

Abstract
Chronic kidney disease is associated with progressive kidney fibrosis, which disrupts normal kidney function. There is a great need for treatments to reduce renal fibrosis. In this issue of the JCI, Ito and colleagues report the development of synthetic ligands of the vitamin D receptor that target the TGF-β–SMAD signaling pathway, which is known to regulate fibrosis-associated gene expression, without inducing VDR-associated genes. These ligands ameliorated renal fibrosis in two different mouse models. This study justifies further investigation of these and related compounds for treatment of humans with chronic kidney disease or other diseases characterized by fibrosis.
TGF-β signaling promotes chronic kidney disease
Chronic kidney disease (CKD) affects approximately 10% of the adult population in the developed world (1). A dominant feature of most forms of CKD is the development of kidney fibrosis, which results in progressive loss of kidney function, enhanced susceptibility to cardiovascular disease, and potentially end stage renal disease. CKD progression occurs even if the original cause of the kidney disease is no longer operant. Despite the worldwide prevalence of CKD, few therapeutic strategies have any impact on the prevention or treatment of fibrotic kidney disease.
One factor with a prominent role in fibrosis development in the kidney and other organs is TGF-β, which signals through the TGF-β–SMAD signaling pathway (2). TGF-β binds to cell surface type I and II serine/threonine receptor kinases, resulting in phosphorylation of SMAD2 and SMAD3, which are then released into the cytosol and bind in a complex with SMAD4. After translocation to the nucleus, the SMAD2/3/4 complex localizes to SMAD-binding elements within the genome to modulate expression of profibrotic and other target genes (Figure 1 and ref. 3). During kidney injury, a major source of TGF-β is proximal tubule epithelial cells, some of which are arrested in cell cycle phase G2/M (4). The release of TGF-β and other factors by the damaged epithelial cells and infiltrating inflammatory cells act in a paracrine fashion to activate interstitial fibroblasts/pericytes. Once activated, these interstitial cells convert to proliferative myofibroblasts and maladaptively deposit extracellular matrix, which leads to interstitial fibrosis (2, 5). Since TGF-β plays a major role in this pathophysiological response, there have been attempts to therapeutically interrupt the TGF-β signaling pathway with either small molecules or antibodies. Some of these approaches are currently in phase I and II clinical trials (6); however, there is still no accepted TGF-β–targeted therapy for kidney fibrosis.
Figure 1. DLAMs interact with the VDR and block TGF-β signaling by preventing binding to the SMAD3-binding element (SMADBE).
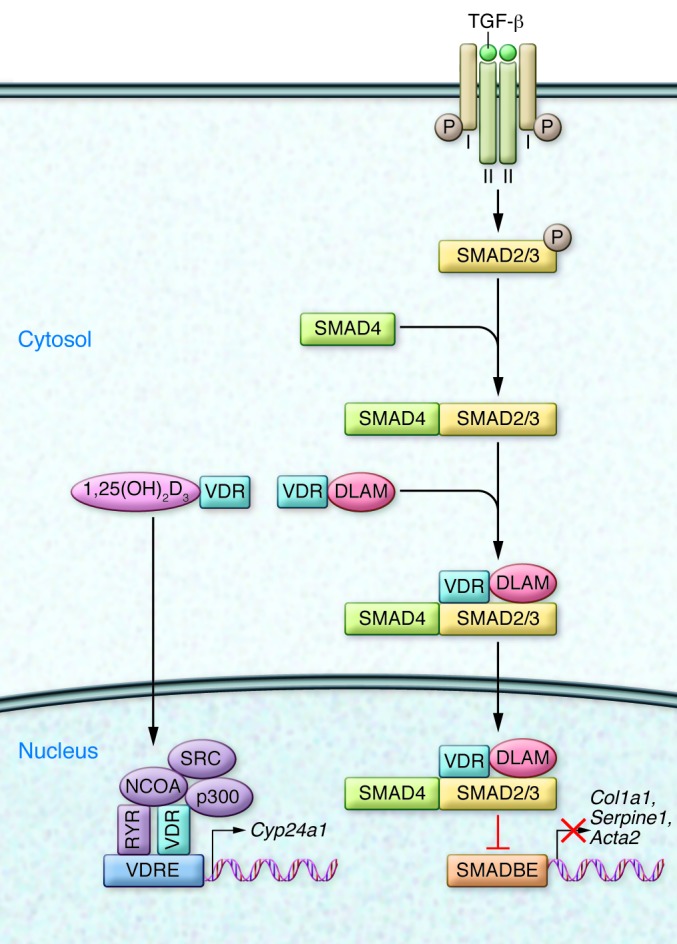
This prevention of binding prevents expression of profibrotic genes. The DLAMs do not affect classical 1,25(OH)2D3-VDR–potentiated actions on genes, such as Cyp24a1, due to interaction with VDRE. Cyp24a1 encodes 1,25(OH)2D3-24-hydroxylase, a mitochondrial enzyme that degrades 1,25(OH)2D3 and hence plays an important role in calcium homeostasis. Normally, TGF-β signals by binding to cell surface type I and type II serine/threonine receptors, which release phosphorylated SMAD3 into the cytosol, where it interacts with SMAD4, translocates to the nucleus, binds to the SMADBE in genes, and upregulates profibrotic gene expression. NCOA, nuclear coactivators; SRC, steroid receptor coactivator.
Vitamin D signaling in the kidney
Interaction of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] with the vitamin D receptor (VDR) modulates the transcription of more than 200 genes (7). The VDR forms a heterodimer with retinoid X receptors (RXRs) that together promote the recruitment of nuclear coactivators and the lysine acetyltransferase CBP/p300, which provides access for the basal transcriptional machinery through histone acetylation (Figure 1 and ref. 8). The VDR is expressed in more than 30 different tissues (9), including the kidney. VDR-RXR heterodimers interact with VDR elements (VDREs) within the genome, which are often in promoter regions of genes. Many 1,25(OH)2D3-regulated genes have pleiotropic roles, including in intestinal calcium/phosphate reabsorption and bone remodeling, which are both important aspects of calcium homeostasis.
A number of epidemiological (10) and interventional (11) studies suggest that vitamin D may mitigate albuminuria or prevent progression of kidney dysfunction. Additionally, many laboratory studies have identified a large number of vitamin D–regulated cellular and tissue processes that could potentially suppress renal fibrosis and benefit kidney structure and function.
Targeting VDR to prevent TGF-β–SMAD–induced kidney fibrosis
In this issue of the JCI, Ito et al. evaluated inhibition of VDR signaling as a therapeutic strategy for suppressing renal fibrosis development (12). They found that 1,25(OH)2D3-bound VDR inhibits the TGF-β–SMAD signaling pathway directly through interaction with SMAD3. In a mouse model of unilateral ureteral obstruction (UUO), which is well known for its rapid development of fibrotic kidney disease, treatment with 1,25(OH)2D3 markedly reduced TGF-β–stimulated profibrotic gene expression and fibrosis development. Fibrosis was inhibited despite the observation that this treatment did not prevent the UUO-induced increase in TGF-β and subsequent increase in SMAD2 and SMAD3 phosphorylation. In murine renal epithelial cells stimulated with TGF-β, 1,25(OH)2D3 treatment did not affect SMAD2 or SMAD3 phosphorylation or their translocation to the nucleus, but did mitigate the upregulation of a number of TGF-β target genes implicated in fibrosis. ChiP assays revealed that the TGF-β–enhanced interaction between SMAD3 and the Serpine1 (encoding plasminogen activator inhibitor–1 [PAI1]) or Acta2 (encoding α-SMA) promoters was markedly suppressed in the presence of 1,25(OH)2D3. Binding of SMAD3 to the SMAD3 DNA-binding element was blocked by the presence of the VDR-ligand binding domain and 1,25(OH)2D3.
Ito and colleagues generated two synthetic analogs of 1,25(OH)2D3 that interact with the VDR and inhibit TGF-β–induced SMAD signaling without activating classical VDR–mediated genes, thus preventing the development of hypercalcemia. The crystal structure of the VDR bound to 1,25(OH)2D3 was previously solved and revealed that 1,25(OH)2D3 associates with helix 12 (H12) to stabilize VDR, which facilitates interactions with coactivators (13). Removal of H12 not only prevented VDR-dependent transcriptional activity, but also abolished inhibition of TGF-β–dependent transcription; therefore, this region of H12 is important for interaction with SMAD3. Since the physiological 1,25(OH)2D3 analog, 1α,25-dihydroxyvitamin D3-26,23-lactone (1,25-lactone), has a γ-butyrolactone ring, the authors proposed that it could affect interaction with the H12 region differently than 1,25(OH)2D3. 1,25-lactone had no effect on TGF-β–induced SMAD3 phosphorylation, but blocked TGF-β signaling; however, this molecule did have some agonistic effect on VDR-mediated transcription of Cyp24a1 [encoding 1,25(OH)2D3-24-hydroxylase]. The authors found that 1,25-lactone in complex with the ligand-binding domain of VDR associates differently with residues His305 and His397 than does 1,25(OH)2D3. Based on the structure of 1,25-lactone and its effect on VDR activity, Ito et al. designed 1α,25-dihydroxyvitamin D3-26,23-lactam (DLAM) derivatives, which are analogs of 1,25-lactone that mimic VDR ligands (14). DLAM candidates were chosen for their ability to alter the configuration of the H12 region.
In the UUO model, the authors found that administration of DLAMs reduced the level of SMAD3-dependant mRNAs, but did not increase Cyp24a1 mRNA levels (12). DLAM treatment reduced mRNA levels of Serpine1, Acta2, and Col1a1 (encoding type I collagen) in the obstructed kidney, and ChIP analysis revealed suppressed binding of SMAD3 to the Serpine1 promoter after DLAM treatment. Thus, TGF-β–SMAD signaling was inhibited by DLAM treatment, but other actions of the VDR were not stimulated (Figure 1). The authors also found that serum calcium levels were unaffected by DLAM treatment.
Evaluation of paracalcitol, a VDR agonist used to treat secondary hyperparathyroidism in CKD and known to produce less hypercalcemia than 1,25(OH)2D3, revealed that unlike with DLAM treatment, Cyp24a1 expression was increased. While paracalcitol decreased mRNA levels of Col1a1 and Acta2, it did not decrease the TGF-β target gene Serpine1, in obstructed kidneys. Furthermore, in contrast to DLAM, paracalcitol increased serum calcium levels considerably. DLAM treatment in a second mouse model of renal fibrosis, folic acid nephropathy, had similar effects to those found in the UUO model. These included decreased levels of Serpine1, Acta2, and Col1a1 mRNA and no change in VDR-dependent mRNAs. Additionally, progressive fibrosis and renal failure were inhibited in DLAM-treated mice.
A recent important related study demonstrated that treatment with a synthetic VDR agonist, calcipotriol, is protective against liver fibrosis and does not cause hypercalcemia (15). Ding et al. found that TGF-β causes redistribution of genome-wide VDR-binding sites in hepatic stellate cells, which are comparable to interstitial fibroblasts/pericytes in the kidney. In the presence of calcipotriol, SMAD3 occupancy on profibotic genes is reduced; therefore, carbon tetrachloride–treated mice exhibit less liver fibrosis in the presence of calcipotriol.
Implications and future directions
The study by Ito et al. (12) provides intriguing promise for kidney fibrosis treatment and prevention. The authors made informative distinctions between TGF-β–dependent effects on profibrotic gene expression and classical 1,25(OH)2D3-VDR–dependent transcription to motivate development of antifibrotic agents that do not cause hypercalcemia in mice. In particular, they applied sophisticated knowledge of the VDR structure and its interaction with 1,25(OH)2D3 and a naturally occurring analog, 1,25-lactone (16), to derive compounds with anti–TGF-β activity that do not promote the expression of VDR-dependent genes. It is possible that these or related agents may serve to reduce fibrosis in humans.
A word of caution is indicated, however. Protective effects of 1,25(OH)2D3 in humans have been reported in large epidemiological studies (17) and may relate to VDR-mediated responses that are unrelated to TGF-β mitigation. 1,25(OH)2D3 has also been reported to have important effects on innate and adaptive immunity as well as protective effects on the cardiovascular system, cancer susceptibility, and neurocognitive function (18). Therefore, an agent specifically targeting the TGF-β pathway may prove ineffective if other VDR-mediated effects of 1,25(OH)2D3 are more important or are necessary to complement the anti–TGF-β effects of treatment. Additionally, it is unknown whether other noncalcemic gene targets of 1,25(OH)2D3 are stimulated by DLAMs, as the specificity of the DLAMs used to block TGF-β signaling have not been evaluated beyond the lack of effects on Cyp24a1 transcription and the calcemic effect in animals.
An interesting aspect of this work is the characterization of the action of the 1,25(OH)2D3 physiological analog 1,25-lactone. It is possible that 1,25-lactone represents an endogenous fibrosis inhibitor in organs, including the kidney, heart, lung, and liver. Could subjects with CKD have reduced levels of 1,25-lactone, which would exacerbate the detrimental profibrotic effects attributed to low levels of 1,25(OH)2D3? Are 1,25-lactone levels lower in African-Americans, who, as a group, have high incidence of CKD and albuminuria and lower levels of circulating vitamin D [25(OH)D] (19)? This study provides an elegant approach for systematically developing therapeutic targets for disease treatment. Is it possible that this general approach to developing targeted ligands will be effective in identifying specific beneficial analogs for other steroid hormones that are important therapeutically. Analogs of glucocorticoids that preserve beneficial effects while minimizing detrimental effects would be of great benefit in a number of diseases, including diseases of the kidney.
Acknowledgments
J.V. Bonventre is supported by NIH awards DK 39773 and DK 72381.
Footnotes
Conflict of interest: The author or his family hold equity interests in Patientkeeper, AMAG, Pacific Biosciences, MediBeacon, Theravance, Sentien, DxNow, and DRP. Joseph V. Bonventre is a consultant for Janssen RND, Keryx, and editor of Seminars In Nephrology. Joseph V. Bonventre holds patents on KIM-1, which have been assigned to Partners Healthcare which has licensed them to Sekisui, Novartis, Johnson and Johnson, BiogenIdec, and a number of research reagent providers. He has received research support from NovoNordisk.
Citation for this article: J Clin Invest. 2013;123(11):4570–4573. doi:10.1172/JCI72748.
See the related article beginning on page 4579.
References
- 1.Eckardt KU, et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet. 2013;382(9887):158–169. doi: 10.1016/S0140-6736(13)60439-0. [DOI] [PubMed] [Google Scholar]
- 2.Kaissling B, Lehir M, Kriz W. Renal epithelial injury and fibrosis. Biochim Biophys Acta. 2013;1832(7):931–939. doi: 10.1016/j.bbadis.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlberg C, Seuter S. A genomic perspective on vitamin D signaling. Anticancer Res. 2009;29(9):3485–3493. [PubMed] [Google Scholar]
- 8.Sutton AL, MacDonald PN. Vitamin D: more than a “bone-a-fide” hormone. Mol Endocrinol. 2003;17(5):777–791. doi: 10.1210/me.2002-0363. [DOI] [PubMed] [Google Scholar]
- 9.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Boer IH, Ioannou GN, Kestenbaum B, Brunzell JD, Weiss NS. 25-Hydroxyvitamin D levels and albuminuria in the Third National Health and Nutrition Examination Survey (NHANES III). Am J Kidney Dis. 2007;50(1):69–77. doi: 10.1053/j.ajkd.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 11.de Zeeuw D, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet. 2010;376(9752):1543–1551. doi: 10.1016/S0140-6736(10)61032-X. [DOI] [PubMed] [Google Scholar]
- 12.Ito I, et al. A nonclassical vitamin D receptor pathway suppresses renal fibrosis. J Clin Invest. doi: 10.1172/JCI67804. doi: 10.1172/JCI67804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5(1):173–179. doi: 10.1016/S1097-2765(00)80413-X. [DOI] [PubMed] [Google Scholar]
- 14.Nakano Y, et al. Practical synthesis and evaluation of the biological activities of 1α,25-dihydroxyvitamin D3 antagonists, 1alpha,25-dihydroxyvitamin D3-26,23-lactams. Designed on the basis of the helix 12-folding inhibition hypothesis. J Med Chem. 2006;49(8):2398–2406. doi: 10.1021/jm050738x. [DOI] [PubMed] [Google Scholar]
- 15.Ding N, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153(3):601–613. doi: 10.1016/j.cell.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishizuka S, Ohba T, Norman AW. Vitamin D: Molecular, Cellular, And Clinical Endocrinology. 1988. 1α,25(OH)2D3-26,23-Lactone is a major metabolite of 1α,25(OH)2D3 under physiological conditions. In: Norman AW, Schaefer K, Grigoleit HG, Herrath DV, eds. ; pp. 143–144. Berlin, Germany: Walter de Gruyter; [Google Scholar]
- 17.Li YC. Vitamin D receptor signaling in renal and cardiovascular research. Semin Nephrol. 2013;33(5):433–447. doi: 10.1016/j.semnephrol.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pludowski P, et al. Vitamin D effects on musculoskeletal health, immunity, autoimmunity, cardiovascular disease, cancer, fertility, pregnancy, dementia and mortality-a review of recent evidence. Autoimmun Rev. 2013;12(10):976–989. doi: 10.1016/j.autrev.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Essien E, Goel N, Melamed ML. Role of vitamin D receptor activation in racial disparities in kidney disease outcomes. Semin Nephrol. 2013;33(5):416–424. doi: 10.1016/j.semnephrol.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]