

Abstract
The DNA damage-responsive protein kinases ATM and ATR phosphorylate SQ/TQ motifs that lie in clusters in most of their in vivo targets. Budding yeast Esc4p contains a cluster of SQ/TQ motifs, suggesting that it might be a target of Mec1p/Tel1p (yeast ATR/ATM). Here it is reported that Esc4p is phosphorylated by Mec1p in response to DNA damage during DNA replication and that cells lacking Esc4p are hypersensitive to DNA damage specifically during S phase. Esc4p is not required for the intra-S-phase checkpoint but is essential for resumption of chromosome replication after DNA damage, and its role in promoting restart appears to be distinct from that of Rad53p. Mutation of Esc4p SQ/TQ motifs phosphorylated by Mec1p or mutation of the BRCT domains of Esc4p also renders cells unable to restart DNA replication after DNA damage and causes hypersensitivity to genotoxins. These results identify Esc4p as an important new S-phase-specific target of Mec1p.
Keywords: ATR, DNA damage, Esc4p, Mec1p
Introduction
Cells have complex surveillance systems aimed at eliminating DNA damage and at rescuing DNA replication forks that stall at sites of damage. If DNA lesions are not immediately repaired, the cell initiates a complex series of events aimed at repairing them as quickly as possible. This helps to prevent changes in the genome that could deregulate cell function (Rouse and Jackson, 2002; Shiloh, 2003). Cells in S phase are at high risk from the deleterious effects of DNA damage (McGlynn and Lloyd, 2002) as DNA adducts can block fork progression and DNA synthesis. Replication fork stalling or replication through nicks or gaps can generate DNA double-strand ends that trigger inappropriate recombination events thereby undermining genome stability (McGlynn and Lloyd, 2002).
In budding yeast, Mec1p and Tel1p are important regulators of a group of proteins that detect, signal and repair DNA damage (Carr, 2002; Rouse and Jackson, 2002). Disruption of MEC1 causes cell lethality, and mec1 mutations cause hypersensitivity to agents that cause DNA damage. Unlike wild-type cells, mec1Δ cells do not slow down cell cycle progression in response to DNA damage (Weinert et al, 1994; Paulovich and Hartwell, 1995). Although telΔ cells are not sensitive to DNA damage, mec1ΔtelΔ cells are more sensitive than mec1Δ cells, indicating that Mec1p and Tel1p may function redundantly (Abraham, 2001). The mammalian orthologues of Mec1p and Tel1p are ATR and ATM, respectively (Abraham, 2001; Shiloh, 2003). Mutations in ATM (ataxia telangiectasia (A-T)-mutated) cause A-T, a debilitating condition characterised by cellular hypersensitivity to double-strand breaks that predisposes sufferers to cancer (Shiloh, 2003). ATR (ATM- and Rad3-related) is an essential gene; loss of ATR is accompanied by a high degree of genome instability (Shiloh, 2003).
Stalled replication forks and DNA lesions trigger Mec1p/Tel1p-dependent phosphorylation of a variety of proteins. Phosphorylation of the Chk1p and Rad53p (Chk2p) protein kinases, for example, increases their activity towards cell cycle regulators (Shiloh, 2003). ATM/ATR phosphorylate Ser/Thr–Gln (SQ/TQ) motifs (Kim et al, 1999; O'Neill et al, 2000) that lie in clusters of between three and six consecutive motifs in most, but not all, of their in vivo targets (Figure 1A). Searching the Saccharomyces Genome Database revealed several proteins with SQ/TQ clusters, including Esc4p (Figure 1B), also known as Yhr154w and RTT107 (Scholes et al, 2001).
Figure 1.
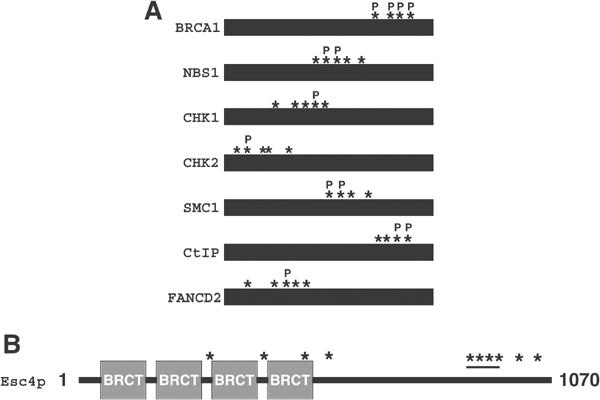
SQ/TQ motifs phosphorylated by ATR/ATM lie in clusters. (A) Schematic representation of a selection of proteins phosphorylated by ATR and/or ATM. Asterisks represent SQ/TQ motifs. ‘P' symbols denote reported in vivo ATM/ATR phosphorylation sites. (B) Schematic representation of Esc4p. SQ/TQ motifs are represented by asterisks and the SQ/TQ cluster between amino acids 743 and 807 is underlined. BRCT domains are also highlighted. One database suggests a cryptic BRCT domain at the C-terminus but this is not shown.
Esc4p has four N-terminal BRCT domains (Figure 1B), small modules of around 100 amino acids found mainly in proteins involved in signalling/repair of DNA damage or cell cycle regulation (Bork et al, 1997; Callebaut and Mornon, 1997). In this light, several studies point to a role for Esc4p in DNA damage responses. Genome-wide analysis demonstrated that cells lacking ESC4, although viable, are hypersensitive to the DNA alkylating agent methylmethanesulphonate (MMS), but not to ultraviolet (UV) or ionising radiation (IR) (Chang et al, 2002; Hanway et al, 2002). Another global screen revealed that deletion of ESC4 kills cells lacking SGS1 (Tong et al, 2001). A global high-throughput protein–protein interaction screen showed that Esc4p interacts with Mms22p, a protein of unknown function that is required for viability in the presence of MMS (Ho et al, 2002). ESC4, along with several components of the Mec1p signalling pathway, was also identified in a screen for genes affecting retrotransposition (Scholes et al, 2001).
Here I describe Mec1p-dependent phosphorylation of Esc4p in response to DNA damage during DNA replication. Esc4p is required to survive DNA damage in S phase but is not required for the intra-S-phase checkpoint. Instead, Esc4p promotes restart of DNA synthesis after DNA damage in S phase but, unlike Rad53p, is not required to prevent irreversible fork catastrophe when forks stall initially. Mutating Mec1p phosphorylation sites in Esc4p results in hypersensitivity to DNA damage and prevents completion of chromosome replication after DNA damage. These findings identify Esc4p as an important new target of Mec1p that promotes genome stability during S phase.
Results
DNA damage triggers Mec1p-dependent Esc4p phosphorylation
To investigate Esc4p phosphorylation, a MYC(13) epitope tag was placed on endogenous Esc4p. Cells were treated with genotoxins, lysed and anti-MYC immunoprecipitates were subjected to Western blot analysis with an antibody that has been reported to recognise phosphorylated SQ/TQ motifs (Schwartz et al, 2002; Lee et al, 2003). As shown in Figure 2A, MYC(13)-Esc4p was recognised by the anti-phospho-SQ/TQ antibody after treating cells with camptothecin (CPT), MMS or hydroxyurea (HU), but not after treatment with IR, UV or the UV-mimetic drug 4-nitroquinoline oxide (4-NQO).
Figure 2.
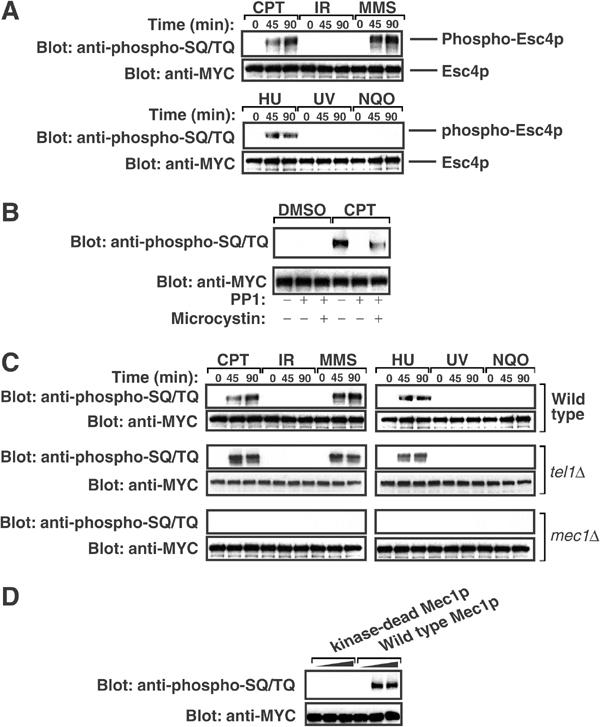
DNA damage stimulates Mec1p-dependent phosphorylation of Esc4p. (A) Strain JRY100 (MYC(13)-ESC4) was grown to mid-log phase in liquid culture and incubated in the presence of CPT (5 μg/ml), MMS (0.02%), HU (50 mM) or 4-NQO (5 μg/ml) for the indicated times. Alternatively, cells were exposed to IR (150 Gy) or UV (50 J/m2) and incubated at 30°C for the indicated times. Native extracts were prepared and aliquots (8 mg) were subjected to immunoprecipitation with anti-MYC antibodies. Most (90%) of each immunoprecipitate was subjected to Western blot analysis with the anti-phospho-SQ/TQ antibody and the remainder was blotted with anti-MYC. (B) Strain JRY100 (MYC(13)-ESC4) was grown to mid-log phase in liquid culture and incubated with DMSO (0.2%) or CPT (5 μg/ml) for 90 min. Native extracts were prepared and anti-MYC immunoprecipitates were washed extensively and either left untreated or incubated with the catalytic subunit of PP1 (5 mU/ml) for 30 min in the presence or absence of the phosphatase inhibitor microcystin-LR (1 μM). Beads were then subjected to Western blot analysis with the indicated antibody as in (A). (C) Strains JRY100 (MYC(13)-ESC4), JRY104 (MYC(13)-ESC4 mec1Δsml1-1) and JRY105 (MYC(13)-ESC4 tel1Δsml1-1) were subjected to the same analysis as in (A). (D) MYC(13)-Esc4p was immunoprecipitated from cell extracts (15 mg), washed extensively and eluted from the affinity resin with c-MYC peptide (EQKLISEEDL; 0.1 mg/ml). The peptide was removed by dialysis. Increasing amounts of wild-type or kinase-dead forms of HA-tagged Mec1p were immunoprecipitated from YLL476.34/2C and YLL593.1.3 cells, respectively (Paciotti et al, 2000), and the beads were incubated with Esc4p in the presence of magnesium chloride (10 mM), manganese chloride (10 mM) and ATP (0.1 mM) for 60 min at 30°C. Esc4p was then subjected to Western blot analysis with indicated antibodies as in (A).
To confirm the specificity of the anti-phospho-SQ/TQ antibody, Esc4p immunoprecipitated from extracts of CPT-treated cells was incubated with protein phosphatase 1 (PP1). This abolished recognition of Esc4p by the anti-phospho-SQ/TQ antibody but not in the presence of the phosphatase inhibitor microcystin-LR (Figure 2B). Similar results were obtained with Esc4p from cells treated with MMS or HU (data not shown). This confirms that Esc4p becomes phosphorylated when DNA damage occurs. The role of Mec1p and Tel1p in Esc4p phosphorylation was examined. Whereas CPT, MMS and HU stimulated Esc4p phosphorylation in wild-type and tel1Δ cells, no detectable SQ/TQ phosphorylation of Esc4p was observed in cells lacking Mec1p (Figure 2C) even after 4 h of treatment (data not shown). This suggested that Esc4p is phosphorylated directly by Mec1p. To test this, MYC(13)-Esc4p from untreated cells was incubated with Mec1p in the presence of Mg2+-ATP. Esc4p incubated with wild-type, but not kinase-dead, Mec1p was recognised by the anti-phospho-SQ/TQ antibody (Figure 2D). Taken together, these data indicate that Esc4p is phosphorylated at one or more SQ/TQ motif in a Mec1p-dependent, Tel1p-independent manner in response to certain types of DNA damage.
Esc4p is required to survive DNA damage specifically during S phase
It was shown previously that cells lacking Esc4p lose viability when chronically exposed to MMS (Chang et al, 2002; Hanway et al, 2002). I compared the ability of esc4Δcells and mec1Δ to tolerate acute treatment with different genotoxins. Cells lacking Esc4p are exquisitely hypersensitive to CPT and to high concentrations of MMS and HU, but not to IR (Figure 3A) or UV light (data not shown) in different genetic backgrounds (data not shown). esc4Δmec1Δ double mutant cells are not more sensitive to CPT, MMS or HU than the most sensitive of the respective single mutants (Figure 3B), suggesting that ESC4 and MEC1 act in the same pathway to promote cell survival in the presence of these agents.
Figure 3.
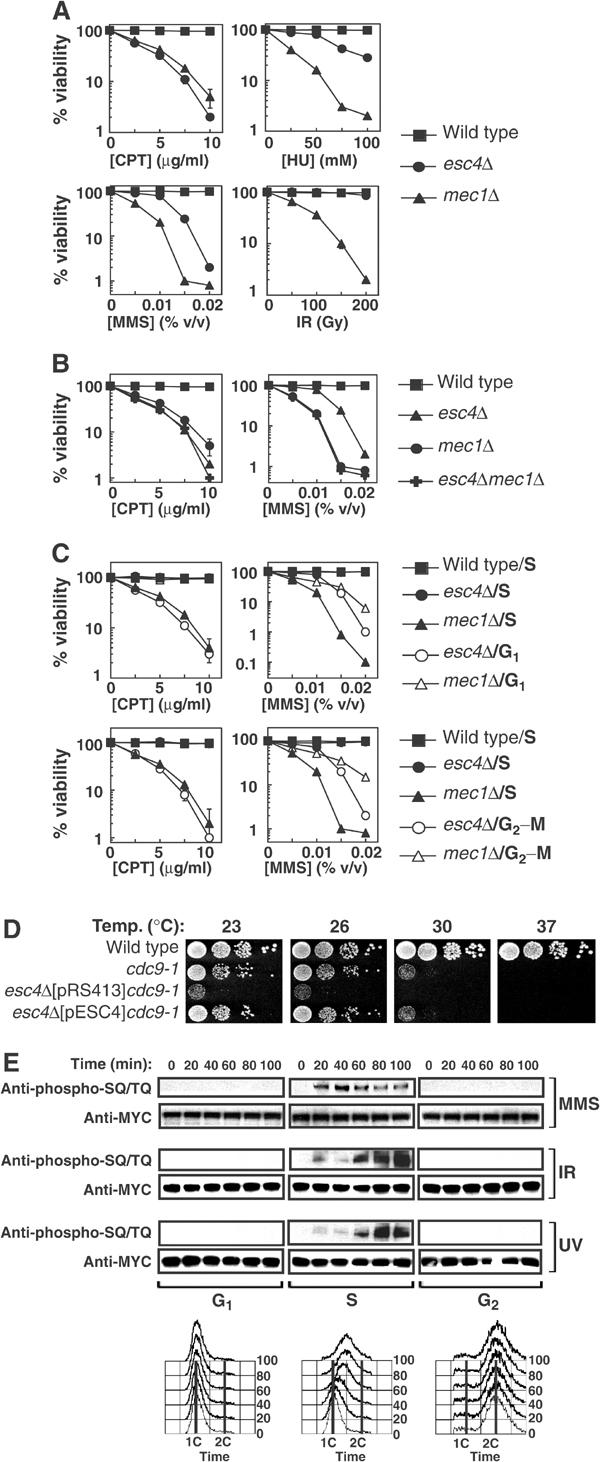
Esc4p responds to DNA damage specifically during DNA replication. (A, B) Strains W303-1A (wild-type), JRY99 (esc4Δ), U960-5C (mec1Δ sml1-1) (A, B) and JRY103 (esc4Δmec1Δ sml1-1) (B) were grown to early log phase in liquid culture. The relevant drug was added at the indicated concentrations in the case of CPT, MMS and HU, and the cells were grown at 30°C for a further 3.5 h. Cell suspensions were diluted 100-fold, plated on YPD-agar and incubated for 3 days at 30°C before counting viable colonies. In the case of IR, diluted cells were irradiated with a Cs137 source at a delivery rate of 2.8 Gy/min, before spreading on YPD-agar. The average of five independent experiments is shown. (C) Strains W303-1A (wild-type), JRY99 (esc4Δ) and U960-5C (mec1Δsml1Δ) were grown to early log phase and arrested in G1 by addition of alpha-factor (5 μg/ml). Cells were then released from arrest into fresh medium (‘S') and incubated for 10 min at 30°C before addition of CPT or MMS at the indicated concentrations for 120 min. Alternatively, cells were incubated in fresh medium containing alpha-factor to hold cells in G1 during treatment with CPT and MMS (‘G1'). In other experiments, cells were held at the G2–M boundary by incubation with nocodazole (15 μg/ml) for 1.5 h before addition of CPT or MMS in the continued presence of nocodazole (‘G2–M'). After exposure to genotoxins, cells were washed and spread on YPD-agar and colonies were counted 3 days later. (D) Strains A364a (wild-type), DLY1080 (cdc9-1) and JRY102 (esc4Δcdc9-1) transformed with pRS413 (empty vector) or with pESC4 were grown to mid-log phase and 10-fold serial dilutions were spotted on YPD-agar and incubated at the indicated temperature for 2–4 days before the number of viable colonies was determined. (E) Cells expressing MYC(13)-Esc4p (strain JRY100) were grown to mid-log phase, arrested in G1 and released into S phase in the presence of MMS (0.02% v/v) for the times indicated or maintained in alpha-factor to hold cells in G1 during MMS treatment. Alternatively, cells were arrested at the G2–M boundary by addition of nocodazole and held there during treatment with MMS. Cells were also exposed to UV light (50 J/m2) or IR (150 Gy) and incubated at 30°C for the times indicated while held in G1 or at G2–M or released into S phase. Native extracts were prepared and anti-MYC immunoprecipitates were subjected to Western blot analysis. Samples of cells were also fixed in 70% ethanol and subjected to FACS analysis.
Hypersensitivity to CPT, MMS and HU, but not UV and IR, is usually indicative of a defect in DNA damage responses during S phase. CPT inhibits DNA topoisomerase I (Top1p), an enzyme that nicks DNA at regions of excess positive supercoiling (Wang, 2002). Top1p binds covalently to one end of the nick and CPT traps this DNA–Top1p intermediate, preventing religation. Formation of CPT–Top1p–DNA adducts is reversible but double-strand breaks are generated when replication forks collide with these structures (Wang, 2002). The major lesion, N3-methlyadenine, induced by MMS potently impedes progression of DNA replication forks (Beranek, 1990). HU also stalls replication forks by depleting cellular dNTP pools. Consequently, I tested whether the role of Esc4p is restricted to S phase. As shown in Figure 3C (top panels), release from G1 into S phase in the presence of CPT or MMS results in loss of viability of esc4Δ or mec1Δ cells but not of wild-type cells. However, holding cells in G1 with alpha-factor (Figure 3C, top panels) or at the G2–M boundary with nocodazole (Figure 3C, bottom panels) prevented CPT- or MMS-induced loss of viability of esc4Δ cells. As reported previously (Tercero and Diffley, 2001), holding mec1Δ cells outside of S phase only partly rescued MMS-induced loss of viability (Figure 3C, right panels).
When cells harbouring a conditional allele of DNA ligase I (cdc9-1) are grown at the nonpermissive temperature, Okazaki fragments generated during DNA replication are not efficiently ligated (Johnston and Nasmyth, 1978), resulting in Mec1p-dependent cell cycle arrest (Weinert and Hartwell, 1993). Disruption of ESC4 in cells harbouring the cdc9-1 mutation results in a significant reduction in growth rate at the permissive temperature (Figure 3D) and these cells lose viability after less than 10 generations (data not shown). Shifting cdc9–1esc4Δ cells to higher temperatures resulted in a greater degree of cell death compared with the cdc9-1 single mutant (Figure 3D).
The cell cycle dependence of DNA damage-stimulated Esc4p phosphorylation was also examined. When cells were held in G1 or at the G2–M boundary during treatment with MMS, IR or UV, no increase in Esc4p SQ/TQ phosphorylation was observed (Figure 3E). In contrast, when cells were released from G1 arrest into S phase, Esc4p became rapidly phosphorylated in response to all of these agents (Figure 3E). This was surprising because Esc4p immunoprecipitated from extracts of asynchronously growing cells treated with UV or IR was not recognised by the anti-SQ/TQ antibody (Figure 2A; see Discussion). Taken together, these data indicate that Esc4p plays an important role in responding to DNA damage during S phase.
Esc4p is required for completion of chromosome replication after DNA damage but not for cell cycle arrest
The above results prompted investigation of which aspect(s) of the DNA damage response is defective in esc4Δ cells. DNA lesions and nucleotide depletion cause a Mec1p-dependent decrease in the rate of bulk DNA synthesis, often referred to as the intra-S-phase checkpoint (Paulovich and Hartwell, 1995; Paulovich et al, 1997). It has been reported that Esc4p is not required for this checkpoint (Chang et al, 2002), in agreement with experiments carried out in this laboratory (data not shown). One group has reported that esc4Δ cells move even more slowly through S phase than wild-type cells in MMS (Chang et al, 2002). It was also reported that Esc4p does not function in the same epistasis group as Rad9p or Rad24p, which are required for the DNA damage checkpoint (Hanway et al, 2002). Consistent with these observations, I found that Mec1p-mediated phosphorylation and activation of Rad53p, required for the intra-S-phase checkpoint (Schwartz et al, 2002; Lee et al, 2003), occurs independently of Esc4p. As shown in Figure 4A, Rad53p became phosphorylated (blot panels) and activated (column panels) when wild-type cells or esc4Δ cells were released from G1 into S phase in the presence of MMS or HU and weakly in the presence of CPT. Thus, unlike Mec1p (Figure 4A), Esc4p is not required for genotoxin-induced activation of Rad53p or for the intra-S-phase checkpoint.
Figure 4.
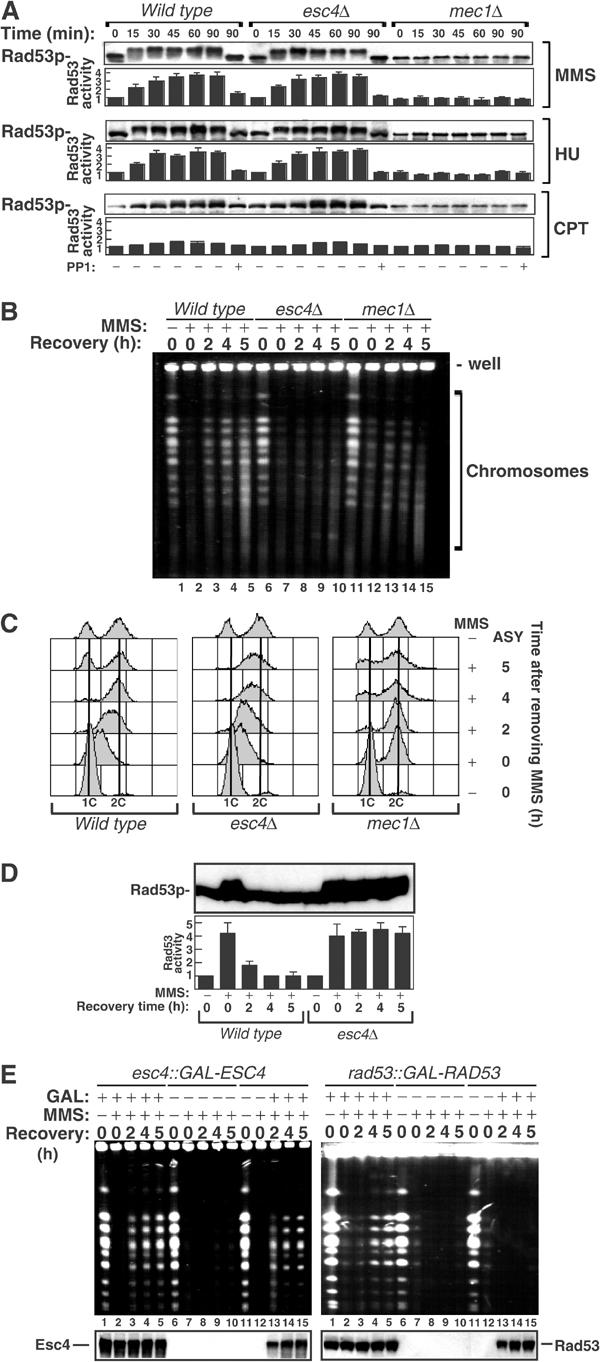
Esc4p is required for resumption of chromosome replication after DNA damage. (A) Strains W303-1A, JRY99 (esc4Δ) and U960-5C (mec1Δsml1Δ) were grown to mid-log phase in liquid culture, arrested in G1 with alpha-factor and released from arrest into fresh medium. After incubation for 10 min at 30°C, MMS (0.006%), CPT (5 μg/ml) or HU (20 mM) was added. After the times indicated, cells were lysed and extracts were subjected to Western blotting with anti-Rad53p antibodies (blot panels). Alternatively, histone H1 kinase activity in anti-Rad53p immunoprecipitates was measured (column panels). At the 90 min time point, an aliquot of each extract was treated with PP1 before Western blotting or immunoprecipitation. (B) Strains W303-1A, JRY99 (esc4Δ) and U960-5C (mec1Δsml1Δ) were grown to mid-log phase in liquid culture, arrested in G1 with alpha-factor (lanes 1, 6 and 11) and released from arrest into fresh medium for 10 min at 30°C before addition of MMS (0.033%). After 60 min in MMS (lanes 2, 7 and 12), cells were washed in fresh medium containing sodium thiosulphate (2.5% w/v), then in YPD and incubated in YPD at 30°C. Samples (108 cells) were taken at each stage and at 2, 4 and 5 h later. Chromosomes were prepared, subjected to PFGE and stained with ethidium bromide as described in Materials and methods. (C) Same as (B), except that fixed cells were subjected to FACS analysis as described in Materials and methods. ASY denotes asynchronously growing cells. (D) Same as (B) except that cells were not fixed but were lysed and extracts were subjected to Western blotting with anti-Rad53p antibodies (upper panel). Alternatively, histone H1 kinase activity in anti-Rad53p immunoprecipitates was measured (lower panel). (E) Strains JRY113 (esc4::GAL-ESC4) or JRY114 (rad53::GAL-RAD53) were subjected to the same analysis as described in (B) with the following modifications. Cells were grown to mid-log phase in YPD-raffinose and then switched to YPD-galactose (GAL; lanes 1–5) or glucose (lanes 6–10) for alpha-factor arrest and the rest of the experiment. Alternatively, cells were grown to mid-log phase in YPD-raffinose, maintained in this medium during G1 arrest and MMS treatment and switched to YPD-galactose after MMS had been quenched and washed away (lanes 11–15). The lower panels show Western blot analysis of cell extracts to monitor expression of Esc4p (left panel) and Rad53p (right panel) during the experiment.
The above data indicate that the genotoxin hypersensitivity of cells lacking Esc4p is not due to a checkpoint defect and prompted examination of recovery of DNA replication after DNA damage. Cells were arrested in G1 and released into S phase in the presence of MMS. After 60 min, MMS was removed and cells were incubated in fresh medium to allow repair/recovery. Chromosomes were prepared from wild-type, esc4Δ and mec1Δ cells at different times during recovery and separated by pulsed field gel electrophoresis (PFGE). These gels resolve the 16 yeast chromosomes into a characteristic ladder of bands following ethidium bromide staining (Figure 4B, lanes 1, 6 and 11). However, incompletely replicated chromosomes fail to enter a pulsed field gel because of the presence of forks and replication bubbles that impede migration (Hennessy et al, 1991; Desany et al, 1998). When wild-type cells are released into S phase in the presence of MMS, chromosomes are not completely replicated 60 min later (data not shown) and consequently do not enter the pulsed field gel (Figure 4B, lanes 2, 7 and 12). When MMS is removed and cells are allowed to recover, damage is repaired and DNA synthesis is completed in wild-type cells enabling chromosomes to re-enter the gel eventually (Figure 4B, lanes 3–5). However, chromosomes from cells lacking Esc4p do not re-enter the pulsed field gels even 5 h after recovery (Figure 4B, lanes 8–10). Similar results were obtained with cells lacking Mec1p (Figure 4B, lanes 11–15) and when MMS was replaced by HU (data not shown). To confirm these data, bulk DNA synthesis was measured by FACS analysis during recovery from DNA damage. When MMS is removed from S-phase cells lacking Esc4p, DNA replication continues, but more slowly than in wild-type cells (Figure 4C). After 4 h of recovery, however, when wild-type cells have completed DNA replication, cells lacking Esc4p are still in late S phase and do not progress further (Figure 4C). Thus, cells are stuck in late S phase, confirming that Esc4p is required for completion of bulk DNA synthesis after DNA damage.
Recovery of stalled replication forks and repair of DNA damage are accompanied by downregulation of Mec1p signalling and inactivation of Rad53p (Pellicioli et al, 1999; Leroy et al, 2003). As shown in Figure 4D, the levels of phosphorylated (upper panel), active (lower panel) Rad53p decrease dramatically 2 h after removal of MMS from wild-type cells, and after 4 h Rad53p phosphorylation is undetectable and its activity is low (Figure 4D). However, in cells lacking Esc4p, Rad53p is still highly phosphorylated and active at least 5 h after removal of MMS (Figure 4D), suggesting the continued presence of aberrant structures, maybe stalled forks, that trigger Mec1p signalling.
Rad53p, like Esc4p, is required for resumption of DNA synthesis in cells released from HU (Desany et al, 1998) and this reflects its role in preventing irreversible catastrophe when replication forks stall (Tercero et al, 2003). Treatment of rad53 cells with MMS or HU causes forks fired from early origins to terminate and this is accompanied by the accumulation of unusual structures in the vicinity of the fork (Lopes et al, 2001; Sogo et al, 2002). This cannot be reversed: if Rad53p protein expression is induced in rad53 cells after MMS has been removed, then DNA synthesis cannot be resumed (Tercero et al, 2003). To investigate if the same is true for Esc4p, a yeast strain was made in which the ESC4 promoter was replaced with the GAL1, 10 promoter allowing expression of Esc4p at specific times. Cells were released from G1 into S phase in the presence of MMS for 60 min. MMS was then quenched and washed away and cells were allowed to recover. When galactose (GAL) was included in the culture medium at all steps, Esc4p was expressed and chromosome replication resumed normally when MMS was removed (Figure 4E, left panel, lanes 1–5). In contrast, when GAL was absent, Esc4p expression was repressed and chromosome replication did not recover (Figure 4E, left panel, lanes 6–10). Strikingly, when cells were switched to GAL only after MMS had been quenched and washed away, chromosome replication still recovered (Figure 4E, left panel, lanes 11–15). In contrast, when a similar analysis was carried out in cells with a GAL-regulatable Rad53p, switching cells to GAL after MMS had been quenched and washed away did not allow resumption of chromosome replication (Figure 4E, right panel), consistent with a previous report (Tercero et al, 2003). These results indicate that Esc4p plays a late role, distinct from that of Rad53p, in promoting restart of stalled replication forks. Consistent with this, none of the unusual fork structures observed in rad53Δ cells were seen in esc4Δ cells after treatments with HU or MMS (C Cotta Ramusino and M Foiani, personal communication).
Involvement of Esc4p in genome repair and stability
Cells rely heavily on Rad52p-dependent recombination between sister chromatids to rescue stalled forks and to repair strand breaks that occur in the vicinity of the replisome (Kadyk and Hartwell, 1992; Zou and Rothstein, 1997). A role for Esc4p in recombinational repair was examined by measuring rates of sister chromatid exchange (SCE) in S-phase cells before and after DNA damage. In this system, His+ recombinants are produced as a result of recombination between two truncated his3 fragments at the TRP1 locus (Figure 5A; Fasullo et al, 2001). When wild-type cells were released from G1 into S phase in the presence of CPT or MMS, the rate of SCE was 15- and 29-fold higher than in untreated cells, respectively (Figure 5A). In esc4Δ cells, CPT and MMS caused a 16- and 55-fold induction, respectively, in the rate of SCE (Figure 5A) compared with untreated esc4Δ cells. These differences are small and are unlikely to explain fully the complete lack of recovery of chromosome replication after fork stalling in esc4Δ cells. To investigate further, RAD52 was disrupted in these cells. rad52Δ cells were more sensitive to CPT (Figure 5B) or MMS (data not shown; Hanway et al, 2002) than esc4Δ cells, and esc4Δrad52Δ double mutant cells were found to be considerably more sensitive to CPT than rad52Δ cells (Figure 5B). These results indicate that the role of Esc4p in enabling completion of DNA replication after DNA damage is, at least partly, distinct from that of Rad52p.
Figure 5.
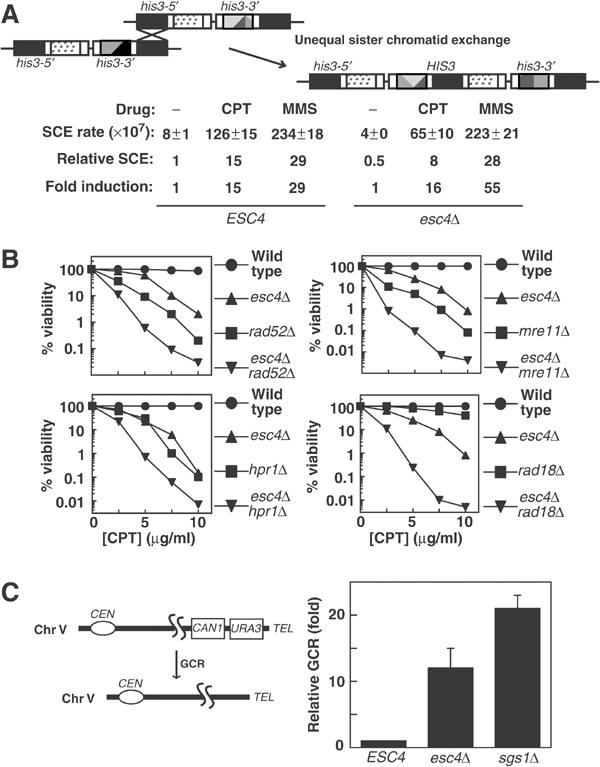
Involvement of Esc4p in genome repair and stability. (A) Schematic diagram of the assay used to measure unequal SCE. To measure DNA damage-stimulated SCE, strains YB163 (ESC4) and JRY106 (esc4Δ) were arrested in G1 and released into fresh medium containing MMS (0.033% v/v) or CPT (7.5 μg/ml). After 60 min, cells were washed and plated on YPD-agar and on plates lacking histidine. The rate of SCE is expressed as the number of His+ cells per colony forming unit. ‘Relative SCE' indicates the levels of SCE relative to those in untreated wild-type cells and ‘fold induction' indicates the levels of SCE relative to untreated cells in each individual strain. Five independent experiments were carried out and the average ±standard error of the mean (s.e.m.) is shown. (B) The indicated yeast strains were grown to early log phase and CPT was added at the concentrations indicated for 4 h. Cell suspensions were diluted 100-fold, plated on YPD-agar and incubated for 3 days at 30°C before counting viable colonies. (C) The relative level of GCR in strains RDKY3615 (ESC4), JRY108 (esc4Δ) and JRY109 (sgs1Δ) was assessed as described previously (Chen and Kolodner, 1999).
The protein most similar to Esc4p in fission yeast is Brc1. brc1 was identified as a multicopy suppressor of the UV hypersensitivity associated with loss of Rad18 (Verkade et al, 1999), a protein that is structurally related to the SMC family of proteins. Rad18 participates in the RAD6 postreplication repair pathway (Prakash, 1981). Disruption of ESC4 in rad18Δ cells results in much greater sensitivity to CPT than that seen in the single mutants (Figure 5B), again indicating that the function of Esc4p overlaps with, but is distinct from, the RAD6 pathway. Similar results were reported previously using MMS (Hanway et al, 2002). Cells lacking components of the Mre11p–Rad50p–Xrs2p (MRX) complex show a high degree of sensitivity to CPT, and disruption of Esc4p in mre11Δ cells (Figure 5B) or rad50Δ (data not shown) cells dramatically increases CPT hypersensitivity, indicating that the function of Esc4p is similar to, but at least partly distinct from that of the MRX complex. Similarly, disruption of HPR1, which encodes a TopI-like protein involved in regulating recombination, renders cells hypersensitive to CPT (Merker and Klein, 2002). However, esc4Δhpr1Δ cells are much more sensitive to CPT than the respective single mutants (Figure 5B).
Mutations in DNA damage response genes often lead to gross chromosomal rearrangements (GCR) similar to those often seen in cancer cells (Chen and Kolodner, 1999). In yeast, GCR can be easily measured by monitoring the rate of simultaneous loss of two markers at the end of chromosome V (Figure 5C, left panel) (Chen and Kolodner, 1999). Disruption of ESC4 causes an approximately 12-fold increase in GCR levels in cells that had not been exposed to exogenous agents that damage DNA, compared with a 21-fold increase in cells lacking Sgs1p (Figure 5C, right panel), a DNA helicase that regulates recombination (Hickson, 2003). Thus, loss of Esc4p causes genome instability presumably due to defects in dealing with endogenous DNA damage during S phase.
Phosphorylation of Esc4p by Mec1p is important for Esc4p function
Experiments were carried out to determine which of the SQ/TQ motifs in Esc4p are phosphorylated by Mec1p. N-terminally MYC-tagged Esc4p, under the control of the ESC4 promoter, was expressed in esc4Δ cells from a low-copy ARS/CEN plasmid, pESC4. The Ser or Thr residue in each individual SQ/TQ motif in pESC4 was mutated to Ala, and Esc4p mutants were introduced into esc4Δ cells. Each of the single SQ/TQ mutants was phosphorylated with the same efficiency after DNA damage as wild-type Esc4p and each single mutant fully restored the ability of esc4Δ cells to grow in the presence of high concentrations of CPT, MMS or HU (data not shown). This suggested that two or more SQ/TQ motifs in Esc4p are phosphorylated by Mec1p. To test this, combinations of SQ/TQ motifs were mutated (Figure 6A). In one mutant (MUT1) the Ser/Thr residues in all four motifs in the SQ/TQ cluster (Ser743, Thr758, Thr773 and Ser806) were mutated to Ala, while in another mutant (MUT2) the Ser or Thr residues at Ser255, Ser361, Ser390 and Thr502 were all mutated (Figure 6A). As shown in Figure 6B, Esc4p-wild type and Esc4p-MUT2 were efficiently phosphorylated after treatment of cells with CPT or MMS. In contrast, Esc4p-MUT1 showed no detectable DNA damage-induced SQ/TQ phosphorylation, suggesting that Ser743, Thr758, Thr773 and Ser806, or a subset of these residues, are phosphorylated in response to DNA damage. Consistent with this, Esc4p-wild type, but not Esc4p-MUT1, was recognised by the anti-phospho-SQ/TQ antibody after incubation with Mec1p and Mg2+-ATP in vitro (data not shown). Strikingly, whereas Esc4p-wild type and Esc4p-MUT2 fully rescued the inability of these cells to grow in the presence of high concentrations of MMS or CPT, Esc4p-MUT1 did not (Figure 6B, lowest panel). These results show that phosphorylation of Esc4p by Mec1p is essential for Esc4p function.
Figure 6.
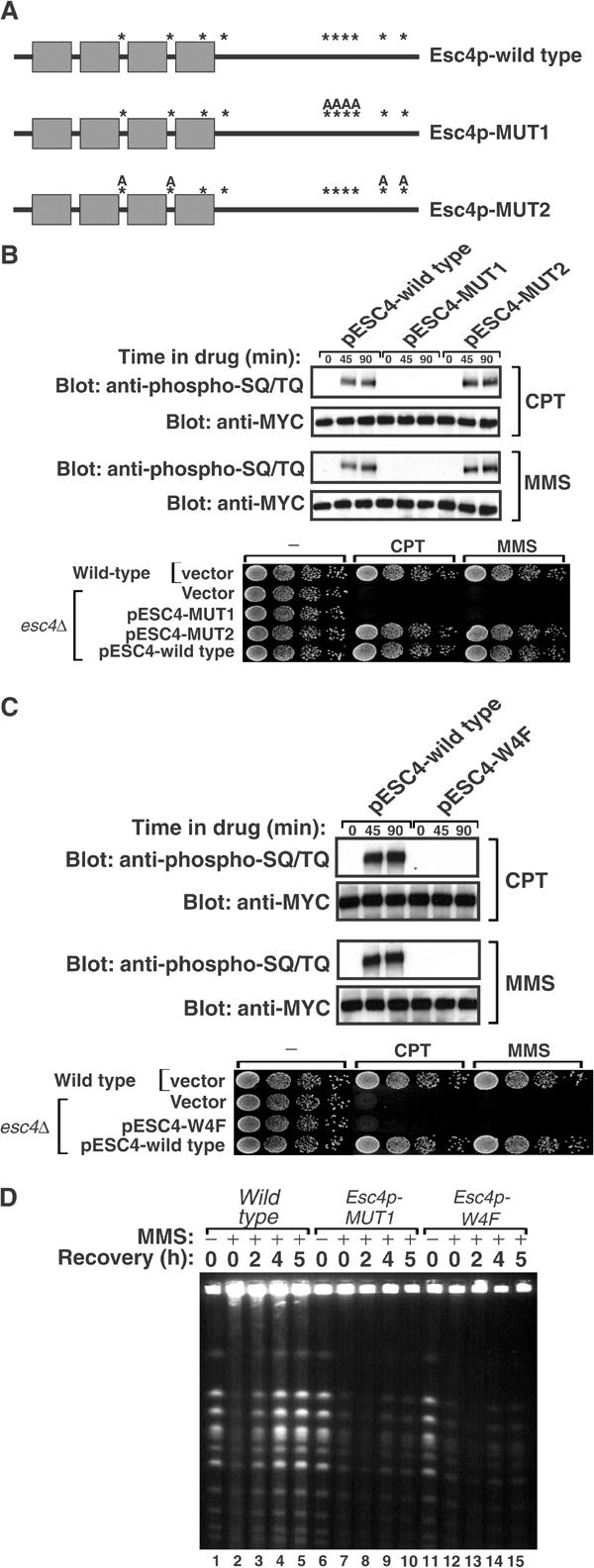
Mec1p-dependent phosphorylation of Esc4p is required for resumption of DNA synthesis after DNA damage. (A) Schematic representation of Esc4p-wild type, Esc4p-MUT1 and Esc4p-MUT2. SQ/TQ motifs are represented by asterisks. In Esc4p-MUT1, Ser743, Thr758, Thr773 and Ser806 are all mutated to Ala (‘A') residues. In Esc4p-MUT2, Ser255, Ser361, Ser390 and Thr502 are all mutated to Ala. (B) esc4Δ cells (strain JRY99) transformed with pESC4-wild type, pESC4-MUT1 or pESC4-MUT2 were grown to mid-log phase in liquid culture and incubated with CPT (5 μg/ml) or MMS (0.02%) for the times indicated. Native extracts were prepared and Esc4p was immunoprecipitated with anti-MYC antibodies and subjected to Western blot analysis with the indicated antibodies (top panels). Alternatively, 10-fold serial dilutions of cells were spotted on agar plates lacking histidine and containing CPT (5 μg/ml), MMS (0.02% v/v) or no drug and incubated at 30°C for 3 days (bottom panel). (C) Same as (B), except that esc4Δ cells transformed with pESC4-wild type or pESC4-W4F were used. (D) Same as described in the legend to Figure 4B except that esc4Δ cells were transformed with pESC4-wild type or pESC4-W4F.
BRCT domains of Esc4p are essential for its phosphorylation
BRCT domains possess five blocks of sequence similarity (Bork et al, 1997). The most highly conserved motif, in block D, is based around a conserved aromatic residue—Trp, Phe or Tyr. This corresponds to Trp98, Trp208, Trp347 and Trp448 in BRCT domains 1–4, respectively, of Esc4p. Each of these Trp residues was mutated to Phe in pESC4 and the resulting mutant (Esc4p-W4F) was introduced into esc4Δ cells. As shown in Figure 6C (top panels), Esc4p-W4F was expressed at the same levels as Esc4p-wild type but was not phosphorylated in response to CPT, MMS or HU. In contrast, Esc4p-W4F incubated with Mec1p in vitro was recognised by the anti-phospho-SQ/TQ antibody to the same extent as Esc4p-wild type (data not shown), indicating that lack of phosphorylation of Esc4p-W4F in vivo is not because it cannot be recognised by Mec1p. pESC4-W4F could not rescue the hypersensitivity of esc4Δ cells to CPT or MMS (Figure 6C). Thus, functional BRCT domains are required for phosphorylation of Esc4p when DNA is damaged. Strikingly, cells harbouring Esc4p-MUT1 or Esc4p-W4F cannot resume chromosome replication after transient exposure to MMS (Figure 6D). These results show that the BRCT domains of Esc4p are important for its phosphorylation by Mec1p and for Esc4p function.
Discussion
Esc4p is an S-phase-specific target of Mec1p
Screening the Saccharomyces Genome Database for proteins that contain a cluster of SQ/TQ motifs revealed Esc4p as a potential target of Mec1p/Tel1p (Figure 1B). Mec1p can phosphorylate Esc4p in vitro and Esc4p becomes phosphorylated in response to DNA damage at SQ/TQ motifs in a manner that depends on Mec1p, but not Tel1p (Figure 2). Intriguingly, Esc4p phosphorylation is restricted to S phase (Figure 3E), and at present the molecular basis for this is not known. Maybe Esc4p associates with the replisome, either constitutively or inducibly, possibly mediated by its BRCT domains. Translocation of Mec1p to stalled replication forks (Rouse and Jackson, 2002; Katou et al, 2003) would then juxtapose Mec1p with Esc4p, enabling phosphorylation of the latter and this is currently under investigation. Alternatively, it may be that Esc4p phosphorylation is actively suppressed outside of S phase or actively ‘licensed' during DNA replication.
Although treatment of asynchronously growing cells with UV or IR does not cause detectable levels of Esc4p phosphorylation (Figure 2A) surprisingly, these agents stimulate Esc4p phosphorylation in S phase cells (Figure 3E). CPT and HU do not induce DNA damage until cells reach S phase and cells accumulate there, allowing high levels of Esc4p phosphorylation in asynchronous cultures. However, it is likely that in asynchronous cultures, UV- and IR-induced cell cycle checkpoints prevent G1 or G2/M cells from entering S phase thereby restricting Esc4p phosphorylation to the small proportion of cells in S phase in these cultures. Consistent with this, although esc4Δ cells are not sensitive to UV or IR, esc4Δrad9Δ cells are much more sensitive to both these agents than rad9Δ cells (data not shown).
Mutation of all four motifs in the SQ/TQ cluster (Ser743, Thr758, Thr773 and Ser806; Figure 6A) abolished Mec1p-dependent Esc4p phosphorylation, whereas mutation of a combination of motifs outside of this cluster (Ser255, Ser361, Ser390 and Thr502 together) had no effect (Figure 6). This suggests that Mec1p phosphorylates Esc4p at two or more of Ser743, Thr758, Thr773 and Ser806 but does not prove it since mutation of these four residues might, for example, change the conformation of Esc4p. 32P-labelling of cells enables identification of in vivo phosphorylation sites; however, Esc4p is present at very low abundance in cells (data not shown), making this type of analysis difficult. Phospho-specific antibodies against individual SQ/TQ motifs will be required to examine unambiguously phosphorylation of Esc4p in response to DNA damage.
Phosphorylation of Esc4p by Mec1p is required to survive high levels of DNA damage in S phase
Disruption of ESC4 (Figure 3A) or mutation of SQ/TQ motifs that prevent Mec1p-mediated Esc4p phosphorylation (Figure 6B) causes hypersensitivity to S-phase-specific DNA damage, and preventing entry to S phase fully rescues the hypersensitivity of esc4Δ cells to all of these agents (Figure 3C; data not shown). Thus Esc4p phosphorylation is required to tolerate DNA damage primarily during DNA replication. However, esc4Δ cells are viable under laboratory conditions, and the time taken for these cells to transit S phase is indistinguishable from wild-type cells (data not shown). This suggests that Esc4p, unlike Mec1p, is not required to survive the low levels of DNA damage that occur in most cells during DNA replication. It may be that Esc4p is only required when the number of DNA lesions rises above a critical threshold, or when DNA repair capacity is suboptimal. Alternatively, it may be that Esc4p functions redundantly with other proteins in responding to background levels of DNA damage. It is interesting to note that cells lacking ESC4 have elevated levels of genome instability in the absence of exogenous sources of DNA damage (Figure 6), albeit at much lower levels than in cells lacking Mec1p (Myung et al, 2001). This might reflect small proportions of esc4Δ cells that at some point sustain a high level of DNA damage during DNA replication. The mammalian protein with highest sequence similarity to Esc4p is PTIP (Pax2 transactivation domain interacting protein; data not shown). A fragment of human PTIP was recently shown to form foci after DNA damage (Manke et al, 2003). Work in this lab has shown that, as with Esc4p, reducing PTIP expression in cells causes major problems in dealing with DNA damage in S phase (P Jowsey and J Rouse, unpublished).
How does Esc4p enable completion of chromosome replication after DNA damage?
When esc4Δ cells are exposed to MMS or HU (data not shown) in early S phase, the intra-S-phase checkpoint is activated but chromosome replication cannot be completed when MMS is removed (Figure 5C). The same is true for rad53Δ cells (Desany et al, 1998), where unusual DNA structures accumulate in the vicinity of a replication fork in HU or MMS (Lopes et al, 2001; Sogo et al, 2002). These structures are not observed in cells lacking Esc4p, however (C Cotta-Ramusino and M Foiani, personal communication). DNA synthesis cannot be resumed if Rad53p is induced in rad53Δ cells after MMS is removed (Tercero et al, 2003), probably because the aberrant fork structures described above prevent this. In contrast, induction of Esc4p after MMS is removed from cells allows completion of DNA synthesis, suggesting that Esc4p acts later than, and at a point distinct from, Rad53p. One possibility is that Esc4p is involved directly in switching off the intra-S-phase checkpoint after DNA repair/bypass, but preliminary experiments carried out in this laboratory suggest that this is not the case.
In prokaryotes, it is well established that recombination is essential for restarting stalled replication forks (Marians, 2000). Levels of DNA damage-induced SCE in esc4Δ cells are similar to those seen in wild-type cells. Also, disruption of Esc4p in cells lacking Rad52p, Mre11p and Rad50p causes much greater sensitivity to DNA damage than seen in the respective single mutants (Figure 5B). However, a role for Esc4p in somehow facilitating DNA repair cannot be excluded. A screen for multicopy suppressors of the DNA damage sensitivity of esc4Δ cells identified several such genes (Supplementary Table 2). RAD10 has been implicated in the cleavage of CPT adducts to allow strand invasion during recombination. HPR1, a protein with similarity to Top1p, has also been shown to regulate recombination (Merker and Klein, 2002), and SMC1 (a known target of ATM in mammals; Kim et al, 2002) and BRN1 are regulators of sister chromatid cohesion/segregation (Lavoie et al, 2000). It will be interesting to investigate if Esc4p plays a role in making the correct recombination partner choice. Dot1p, a histone H3 methyltransferase (Khan and Hampsey, 2002), was also isolated as a multicopy suppressor of esc4Δ, suggesting that Esc4p regulates chromatin. It will be interesting to investigate if chromatin structure at stalled replication forks is modulated by Esc4p to facilitate recombination and/or fork restart. It is possible that Esc4p regulates origin-independent DNA synthesis in a manner analogous to the prokaryotic PriA protein, which assembles DNA replication proteins onto certain branched DNA recombination intermediates (Marians, 2000). However, such a mechanism for fork restart has not yet been described in eukaryotes.
Materials and methods
Full details of all plasmids, antibodies, yeast strains, and materials and methods not described in the text are given in Supplementary Data.
Cell extracts, Western blotting and immunoprecipitation
Preparation of native cell extracts, preparation of extracts for Western blot analysis by the TCA lysis method and all immunoprecipitations were carried out as described previously (Rouse and Jackson, 2000). In experiments where MYC(13)-Esc4p was immunoprecipitated and subjected to Western blot analysis, approximately 90% of each precipitate was blotted with anti-SQ/TQ antibody and the remaining 10% was blotted with anti-MYC.
Analysis of chromosomes by pulsed field gel electrophoresis
Cells were grown to early log-phase (OD600 0.5) in YPD at 30°C and arrested in G1 by addition of alpha-factor (5 μg/ml). When budded cells accounted for less than 5% of the population, cells were released from arrest by filtration and extensive washing and incubated in YPD for 10 min before addition of MMS (0.033%). After 60 min in MMS, cells were filtered, washed extensively with YPD containing 2.5% (w/v) sodium thiosulphate and incubated in YPD at 30°C. At the times indicated, 1 × 108 cells were removed and fixed in 70% ethanol at 4°C overnight before preparation of chromosomes, exactly as described in the CHEF DRII instruction manual (BioRad). PFGE was carried out using BioRad CHEF DRII apparatus at 14°C in a 1% agarose gel (pulsed field-certified, BioRad) in 0.5 × TBE for 27 h at 6 V/cm using a 120° included angle with a 6.8–158 s switch-time ramp. Gels were stained with 1 μg/ml ethidium bromide for 30 min and washed for 2 h in water before DNA was visualised.
Supplementary Material
Supplementary Data Table 1
Supplementary Data
Acknowledgments
I am grateful to Daniel Durocher and members of the Rouse laboratory, especially Paul Jowsey and Sonja Flott, for critical reading of the manuscript and for helpful suggestions. I thank David Lydall, Michael Fasullo, Richard Kolodner and Steve McKnight for providing yeast strains and plasmids. I am grateful to the protein production team in the Division of Signal Transduction Therapy in this Institute for PP1 and to the DNA Sequencing Service in this Institute. I also thank Hilary-Kay Young at the University of Dundee for providing the PFGE apparatus. I am grateful to Cecilia Cotta-Ramusino and Marco Foiani for 2D gel analysis of ARS305 in esc4Δ cells and for helpful suggestions. This work was funded by the Medical Research Council, UK.
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Beranek DT (1990) Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res 231: 11–30 [DOI] [PubMed] [Google Scholar]
- Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV (1997) A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J 11: 68–76 [PubMed] [Google Scholar]
- Callebaut I, Mornon JP (1997) From BRCA1 to RAP1: a widespread BRCT module closely associated with DNA repair. FEBS Lett 400: 25–30 [DOI] [PubMed] [Google Scholar]
- Carr AM (2002) DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair (Amst) 1: 983–994 [DOI] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW (2002) A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci USA 99: 16934–16939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kolodner RD (1999) Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet 23: 81–85 [DOI] [PubMed] [Google Scholar]
- Desany BA, Alcasabas AA, Bachant JB, Elledge SJ (1998) Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev 12: 2956–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasullo M, Giallanza P, Dong Z, Cera C, Bennett T (2001) Saccharomyces cerevisiae rad51 mutants are defective in DNA damage-associated sister chromatid exchanges but exhibit increased rates of homology-directed translocations. Genetics 158: 959–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanway D, Chin JK, Xia G, Oshiro G, Winzeler EA, Romesberg FE (2002) Previously uncharacterized genes in the UV- and MMS-induced DNA damage response in yeast. Proc Natl Acad Sci USA 99: 10605–10610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy KM, Lee A, Chen E, Botstein D (1991) A group of interacting yeast DNA replication genes. Genes Dev 5: 958–969 [DOI] [PubMed] [Google Scholar]
- Hickson ID (2003) RecQ helicases: caretakers of the genome. Nat Rev Cancer 3: 169–178 [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180–183 [DOI] [PubMed] [Google Scholar]
- Johnston LH, Nasmyth KA (1978) Saccharomyces cerevisiae cell cycle mutant cdc9 is defective in DNA ligase. Nature 274: 891–893 [DOI] [PubMed] [Google Scholar]
- Kadyk LC, Hartwell LH (1992) Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132: 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424: 1078–1083 [DOI] [PubMed] [Google Scholar]
- Khan AU, Hampsey M (2002) Connecting the DOTs: covalent histone modifications and the formation of silent chromatin. Trends Genet 18: 387–389 [DOI] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 274: 37538–37543 [DOI] [PubMed] [Google Scholar]
- Kim ST, Xu B, Kastan MB (2002) Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev 16: 560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie BD, Tuffo KM, Oh S, Koshland D, Holm C (2000) Mitotic chromosome condensation requires Brn1p, the yeast homologue of Barren. Mol Biol Cell 11: 1293–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Schwartz MF, Duong JK, Stern DF (2003) Rad53 phosphorylation site clusters are important for rad53 regulation and signaling. Mol Cell Biol 23: 6300–6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy C, Lee SE, Vaze MB, Ochsenbien F, Guerois R, Haber JE, Marsolier-Kergoat MC (2003) PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol Cell 11: 827–835 [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, Yaffe MB (2003) BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302: 636–639 [DOI] [PubMed] [Google Scholar]
- Marians KJ (2000) PriA-directed replication fork restart in Escherichia coli. Trends Biochem Sci 25: 185–189 [DOI] [PubMed] [Google Scholar]
- McGlynn P, Lloyd RG (2002) Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol 3: 859–870 [DOI] [PubMed] [Google Scholar]
- Merker RJ, Klein HL (2002) hpr1Delta affects ribosomal DNA recombination and cell life span in Saccharomyces cerevisiae. Mol Cell Biol 22: 421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Datta A, Kolodner RD (2001) Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 104: 397–408 [DOI] [PubMed] [Google Scholar]
- O'Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees-Miller SP, Abraham RH, Lai JH, Hill D, Shiloh Y, Cantley LC, Rathbun GA (2000) Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem 275: 22719–22727 [DOI] [PubMed] [Google Scholar]
- Paciotti V, Clerici M, Lucchini G, Longhese MP (2000) The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev 14: 2046–2059 [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Hartwell LH (1995) A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82: 841–847 [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Margulies RU, Garvik BM, Hartwell LH (1997) RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics 145: 45–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M (1999) Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18: 6561–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash L (1981) Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol Gen Genet 184: 471–478 [DOI] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2000) LCD1: an essential gene involved in checkpoint control and regulation of the MEC1 signalling pathway in Saccharomyces cerevisiae. EMBO J 19: 5801–5812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2002) Interfaces between the detection, signaling, and repair of DNA damage. Science 297: 547–551 [DOI] [PubMed] [Google Scholar]
- Scholes DT, Banerjee M, Bowen B, Curcio MJ (2001) Multiple regulators of Ty1 transposition in Saccharomyces cerevisiae have conserved roles in genome maintenance. Genetics 159: 1449–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF (2002) Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell 9: 1055–1065 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- Tercero JA, Diffley JF (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412: 553–557 [DOI] [PubMed] [Google Scholar]
- Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336 [DOI] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368 [DOI] [PubMed] [Google Scholar]
- Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O'Connell MJ (1999) Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol Biol Cell 10: 2905–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC (2002) Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3: 430–440 [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH (1993) Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics 134: 63–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH (1994) Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 8: 652–665 [DOI] [PubMed] [Google Scholar]
- Zou H, Rothstein R (1997) Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell 90: 87–96 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data Table 1
Supplementary Data