approach to lipidomics. J Lipid Res. 2010;51(8):2153–2170. doi: 10.1194/jlr.M004796. [DOI] [PMC free article] [PubMed] [Google Scholar]